

Abstract
Background
Aging is accompanied by increasing vulnerability to pathologies such as atherosclerosis (ATH) and Alzheimer disease (AD). Are these different pathologies, or different presentations with a similar underlying pathoetiology?
Discussion
Both ATH and AD involve inflammation, macrophage infiltration, and occlusion of the vasculature. Allelic variants in common genes including APOE predispose to both diseases. In both there is strong evidence of disease association with viral and bacterial pathogens including herpes simplex and Chlamydophila. Furthermore, ablation of components of the immune system (or of bone marrow-derived macrophages alone) in animal models restricts disease development in both cases, arguing that both are accentuated by inflammatory/immune pathways. We discuss that amyloid β, a distinguishing feature of AD, also plays a key role in ATH. Several drugs, at least in mouse models, are effective in preventing the development of both ATH and AD. Given similar age-dependence, genetic underpinnings, involvement of the vasculature, association with infection, Aβ involvement, the central role of macrophages, and drug overlap, we conclude that the two conditions reflect different manifestations of a common pathoetiology.
Mechanism
Infection and inflammation selectively induce the expression of cholesterol 25-hydroxylase (CH25H). Acutely, the production of ‘immunosterol’ 25-hydroxycholesterol (25OHC) defends against enveloped viruses. We present evidence that chronic macrophage CH25H upregulation leads to catalyzed esterification of sterols via 25OHC-driven allosteric activation of ACAT (acyl-CoA cholesterol acyltransferase/SOAT), intracellular accumulation of cholesteryl esters and lipid droplets, vascular occlusion, and overt disease.
Summary
We postulate that AD and ATH are both caused by chronic immunologic challenge that induces CH25H expression and protection against particular infectious agents, but at the expense of longer-term pathology.
Keywords: Atherosclerosis, Alzheimer, APOE, Infection, Inflammation, Cholesterol, 25-hydroxycholesterol
Background
Better nutrition and lifestyle changes make important contributions to extending human lifespan, but new morbidities are encountered with aging, notably AD and ATH. At first sight these appear to be different conditions. In the present debate we address whether the two conditions are different, or instead share a common etiology. We build upon a previous debate – Ill or Just Old? – and agree with Izaks and Westendorp that ‘we should investigate the risk factors (component causes) of diseases in the latter part of life’ [1]. The discussion here commences with age-related risk factors, genetic predispositions, animal models, and the central involvement of the vasculature and inflammation. We then extend the discussion to infection, amyloid β, animal models, infection, drugs, and the central signaling role of cholesterol derivatives. We suggest that both conditions result from an inflammatory disorder as a result of an infectious condition, both crucially linked to sterol metabolism and innate immunity, leading to vascular occlusion.
Discussion
Disease characteristics
AD is the main form of dementia (~70%) in Western countries, and is characterized by the presence in postmortem brain of extracellular amyloid plaques composed of ‘Aβ’ generated by the aggregation of toxic peptide fragments of the Alzheimer precursor protein, APP, and intraneuronal deposition of highly phosphorylated filamentous aggregates (neurofibrillary tangles, NFT) of the microtubule-associated protein Tau. Onset is typically above age 70 (Figure 1).
Figure 1.
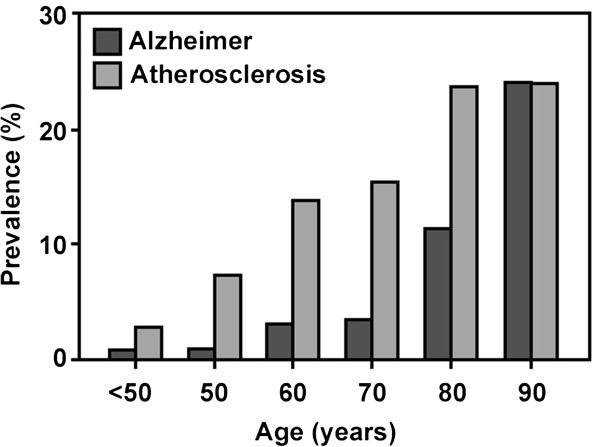
Age-dependence of Alzheimer disease and atherosclerotic vascular disease. Compiled from multiple sources including [2-5].
By contrast, ATH (from Greek athera, ‘gruel’: gruel-like deposits, and sclerosis, ‘hardening’), also known as arteriosclerotic vascular disease, is not a unitary disorder, and instead ranges from primary arterial atheroma – inflammation and accumulation of cholesterol-laden macrophages in the walls of major arteries – to ‘plaque’ formation and inflammation in the arterial wall [6], leading to progressive occlusion, with consequent risk of myocardial infarction or cerebral stroke because plaque rupture can provoke thrombosis. Disease development is accompanied by disruption of the endothelial cell layer, vascular smooth muscle cell migration, and matrix calcification. Onset is a little earlier than for AD, but ultimately affects a similar proportion of the elderly (Figure 1).
The ongoing rise in both AD and ATH has been ascribed, rightly or wrongly, to the increasing adoption of a Western sedentary lifestyle accompanied by a diet rich in fats and sugars. Both disorders are essentially unknown in children and young adults, with onset in later life (Figure 1).
Vascular involvement
At first glance the two diseases would appear to be distinct, with ATH being characterized by cholesterol-rich deposits in arterial walls and AD by neuronal loss, NFT, and amyloid plaque formation. However, there is increasing evidence that AD is also associated with vascular dysfunction. Although the structure of cerebral arteries and arterioles differs somewhat from that of the major blood vessels, they are similarly dependent on endothelial and smooth muscle cells [7].
Studies in AD mouse models have confirmed that disease development is associated with deposits in the cerebral arterial vasculature [8]. In patients, extracellular deposits of amyloid in AD brain are principally associated with the cerebral arterial vasculature, and deposit density declines with distance from the larger vessels [9,10]. It has been postulated that dysfunction of vascular endothelial cells lining brain blood vessels plays a central role in precipitating neuronal death [11].
Brain scanning revealed that AD is associated with decreased cerebral blood flow [12,13], as also seen in AD mouse models [14]. Roher [15] examined cerebral arteries from confirmed AD cases and age-matched non-demented controls. In addition to plaques and tangles, it was found that AD cases displayed a degree of cerebral artery (circle of Willis) occlusion that was significantly greater than in controls (Figure 2), and there was a positive correlation between the degree of arterial stenosis and NFT score [15]. This finding was confirmed in a study by Hofman et al. who examined AD patients and controls for markers of atherosclerosis including vessel wall thickness as assessed by ultrasonography. All markers of ATH were over-represented in AD patients versus controls, and the odds ratio for AD in those with significant ATH versus those without was 3.0 (CI 1.5–6) [16]. Since then the lead findings have been widely confirmed [17-19]; the link between intracranial atherosclerosis and AD is not an artifact of diagnostic misclassification [20].
Figure 2.
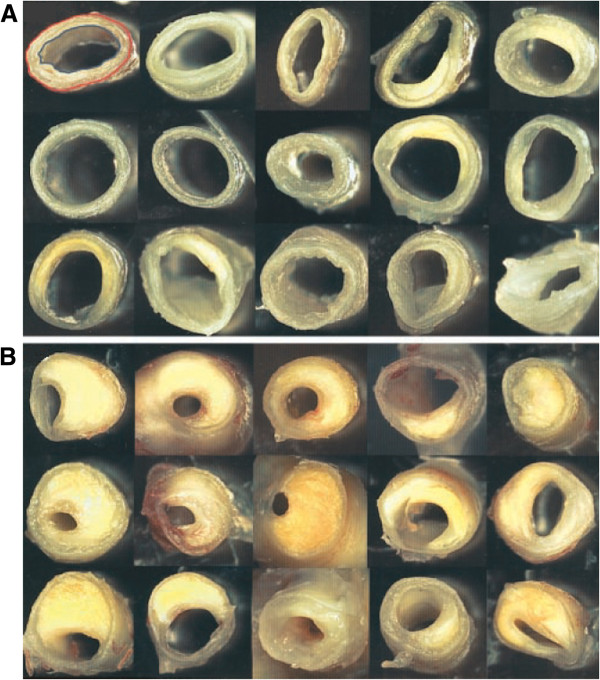
Occlusions of brain blood vessels (‘circle of Willis’) in controls and AD. Panel (A) shows cerebral arteries from non-demented elderly individuals, whereas Panel (B) shows arteries from AD patients showing atheromatous plaque deposition. Figure reproduced, with permission, from [15].
The recent Baltimore Longitudinal Study of Aging (BLSA) found that individuals with (non-brain) coronary or aortic ATH per se are not at increased risk of AD. However, intracranial atherosclerosis was confirmed as a strong risk factor for dementia [21].
It remains possible that AD might encompass two distinct conditions: a major class with involvement of the cerebral vasculature, and a minor class in which no such involvement is apparent. However, this is unclear. Ellis et al. provide evidence that the major class of AD (83%) is associated with brain angiopathy [22]. The second most common classification (15%) of senile dementia, cerebral amyloid angiopathy (CAA/vascular dementia), is primarily associated with amyloid-positive lesions of the cerebral vasculature, and has substantial overlaps with both ATH and AD [23,24]. Further studies are needed on the subclassification of AD-related senile dementias according to type of vascular involvement. However, the combined evidence demonstrates that the large majority of clinically diagnosed AD cases display significant vascular involvement.
In sum, the major forms of both AD and ATH are associated with vascular wall thickening and blood vessel occlusion. The predominant localizations differ (major arteries in ATH, cerebral arterial vasculature in AD); the pathways leading to disease may also differ. In ATH, vascular deposits impair heart function and are at significant risk of entering the circulation, leading to stroke. In AD, brain hypoperfusion has been causally associated with disease [25]. We surmise that thickening of the cerebrovasculature leads to impaired O2 and nutrient delivery to the brain, predisposing to neuronal loss (Figure 3). These pathways are not necessarily independent: ATH alone might compromise cerebral O2/nutrient supply and, conversely, AD-like processes in crucial brain regions could deregulate the cardiovascular system.
Figure 3.
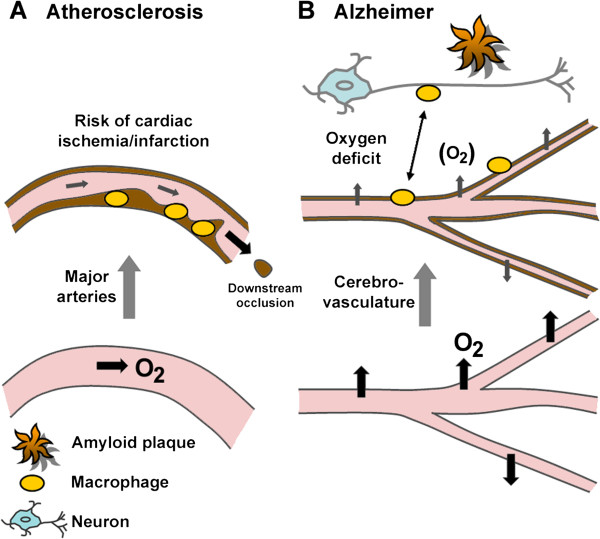
Differential contribution of vessel wall thickening (dark-brown coloration) to disease. (A) Atherosclerosis is a chronic inflammatory condition characterized by the accumulation of cholesterol-laden macrophages (foam cells) in arterial walls, partial occlusion and, when the plaques rupture, risk of myocardial infarction and stroke. Partial occlusion compromises oxygen supply to other tissues. (B) In Alzheimer disease neuronal loss is accompanied by thickening of brain vessel walls, recruitment of macrophages, and the formation of amyloid deposits of Aβ in the vicinity of the cerebrovasculature. Macrophages are implicated in shuttling Aβ between amyloid deposits and vessel walls. Mechanisms underlying neuronal loss are not understood, but impaired trans-vessel oxygen and glucose delivery, and reduced removal of toxic metabolites, may predispose to neuronal death; impairment of blood–brain barrier function may also contribute.
Overall, the evidence suggests that AD and ATH represent a spectrum of related conditions, with vascular involvement as a common predisposing factor, although the site of vascular involvement differs between the two diseases. We next dwell on the different genetic risk factors and how they cast light on the relationship between AD and ATH.
Genetic predisposition
Polymorphic loci associated with both diseases
If there is an overlap between AD and ATH it would be expected that risk alleles would be shared between the two diseases. Hyperlipidemia is a risk factor for both diseases, and mutations leading to hyperlipidemia are major risk factors for ATH (not reviewed). In AD the situation is complicated because individuals with hypercholesterolemia generally die at a younger age; but, for example, early signs of cognitive impairment are seven-fold increased in patients with hyperlipidemia due to low-density lipoprotein receptor (LDLR) mutations [26].
Polymorphisms associated with disordered lipid metabolism showing evidence of bias in both diseases include a gene cluster on chromosome 2q14-21: bridging integrator 1/amphiphysin II, BIN1 (2q14) – cytochrome P450, family 27, subfamily C, polypeptide 1 (potential cholesterol hydroxylase), CYP27C1 (2q14.3) – and excision repair cross-complementing repair deficiency complementation group 3, ERCC3 (2q21), over 0.3 Mb. Genome-wide association studies (GWAS) of lipid metabolic disorders have highlighted further associations with polymorphisms in the genes cholesteryl ester transfer protein, plasma, CETP (16q21); clusterin, CLU (8p21-p12); complement component 3b/4b receptor 1, CR1 (1q32); and low-density lipoprotein receptor, LDLR (19p13.2) (for reviews and databases see [27-30] and the Catalog of Published Genome-Wide Studies; http://www.genome.gov/gwastudies/).
However, interpreting the link between genes and disease is difficult because (i) GWAS studies require polymorphisms in the population, and genes lacking polymorphisms are therefore not identified, and (ii) it is difficult to dissociate direct causality from indirect association. The exception is the gene encoding apolipoprotein E, APOE (19q13.2).
The APOE locus: one candidate gene or several?
Multiple GWAS studies have firmly highlighted alleles in and around the APOE locus as risk factors for both diseases (http://www.genome.gov/gwastudies/).
The APOE gene is located within a tight cluster of genes at chromosome 19q13: poliovirus receptor-related 2/herpesvirus entry mediator B/nectin-2, PVRL2 (19q13.2) – translocase of outer mitochondrial membrane 40, TOMM40 (19q13) – APOE (19q13.2) – apolipoproteins C-I, C-II, C-IV, APOC1/C2/C4 (all 19q13.2) – and cleft lip and palate associated transmembrane protein 1, CLPTM1 (19q13.3), over a distance of 0.1 Mb. Although work on APOE alleles has dominated the field (discussed below), linkage disequilibrium between SNPs in different genes suggests that genes other than APOE, notably PVRL2, TOMM40, and APOC1, may influence the development of AD [31]. Attention has focused on an intronic poly(T) polymorphism in different TOMM44 alleles. Although some studies found no association between disease (AD and/or ATH) and TOMM40 variants [32,33], others reported associations, but in opposite directions [34-36], a possible indication of population-specific risk factors. More detailed analysis [37] indicates that there are several different allelic variants in this poly(T) region, and some appear to associate with age of AD onset. Mice knocked out for another component of the TOMM complex, TOMM5, display a complex inflammatory lung phenotype [38], but possible predisposition to age-related disease was not studied. There also appears to be complex transcriptional interplay between APOE and TOMM44[39].
Overall, the role of APOE in both AD and ATH has been confirmed independently by multiple studies and by transgenic modeling (below), but it remains open whether linked genes, possibly TOMM40, also contribute to the pathoetiology of AD and/or ATH [40].
Role of APOE
The ϵ4 allele at the APOE locus is a major risk factor for both diseases. APOE protein is a lipid transport molecule that circulates in the blood in a complex with lipid-rich lipoprotein particles that transport largely insoluble cholesterol. Lipoproteins, named on the basis of density (principally low-density lipoprotein, LDL; high-density lipoprotein, HDL; and very low density lipoprotein, VLDL), consist of phospholipids, cholesterol esters, and cholesterols, organized into 20–50 nm micelles with apolipoproteins at their surfaces.
Although detailed summary would be out of place here, it is generally held that LDL and VLDL mediate cholesterol transport between the liver and peripheral tissues [41-44]. The principal apolipoprotein is APOB100, and both APOB100 and APOE bind to the cellular LDL receptor to facilitate cellular uptake. APOE binding to LDLR (and the LDLR-related receptor, LRP1; the VLDL receptor, and the APOE receptor 2) thereby plays a role in cholesterol delivery. Conversely, HDL particles predominantly contain APOA1, A2, C, and E, and mediate reverse cholesterol transport from peripheral tissues to the liver for secretion into bile [45], and hepatic uptake is largely mediated by APOA1 binding to specific receptors on hepatocytes. APOE is a minor but crucial lipoprotein component in much of the body, but is the major apolipoprotein in cerebrospinal fluid.
There is a third role, where APOE mediates hepatic uptake of intestinally-derived remnant lipoproteins. Cell-surface heparan sulfate proteoglycans (HSPG) appear to function as a receptor for APOE [46]. In all three roles, APOE is likely to govern export of cholesterol from the cell, and thus the deposition of cholesterols in lipid-rich intracellular aggregates in the vascular wall. Allelic variants of APOE alter the function of the protein in several ways.
APOE variants in AD and ATH
There are three principal alleles at the apolipoprotein E locus, APOE (19q13.2): ϵ2 (cys112, cys158), ϵ3 (cys112, arg158), and ϵ4 (arg112, arg158), giving six different genotypes in human populations, with some further minor variants (reviewed in [47-49]); homozygosity for ϵ4 is the greatest risk factor for both AD and ATH, with risk ratios declining generally ϵ4 > ϵ3 > ϵ2.
The allelic differences affect APO structure and function. APOE protein contains two structural domains, the N-terminal receptor-binding domain, and the C-terminal lipid-binding domain, separated by a hinge region ([50] for review). Both polymorphic sites are within the domain (amino acids 1–191) that includes the receptor-binding site.
These changes affect receptor binding. APOE3 shows reduced receptor binding compared to APOE4, and APOE2 is very markedly impaired in LDLR binding (50-fold reduction), although it can still bind to HSPG for hepatic clearance of remnant lipoproteins. APOE4 protein is also more susceptible to unfolding than E3 or E2 [51]. In addition, the polymorphic forms affect lipoprotein association. Notably, APOE2 and APOE3 bind preferentially to HDL particles, whereas APOE4 binds preferentially to VLDL [52-55].
At a functional level, APOE3 promotes markedly greater cholesterol efflux than APOE4 [56-58]. In part this may reflect APOE-mediated changes in the expression of the gene ATP-binding cassette, subfamily A, member 1, ABCA1 (9q31.1), a locus identified by GWAS. ABCA1, the key sterol transporter in many tissues, is thought to catalytically ‘flop’ sterols from one cellular membrane to another, and thus to play a crucial role in transport of sterols out of the cell, with highest activity for side-chain oxidized cholesterols [59]. APOE4 was reported to be impaired, versus APOE3, in upregulating ABCA1 expression and cholesterol efflux from lipid-laden macrophages [60]. Thus APOE4, versus APOE2/3, is likely to enhance intracellular cholesterol accumulation, a feature of ATH lesions.
Fragments of APOE, like Aβ, can be toxic. Similarly to APP, APOE undergoes cleavage, and APOE4 is more susceptible to cleavage than APOE3 [61]. The resulting fragments can cause AD-like neurotoxicity in mouse models [62] and the lipid-binding region of APOE is required for this toxicity [63]. The mechanism and relevance remain unknown.
APOE plays diverse regulatory roles: infection and inflammation
APOE is not a mere cholesterol transporter and is thought to play further roles in tissue repair, immunity, inflammation, and infection [48,64]. APOE polymorphisms affect not only the function of protein in cholesterol transport but also other processes including infection and immunity (below) and tissue repair. For example, APOE4 was shown to be less effective than either E2 or E3 in promoting neuronal repair [65] but the underlying mechanisms are not understood. What remains open to debate, however, is the exact biochemical process(es) influenced by the polymorphisms that impact upon the risk of ATH or AD development – and indeed whether they are similar in the two diseases or act independently. Studies in animal models are beginning to unravel potentially separable roles of APOE and co-culprits in the two diseases.
Familial disease and transgenic models
No causal and highly-penetrant single gene mutations are known in ATH; modeling the involvement of APOE in transgenic mice has generally relied on the use of knockout mice. Mice knocked out for APOE (Apoe−/−), particularly when fed with a high-fat diet, develop atherosclerotic lesions similar to those seen in human ATH [66]. In addition, mice deficient in the APOE-binding LDL receptor (Ldlr−/−) develop ATH [67], further accentuated by humanized APOB [68], suggesting that differential APOE binding to LDLR may underlie the role of APOE polymorphisms in ATH development. A caveat remains, however, because it is not known whether Apoe knockout affects the function of neighboring genes whose transcriptional control overlaps with that of Apoe.
In AD, well-known (but rare) autosomal dominant mutations are known to cause familial disease. Mutations in the gene amyloid beta precursor protein, APP (21q21.3), encoding the precursor to Aβ peptide, are found in many cases of familial AD, notably in a Swedish pedigree that contains a double-replacement within APP protein (Lys595 to Asn plus Met596 to Leu) that facilitates disease-specific cleavage [69], leading to pathogenic production of Aβ peptide and the deposition in brain of amyloid plaques at an early age. Mutations in the genes presenilin 1, PSEN1 (14q24.3) and presenilin 2, PSEN2 (1q31-q42), encoding key components of the APP processing machinery, have been found in several cohorts of familial AD [30]. These findings reinforce the tight linkage between abnormal APP processing, Aβ deposition, and AD development.
Single-gene mutations of this type lend themselves to modeling in transgenic animals, and for many years AD research has dwelt on the expression, in mouse brain, of abnormal AD-associated mutant forms of APP and or PSEN1/2. Mice expressing the Swedish variant of APP [70-72] show deposition of aggregated Aβ, and learning and memory deficits. However, transgenic mice overexpressing mutant AD-related APP (APPAD mice) are likely to reiterate only some aspects of the human disease because (i) APP mutations are rare in sporadic AD, and (ii) Aβ is unlikely to be an essential component of sporadic AD (see later), although it clearly plays a role. Nevertheless, most work in the field has employed APPAD animals as the best available model of AD.
Alzheimer precursor protein (APP) modulates both AD and ATH
AD is characterized by cerebral Aβ deposits and NFT, whereas pathologic vascular occlusion is the hallmark of ATH. However, we see again evidence of a molecular spectrum encompassing both diseases. It is notable that APOE binds to Aβ and facilitates uptake [73]; APOE4 enhances Aβ production more than APOE3, and synergizes with Aβ toxicity [74,75]. In AD, Aβ is associated with macrophages and the cerebrovasculature, notably in CAA (Figure 4), and reduced cerebral blood flow was seen in AD mice brain [14]. Macrophages ingesting Aβ have been implicated in shuttling Aβ between blood vessels and neurons [77].
Figure 4.

Aβ in ATH plaque and in AD and senile cerebral amyloid angiopathy (CAA) macrophages and vessels. (A) ATH. Colocalization of iNOS-expressing macrophages with Aβ and platelets in advanced human atherosclerotic plaque. The panel shows double immunohistochemical stain for Aβ (red) and iNOS-positive macrophages (brown), showing close colocalization (arrows). Panel adapted, with permission, from [76]. (B) Aβ (red) in AD neurons and perivascular macrophages (green, anti-CD68 staining, a macrophage marker). Panel adapted, with permission, from [77](C) Aβ (red) in vessel walls in CAA (green staining: tissue transglutaminase, an extracellular matrix remodeling protein). Panel adapted, with permission, from [78].
APP and its toxic fragment, Aβ, are also implicated in ATH. Serum Aβ levels are reported to be elevated in stroke patients [79] and Aβ can exert toxic effects on the vascular endothelium [80,81]. Human ATH lesions have been demonstrated to contain Aβ [82] (Figure 4). Expression of AD-related APP in a strain of ATH-prone mice led to aortic atherosclerosis [83], and atherosclerotic lesions in Apoe knockout mice were significantly increased by overexpression of AD-related mutant APP (APPAD mice) [84].
Conversely, genetic knockout of APP function reduced ATH plaque size by up to 90% in ATH-prone Apoe−/− animals [85], confirming that APP plays a prominent role in ATH.
The APOE paradox: gene knockouts reveal contrasting roles of APOE in AD and ATH models
Shared involvement of the vasculature and APP/Aβ in both ATH and AD, together with common risk loci identified by GWAS, underscores the molecular overlaps between the two conditions. Nevertheless, a central factor in both diseases is APOE4, and extensive studies have been carried out in transgenic and knockout mice in the attempt to unravel the molecular role of APOE.
Disease processes in both AD and ATH are accelerated in APOE4 individuals, whereas E2 and E3 offer a measure of protection. However, the biochemical underpinnings that lead to disease remain obscure. One hypothesis might be that APOE4 protein accelerates uptake of cholesterol-rich particles by the vasculature, leading to more rapid disease progression. Indeed, compared to APOE4, the protective APOE2 and APOE3 proteins show reduced receptor binding.
However, genetic knockout of APOE in mice accelerates ATH [66,67], arguing against this interpretation. Similar findings were reported in mice deficient in LDLR [68], demonstrating that APOE/LDLR-mediated cholesterol export is protective against ATH.
Strikingly different observations have been made in AD models. When crossed onto an Apoe−/− background no amyloid deposits were found in any brain region of transgenic APPAD mice [86]. Vascular pathology was seen in two different lines of APPAD transgenic mice, but when the lines were crossed to APOE knockout animals vascular Aβ pathology was abolished in both types of APPADApoe−/− mice, even in very elderly animals [87]. Reduced AD-like pathology in Apoe knockout mice has been confirmed [88].
This result is a paradox because APOE4 confers greatest susceptibility to both disorders. It is unknown how APOE knockout can accentuate ATH in mouse models, but reduce AD development. We conclude that APOE protein function normally protects against ATH, but co-contributes to AD, suggesting a bifurcation of the pathways leading to ATH versus AD. A potential difficulty is that mice do not reiterate all aspects of either disease, and Aβ alone is not an accurate proxy for human AD. Although true human APOE deficiency is associated with marked risk of ATH [89], it remains unknown whether such deficiency in human protects against, or accelerates, AD development.
Other gene knockouts – parallel effects on ATH and AD
Although knockout of the Apoe gene differentially affects disease development in ATH and AD mouse models, this was not found for other genes studied. Other knockouts generally influence disease onset/progression similarly for ATH and AD (Table 1). The role of LDLR in AD pathology remains somewhat unclear because LDLR knockout appeared not to affect disease development in one AD model [107] whereas there was a significant increase in Aβ deposition in other APPADLdlr−/− mice [108], as confirmed [109], and, unlike APOE, elimination of LDLR appears to increase the severity of both AD and ATH in the relevant mouse models.
Table 1.
Gene knockouts in mice and disease progression in ATH and AD models
| Gene a | ATH | Refs | AD | Refs |
|---|---|---|---|---|
|
Abca1 |
↑↑b |
[90,91] |
↑↑ |
[92] |
|
Acat1
c,d
|
↓↑ |
[93-96] |
↓↓ |
[97] |
|
Acat2
c,d
|
↓↓ |
[98,99] |
n/ae |
|
|
Apoe |
↑↑ |
See textf |
↓↓ |
See texte |
|
App |
↓↓ |
[85]; conversely, App overexpression amplifies ATH (see text) |
↓↓* |
*By inference; the production of Aβ (processing product of human APP) is central to AD pathology |
|
Clu1 |
↓↓ |
[100] |
↓↓↑? |
[101,102] |
|
Cyp7b1 |
↑↑ |
[103] |
↑↑** |
**By inference; CYP7B1 expression is downregulated in AD patient brain [104] |
|
Ifngr1 |
↓↓ |
[105] |
↓↓ |
[106] |
|
Ldlr |
↑↑ |
[67,68] |
↑↑ |
[107-109] |
| Other immune system components | ↓↓g | See text | ↓↓(↑)f | See text |
aGene names: Apoe, apolipoprotein E; App, amyloid beta precursor protein; Abca1, ATP-binding cassette, sub-family A (ABC1), member 1 (cholesterol efflux regulatory protein); Acat1/2, acyl-CoA cholesterol acyltransferases 1/2 [SOAT1/2; see note (d) on nomenclature]; Clu1, clustered mitochondria (CluA/CLU1) homolog; Cyp7b1, cytochrome P450 7B1 (sterol and steroid 7α-hydroxylase); Ifngr1, interferon γ receptor 1; Ldlr, low density lipoprotein receptor.
b↑↑, Increased disease development; ↓↓, reduced disease development.
cThe biology of ACAT1 versus ACAT2 differs between mouse and human [110,111].
dNote concerning nomenclature: acyl-CoA cholesterol acyltransferases 1/2 (ACAT1/2) are more properly known as sterol O-acyltransferases 1/2 (SOAT1/2) and the name ACAT conflicts with the official symbol for a different enzyme (acetyl-CoA acetyltransferase 1/2; ACAT1/2); see http://www.ncbi.nlm.nih.gov/gene/6646 and http://www.ncbi.nlm.nih.gov/gene/8435. However, because the literature largely continues to use ACAT1/2 (rather than SOAT1/2) for acyl-CoA cholesterol acyltransferases 1/2 this usage is followed here.
eData not available.
fThe ApoE paradox. APOE4 is a risk factor for both AD and ATH, but knockout promotes ATH but reduces AD in mouse models.
gSome data are contradictory; text for details.
*By inference; the production of Aβ (processing product of human APP) is central to AD pathology.
**By inference; CYP7B1 expression is downregulated in AD patient brain [104].
One may conclude that several common genes act in parallel to predispose to both disorders, but that there is subtle divergence in the molecular pathways leading to one or other disease, notably at the level of APOE. This presents a conundrum that is not yet understood because APOE4 is a risk factor for both diseases.
Site of action: the immune system
GWAS studies and animal models have confirmed that key genes are involved in both ATH and AD and, in addition to cholesterol metabolism, these also address inflammation and immunity. The evidence demonstrates that the immune system centrally determines disease outcome in both cases.
Both diseases have an inflammatory component
Inflammatory pathways have been implicated in both ATH and AD. For example, C-reactive protein (CRP) levels are markedly altered in both diseases. CRP, a marker of inflammation induced by interleukins IL-1 and IL-6, binds to phosphocholine, a component of the bacterial cell wall, and has immunomodulatory properties (reviewed in [112]). In ATH, upregulation of CRP has been known for several decades [113]. For example, CRP immunoreactivity was present in 90% of atheromatous plaques but in only 3% of normal specimens [114]. In AD, there is no evidence for systemic CRP upregulation in blood or CSF, but CRP mRNA levels in brain, particularly in hippocampus, an early site of AD pathology, were increased by over 20-fold versus controls [115], pointing to local inflammation in the brain. For more extensive summary on inflammation in AD and ATH the reader is referred to recent reviews [116-119].
Immune downregulation attenuates ATH and AD
Multiple studies confirm the central involvement of the immune system in both diseases and, moreover, that impaired immune function abrogates both diseases. For ATH, M-CSF deficiency resulted in significantly reduced atherogenesis [120]. Song et al.[121] crossed Rag1-deficient mice (that lack mature T and B lymphocytes) with Ldlr−/− mice, generating animals in which ATH lesion development was markedly reduced; similar findings were reported for Rag-1 deficient Apoe−/− mice, although only significantly in males [122]. Mature B cell depletion using a CD20-specific monoclonal antibody induces a significant reduction of ATH in various mouse models of the disease [123]. IFN-γ receptor knockout mice exhibited a substantial reduction in ATH lesion size [105]. Similar findings have been reported for other immune system components (not reviewed).
In AD models there have been some inconsistent findings. For example, deficiency of Ccr2 (chemokine C-C motif receptor 2, a protein expressed principally on microglia) was reported to accelerate disease [124]. However, other findings support the view that immune system downregulation prevents AD development. Knockout of IFN-γ receptor reduced gliosis and amyloid plaques [106], and blockade of TNF-α reduced Aβ-induced cognitive impairments [125]. Ablation of CD14, a key molecule in innate immunity, led to decreased plaque burden [126]. DOCK2 (dedicator of cytokinesis 2) is expressed in brain microglial immune cells and modulates cytokine secretion and phagocytosis; knockout was reported to result in reduced plaque area and size [127].
In both diseases, therefore, inhibition of the immune system generally attenuates disease processes. This argues that activation of the immune system is centrally involved in the pathoetiology of both diseases.
Site of action: the immune system determines disease development
The suggestion that the immune system (and potentially infection; below) are implicated in the pathoetiology of both diseases prompts the speculation that key deficiencies, such as of Apoe or Ldlr, only in immune cells, might alone reiterate the disease phenotype of the animal models.
The techniques are available to address this issue. If a knockout mouse is irradiated, and then transplanted with bone marrow cells from a wild type mouse, the immune system regenerates, producing a mouse in which the immune system alone carries the wild type allele. Conversely, knockout bone can be transplanted into a wild type mouse, producing an animal in which the knockout is only present in bone marrow-derived cells.
Van Eck [128] transplanted Apoe+/+ bone marrow into atherosclerosis-prone Apoe null mice, and observed that bone-marrow transplantation led to a marked reduction in ATH lesions. Herijgers et al.[129] transplanted bone marrow from Ldlr−/− mice into irradiated wild type mice and, despite no significant changes in serum cholesterol or lipoprotein profiles, animals developed atherosclerotic lesions. A similar finding was reported by Fazio et al.[130]. Zhao et al. transplanted bone marrow from ABCA1/SR-BI double knockout mice into ATH-prone mice and reported that this increased disease development, despite an unexpected reduction in circulating cholesterol levels [131].
Similar findings using bone-marrow transplantation have been reported in AD models. Keene et al.[132] transplanted prostaglandin receptor (PTGER2)-deficient (EP2−/−) bone marrow into irradiated APPAD mice, and observed that lesion sizes were substantially reduced in mice receiving knockout bone marrow compared to mice receiving EP2+/+ marrow. Hao et al.[133] performed a similar experiment with bone marrow deficient in myeloid differentiation factor 88, and reported that the deficiency (in bone marrow-derived cells alone) markedly reduced amyloid burden. Wild type bone-marrow transplantation into APPAD mice markedly reduced cerebral pathology and, conversely, mutant (PS1) bone marrow exacerbated disease [134]. In the most recent study, APPAD mice received bone-marrow transplants from mice expressing either human APOE4 or APOE3. Transplantation markedly reduced pathology, but the APOE3 transplants were far more effective [135].
Therefore, for both diseases, at least in mouse models, the genotype of bone marrow-derived cells determines disease development, and not that of the host. This is despite the fact that transplanted knockout animals generally maintain the marked changes in levels of blood cholesterols and lipoproteins characteristic of the host knockout mouse, demonstrating that these systemic changes are not directly responsible for disease development.
Central role of macrophages
Macrophage infiltration and foam cell formation are known to play a central role in ATH disease development (for recent comprehensive review see Moore and Tabas [136]). The situation in AD is more contentious, but the available evidence indicates that, here again, macrophages play the central role.
Macrophage infiltration is a feature of AD brain. Macrophage numbers are dramatically increased in AD brain [137], as seen in HIV-1 encephalitis; infiltration is most abundant in perivascular regions and locates to endothelial tight junctions, Aβ plaques, and macrophages that partially encircle the walls of Aβ-rich CAA [137]. Zaghi et al.[77] demonstrated that, in human AD brain, macrophages strongly home to deposits within and surrounding the brain vasculature where they colocalize with Aβ (Figure 4B).
Studies in mouse genetic model confirm a central role for macrophages in both diseases. In ATH, a human APOE transgene under the control of the macrophage lysozyme promoter was crossed into Ldlr−/− mice; this significantly reduced ATH lesion area [138]. The same finding was reported with macrophage-specific Apoe gene repair in APOE-knockdown mice [139]. Knockouts of PPARγ or LRP1 only in macrophages increased lesion size in ATH-prone mice [140,141].
In AD the situation is complicated because the brain contains both resident brain-specific macrophage-like cells, the microglia, and true macrophages that infiltrate from the circulation. Wegiel et al. have argued that microglia actively promote disease development in APPAD mice and play a pivotal role in amyloid deposition [142,143]. Simard et al.[144] argued instead that bone marrow-derived microglial cells are protective and can remove amyloid deposits. However, Grathwohl et al. used a microglia-specific cell ablation technique in APPAD mice; nearly complete ablation of microglia had no effect on AD disease development [145]. Hawkes and McLaurin [146] argued that infiltrating peripheral macrophages, rather than microglia, play a central role in clearing Aβ deposits.
In a pivotal study, Town et al. used macrophage-specific expression of a dominant-negative form of TGF-β in APPAD mice; expression in microglia was absent. Ablation of macrophage TGF-β signaling markedly inhibited disease development [147], confirming that macrophages alone can play a determinant role in AD disease development. For recent reviews on macrophage recruitment into AD brain see [148,149].
The central involvement of the immune system (and specifically of macrophages) unavoidably prompts the question of whether some infectious component might contribute to disease development in ATH and/or AD. The idea that a common disease condition might have an infectious component is not new. Although initially challenged [150], Warren and Marshall in 1984 observed that biopsy specimens of patients with gastric ulcers contained spiral or curved bacteria [151], and received the Nobel prize for their discovery that Helicobacter pylori is a cause of gastric ulcers.
Transmissibility of AD
AD has features of transmissibility. There is intriguing evidence that pathology spreads progressively through the brain from initial foci. Duff and colleagues [152] report that Tau pathology in human AD brain commences in the entorhinal cortex and spreads trans-synaptically from cell to cell. Similar findings were reported in mice by de Calignon et al.[153] who expressed a mutant form of human Tau that predisposes to AD-like pathology in the entorhinal cortex. Pathology propagated from transgene-expressing neurons to adjacent brain regions lacking any detectable transgene expression.
There is direct evidence of transmissibility. Marmosets do not normally develop AD pathology but, when injected intracerebrally with brain tissue from a patient with early-onset AD, animals developed AD-like amyloid plaques (but no NFT) 6–7 years after inoculation [154]. Non-AD brain tissue failed to transmit disease and the induced degeneration was transmissible to further animals [155].
Further evidence for transmissibility emerges from APP transgenic models in which mice develop AD-like pathology only late in life ([156] for review). Disease onset was remarkably accelerated by inoculation of extracts of human AD brain into young APPAD transgenic mice [157,158]. Using 10% w/v brain homogenates from postmortem AD patients, Meyer-Luehmann et al.[158] demonstrated that inoculation into young transgenic mice induced robust deposition of Aβ, whereas non-AD brain failed to do so. Similar seeding has been reported when brain extracts from older (diseased) APP transgenic mice are injected into young transgenic animals [158-161]. Parallel observations have been reported in transgenic mice and rats that do not alone develop disease [162,163]. Although classic AD-like Tau pathology is generally absent, marking differences between human and murine Tau, NFT were induced when extracts of transgenic mouse AD brain were inoculated into transgenic APPAD mice expressing mutant Tau [164].
Although pretreatment of brain extracts with antibody to Aβ could inhibit seeding, confirming a role for Aβ, experiments using synthetic Aβ peptides (1–40 or 1–42), in either soluble or aggregated forms, failed to transmit infection; neither did oligomeric forms of Aβ prepared from cell cultures overexpressing APP [158]. Failure of synthetic Aβ to transmit disease has been confirmed [155]. Rosen et al.[163] state: ‘At present, there is no evidence that AD per se is transmissible in the same manner as is prion disease’, and others have suggested that a second factor is likely to be required [165].
These experiments need to be interpreted with caution because Aβ deposition is not an accurate proxy for Alzheimer-type dementia; some individuals with extensive amyloid deposits fail to show significant cognitive impairment (reviewed in [166]) and clinical trials to remove amyloid deposits have failed to lead to disease improvement. Equally, in the animal models discussed above there has been no demonstration that the animals suffer from a condition that strictly reproduces human dementia. Despite this caveat, the transmissibility of AD pathology has been widely replicated and, at face value, given the failure of Aβ peptides (either soluble or aggregated) to transmit disease, might suggest that a second agent (possibly infectious) may be required for full transmission.
Evidence for an infectious component to AD
For AD, the idea that microorganisms might participate in senile dementia was first proposed by Fischer in 1907 [167]. More than a century later, a volume of data supporting this hypothesis has begun to accumulate [168-170], and reports have appeared of associations between AD and diverse infectious agents including both viruses and bacteria [171]. Evidence for a causal link between infection and disease is generally based on two types of observations. (i) Statistical association between an infectious agent and clinical disease; however, such associations could be fortuitous, and are often regarded as unconvincing. (ii) A second type of study – intervention – is necessary to demonstrate causation. We therefore address both types of evidence. Although not fully comprehensive, the selection aims to highlight both the diversity (and the inconsistencies) in the literature.
Herpes simplex virus type 1 (HSV-1)
Latent herpesvirus HSV-1 is widespread in the population and virus reactivation is associated with lesions of the skin and the central nervous system (CNS). Mann et al.[172] and Esiri [173] provided the first evidence of HSV-1-immunopositive neurons in AD brain. HSV-1 DNA was detected in brain tissue of 3/3 patients with familial AD but all but one of six age-matched controls were negative [174]. Other studies have suggested that the presence of HSV-1 DNA in AD and control brain samples is unrelated to disease status (e.g. [175]). A complication is that a majority of the population is HSV-1 seropositive. In an alternative approach, Letenneur et al. used IgM seropositivity as a marker of recent herpesvirus activation in a large cohort of healthy elderly. Those who were IgM-positive were significantly more likely to develop AD during the follow-up period of 14 years (relative risk 2.55; 95% CI 1.38–4.72) [176].
Chlamydophila (Chlamydia)
Until recently known as a Chlamydia species, Chlamodophila pneumoniae is an obligate intracellular bacterium associated with respiratory infections of humans and animals. Using PCR and electron microscopy, Balin et al. identified C. pneumonia in 90% of postmortem AD brain samples but in only 5% of control samples [177]. Both typical intracellular and atypical extracellular forms were found in astrocytes, perivascular macrophages, microglia, and neurons; moreover, cells carrying the bacteria colocalized with amyloid plaques and NTFs [178,179].
Spirochetes
This diverse group of double-membrane spiral-shaped bacteria are generally free-living, but are responsible for several important diseases including Lyme disease (principally Borrellia burgdorferi) and syphilis (Treponema pallidum). Rivière et al. reported that several species of oral Treponema were present in brain samples from both AD (14/17 positive) and controls (4/18), but the quantity of bacterial DNA was significantly higher in AD patients [180]. In another study, using a panel of methods, spirochetes were detected in CSF, blood, and brain of 14 AD patients, whereas 13 control samples were all negative [181]. In 3 of 14 samples the bacteria were identified as Borrelia burgdoferi. Spirochetes colocalized with senile plaques and NTFs and were present in vascular walls in association with amyloid deposition [168] (Figure 5).
Figure 5.

Spirochetes in Alzheimer disease brain. (A) Detection in an immature senile plaque using a cocktail of specific antibodies against Borrelia burgdori (dark-brown staining). (B)Borrelia sequences in a mature plaque detected using in situ hybridization using a B. burgdorfi probe. (C) Detection in an amorphous plaque using an antibody to bacterial peptidoglycan. (D) Control brain stained with antibodies against B. Burgdorfi. Arrows in B indicate bodies resembling helical spirochetes. Scale bar, 80 μm for (A, D), 30 μm for (B), and 20 μm for (C). Figure kindly provided by Judith Miklossy, Switzerland.
Helicobacter pylori
Also with a helical or spiral structure, the Gram-negative Spirilla Helicobacter pylori is a causal agent for gastric ulceration and has also been highlighted as a potential risk factor for AD development. It was reported that 88% of AD patients were positive by histology for H. pylori versus 47% of controls [182]. In a group of 53 AD patients, H. pylori infection was significantly associated with reduced cognitive ability and higher CSF Tau [183] and, in the most recent study, in which 600 elderly individuals were followed for a period of 19 years, H. pylori infection determined by serology was found to be a risk factor (risk ratio 1.46) for developing dementia [184]. However, another study failed to find any association between H. pylori infection and AD [185].
Intervention
Infection can precipitate AD-like pathology in animal models (see later), but few studies have addressed possible intervention in the clinic. Kountouras et al.[186] reported that eradication of H. pylori infection was associated with a significant reduction in mortality risk in 46 patients with probable AD (risk ratio, 0.287; 95% CI 0.114–0.725).
Does ATH also have an infectious component?
For over 100 years there have been reports of an association between acute infectious disease, atherosclerosis, and stroke. To our knowledge, direct transmission from primary disease material has not been attempted in ATH models, and (in contrast to AD) there is so far no evidence that ATH can be ‘seeded’ by inoculation of extracts of diseased arteries; we feel such studies may need to be carried out. However, investigations have implicated diverse infectious agents in the pathoetiology of ATH.
Herpesviruses
Benditt et al.[187] detected herpes simplex virus (HSV) by in situ hybridization of aortic samples (Figure 6), but failed to detect DNA sequences of another herpesvirus, cytomegalovirus (CMV); two of four samples with abnormally thick intima media were strongly positive for HSV. Melnick and colleagues [189,190] reported antigens and CMV sequences in association with ATH, and Speir et al.[191] showed that one third of atherosclerotic lesions obtained by coronary atherectomy contained CMV DNA sequences. CMV infection was identified as an independent risk factor for restenosis after coronary angiopathy [192] and CMV-positivity is associated with endothelial dysfunction and an increased atherosclerotic burden [193]. Nieto et al.[194] found a graded and significant relation between the odds of intima media thickening and the level of CMV antibodies. However, Hendrix et al.[188] reported comparable detection frequencies (ca 30–50%) of CMV sequences in arterial samples from both patients with atherosclerosis and non-ATH controls. Because a majority of the population is already positive for CMV and/or HSV subtypes, it is difficult to ascertain whether herpesviruses are bystanders or might potentially be causally implicated in ATH pathology.
Figure 6.
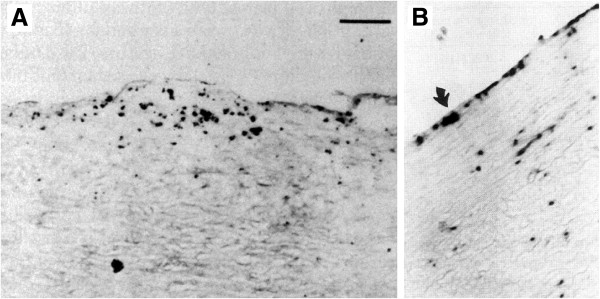
Virus detection in ATH lesions. (A) Herpes simplex virus sequences in a thoracic artery lesion from a patient undergoing coronary bypass surgery detected by in situ hybridization. Scale bar, 25 μm. Panel adapted, with permission, from [187]. (B) Cytomegalovirus sequences in arterial wall from a patient with severe atherosclerosis detected by in situ hybridization. Original magnification 100×. Panel adapted, with permission, from [188].
Chlamydophila
Using electron microscopy, Shor et al.[195] first reported the presence of chlamydia-like structures in seven samples of fatty streaks and atheromatous plaques that were confirmed by immunocytochemistry in five cases. The same group [196] reported C. pneumoniae antigen and sequences in 35–50% of lesions, as confirmed in several follow-up studies [197]. For example, PCR detected C. pneumoniae DNA in 31% of atherosclerotic plaques but in only 2% of normal aortic samples [198]. Chlamydophila is known to be able to infect and replicate within monocyte/macrophages, endothelial cells, and vascular smooth muscle cells (SMC) [199]. Review of all published studies [200] recorded that, overall, 46% of plaques were positive whereas <1% of control arteries were positive. Recent review of the field [201] emphasized great variation between studies, but that there has been a ‘high degree of consistency in the association between C. pneumoniae and arterial atheromatous lesions’.
Spirilla and Spirochetes
There have been intermittent reports of an association between ATH and Spirilla and Spirochetes (H. pylori and the dental pathogen Treponema denticola, respectively). For example, Ameriso et al.[202] reported H. pylori DNA in 20/38 atherosclerotic plaques whereas 0/7 normal arterial samples were positive. However, another study failed to detect H. pylori in ATH plaque, although C. pneumonia was found in 50% [203]. Okuda et al.[204] studied T. denticola sequences and reported that 23% of atherosclerotic lesions were positive by PCR whereas 0/14 control aorta samples were positive.
Porphyromonas gingivalis
P. gingivalis, a Gram-negative anaerobe implicated in peridontal disease, has also been proposed to be involved in other diseases including ATH, diabetes, and rheumatoid arthritis [205]. An association between dental health and cardiovascular disease was first established at the end of the 1980s [206,207]. Indeed, periodontal pathogens including P. gingivalis have been detected in different cardiovascular disease cases including atherosclerotic lesions, aneurysms, and endocarditis [208-210]. As reviewed [211], P. gingivalis has been associated with SMC proliferation and endothelial cell apoptosis. The large INVEST study reported a direct relationship between carotid intima thickness and peridontal bacterial burden [212].
Intervention studies
Given the potential role of bacterial infection in ATH, there have been several trials of antibiotic treatment, but without significant benefit [201,213,214]. However, key bacteria (e.g. C. pneumoniae) can persist for long periods as a latent intracellular infection, and it is unclear what degree of clearance was achieved in these studies. The best available data are from chicken (and from mouse models, below). Infection of chickens with Marek disease virus, a herpesvirus, causes them to develop atherosclerotic lesions that resemble ATH in human [215] (reviewed in [216]).
Infectious agents contribute to AD and ATH
Wild type mice inoculated with C. pneumoniae cultivated from AD brain developed amyloid plaques [217]. HSV-1 infection of cultured neuronal and glial cells leads to a dramatic increase in the intracellular levels of Aβ, and antiviral therapy blocked Aβ production [218]. Infection with neuroadapted mouse hepatitis virus strain JHM was also reported to exacerbate AD-like pathology in a transgenic mouse AD model [219]. Of note, Aβ deposition is a common feature of brain infection with HIV in human [220]. Conversely, immunosuppressive Toxoplasma gondii inhibited disease development in an APPAD mouse model [221].
For ATH, diverse experiments in animal models have demonstrated that inoculated infectious agents such as C. pneumoniae persist in atherosclerotic lesions [222] and accelerate ATH development in susceptible mice. For example, C. pneumoniae infection increased aortic ATH in the Ldlr−/− mouse model [223,224], although this has been disputed (e.g., [225,226]), and infection can stimulate cholesterol-rich foam cell formation [227] and SMC proliferation [228], markers of ATH. Infection of ATH mouse models with Porphyromonas gingivalis[229,230], H. pylori[231], or Streptococcus mutans[232] also accelerated atherogenesis. Similar results have been obtained with viral pathogens. Virus infection of ATH-prone mice promotes atherogenesis, exemplified by mouse gammaherpesvirus-68 [233], influenza virus [234], and CMV [235,236].
Conversely, as with AD, infection with an immunosuppressive pathogen (here Schistosoma mansoni) reduced ATH lesions by 50% in infected mice [237].
Non-specific immune activation predisposes to disease
Infection is not strictly required for atherogenesis. Wright et al.[238] reported that the profile of ATH development was unaffected in Apoe−/− mice additionally carrying the lpsd mutation that renders them unable to respond to bacterial lipopolysaccharide (LPS) – although LPS is only one of multiple stimulators of innate immunity. Germfree Apoe−/− mice, that are held to be free of bacteria, viruses, and fungi, developed atherosclerosis [238]. However, non-specific immune challenge can precipitate disease. LPS injections alone can increase atherosclerotic lesion size [239]. Vliegen et al.[240] compared the effects of inoculation of mouse CMV (MCMV) with ultraviolet light (UV)-treated MCMV. The inactivated virus increased atherosclerotic lesion area and T cell number in the atherosclerotic lesions, whereas only live MCMV infection increased T cell numbers in the internal organs.
Similar findings have been made in AD models. Transmission of disease (see earlier) has been reported but, importantly, heating AD brain extracts to 95°C reduced (45%) but did not eliminate transmission [158], arguing for a non-specific inflammatory effect. In support, systemic immune stimulation with the viral mimetic, poly(I:C), during gestation predisposes to AD-like neuropathology [241]. Prenatally stimulated animals had increased levels of Aβ, hyperphosphorylated Tau, and NFT formation. Lee et al. showed that injection (i.p.) of LPS into mouse models led to increased levels of Aβ and Tau aggregation [242].
In conclusion, in both ATH and AD, there is strong evidence linking disease development to infection, and overwhelming indications that infectious agents home to diseased tissue and aggravate pathology. Nonetheless, one suspects that any one of several agents can accelerate atheroma formation – and local immune cell activation (via either infectious or non-infectious agents) precipitates disease. In short, infectious agents per se may not be required for disease development (with the caveat that several agents are intrinsic to the mammalian genome, e.g., endogenous retroviruses, not reviewed) but, in the absence of other risk factors, transmissible agents are more than likely to play a determining role – as stated by Epstein et al. ‘compelling data indicate that infection does contribute to atherogenesis and to the acute complications of atherosclerosis caused by plaque rupture’ [243].
Focal nature of disease
Both AD and ATH are manifested focally, and this affords a further argument. In both conditions, numerous foci of disease replicate the same pattern of progression at different locations. At the same time, there are significant stretches of tissue which are not affected by the disease, despite the presence of all confounding factors for decades since the beginning of the pathological process. The focal nature excludes somatic mutations or other cell-autonomous defects in the cells forming a solid tissue. Instead, a stochastic element, particularly at the initial stage of the disease, is most plausible, and foci of infection are an obvious contender. Subsequent stages may not require direct pathogen involvement, because local inflammation, once established, may persist via the involvement of activated immune cells.
Drug overlap
If the two disorders have a similar etiology, drugs (other than antibiotics, discussed above) effective in one disorder might be expected to show efficacy in the other. Both diseases are associated with elevated blood cholesterols (even though transplant experiments in mice have demonstrated that this is not causal, above), raising the question of whether blockade of cholesterol synthesis might be used to treat ATH or AD.
Statins reduce body excess of cholesterol by inhibiting a key enzyme in de novo cholesterol synthesis, HMG CoA reductase. No conclusive benefits have been reported in AD [244] whereas, in ATH, some benefits have been reported, notably in the ASTEROID trial of rosuvastatin [245,246], although other trials failed to give unequivocal results [247]. Statins have many side-effects and do not appear to be the panacea one might have hoped for. This makes sense given that, in genetic models of both diseases, transplantation of wild type bone marrow abrogates pathology despite the persistence of host hyperlipidemia.
Curcumin
An aromatic component of the spice turmeric (Curcuma longa), this molecule has been suggested to prevent AD Aβ toxicity. Curcumin can reduce amyloid in vivo in transgenic AD models and remove existing plaques [248-251]. Curcumin also reduces Aβ-mediated blockade of long-term potentiation (LTP) [252], a likely electrophysiological correlate of learning and memory. Trials of curcumin in AD patients have been explored [253] and further studies are ongoing.
Curcumin also exerts protective pharmacologic effects against ATH. In different mouse models, curcumin can potently inhibit ATH disease development [254-256]. Although several possible targets have been discussed, including inhibition of NF-κB (a marker and mediator of innate immunity induction) [257] the precise molecular target(s) for the beneficial effects of curcumin remains unknown.
Resveratrol
Resveratrol is a diphenolic molecule and notably a component of red wine. Intriguingly, resveratrol promotes Aβ clearance in cell culture [258] and protects against Aβ toxicity in culture [259,260] and in adult rats [261]. Similar findings have been reported in transgenic mouse AD models treated with resveratrol [262] or even, perhaps controversially, Cabernet Sauvignon [263]. The molecule is in clinical trials in AD [264].
For ATH, the potential protective activity of resveratrol has been discussed for three decades. Like curcumin, resveratrol has been shown to reduce atheroma formation in different mouse models of atherosclerosis (Apoe−/−Lldl−/− on a high-fat diet) [265-267], in some cases dramatically [268]. Protective effects in hypercholesterolemic rabbits have also been recorded [269], and several clinical trials are ongoing in diverse indications. The specific molecular target is not known but, among other activities, resveratrol has been reported to inhibit ACAT [270].
Acyl-CoA cholesterol acyltransferase (ACAT) inhibitors
ACAT (also known as SOAT, see footnote on nomenclature in Table 1) is a key enzyme catalyzing the esterification of cholesterols. In mouse models, inhibition of the enzyme (ACAT1/2) attenuates both ATH and AD. For ATH, to give only two recent examples, in Apoe−/− mice the inhibitor F1394 retarded ATH plaque progression [271]; similar observations were made with the inhibitor Manzamine A [272]. Knockout studies for ACAT1 and ACAT2 have generally revealed a protective role of gene disruption (although the literature is discordant for ACAT1; Table 1). In AD, the ACAT inhibitor CI-1011 [273] modulates Aβ production [274] and reduces Aβ accumulation in a transgenic model of AD [275,276]. Similar anti-Aβ effects were observed with a second ACAT inhibitor, CP-113,818 [275]. It was recently reported that knockdown of ACAT1 expression in vivo using a viral vector alleviated AD-like pathology in a mouse model [277], confirming that ACAT1 and ACAT2 are both likely drug targets in AD and ATH.
Acetylcholinesterase (AChE) inhibitors
Given well-established deficits in central cholinergic neurotransmission in AD, AChE inhibitors such as donepezil, galantamine, and rivastigmine have been widely trialed in AD – with evidence of efficacy in slowing disease progression (reviewed in [278]). In ATH, perhaps surprisingly, donepezil infusion could attenuate atherogenesis in susceptible mice [279].
The mechanism may not be what we think. Interestingly, the target enzyme AChE reiterates the structure of the catalytic site of a steroid gating enzyme (HSD11B), and molecular design directed to the AChE site yielded HSD11B inhibitors (three for HSD11B1 and four for HSD11B2) [280]. Intriguingly, polymorphisms in the gene encoding the ‘backup’ acetyl choline hydrolyzing enzyme butyrylcholinesterase (BCHE) are reported as risk factors in both ATH [34,281] and AD [282]. Cholesterol hemisuccinate is a weak inhibitor of BChE (IC50 168 μm) but a potent inhibitor of AChE (IC50 0.79 μM) [283]. AChE inhibitors may therefore act, in part, via interference with steroid and sterol metabolism.
AChE is widely expressed at the surface of platelets and red blood cells, macrophages express specific nicotinic acetyl choline receptors, and systemic cholinergic signaling modulates platelet aggregation, macrophage function, and innate immunity (see [284]). Interaction with these pathways, either directly or via CNS effects, could underlie the beneficial effects of AChE inhibition in both ATH and AD.
The overlaps in drug responsiveness between AD and ATH reinforce Roher’s earlier observation that there is ‘an immediate need for prospective clinical trials to assess the efficacy of AD prevention using antiatherosclerotic agents’ [285]. Equally – do other anti-AD drugs combat ATH?
Transcriptome module overlap
Further evidence of commonality between AD and ATH is provided by gene expression analysis. Ray et al.[286] used a systems biology approach to analyze brain RNAs from 20 confirmed AD brains versus 13 controls. 1600 genes differentially expressed in AD were identified and classified according to functional module. The two predominant modules, confirmed by functional annotation clustering, were (i) AD/neurodegeneration, as expected, but also (ii) cardiovascular/coronary artery disease [286]. The authors concluded that many pathways are common to both diseases; their results provide strong support for a mechanistic linkage between AD and ATH.
Mechanisms – inflammation, cholesterol metabolism, immunosterols
Both diseases are underpinned by genes affecting cholesterol transport/metabolism and immunity. Immunostimulation precipitated by infectious agents or specific components such as LPS can increase, sometimes dramatically, the development of ATH or AD in animal models. Equally compelling are the data that the immune system, notably macrophages, is centrally involved in the disease processes that culminate in local inflammation, the formation of cholesterol-loaded foam cells, and vascular occlusion. These observations point to a direct link between infection and cholesterol metabolism, as borne out by studies on APOE.
APOE and infection
APOE alleles, encoding a key lipid transport molecule, play a crucial determining role in the outcome of viral and bacterial infection. In mouse models, APOE modulates infection by HSV-1 [287], Chlamydophila[288], Klebsiella pneumoniae[289], Listeria monocytogenes[290] and Leishmania[291]. In Apoe−/− transgenic mice expressing human APOE, APOE3/APOE4 genotype has a marked influence on HSV-1 propagation and latency – APOE4-expressing mice challenged with HSV-1 had very high levels of virus in brain compared to APOE3-expressing or knockout mice [287,292]. Effects of LDLR mutation on Toxoplasma disease were also reported [293].
Similar findings have been made in human. The APOE4 allele is reported to accelerate HIV proliferation whereas, by comparison, APOE3 is protective [294]. Numbers of Chlamydophila-infected cells and bacterial load were significantly higher in homozygous APOE4 patients than in APOE2 or APOE3 carriers [295]. Conversely, for malaria, APOE3/4 afford protection and APOE2 homozygotes are most susceptible [296].
Thus, for some pathogens (e.g., HIV, HSV-1, Chlamydophila) APOE4 predisposes to disease, whereas for others (e.g., malaria) APOE4 is protective. Understanding how APOE allelic variants can differentially confer susceptibility to one disease, but resistance to another, is clearly of fundamental importance. This raises a central question – how do APOE and cholesterol metabolism relate, at a molecular level, to infection and immunity?
Cholesterol signaling
Adequate cholesterol supply can play an important role in the assembly of the membrane components required by many (but not all) pathogens; some infectious agents are known to exploit transport and uptake mechanisms such as those mediated by APOE and LDLR. However, evidence is now emerging that cholesterol and its oxysterol derivatives play potent roles as specific signaling molecules in the immune system.
Cholesterol itself is poorly soluble, but is prone to spontaneous and enzyme-mediated oxidation at the 7, 11, 5–6, 22/24/25/27 positions [297] that increase mobility: for example, efflux of oxidized cholesterol from macrophages is ~50× faster than of cholesterol itself [298]. Side-chain oxidation is also required for export from the brain.
Oxidized cholesterols are potent signaling molecules. One major pathway is via the liver X receptors (LXR). Although prominently expressed in the liver, where LXRα and LXRβ regulate bile acid synthesis and metabolism/excretion (reviewed in [299]), in peripheral tissues receptor activation feeds back to repress cholesterol synthesis and promotes export of excess cholesterol to the liver and bile for excretion. The best natural LXR ligands are cholesterols oxidized at the 22, 24 and/or 25 positions [300], although 27-hydroxycholesterol (27OHC) has been argued to be the endogenous ligand [301,302].
Specific role of ‘immunosterol’ 25OHC in innate immunity
Elevated levels of oxysterols are reported in ATH lesions, notably 27OHC and 7αOHC [303], and could contribute to atherogenesis (reviewed in [304]); anomalies found in AD brain include oxysterols, cholesterol precursors, and steroids (e.g., [305-308]. However, new evidence from Ghazal’s group [309] has emerged that one molecule, 25OHC, plays a specific role in regulating responses to pathogens.
Several enzymes are known to catalyze the formation of 25OHC, notably the promiscuous enzyme CYP3A [310], CYP27 that catalyzes 24, 25, and 27 hydroxylation [311], and CYP46 that predominantly catalyzes 24S-hydroxylation but also 25-hydroxylation [312]. By contrast, in macrophages (centrally implicated in both ATH and AD), 25OHC synthesis appears to be principally mediated by the key enzyme cholesterol 25-hydroxylase (CH25H).
CH25H is a most unusual enzyme. Unlike classic heme-dependent P450 enzymes (CYPs), that widely catalyze the hydroxylation of hydrophobic compounds including sterols and steroids, CH25H, located in the endoplasmic reticulum, is a di-iron enzyme [313] whose ancestry goes back to yeast. Whereas many CYP enzymes are promiscuous in substrate specificity and site of modification, CH25H appears to be specific for 25-hydroxylation of cholesterol. We have here a glimpse of intriguing evolutionary constraints that deserve to be followed up.
Macrophage CH25H expression is specifically upregulated by inducers of innate immunity, including LPS, poly (I:C), lipoteichoic acid, specific Toll-like receptor (TLR) agonists, and by IFN-α or IFN-β [257,314,315] (Figure 7A). In mice, intraperitoneal administration of a TLR agonist led to marked upregulation of CH25H mRNA, most strikingly in liver (200×), brain (25×), and heart (50×), with a fivefold increase in serum 25OHC levels [257]. Brain expression of CH25H was induced by i.p. LPS injection into mouse [319], and intravenous injection of LPS in healthy human volunteers resulted in 2–3-fold increase in plasma 25OHC [314].
Figure 7.

25OHC induction by immunostimulation or infection promotes cholesterol esterification and foam cell formation. (A) 25OHC is the only oxysterol induced by interferon (IFN) treatment or infection; panel from [309], with permission. (B) 25OHC stimulates cholesterol esterification in intact cells, from [316], with permission. (C) 25OHC is an allosteric effector of ACAT activity in insect cells expressing human ACAT, from [317] with permission; 7KC, 7-ketocholesterol; 6KCh, 6-ketocholestanol; 7αHC, 7α-hydroxycholesterol. (D) Treatment of bone marrow-derived macrophages with low-dose (ca. 0.1 μM) 25OHC, but not cholesterol itself, induces lipid body formation (BODIPY 493/503 fluorescence); scale bar 10 μm. From [318], with permission.
The specificity of the induction is striking. Ghazal’s group [309] screened IFN (β or γ)-treated macrophages by liquid chromatography and mass spectrometry (LC-MS) for all intracellular and secreted oxysterols, revealing 25OHC as the sole oxysterol produced in response to stimulation (Figure 7A). Similar CH25H upregulation and 25OHC synthesis in macrophages was observed in response to viral infection (mouse CMV) [309]. This identifies 25OHC as a specific signaling molecule in response to stimulation of innate immunity.
In this context it is intriguing to note that curcumin, a drug with potential activity against both AD and ATH (see earlier), has been shown able to abolish macrophage expression of CH25H following induction of innate immunity [257]; this could underlie its beneficial affects.
Upregulation of CH25H enzyme plays a protective role. Blanc et al.[309] demonstrated that 25OHC supplementation in the sub-micromolar range (generally 0.1–1 μM) induces a broad-specificity antiviral response, inhibiting infections by influenza virus, HSV-1, varicella zoster, and murine γ herpesvirus 68. Similar potent antiviral effects were demonstrated by Cheng’s group for vesicular stomatitis virus (VSV), HIV, and a range of acutely pathogenic viruses [320]. Inhibition of mouse CMV proliferation by 25OHC has been confirmed [321]. Although both 27OHC and 24(S)25-epoxycholesterol do display significant antiviral effects [309], these molecules were entirely absent from stimulated macrophages in the screen of Blanc et al.
In further investigation of the mechanism, 25OHC was found to interfere with virus proliferation, and virus plaque size in cell culture was diminished in the presence of 25OHC [309]. It was reported that 25OHC inhibits membrane fusion between virus and cell [320] and, importantly, 25OHC had no effect on the non-enveloped viruses adenovirus 5 or 19a [309]. These findings argue that induction of CH25H in response to infection {or by LPS, poly(I:C), or IFN} protects against viral infection by blocking specific membrane steps in virus entry or maturation. Effects on bacterial pathogens have not been tested, but induction in response to LPS suggests that 25OHC could potentially have antibacterial effects.
What is the target for 25OHC?
This work raises the issue of whether there is a specific receptor for 25OHC, or whether 25OHC non-specifically interferes with membrane assembly and fusion processes important for pathogen infection and replication. Blanc et al.[309] suggest that 25OHC at higher concentrations (>1 μm) may have non-specific effects. However, at submicromolar concentrations they report that, unlike 25OHC, a structural enantiomer, ‘ent-25HC’, was inactive (although some activity was observed at high concentration) in an antiviral assay, and concluded that the antiviral effects of low-concentration 25OHC are mediated by specific receptor binding.
How does 25OHC mediate antiviral effects? An effect via LXR and cholesterol metabolism per se seems unlikely because mice lacking CH25H enzyme regulate cholesterol normally [257,322,323]. Both 25OHC and 27OHC suppressed IgA production (a correlate of induction of innate immunity) whereas selective LXR ligands were inactive [257]. Synthetic LXR ligands were devoid of antiviral effects [309]. These studies argue that the protective effects of 25OHC are not mediated by LXR.
One possibility is that 25OHC is onward metabolized by the widely expressed oxysterol-metabolizing enzyme CYP7B1 [324] to 7α,25OHC. This molecule is a selective ligand of the G protein-coupled receptor EBI2, and experiments in knockout mice confirm that this route plays a role in modulating antigen-specific immunity. Knockout of either EBI2 or CH25H produced defects in activated B cell migration [322], and mice deficient in either CYP7B1 or CH25H display defective T cell-dependent responses [325]. However, this pathway is unlikely to explain the potent antiviral effects of 25OHC because only traces of 7α,25OHC were detected in the screen of Blanc et al.[309].
The specific receptor for 25OHC therefore remains unknown, although the molecule is known to bind with high affinity to oxysterol binding proteins OSBP1 and OSBP2 [326,327], and OSBP–sterol binding has been argued to play a specific regulatory role [328], notably in modulating OSBP subcellular localization [329]. 25OHC binding to OSBPs may interfere with essential intracellular targeting and delivery of pathogen components (specific intracellular trafficking pathways will differ between pathogens). OSBPs have been implicated in both AD and ATH [330,331]; oxysterol binding to OSBPs in macrophages is thought to play a direct role in atheromatous plaque formation [332] and macrophage expression of OSBP2 (OSBP-L1) enhances ATH in susceptible mice [331]. The estrogen receptor ERα remains a further contender as a target for 25OHC [333]. However, given the diverse variety of cellular binding sites for cholesterols [334], unraveling the specific molecular targets underlying the antiviral effects of 25OHC will be challenging.
25OHC is implicated in both ATH and AD – the role of ACAT
In support of a role of CH25H enzyme in both diseases, GWAS studies have implicated the gene cluster cholesterol 25-hydroxylase, CH25H (10q23) – lipase A, lysosomal acid, cholesterol esterase, LIPA (10q23.2-q23.3) in both ATH [335,336] and AD [337,338]. The evolutionarily-conserved linkage between CH25H and LIPA, less than 20 kb in all mammalian species examined [339], is intriguing and suggestive because LIPA (lipase A, or cholesteryl ester hydrolase) is the primary enzyme responsible for de-esterification of cholesterol (see e.g., [340]).
Immunostimulation leads to induction of CH25H expression and local production of 25OHC. In AD, increasing expression of CH25H in temporal lobe regions of AD brain correlates with Braak (NFT) staging of disease progression [337]. No studies have been reported in human ATH, but elevated levels of 25OHC have been reported in lungs of patients with chronic obstructive pulmonary disease [341], another condition associated with chronic infection [342].
Chronic overexpression of CH25H is a powerful contender as the culprit for triggering disease pathology because, as first reported by Goldstein and Brown, it has been known for almost 40 years that 25OHC stimulates cholesterol esterification [316] (Figure 7B). Intracellular cholesterol esterification is catalyzed almost exclusively by ACAT (ACAT1/2) that adds a long-chain fatty acid to the cholesterol 3β-hydroxy group, and CH25H overexpression and 25OHC synthesis are known to promote ACAT activity, cholesteryl ester formation, and the generation of foam cells.
Compared to cholesterol itself, oxysterols are highly mobile, but once the 3β-hydroxy group has been attached to a long-chain fatty acid (principally oleic acid, C18; but also palmitic acid, C16 and other similar fatty acids) the molecule becomes insoluble and prone to aggregation. Although the major sterol in advanced ATH plaque appears to be 27OHC (>80% esterified) [303,304], and not apparently esters of 25OHC itself, this may be explained by the fact that 25OHC acts here, not as a substrate, but as an allosteric activator of intracellular esterification.
Work over many years, notably by Chang’s group, has revealed that ACAT enzymes contain two binding sites: (A) the allosteric regulatory site, and (B) the catalytic site. Once a sterol is bound to the A site the enzyme becomes highly active, with promiscuous substrate specificity for a wide range of sterols and even some steroids. Adding 25OHC to the culture medium caused a 20–60-fold increase in sterol esterification without change in enzyme content [317] (Figure 7B). Crucially, 25OHC is the most effective positive allosteric effector of ACAT; the enzyme is only poorly activated by close analogs such as 7-ketocholesterol, 6-ketocholestanol, 7α-OHC, cholate, or cholesterol itself [317,343] (Figure 7C). 25OHC activation of ACAT takes place in multiple cell types including macrophages [344] and neuronal cells [345]. In addition, it has been suggested that 25OHC drives intracellular redistribution of cholesterols to the endoplasmic reticulum [346], where ACAT is located; this could afford a second mechanism underpinning the enhancement of esterification. Therefore, by these routes 25OHC triggers the conversion of the intracellular pool of cholesterols (including 27OHC) into insoluble cholesteryl esters, and thus prevents their export (e.g., [347]). Induction of cholesterol esterification by 25OHC was recently confirmed [321].
In further confirmation of its key role, 25OHC has been shown to promote macrophage foam cell formation in mouse cell culture [318] (Figure 7D). It is interesting that 3 μM 25OHC is typically used to stimulate cholesterol esterification, whereas foam cell formation was reported to take place even at a 10-fold lower concentration [318]; ACAT is thus a further contender as a ‘receptor’ for 25OHC.
The data suggest that chronic overexpression of CH25H is causally associated with disease. In direct support, CH25H gene expression is repressed by transcription factor ATF3 – knockout of ATF3 in susceptible mice led to a marked increase in 25OHC levels and foam cell formation [318]. Ablation of CYP7B1 (that redirects 25OHC by conversion to the EBI2 ligand 7α,25OHC) increased ATH severity in mice, pointing to a specific predisposing effect of 25OHC itself [103]. Circumstantially, the finding that the expression of CYP7B1, that efficiently metabolizes 25OHC [324], is downregulated in AD brain [104] is consistent with the interpretation that excess 25OHC contributes to AD.
The finding that ACAT inhibition alleviates disease development in models of both ATH and AD (see earlier) confirms that ACAT-mediated sterol esterification, driven by 25OHC, is a key contributor to disease progression. This and other evidence argues that other atherosclerotic features (including disruption of endothelial/smooth muscle cell function and matrix calcification) are downstream consequences.
We suggest that chronic production of 25OHC in macrophages leads to ACAT-mediated sterol esterification, the accumulation of insoluble cholesteryl esters, foam cell formation, and vascular occlusion.
Hypothesis: inflammation, macrophages, oxysterols, vasculature
We propose a model in which immune stimulation by pathogens (or components/autoantigens) leads to local induction of CH25H in macrophages. Synthesis of 25OHC confers broad-spectrum inhibition of the growth of enveloped viruses, many of which preferentially target macrophages, and potentially modulates the growth of other pathogens including parasites and intracellular bacteria (Figure 8).
Figure 8.
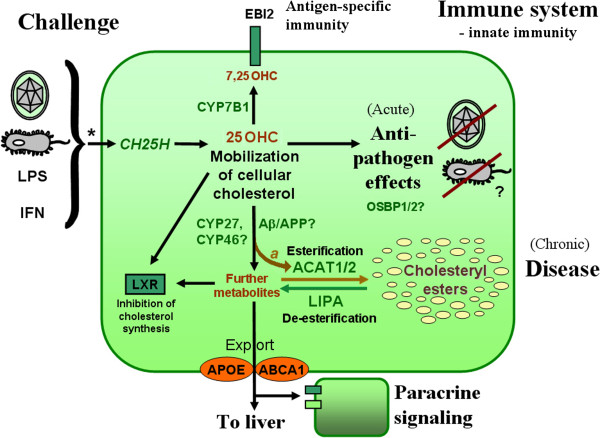
The foam cell hub (simplified). A model for disease development in ATH; similar processes are argued to operate in AD. Only key pathways are depicted. Challenge by infection or proinflammatory agents leads to induction of IFN signaling (*; both IFN-α and IFN-γ. For the specific pathways involved see [257,309] and references therein), upregulation of macrophage CH25H gene expression, and catalytic conversion of part of the cellular cholesterol pool to 25OHC. Acutely, this molecule exerts anti-pathogen effects via an unknown receptor. On chronic inflammatory exposure, hydroxylated cholesterols accumulate, are removed to insoluble intracellular inclusions by ACAT-mediated esterification (allosterically modulated by oxysterols, a), thereby leading to foam cell formation and overt disease. Oxidation by APP/Aβ may divert or enhance this process. APOE and ABCA1 are shown at the periphery to emphasize their role in export. Other key components from Table 1 (not shown in the figure) include LDLR, a trans-cellular receptor for cholesterol mobilization, and CLU, a protein with diverse functions in complement-mediated cell lysis, apoptosis, and lipid transport. Abbreviations: ABCA1, ATP-binding cassette, sub-family A (ABC1), member 1 (cholesterol efflux protein); ACAT, acyl-CoA cholesterol acyltransferase (also known as SOAT); APOE, apolipoprotein E; APP, amyloid-β (A4) precursor protein (Alzheimer precursor protein); CH25H, cholesterol 25-hydroxylase; CLU, clusterin (apolipoprotein J; complement cytolysis inhibitor); CYP27, cytochrome P450 27A1 (sterol 27-hydroxylase); CYP46, cytochrome P450 46A1 [cholesterol 24(S)-hydroxylase]; CYP7B1, cytochrome P450 7B1 (sterol and steroid 7α hydroxylase); EBI2, Epstein–Barr virus (EBV)-induced G protein-coupled receptor 2, IFN, interferon; LPS, lipopolysaccharide; LDLR, low-density lipoprotein receptor; LXR, liver X receptor; 25OHC, 25-hydroxycholesterol.
The downside of this anti-pathogen reaction is, by implication, that chronic production of 25OHC leads to accumulation of cholesteryl esters via the action of ACAT, the formation of fatty inclusions in macrophages, foam cell formation, and vascular occlusion (Figure 3). In turn, foam cells lose mobility and reside in situ for extended periods of time, themselves producing proinflammatory stimuli that amplify the pathology, leading to the formation of further atheromatous occlusions in the heart (ATH) or brain (AD) vasculature and overt disease.
The role of APP/Aβ
There is evidence that APP and Aβ, implicated in both AD and ATH, are themselves involved in oxysterol metabolism. In AD plaques Aβ colocalizes with deposits of APOE and cholesterol [348]. It was first reported that Aβ catalyzes the formation of 7β-OHC, and in this reaction Aβ was 200-fold more active than APP [349]. More recently evidence has emerged that Aβ has predominant cholesterol 3-oxidase activity [350,351], particularly in the presence of divalent cations such as Cu2+ (a known contributor to AD-like pathology in relevant models). Significantly elevated levels of 3-oxidized cholesterols were reported in brains of APPAD mice and in postmortem brain tissue of AD patients [351]. 3- (or 7-) oxidation has potential to modulate the accumulation of cholesteryl esters in foam cells, although this remains to be demonstrated formally. APOE binding to Aβ [73] may also contribute but the role in disease is unknown.
However, the central involvement of Aβ remains contentious [166]. Aβ does not correlate precisely with clinical diagnosis of AD (reviewed in [352]), and the failure of Aβ immunotherapy to ameliorate AD pathology in clinical trials (not reviewed here) suggests that Aβ is not central, and is instead subservient to CH25H.
This has been confirmed via an unusual route. Papassotiropoulos et al.[337] report that, in large cohort of individuals with confirmed AD, specific CH25H haplotypes were associated with a complete absence of postmortem brain Aβ deposits, despite all other aspects of AD pathology. This argues that inflammation and CH25H activation are upstream to Aβ in the chain of events leading to AD.
The downstream induction of Aβ by infection/inflammation raises the question of whether Aβ might in fact be a protective (microbe-sequestrating or inactivating) response rather than a cause of pathology. In support, Aβ was shown to have potent antimicrobial properties, reducing the growth of E. coli by up to 200-fold in vitro, with activity against both Gram-positive and Gram-negative bacteria, and also against the fungus Candida albicans[353]. In the same study, brain homogenates from AD patients showed elevated antimicrobial activity versus control homogenates. It is not yet known whether Aβ-catalyzed sterol metabolism plays a role in this phenomenon. It is notable that reduction of Aβ in AD patients was associated with increased risk of infection [354] and, in both of two large clinical trials of anti-Aβ immunization in AD, 6% of patients developed encephalitis of unknown origin (reviewed in [355]), consistent with a potential defensive role of Aβ.
The role of APOE
Given the role of APOE in modulating disease (see above), by pathways that are only beginning to be understood, Robert Mahley argued that the diversification of APOE function, with three alleles prominent in the human population, has been driven by infectious disease. APOE4 is probably the ancestral allele, and is most similar to homologs in other species [356], and subsequent selection pressure is thought to have led to the emergence of APOE2 and APOE3[50].
The maintenance of multiple functionally different alleles in a population requires divergent selection – for which sickle-cell anemia affords the best precedent. Briefly, the hemoglobin S allele of β-globin is generally deleterious, but has a protective effect against malaria infection, and therefore in malaria-endemic areas the deleterious allele is maintained at high levels in the population [357].
This is paralleled by APOE, where APOE4 alleles are protective against some infectious diseases (e.g., malaria) but increase the proliferation of other pathogens (e.g., HSV-1, HIV) (see earlier). As stated by Mahley: ‘A cataclysmic event in human history driving the evolution of apoE4 to apoE3 to apoE2 could have been an infectious disease, such as the Great Plague, which killed 30–50% of Europeans in the 14th century, or smallpox’ [50].
We have argued here that chronic immune stimulation, leading to 25OHC production, contributes to the development of both ATH and AD. In both cases the APOE4 allele is a major risk factor, and APOE4 can be associated with an increased load of specific pathogens that have been implicated in disease processes.
However, the exact mechanism is unknown. APOE4 is relatively poor at exporting cholesterols, and therefore would be expected to (i) increase intracellular immunosterols, thereby diminishing the proliferation of immunosterol-sensitive pathogens; and (ii) foster lipid droplet formation and vascular occlusion.
This is clearly an oversimplification, because 25OHC in cell culture inhibits several enveloped viruses (e.g., HSV-1 and HIV) whereas, in vivo, APOE4 increases their proliferation. This presents another conundrum that will only be resolved once we begin to understand the full spectrum of immunosterols {25OHC, 27OHC, 24(S)OHC, 24(S),25-epoxycholesterol, and others including cholesterol precursors and 7α derivatives}, their targets, and how they inhibit the proliferation of some pathogens while potentially enhancing others. Nevertheless, the evidence causally implicates CH25H in response to infection/inflammation as a triggering factor for cholesterol mobilization, esterification, and foam cell formation. Outstanding questions are summarized in Table 2.
Table 2.
Outstanding questions
| 1 |
What is the molecular target for 25OHC? |
| 2 |
Does the 'immunosterol' 25OHC also have anti-pathogen effects against bacteria and parasites? |
| 3 |
Do other immunosterols, including 27OHC, 24(S)OHC, 24(S),25-epoxycholesterol, as well as derivatives of cholesterol precursors, operate by the same or different pathways? Do they confer protection to different types of pathogens? |
| 4 |
How does APOE modulate infection? Is there a specific interaction between different polymorphic forms of APOE and oxysterols including 25OHC (or does it act via a partner such as APP/Aβ cholesterol oxidation)? |
| 5 |
Is Aβ production a cause of disease, or is it a defense mechanism? |
| 6 |
Does infection/inflammation in brain microglia also upregulate CH25H expression? |
| 7 |
How do we explain the 'APOE paradox' – that APOE4 is associated with highest risk of both AD and ATH, but Apoe knockout in mouse models accelerates ATH but delays AD development. |
| 8 |
How do we explain the striking age-related increase in both AD and ATH incidence? Is it due to lifetime changes in circulating steroids/sterols? |
| 9 | Are other age-dependent diseases caused by a similar infection/inflammation/occlusion cascade? |
Concluding remarks
Brain versus body: why do some individuals develop AD, others ATH?
There are significant differences between brain and body; these could explain potential differences in the outcome of systemic infection and inflammation.
First, the brain differs from the body in cholesterol metabolism. In addition to being a net exporter of cholesterol, the CNS produces a brain-specific sterol, 24(S)OHC, known as ‘cerebrosterol’ [358]. The enzyme responsible, CYP46A1, is predominantly expressed in the brain [312] – most highly in areas affected by AD [359] – and polymorphisms in the gene cytochrome P450, family 46, subfamily A, polypeptide 1, CYP46A1 (14q32.1), have been associated with risk of AD development (e.g. [360]). It is not known whether 24(S)OHC has specific immunoregulatory effects, but it is certainly plausible to suggest that that activity of CYP46 (also itself a 25-hydroxylase, see earlier) will impact upon the production and effects of 25-hydroxycholesterols, noting that 24(S),25-dihydroxycholesterol resolves to 24(S),25-epoxycholesterol, a further immunosterol with potent biological effects including inhibition of virus proliferation [309].
Second, some infectious agents home selectively to the brain and propagate therein. Examples include encephalitis viruses, poliovirus, rabies virus, and different members of the herpesvirus family. It is possible AD reflects infection and inflammation in association with the cerebrovasculature, whereas ATH is the result of infections propagating in the peripheral vasculature.
Third, the brain immune system differs from that in other tissues. The brain contains its own specialized macrophage-like cells, the microglia, that share with macrophages the properties of self-renewal, mobility, cytokine- and chemokine-responsiveness, antigen presentation, and phagocytosis, although the brain contains both typical macrophages and microglia. Brain microglia could potentially contribute to AD development, but the evidence is inconclusive.
Fourth, microtubule-associated protein Tau is particularly abundant in CNS neurons, and AD is associated with intracellular aggregates (NFT) of Tau. APOE3 binds avidly to Tau whereas APOE4 shows no significant binding. This interaction may modulate AD development [361] but only make a minor contribution to ATH.
Causes, cures, and age-dependence
In both ATH and AD we see central involvement of vascular pathology, clinical association with common predisposing genes/alleles, association with infection, disease enhancement by immune stimulation, the central role of bone marrow-derived cells, principally macrophages, the involvement of Aβ, drug overlap, and cholesterol involvement. These findings suggest that the two diseases share a common pathoetiology. A clear hypothesis is emerging in which chronic infection/inflammation in the vasculature leads, in the short term, to blockade of pathogen proliferation – but in the longer term to 25OHC-driven fatty droplet accumulation and vascular occlusion. Is a battle being played out in our vasculature, where the accumulation of cholesteryl esters, precipitating ATH or AD, is the price of surviving the onslaught of an invading pathogen?
Selective inhibitors of CH25H and/or ACAT may therefore have clinical potential in both diseases; indeed, some widely-available dietary components (with likely activity in these pathways) are under investigation as possible protective agents. Conversely, preventive measures against pathogen infection (e.g., vaccination) are unlikely to be productive, not only because of the diversity of potentially causal agents but also because, for several pathogens, a substantial proportion of the population is already positive from childhood onwards.
In addition, remarkably, in some cases early infection could even be beneficial. Barnton et al.[362] report that latent infection of mice with herpes viruses (mouse gammaherpesvirus 68 or CMV) confers resistance to bacterial infection, with a 100–1000-fold reduction in the replication of Listeria monocytogenes or Yersinia pestis (but without effect on a viral pathogen, West Nile virus). Resistance was not antigen-specific, and required chronic IFN-γ production, suggesting that similar pathways (Figure 8) may be exploited by latent viruses – providing a first indication that bifurcation of immunosterol pathways might potentially foster one pathogen at the expense of another.
The striking age-dependence of disease is not understood. For many pathogens, infections are acquired in early childhood and remain stable, but for others there is a clear age-related increase in seropositivity rates (not reviewed). A recent exciting development is that macrophages show an age-related decline in the expression of the key cholesterol efflux protein, ABCA1 [363], implicated in both ATH and AD (see earlier and Table 1). However, given that age-related phenomena are controlled systemically rather than being cell-intrinsic (e.g., [364]), a more attractive hypothesis might be that the well-documented lifetime declines in tissue levels of steroidal molecules such as 7-dehydrocholesterol (a precursor of vitamin D), allopregnenolone, and dehydroepiandrosterone (DHEA), that interact with the self-same sterol pathways discussed here, contribute to disease processes. Indeed, protective effects of vitamin D and DHEA in both ATH and AD models have been reported; lifetime changes in androgens and estrogens may also play a role.
Ill or just old?
This debate has highlighted many lines of evidence that two major age-related diseases are likely to be precipitated by infection and inflammation, and are not merely a consequence of age per se. However – in the terms of Izaks and Westendorp [1] – infection/inflammation is a ‘component cause’, but not a ‘sufficient cause’. There are undoubtedly good reasons to suspect that a systemic age-related component so far unidentified contributes to both diseases.
This is an exciting area of research, and one hopes that the next years will bring a new understanding of why some individuals develop AD, others ATH, whereas many avoid both conditions. There are implications for drug development, and drugs with moderate efficacy in either ATH or AD warrant testing in the other disorder. Further research will be necessary to unravel the molecular underpinnings that link infection, oxysterols, and the age-relatedness of these two major diseases.
Summary
1. We propose that ATH and AD share a common infectious/inflammatory pathoetiology.
2. Both have similar age-dependence, vascular pathology, genetic underpinnings including the central role of APOE, Aβ involvement, and association with infection. Both diseases are dependent on bone marrow-derived cells, principally macrophages, and show overlapping drug responsiveness.
3. We postulate a causal mechanism. (i) Infection and inflammation, notably in macrophages associated with the vasculature, induce the expression of CH25H. (ii) Acutely, the enzymatic product 25OHC provides protection against pathogen propagation; chronically, macrophage 25OHC activates ACAT, leading to cholesterol esterification, lipid droplet formation, and ultimately vascular occlusion.
4. We infer that, in individuals with ATH or AD, they are neither merely ‘Ill’ nor ‘Old’, but instead both acquired infection/inflammation and endocrine aging are likely to play a joint role in causing these age-related diseases.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
RL was responsible for the concept, SS researched genetic underpinnings and links to infection, and YK investigated links with vasculature occlusion. All authors contributed to writing the manuscript, and all have read and approved the final version.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Richard Lathe, Email: rlathe@pieta-research.org.
Alexandra Sapronova, Email: sashulka.s@gmail.com.
Yuri Kotelevtsev, Email: ykotelev@staffmail.ed.ac.uk.
Acknowledgments
This work was funded, in part, by a Program 220 Megagrant of the Russian Ministry of Science and Education for international scholars (to Y.K.). We thank the European Network for Oxysterol Research (ENOR) for simulating discussions and networking, Judith Miklossy (Switzerland) for providing the graphics in Figure 4, and Peter Ghazal (UK), Luigi Iuliano (Italy), and John and Linda Mullins (UK) for constructive comments on the manuscript. We also thank expert reviewers for insightful contributions and suggestions. Fully comprehensive review of this very wide area of endeavor is scarcely feasible in a single work; we welcome key omissions being brought to our attention and the views of the wider community.
References
- Izaks GJ, Westendorp RG. Ill or just old? Towards a conceptual framework of the relation between ageing and disease. BMC Geriatr. 2003;14:7. doi: 10.1186/1471-2318-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS. et al. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation. 2011;14:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LS, Haq N, Mahmood S, Hoeksema L. Atherosclerotic cardiovascular disease screening in adults: American College Of Preventive Medicine position statement on preventive practice. Am J Prev Med. 2011;14:381–10. doi: 10.1016/j.amepre.2010.11.021. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;14:a006239. doi: 10.1101/cshperspect.a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KY, Wang W, Wu JJ, Liu L, Theodoratou E, Car J, Middleton L, Russ TC, Deary IJ, Campbell H, Wang W, Rudan I. Epidemiology of Alzheimer’s disease and other forms of dementia in China, 1990–2010: a systematic review and analysis. Lancet. 2013;14:2016–2023. doi: 10.1016/S0140-6736(13)60221-4. [DOI] [PubMed] [Google Scholar]
- Lusis AJ. Atherosclerosis. Nature. 2000;14:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;14:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Robbins EM, Betensky RA, Domnitz SB, Purcell SM, Garcia-Alloza M, Greenberg C, Rebeck GW, Hyman BT, Greenberg SM, Frosch MP, Bacskai BJ. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci. 2006;14:365–371. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RA. Classic beta-amyloid deposits cluster around large diameter blood vessels rather than capillaries in sporadic Alzheimer’s disease. Curr Neurovasc Res. 2006;14:289–294. doi: 10.2174/156720206778792948. [DOI] [PubMed] [Google Scholar]
- Armstrong RA. A spatial pattern analysis of beta-amyloid (Abeta) deposition in the temporal lobe in Alzheimer’s disease. Folia Neuropathol. 2010;14:67–74. [PubMed] [Google Scholar]
- Vagnucci AH Jr, Li WW. Alzheimer’s disease and angiogenesis. Lancet. 2003;14:605–608. doi: 10.1016/S0140-6736(03)12521-4. [DOI] [PubMed] [Google Scholar]
- Jagust WJ, Budinger TF, Reed BR. The diagnosis of dementia with single photon emission computed tomography. Arch Neurol. 1987;14:258–262. doi: 10.1001/archneur.1987.00520150014011. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Mueller ST, Walshe TM, English RJ, Holman BL. Cerebral perfusion imaging in Alzheimer’s disease. Use of single photon emission computed tomography and iofetamine hydrochloride I 123. Arch Neurol. 1987;14:165–168. doi: 10.1001/archneur.1987.00520140035014. [DOI] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis. 2002;14:61–68. doi: 10.1006/nbdi.2001.0460. [DOI] [PubMed] [Google Scholar]
- Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD, Sue LI, Beach TG. Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer’s disease. Arterioscler Thromb Vasc Biol. 2003;14:2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44. [DOI] [PubMed] [Google Scholar]
- Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van HF, van Duijn CN, Van BC, Grobbee DE. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet. 1997;14:151–154. doi: 10.1016/S0140-6736(96)09328-2. [DOI] [PubMed] [Google Scholar]
- Stewart R. Cardiovascular factors in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1998;14:143–147. doi: 10.1136/jnnp.65.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS. Cerebrovascular disease and dementia. Br J Radiol. 2007;14:S121–S127. doi: 10.1259/bjr/75681080. [DOI] [PubMed] [Google Scholar]
- Wendell CR, Waldstein SR, Ferrucci L, O’Brien RJ, Strait JB, Zonderman AB. Carotid atherosclerosis and prospective risk of dementia. Stroke. 2012;14:3319–3324. doi: 10.1161/STROKEAHA.112.672527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, Walker DG, Roher AE. Circle of Willis atherosclerosis: association with Alzheimer’s disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 2007;14:13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, Obrien RJ. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann Neurol. 2010;14:231–240. doi: 10.1002/ana.22055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology. 1996;14:1592–1596. doi: 10.1212/WNL.46.6.1592. [DOI] [PubMed] [Google Scholar]
- Grau-Slevin M, Arboix A, Gaffney J, Slevin M. The role of small vessel disease in development of Alzheimer’s disease. Neural Reg Res. 2010;14:310–320. [Google Scholar]
- Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999;14(Suppl 3):S115–S123. doi: 10.1097/00002093-199912003-00017. [DOI] [PubMed] [Google Scholar]
- Roher AE, Debbins JP, Malek-Ahmadi M, Chen K, Pipe JG, Maze S, Belden C, Maarouf CL, Thiyyagura P, Mo H, Hunter JM, Kokjohn TA, Walker DG, Kruchowsky JC, Belohlavek M, Sabbagh MN, Beach TG. Cerebral blood flow in Alzheimer’s disease. Vasc Health Risk Manag. 2012;14:599–611. doi: 10.2147/VHRM.S34874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon D, Quintana M, Mata P, Alonso R, Benavent J, Cruz-Sanchez F, Gich J, Pocovi M, Civeira F, Capurro S, Bachman D, Sambamurti K, Nicholas J, Pappolla MA. Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am J Med. 2010;14:267–274. doi: 10.1016/j.amjmed.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;14:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- Liu H, Liu W, Liao Y, Cheng L, Liu Q, Ren X, Shi L, Tu X, Wang QK, Guo AY. CADgene: a comprehensive database for coronary artery disease genes. Nucleic Acids Res. 2011;14:D991–D996. doi: 10.1093/nar/gkq1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeper NJ, Kullo IJ, Cooke JP. Genetics of peripheral artery disease. Circulation. 2012;14:3220–3228. doi: 10.1161/CIRCULATIONAHA.111.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;14:a006296. doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, Hosford D, Barnes MR, Briley JD, Borrie M, Coletta N, Delisle R, Dhalla D, Ehm MG, Feldman HH, Fornazzari L, Gauthier S, Goodgame N, Guzman D, Hammond S, Hollingworth P, Hsiung GY, Johnson J, Kelly DD, Keren R, Kertesz A, King KS, Lovestone S, Loy-English I, Matthews PM. et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;14:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- Jun G, Vardarajan BN, Buros J, Yu CE, Hawk MV, Dombroski BA, Crane PK, Larson EB, Mayeux R, Haines JL, Lunetta KL, Pericak-Vance MA, Schellenberg GD, Farrer LA. Comprehensive search for Alzheimer disease susceptibility loci in the APOE region. Arch Neurol. 2012;14:1270–1279. doi: 10.1001/archneurol.2012.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelen J, Beekman M, Uh HW, Helmer Q, Kuningas M, Christiansen L, Kremer D, van der Breggen R, Suchiman HE, Lakenberg N, van den Akker EB, Passtoors WM, Tiemeier H, van HD, de Craen AJ, Rivadeneira F, de Geus EJ, Perola M, van der Ouderaa FJ, Gunn DA, Boomsma DI, Uitterlinden AG, Christensen K, van Duijn CM, Heijmans BT, Houwing-Duistermaat JJ, Westendorp RG, Slagboom PE. Genome-wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell. 2011;14:686–698. doi: 10.1111/j.1474-9726.2011.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelberg RP, Ferreira MA, Henders AK, Heath AC, Madden PA, Montgomery GW, Martin NG, Whitfield JB. Genetic variants in LPL, OASL and TOMM40/APOE-C1-C2-C4 genes are associated with multiple cardiovascular-related traits. BMC Med Genet. 2011;14:123. doi: 10.1186/1471-2350-12-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;14:375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Nowotny P, Kauwe JS, Ridge PG, Mayo K, Bertelsen S, Hinrichs A, Fagan AM, Holtzman DM, Morris JC, Goate AM. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Arch Neurol. 2011;14:1013–1019. doi: 10.1001/archneurol.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruszak A, Peplonska B, Safranow K, Chodakowska-Zebrowska M, Barcikowska M, Zekanowski C. TOMM40 rs10524523 polymorphism’s role in late-onset Alzheimer’s disease and in longevity. J Alzheimers Dis. 2012;14:309–322. doi: 10.3233/JAD-2011-110743. [DOI] [PubMed] [Google Scholar]
- Vogel P, Read RW, Rehg JE, Hansen GM. Cryptogenic organizing pneumonia in Tomm5−/− mice. Vet Pathol. 2013;14:65–75. doi: 10.1177/0300985812450723. [DOI] [PubMed] [Google Scholar]
- Bekris LM, Lutz F, Yu CE. Functional analysis of APOE locus genetic variation implicates regional enhancers in the regulation of both TOMM40 and APOE. J Hum Genet. 2012;14:18–25. doi: 10.1038/jhg.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferencz B, Karlsson S, Kalpouzos G. Promising genetic biomarkers of preclinical Alzheimer’s disease: the influence of APOE and TOMM40 on brain integrity. Int J Alzheimers Dis. 2012;14:421452. doi: 10.1155/2012/421452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram JF, Yokoyama S. Apolipoprotein-mediated removal of cellular cholesterol and phospholipids. J Lipid Res. 1996;14:2473–2491. [PubMed] [Google Scholar]
- Grundy SM. Low-density lipoprotein, non-high-density lipoprotein, and apolipoprotein B as targets of lipid-lowering therapy. Circulation. 2002;14:2526–2529. doi: 10.1161/01.CIR.0000038419.53000.D6. [DOI] [PubMed] [Google Scholar]
- Ohashi R, Mu H, Wang X, Yao Q, Chen C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM. 2005;14:845–856. doi: 10.1093/qjmed/hci136. [DOI] [PubMed] [Google Scholar]
- Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;14:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund-Katz S, Phillips MC. High density lipoprotein structure-function and role in reverse cholesterol transport. Subcell Biochem. 2010;14:183–227. doi: 10.1007/978-90-481-8622-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;14:1–16. [PubMed] [Google Scholar]
- Corbo RM, Scacchi R. Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a ‘thrifty’ allele? Ann Hum Genet. 1999;14:301–310. doi: 10.1046/j.1469-1809.1999.6340301.x. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;14:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- Seripa D, D’Onofrio G, Panza F, Cascavilla L, Masullo C, Pilotto A. The genetics of the human APOE polymorphism. Rejuvenation Res. 2011;14:491–500. doi: 10.1089/rej.2011.1169. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;14(Suppl):S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. J Biol Chem. 2002;14:50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- Weisgraber KH. Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112. J Lipid Res. 1990;14:1503–1511. [PubMed] [Google Scholar]
- Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem. 1994;14:22358–22365. [PubMed] [Google Scholar]
- Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J Biol Chem. 1996;14:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- Nguyen D, Dhanasekaran P, Nickel M, Nakatani R, Saito H, Phillips MC, Lund-Katz S. Molecular basis for the differences in lipid and lipoprotein binding properties of human apolipoproteins E3 and E4. Biochemistry. 2010;14:10881–10889. doi: 10.1021/bi1017655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeren J, Grewal T, Laatsch A, Becker N, Rinninger F, Rye KA, Beisiegel U. Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem. 2004;14:55483–55492. doi: 10.1074/jbc.M409324200. [DOI] [PubMed] [Google Scholar]
- Gong JS, Morita SY, Kobayashi M, Handa T, Fujita SC, Yanagisawa K, Michikawa M. Novel action of apolipoprotein E (ApoE): ApoE isoform specifically inhibits lipid-particle-mediated cholesterol release from neurons. Mol Neurodegener. 2007;14:9. doi: 10.1186/1750-1326-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minagawa H, Gong JS, Jung CG, Watanabe A, Lund-Katz S, Phillips MC, Saito H, Michikawa M. Mechanism underlying apolipoprotein E (ApoE) isoform-dependent lipid efflux from neural cells in culture. J Neurosci Res. 2009;14:2498–2508. doi: 10.1002/jnr.22073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam SP, Mok L, Chimini G, Vasa M, Deeley RG. ABCA1 mediates high-affinity uptake of 25-hydroxycholesterol by membrane vesicles and rapid efflux of oxysterol by intact cells. Am J Physiol Cell Physiol. 2006;14:C490–C502. doi: 10.1152/ajpcell.00055.2006. [DOI] [PubMed] [Google Scholar]
- Okoro EU, Zhao Y, Guo Z, Zhou L, Lin X, Yang H. Apolipoprotein E4 is deficient in inducing macrophage ABCA1 expression and stimulating the Sp1 signaling pathway. PLoS One. 2012;14:e44430. doi: 10.1371/journal.pone.0044430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A. 2003;14:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, Dee FJ, Wyss-Coray T, Buttini M, Mucke L, Mahley RW, Huang Y. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;14:2527–2534. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, ran MT, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;14:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;14:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. Apolipoprotein E: from atherosclerosis to Alzheimer’s disease and beyond. Curr Opin Lipidol. 1999;14:207–217. doi: 10.1097/00041433-199906000-00003. [DOI] [PubMed] [Google Scholar]
- Breslow JL. Mouse models of atherosclerosis. Science. 1996;14:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. 1994;14:1885–1893. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell-Braxton L, Veniant M, Latvala RD, Hirano KI, Won WB, Ross J, Dybdal N, Zlot CH, Young SG, Davidson NO. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med. 1998;14:934–938. doi: 10.1038/nm0898-934. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;14:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;14:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer’s disease mutations and a “humanized” Abeta sequence. J Biol Chem. 1996;14:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;14:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, Zlokovic B, Smith JD, Ladu MJ, Rostagno A, Frangione B, Ghiso J. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem J. 2000;14(Pt 2):359–365. [PMC free article] [PubMed] [Google Scholar]
- Ji ZS, Miranda RD, Newhouse YM, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem. 2002;14:21821–21828. doi: 10.1074/jbc.M112109200. [DOI] [PubMed] [Google Scholar]
- Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci U S A. 2005;14:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Meyer GR, de Cleen DM, Cooper S, Knaapen MW, Jans DM, Martinet W, Herman AG, Bult H, Kockx MM. Platelet phagocytosis and processing of beta-amyloid precursor protein as a mechanism of macrophage activation in atherosclerosis. Circ Res. 2002;14:1197–1204. doi: 10.1161/01.RES.0000020017.84398.61. [DOI] [PubMed] [Google Scholar]
- Zaghi J, Goldenson B, Inayathullah M, Lossinsky AS, Masoumi A, Avagyan H, Mahanian M, Bernas M, Weinand M, Rosenthal MJ, Espinosa-Jeffrey A, de VJ, Teplow DB, Fiala M. Alzheimer disease macrophages shuttle amyloid-beta from neurons to vessels, contributing to amyloid angiopathy. Acta Neuropathol. 2009;14:111–124. doi: 10.1007/s00401-008-0481-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jager M, van der Wildt B, Schul E, Bol JG, van Duinen SG, Drukarch B, Wilhelmus MM. Tissue transglutaminase colocalizes with extracellular matrix proteins in cerebral amyloid angiopathy. Neurobiol Aging. 2013;14:1159–1169. doi: 10.1016/j.neurobiolaging.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Lee PH, Bang OY, Hwang EM, Lee JS, Joo US, Mook-Jung I, Huh K. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J Neural Transm. 2005;14:1371–1379. doi: 10.1007/s00702-004-0274-0. [DOI] [PubMed] [Google Scholar]
- Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. beta-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;14:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- Townsend KP, Obregon D, Quadros A, Patel N, Volmar C, Paris D, Mullan M. Proinflammatory and vasoactive effects of Abeta in the cerebrovasculature. Ann N Y Acad Sci. 2002;14:65–76. doi: 10.1111/j.1749-6632.2002.tb04799.x. [DOI] [PubMed] [Google Scholar]
- Kokjohn TA, van Vickle GD, Maarouf CL, Kalback WM, Hunter JM, Daugs ID, Luehrs DC, Lopez J, Brune D, Sue LI, Beach TG, Castano EM, Roher AE. Chemical characterization of pro-inflammatory amyloid-beta peptides in human atherosclerotic lesions and platelets. Biochim Biophys Acta. 1812;14:1508–1514. doi: 10.1016/j.bbadis.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Cao D, Garber DW, Kim H, Fukuchi K. Association of aortic atherosclerosis with cerebral beta-amyloidosis and learning deficits in a mouse model of Alzheimer’s disease. Am J Pathol. 2003;14:2155–2164. doi: 10.1016/S0002-9440(10)63572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibolla G, Norata GD, Meda C, Arnaboldi L, Uboldi P, Piazza F, Ferrarese C, Corsini A, Maggi A, Vegeto E, Catapano AL. Increased atherosclerosis and vascular inflammation in APP transgenic mice with apolipoprotein E deficiency. Atherosclerosis. 2010;14:78–87. doi: 10.1016/j.atherosclerosis.2009.10.040. [DOI] [PubMed] [Google Scholar]
- van de Parre TJ, Guns PJ, Fransen P, Martinet W, Bult H, Herman AG, De Meyer GR. Attenuated atherogenesis in apolipoprotein E-deficient mice lacking amyloid precursor protein. Atherosclerosis. 2011;14:54–58. doi: 10.1016/j.atherosclerosis.2011.01.032. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999;14:15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Taylor JW, Demattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM. Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci. 2003;14:7889–7896. doi: 10.1523/JNEUROSCI.23-21-07889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson LN, Arendash GW, Leighty RE, Costa DA, Low MA, Garcia MF, Cracciolo JR, Rojiani A, Wu X, Bales KR, Paul SM, Potter H. Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol Aging. 2004;14:1153–1167. doi: 10.1016/j.neurobiolaging.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Schaefer EJ, Gregg RE, Ghiselli G, Forte TM, Ordovas JM, Zech LA, Brewer HB Jr. Familial apolipoprotein E deficiency. J Clin Invest. 1986;14:1206–1219. doi: 10.1172/JCI112704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eck M, Bos IS, Kaminski WE, Orso E, Rothe G, Twisk J, Bottcher A, Van Amersfoort ES, Christiansen-Weber TA, Fung-Leung WP, Van Berkel TJ, Schmitz G. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc Natl Acad Sci U S A. 2002;14:6298–6303. doi: 10.1073/pnas.092327399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers B, Zhao Y, Hoekstra M, Hildebrand RB, Ye D, Meurs I, Van Berkel TJ, Van EM. Augmented atherogenesis in LDL receptor deficient mice lacking both macrophage ABCA1 and ApoE. PLoS One. 2011;14:e26095. doi: 10.1371/journal.pone.0026095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;14:43224–43235. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- Yagyu H, Kitamine T, Osuga J, Tozawa R, Chen Z, Kaji Y, Oka T, Perrey S, Tamura Y, Ohashi K, Okazaki H, Yahagi N, Shionoiri F, Iizuka Y, Harada K, Shimano H, Yamashita H, Gotoda T, Yamada N, Ishibashi S. Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J Biol Chem. 2000;14:21324–21330. doi: 10.1074/jbc.M002541200. [DOI] [PubMed] [Google Scholar]
- Accad M, Smith SJ, Newland DL, Sanan DA, King LE Jr, Linton MF, Fazio S, Farese RV Jr. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J Clin Invest. 2000;14:711–719. doi: 10.1172/JCI9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV Jr. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest. 2001;14:163–171. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YR, Dove DE, Major AS, Hasty AH, Boone B, Linton MF, Fazio S. Reduced ABCA1-mediated cholesterol efflux and accelerated atherosclerosis in apolipoprotein E-deficient mice lacking macrophage-derived ACAT1. Circulation. 2005;14:2373–2381. doi: 10.1161/01.CIR.0000164236.19860.13. [DOI] [PubMed] [Google Scholar]
- Bryleva EY, Rogers MA, Chang CC, Buen F, Harris BT, Rousselet E, Seidah NG, Oddo S, LaFerla FM, Spencer TA, Hickey WF, Chang TY. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci U S A. 2010;14:3081–3086. doi: 10.1073/pnas.0913828107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner EL, Tow B, Buhman KK, Wilson M, Sanan DA, Rudel LL, Farese RV Jr. Deficiency of acyl CoA:cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 2003;14:1262–1267. doi: 10.1073/pnas.0336398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TA III, Kelley K, Wilson MD, Sawyer JK, Rudel LL. Dietary fat-induced alterations in atherosclerosis are abolished by ACAT2-deficiency in ApoB100 only, LDLr−/− mice. Arterioscler Thromb Vasc Biol. 2007;14:1396–1402. doi: 10.1161/ATVBAHA.107.142802. [DOI] [PubMed] [Google Scholar]
- Hamada N, Miyata M, Eto H, Ikeda Y, Shirasawa T, Akasaki Y, Miyauchi T, Furusho Y, Nagaki A, Aronow BJ, Tei C. Loss of clusterin limits atherosclerosis in apolipoprotein E-deficient mice via reduced expression of Egr-1 and TNF-alpha. J Atheroscler Thromb. 2011;14:209–216. doi: 10.5551/jat.5819. [DOI] [PubMed] [Google Scholar]
- Demattos RB, O’Dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, Paul SM, Aronow BJ, Holtzman DM. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2002;14:10843–10848. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB, Cirrito JR, Parsadanian M, May PC, O’Dell MA, Taylor JW, Harmony JA, Aronow BJ, Bales KR, Paul SM, Holtzman DM. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;14:193–202. doi: 10.1016/S0896-6273(03)00850-X. [DOI] [PubMed] [Google Scholar]
- Umetani M, Mangelsdorf DJ, Shaul PW. 27-Hydroxycholesterol, the first identified endogenous SERM, promotes atherogenesis in mice. Circulation. 2010;14:A18427. doi: 10.1016/j.tem.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau JL, Rasmuson S, Andrew R, Graham M, Noble J, Olsson T, Fuchs E, Lathe R, Seckl JR. Dehydroepiandrosterone 7-hydroxylase CYP7B: predominant expression in primate hippocampus and reduced expression in Alzheimer’s disease. Neuroscience. 2003;14:307–314. doi: 10.1016/S0306-4522(03)00438-X. [DOI] [PubMed] [Google Scholar]
- Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;14:2752–2761. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;14:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer JD, Demattos RB, McCormick LM, O’Dell MA, Spinner ML, Bales KR, Paul SM, Sullivan PM, Parsadanian M, Bu G, Holtzman DM. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J Biol Chem. 2005;14:25754–25759. doi: 10.1074/jbc.M502143200. [DOI] [PubMed] [Google Scholar]
- Cao D, Fukuchi K, Wan H, Kim H, Li L. Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice. Neurobiol Aging. 2006;14:1632–1643. doi: 10.1016/j.neurobiolaging.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Katsouri L, Georgopoulos S. Lack of LDL receptor enhances amyloid deposition and decreases glial response in an Alzheimer’s disease mouse model. PLoS One. 2011;14:e21880. doi: 10.1371/journal.pone.0021880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiner VL, Cases S, Myers HM, Sande ER, Bellosta S, Schambelan M, Pitas RE, McGuire J, Herz J, Farese RV Jr. Disruption of the acyl-CoA:cholesterol acyltransferase gene in mice: evidence suggesting multiple cholesterol esterification enzymes in mammals. Proc Natl Acad Sci U S A. 1996;14:14041–14046. doi: 10.1073/pnas.93.24.14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme A: cholesterol acyltransferases. Am J Physiol Endocrinol Metab. 2009;14:E1–E9. doi: 10.1152/ajpendo.90926.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black S, Kushner I, Samols D. C-reactive Protein. J Biol Chem. 2004;14:48487–48490. doi: 10.1074/jbc.R400025200. [DOI] [PubMed] [Google Scholar]
- Paffen E, DeMaat MP. C-reactive protein in atherosclerosis: A causal factor? Cardiovasc Res. 2006;14:30–39. doi: 10.1016/j.cardiores.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Reynolds GD, Vance RP. C-reactive protein immunohistochemical localization in normal and atherosclerotic human aortas. Arch Pathol Lab Med. 1987;14:265–269. [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL. Human neurons generate C-reactive protein and amyloid P: upregulation in Alzheimer’s disease. Brain Res. 2000;14:80–89. doi: 10.1016/S0006-8993(00)02970-X. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation of the brain in Alzheimer’s disease: implications for therapy. J Leukoc Biol. 1999;14:409–415. doi: 10.1002/jlb.65.4.409. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I. et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;14:383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;14:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;14:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao JH, Tripathi J, Mishra NK, Cai Y, Tripathi S, Wang XP, Imes S, Fishbein MC, Clinton SK, Libby P, Lusis AJ, Rajavashisth TB. Role of macrophage colony-stimulating factor in atherosclerosis: studies of osteopetrotic mice. Am J Pathol. 1997;14:1687–1699. [PMC free article] [PubMed] [Google Scholar]
- Song L, Leung C, Schindler C. Lymphocytes are important in early atherosclerosis. J Clin Invest. 2001;14:251–259. doi: 10.1172/JCI200111380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida HA, Kristo F, Rateri DL, Lu H, Charnigo R, Cassis LA, Daugherty A. Total lymphocyte deficiency attenuates AngII-induced atherosclerosis in males but not abdominal aortic aneurysms in apoE deficient mice. Atherosclerosis. 2010;14:399–403. doi: 10.1016/j.atherosclerosis.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, Taleb S, Van VE, Esposito B, Vilar J, Sirvent J, Van SJ, Tedgui A, Tedder TF, Mallat Z. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. 2010;14:1579–1587. doi: 10.1084/jem.20100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;14:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Medeiros R, Prediger RD, Passos GF, Pandolfo P, Duarte FS, Franco JL, Dafre AL, Di GG, Figueiredo CP, Takahashi RN, Campos MM, Calixto JB. Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J Neurosci. 2007;14:5394–5404. doi: 10.1523/JNEUROSCI.5047-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Reed QW, Cramer PE, Landreth GE. Deletion of CD14 attenuates Alzheimer’s disease pathology by influencing the brain’s inflammatory milieu. J Neurosci. 2010;14:15369–15373. doi: 10.1523/JNEUROSCI.2637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino PJ, Yang Y, Li X, Hemingway JF, Cherne MK, Khademi SB, Fukui Y, Montine KS, Montine TJ, Keene CD. Ablation of the microglial protein DOCK2 reduces amyloid burden in a mouse model of Alzheimer’s disease. Exp Mol Pathol. 2013;14:366–371. doi: 10.1016/j.yexmp.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eck M, Herijgers N, Yates J, Pearce NJ, Hoogerbrugge PM, Groot PH, Van Berkel TJ. Bone marrow transplantation in apolipoprotein E-deficient mice. Effect of ApoE gene dosage on serum lipid concentrations, (beta)VLDL catabolism, and atherosclerosis. Arterioscler Thromb Vasc Biol. 1997;14:3117–3126. doi: 10.1161/01.ATV.17.11.3117. [DOI] [PubMed] [Google Scholar]
- Herijgers N, Van EM, Groot PH, Hoogerbrugge PM, van Berkel TJ. Low density lipoprotein receptor of macrophages facilitates atherosclerotic lesion formation in C57Bl/6 mice. Arterioscler Thromb Vasc Biol. 2000;14:1961–1967. doi: 10.1161/01.ATV.20.8.1961. [DOI] [PubMed] [Google Scholar]
- Fazio S, Babaev VR, Burleigh ME, Major AS, Hasty AH, Linton MF. Physiological expression of macrophage apoE in the artery wall reduces atherosclerosis in severely hyperlipidemic mice. J Lipid Res. 2002;14:1602–1609. doi: 10.1194/jlr.M200108-JLR200. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Pennings M, Hildebrand RB, Ye D, Calpe-Berdiel L, Out R, Kjerrulf M, Hurt-Camejo E, Groen AK, Hoekstra M, Jessup W, Chimini G, Van Berkel TJ, Van EM. Enhanced foam cell formation, atherosclerotic lesion development, and inflammation by combined deletion of ABCA1 and SR-BI in Bone marrow-derived cells in LDL receptor knockout mice on western-type diet. Circ Res. 2010;14:e20–e31. doi: 10.1161/CIRCRESAHA.110.226282. [DOI] [PubMed] [Google Scholar]
- Keene CD, Chang RC, Lopez-Yglesias AH, Shalloway BR, Sokal I, Li X, Reed PJ, Keene LM, Montine KS, Breyer RM, Rockhill JK, Montine TJ. Suppressed accumulation of cerebral amyloid beta peptides in aged transgenic Alzheimer’s disease mice by transplantation with wild-type or prostaglandin E2 receptor subtype 2-null bone marrow. Am J Pathol. 2010;14:346–354. doi: 10.2353/ajpath.2010.090840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Liu Y, Liu S, Walter S, Grimm MO, Kiliaan AJ, Penke B, Hartmann T, Rube CE, Menger MD, Fassbender K. Myeloid differentiation factor 88-deficient bone marrow cells improve Alzheimer’s disease-related symptoms and pathology. Brain. 2011;14:278–292. doi: 10.1093/brain/awq325. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Obregon D, Hou H, Giunta B, Ehrhart J, Fernandez F, Mori T, Nikolic W, Zhao Y, Morgan D, Town T, Tan J. Mutant presenilin-1 deregulated peripheral immunity exacerbates Alzheimer-like pathology. J Cell Mol Med. 2011;14:327–338. doi: 10.1111/j.1582-4934.2009.00962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Cudaback E, Jorstad NL, Hemingway JF, Hagan CE, Melief EJ, Li X, Yoo T, Khademi SB, Montine KS, Montine TJ, Keene CD. APOE3, but not APOE4, bone marrow transplantation mitigates behavioral and pathological changes in a mouse model of Alzheimer disease. Am J Pathol. 2013;14:905–917. doi: 10.1016/j.ajpath.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;14:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood–brain barrier. Eur J Clin Invest. 2002;14:360–371. doi: 10.1046/j.1365-2362.2002.00994.x. [DOI] [PubMed] [Google Scholar]
- Tennert C, Teupser D, Mueller MA, Wilfert W, Renner-Muller I, Stein O, Stein Y, Sippel AE, Wolf E, Thiery J. Effect of macrophage ApoE on atherosclerosis in LDL-receptor deficient mice. Biochem Biophys Res Commun. 2007;14:574–579. doi: 10.1016/j.bbrc.2007.07.067. [DOI] [PubMed] [Google Scholar]
- Gaudreault N, Kumar N, Olivas VR, Eberle D, Rapp JH, Raffai RL. Macrophage-specific apoE gene repair reduces diet-induced hyperlipidemia and atherosclerosis in hypomorphic Apoe mice. PLoS One. 2012;14:e35816. doi: 10.1371/journal.pone.0035816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;14:1647–1653. doi: 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ Res. 2007;14:670–677. doi: 10.1161/01.RES.0000260204.40510.aa. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging. 2001;14:49–61. doi: 10.1016/S0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Imaki H, Wang KC, Wegiel J, Rubenstein R. Cells of monocyte/microglial lineage are involved in both microvessel amyloidosis and fibrillar plaque formation in APPsw tg mice. Brain Res. 2004;14:19–29. doi: 10.1016/j.brainres.2004.06.058. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;14:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;14:1361–1363. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;14:1261–1266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T. Macrophages in Alzheimer’s disease: the blood-borne identity. J Neural Transm. 2010;14:961–970. doi: 10.1007/s00702-010-0422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai-Zadeh K, Gate D, Gowing G, Town T. How to get from here to there: macrophage recruitment in Alzheimer’s disease. Curr Alzheimer Res. 2011;14:156–163. doi: 10.2174/156720511795256017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall BJ. In: Helicobacter pylori: Physiology and Genetics. Mobley HLT, Mendz GL, Hazell SL, editor. Washington DC: ADM Press; 2001. One hundred years of discovery and rediscovery of Helicobacter pylori and its association with peptic ulcer disease. Chapter 3. [PubMed] [Google Scholar]
- Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;14:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;14:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;14:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker HF, Ridley RM, Duchen LW, Crow TJ, Bruton CJ. Induction of beta (A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol Neurobiol. 1994;14:25–39. doi: 10.1007/BF02778005. [DOI] [PubMed] [Google Scholar]
- Ridley RM, Baker HF, Windle CP, Cummings RM. Very long term studies of the seeding of beta-amyloidosis in primates. J Neural Transm. 2006;14:1243–1251. doi: 10.1007/s00702-005-0385-2. [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;14:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci. 2000;14:3606–3611. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;14:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, Roth K, Aguzzi A, Staufenbiel M, Walker LC, Jucker M. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A. 2009;14:12926–12931. doi: 10.1073/pnas.0903200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;14:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J Neurosci. 2011;14:14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Duran-Aniotz C, Castilla J, Estrada LD, Soto C. De novo induction of amyloid-beta deposition in vivo. Mol Psychiatry. 2012;14:1347–1353. doi: 10.1038/mp.2011.120. [DOI] [PubMed] [Google Scholar]
- Rosen RF, Fritz JJ, Dooyema J, Cintron AF, Hamaguchi T, Lah JJ, LeVine H III, Jucker M, Walker LC. Exogenous seeding of cerebral beta-amyloid deposition in betaAPP-transgenic rats. J Neurochem. 2012;14:660–666. doi: 10.1111/j.1471-4159.2011.07551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am J Pathol. 2007;14:2012–2020. doi: 10.2353/ajpath.2007.070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, LeVine H III, Mattson MP, Jucker M. Inducible proteopathies. Trends Neurosci. 2006;14:438–443. doi: 10.1016/j.tins.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;14:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Fischer O. Miliare Nekrosen mit drusigen Wucherungen der Neurofibrillen, eine regelmässige Veränderung der Hirnrinde bei seniler Demenz. Monatschr f Psychiat Neurol. 1907;14:372. [Google Scholar]
- Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med. 2011;14:e30. doi: 10.1017/S1462399411002006. [DOI] [PubMed] [Google Scholar]
- Shima K, Kuhlenbaumer G, Rupp J. Chlamydia pneumoniae infection and Alzheimer’s disease: a connection to remember? Med Microbiol Immunol. 2010;14:283–289. doi: 10.1007/s00430-010-0162-1. [DOI] [PubMed] [Google Scholar]
- De CG, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R, Garaci E, Grassi C, Palamara AT. Infectious agents and neurodegeneration. Mol Neurobiol. 2012;14:614–638. doi: 10.1007/s12035-012-8320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo K, van RR, Verhoeff NP. Alzheimer’s disease and infection: do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement. 2009;14:348–360. doi: 10.1016/j.jalz.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Mann DMA, Tates PP, Davides JS, Hawkes J. Viruses, Parkinsonism and Alzheimer’s Disease. J Neurol Neurosurg Psychiat. 1981;14:651. doi: 10.1136/jnnp.44.7.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri MM. Viruses and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1982;14:759–760. doi: 10.1136/jnnp.45.8.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori I, Kimura Y, Naiki H, Matsubara R, Takeuchi T, Yokochi T, Nishiyama Y. Reactivation of HSV-1 in the brain of patients with familial Alzheimer’s disease. J Med Virol. 2004;14:605–611. doi: 10.1002/jmv.20133. [DOI] [PubMed] [Google Scholar]
- Walker DG, O’Kusky JR, McGeer PL. In situ hybridization analysis for herpes simplex virus nucleic acids in Alzheimer disease. Alzheimer Dis Assoc Disord. 1989;14:123–131. doi: 10.1097/00002093-198903030-00001. [DOI] [PubMed] [Google Scholar]
- Letenneur L, Peres K, Fleury H, Garrigue I, Barberger-Gateau P, Helmer C, Orgogozo JM, Gauthier S, Dartigues JF. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: a population-based cohort study. PLoS One. 2008;14:e3637. doi: 10.1371/journal.pone.0003637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balin BJ, Gerard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, Whittum-Hudson JA, Hudson AP. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol. 1998;14:23–42. doi: 10.1007/s004300050071. [DOI] [PubMed] [Google Scholar]
- Gerard HC, Dreses-Werringloer U, Wildt KS, Deka S, Oszust C, Balin BJ, Frey WH, Bordayo EZ, Whittum-Hudson JA, Hudson AP. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol Med Microbiol. 2006;14:355–366. doi: 10.1111/j.1574-695X.2006.00154.x. [DOI] [PubMed] [Google Scholar]
- Hammond CJ, Hallock LR, Howanski RJ, Appelt DM, Little CS, Balin BJ. Immunohistological detection of Chlamydia pneumoniae in the Alzheimer’s disease brain. BMC Neurosci. 2010;14:121. doi: 10.1186/1471-2202-11-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere GR, Riviere KH, Smith KS. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol. 2002;14:113–118. doi: 10.1046/j.0902-0055.2001.00100.x. [DOI] [PubMed] [Google Scholar]
- Miklossy J. Alzheimer’s disease–a spirochetosis? Neuroreport. 1993;14:841–848. doi: 10.1097/00001756-199307000-00002. [DOI] [PubMed] [Google Scholar]
- Kountouras J, Tsolaki M, Gavalas E, Boziki M, Zavos C, Karatzoglou P, Chatzopoulos D, Venizelos I. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology. 2006;14:938–940. doi: 10.1212/01.wnl.0000203644.68059.5f. [DOI] [PubMed] [Google Scholar]
- Roubaud-Baudron C, Krolak-Salmon P, Quadrio I, Megraud F, Salles N. Impact of chronic Helicobacter pylori infection on Alzheimer’s disease: preliminary results. Neurobiol Aging. 2012;14:1009. doi: 10.1016/j.neurobiolaging.2011.10.021. [DOI] [PubMed] [Google Scholar]
- Roubaud-Baudron C, Letenneur L, Langlais A, Buissonniere A, Megraud F, Dartigues JF, Salles N. Does Helicobacter pylori infection increase incidence of dementia? The Personnes Agées QUID Study. J Am Geriatr Soc. 2013;14:74–78. doi: 10.1111/jgs.12065. [DOI] [PubMed] [Google Scholar]
- Shiota S, Murakami K, Yoshiiwa A, Yamamoto K, Ohno S, Kuroda A, Mizukami K, Hanada K, Okimoto T, Kodama M, Abe K, Yamaoka Y, Fujioka T. The relationship between Helicobacter pylori infection and Alzheimer’s disease in Japan. J Neurol. 2011;14:1460–1463. doi: 10.1007/s00415-011-5957-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kountouras J, Boziki M, Gavalas E, Zavos C, Deretzi G, Chatzigeorgiou S, Katsinelos P, Grigoriadis N, Giartza-Taxidou E, Venizelos I. Five-year survival after Helicobacter pylori eradication in Alzheimer disease patients. Cogn Behav Neurol. 2010;14:199–204. doi: 10.1097/WNN.0b013e3181df3034. [DOI] [PubMed] [Google Scholar]
- Benditt EP, Barrett T, McDougall JK. Viruses in the etiology of atherosclerosis. Proc Natl Acad Sci U S A. 1983;14:6386–6389. doi: 10.1073/pnas.80.20.6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix MG, Dormans PH, Kitslaar P, Bosman F, Bruggeman CA. The presence of cytomegalovirus nucleic acids in arterial walls of atherosclerotic and nonatherosclerotic patients. Am J Pathol. 1989;14:1151–1157. [PMC free article] [PubMed] [Google Scholar]
- Melnick JL, Petrie BL, Dreesman GR, Burek J, McCollum CH, DeBakey ME. Cytomegalovirus antigen within human arterial smooth muscle cells. Lancet. 1983;14:644–647. doi: 10.1016/s0140-6736(83)92529-1. [DOI] [PubMed] [Google Scholar]
- Petrie BL, Melnick JL, Adam E, Burek J, McCollum CH, DeBakey ME. Nucleic acid sequences of cytomegalovirus in cells cultured from human arterial tissue. J Infect Dis. 1987;14:158–159. doi: 10.1093/infdis/155.1.158. [DOI] [PubMed] [Google Scholar]
- Speir E, Modali R, Huang ES, Leon MB, Shawl F, Finkel T, Epstein SE. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science. 1994;14:391–394. doi: 10.1126/science.8023160. [DOI] [PubMed] [Google Scholar]
- Zhou YF, Leon MB, Waclawiw MA, Popma JJ, Yu ZX, Finkel T, Epstein SE. Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N Engl J Med. 1996;14:624–630. doi: 10.1056/NEJM199608293350903. [DOI] [PubMed] [Google Scholar]
- Grahame-Clarke C, Chan NN, Andrew D, Ridgway GL, Betteridge DJ, Emery V, Colhoun HM, Vallance P. Human cytomegalovirus seropositivity is associated with impaired vascular function. Circulation. 2003;14:678–683. doi: 10.1161/01.CIR.0000084505.54603.C7. [DOI] [PubMed] [Google Scholar]
- Nieto FJ, Adam E, Sorlie P, Farzadegan H, Melnick JL, Comstock GW, Szklo M. Cohort study of cytomegalovirus infection as a risk factor for carotid intimal-medial thickening, a measure of subclinical atherosclerosis. Circulation. 1996;14:922–927. doi: 10.1161/01.CIR.94.5.922. [DOI] [PubMed] [Google Scholar]
- Shor A, Kuo CC, Patton DL. Detection of Chlamydia pneumoniae in coronary arterial fatty streaks and atheromatous plaques. S Afr Med J. 1992;14:158–161. [PubMed] [Google Scholar]
- Kuo CC, Gown AM, Benditt EP, Grayston JT. Detection of Chlamydia pneumoniae in aortic lesions of atherosclerosis by immunocytochemical stain. Arterioscler Thromb. 1993;14:1501–1504. doi: 10.1161/01.ATV.13.10.1501. [DOI] [PubMed] [Google Scholar]
- Gibbs RG, Carey N, Davies AH. Chlamydia pneumoniae and vascular disease. Br J Surg. 1998;14:1191–1197. doi: 10.1046/j.1365-2168.1998.00861.x. [DOI] [PubMed] [Google Scholar]
- Kaplan M, Yavuz SS, Cinar B, Koksal V, Kut MS, Yapici F, Gercekoglu H, Demirtas MM. Detection of Chlamydia pneumoniae and Helicobacter pylori in atherosclerotic plaques of carotid artery by polymerase chain reaction. Int J Infect Dis. 2006;14:116–123. doi: 10.1016/j.ijid.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Deniset JF, Cheung PK, Dibrov E, Lee K, Steigerwald S, Pierce GN. Chlamydophila pneumoniae infection leads to smooth muscle cell proliferation and thickening in the coronary artery without contributions from a host immune response. Am J Pathol. 2010;14:1028–1037. doi: 10.2353/ajpath.2010.090645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalayoglu MV, Libby P, Byrne GI. Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. JAMA. 2002;14:2724–2731. doi: 10.1001/jama.288.21.2724. [DOI] [PubMed] [Google Scholar]
- Watson C, Alp NJ. Role of Chlamydia pneumoniae in atherosclerosis. Clin Sci (Lond) 2008;14:509–531. doi: 10.1042/CS20070298. [DOI] [PubMed] [Google Scholar]
- Ameriso SF, Fridman EA, Leiguarda RC, Sevlever GE. Detection of Helicobacter pylori in human carotid atherosclerotic plaques. Stroke. 2001;14:385–391. doi: 10.1161/01.STR.32.2.385. [DOI] [PubMed] [Google Scholar]
- Blasi F, Denti F, Erba M, Cosentini R, Raccanelli R, Rinaldi A, Fagetti L, Esposito G, Ruberti U, Allegra L. Detection of Chlamydia pneumoniae but not Helicobacter pylori in atherosclerotic plaques of aortic aneurysms. J Clin Microbiol. 1996;14:2766–2769. doi: 10.1128/jcm.34.11.2766-2769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K, Ishihara K, Nakagawa T, Hirayama A, Inayama Y, Okuda K. Detection of Treponema denticola in atherosclerotic lesions. J Clin Microbiol. 2001;14:1114–1117. doi: 10.1128/JCM.39.3.1114-1117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southerland JH, Taylor GW, Moss K, Beck JD, Offenbacher S. Commonality in chronic inflammatory diseases: periodontitis, diabetes, and coronary artery disease. Periodontol 2000. 2006;14:130–143. doi: 10.1111/j.1600-0757.2005.00138.x. [DOI] [PubMed] [Google Scholar]
- Mattila KJ, Nieminen MS, Valtonen VV, Rasi VP, Kesaniemi YA, Syrjala SL, Jungell PS, Isoluoma M, Hietaniemi K, Jokinen MJ. Association between dental health and acute myocardial infarction. BMJ. 1989;14:779–781. doi: 10.1136/bmj.298.6676.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesche WJ, Schork A, Terpenning MS, Chen YM, Kerr C, Dominguez BL. The relationship between dental disease and cerebral vascular accident in elderly United States veterans. Ann Periodontol. 1998;14:161–174. doi: 10.1902/annals.1998.3.1.161. [DOI] [PubMed] [Google Scholar]
- Kurihara N, Inoue Y, Iwai T, Umeda M, Huang Y, Ishikawa I. Detection and localization of periodontopathic bacteria in abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. 2004;14:553–558. doi: 10.1016/j.ejvs.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Zaremba M, Gorska R, Suwalski P, Kowalski J. Evaluation of the incidence of periodontitis-associated bacteria in the atherosclerotic plaque of coronary blood vessels. J Periodontol. 2007;14:322–327. doi: 10.1902/jop.2006.060081. [DOI] [PubMed] [Google Scholar]
- Wada K, Kamisaki Y. Roles of oral bacteria in cardiovascular diseases–from molecular mechanisms to clinical cases: Involvement of Porphyromonas gingivalis in the development of human aortic aneurysm. J Pharmacol Sci. 2010;14:115–119. doi: 10.1254/jphs.09R22FM. [DOI] [PubMed] [Google Scholar]
- Chatzidimitriou D, Kirmizis D, Gavriilaki E, Chatzidimitriou M, Malisiovas N. Atherosclerosis and infection: is the jury still not in? Future Microbiol. 2012;14:1217–1230. doi: 10.2217/fmb.12.87. [DOI] [PubMed] [Google Scholar]
- Desvarieux M, Demmer RT, Jacobs DR Jr, Rundek T, Boden-Albala B, Sacco RL, Papapanou PN. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST) J Hypertens. 2010;14:1413–1421. doi: 10.1097/HJH.0b013e328338cd36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawhorter SD, Lauer MA. Is atherosclerosis an infectious disease? Cleve Clin J Med. 2001;14:449–458. doi: 10.3949/ccjm.68.5.449. [DOI] [PubMed] [Google Scholar]
- Andraws R, Berger JS, Brown DL. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: a meta-analysis of randomized controlled trials. JAMA. 2005;14:2641–2647. doi: 10.1001/jama.293.21.2641. [DOI] [PubMed] [Google Scholar]
- Minick CR, Fabricant CG, Fabricant J, Litrenta MM. Atheroarteriosclerosis induced by infection with a herpesvirus. Am J Pathol. 1979;14:673–706. [PMC free article] [PubMed] [Google Scholar]
- Hajjar DP. Warner-Lambert/Parke-Davis Award Lecture. Viral pathogenesis of atherosclerosis. Impact of molecular mimicry and viral genes. Am J Pathol. 1991;14:1195–1211. [PMC free article] [PubMed] [Google Scholar]
- Little CS, Hammond CJ, MacIntyre A, Balin BJ, Appelt DM. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging. 2004;14:419–429. doi: 10.1016/S0197-4580(03)00127-1. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007;14:95–100. doi: 10.1016/j.neulet.2007.09.077. [DOI] [PubMed] [Google Scholar]
- Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, LaFerla FM. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;14:2811–2822. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS. 2005;14:407–411. doi: 10.1097/01.aids.0000161770.06158.5c. [DOI] [PubMed] [Google Scholar]
- Jung BK, Pyo KH, Shin KY, Hwang YS, Lim H, Lee SJ, Moon JH, Lee SH, Suh YH, Chai JY, Shin EH. Toxoplasma gondii infection in the brain inhibits neuronal degeneration and learning and memory impairments in a murine model of Alzheimer’s disease. PLoS One. 2012;14:e33312. doi: 10.1371/journal.pone.0033312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed TC, Kuo C, Grayston JT, Campbell LA. Murine models of Chlamydia pneumoniae infection and atherosclerosis. J Infect Dis. 1997;14:883–890. doi: 10.1086/513986. [DOI] [PubMed] [Google Scholar]
- Liu L, Hu H, Ji H, Murdin AD, Pierce GN, Zhong G. Chlamydia pneumoniae infection significantly exacerbates aortic atherosclerosis in an LDLR−/− mouse model within six months. Mol Cell Biochem. 2000;14:123–128. doi: 10.1023/A:1026531506202. [DOI] [PubMed] [Google Scholar]
- Hauer AD, de VP, Peterse N, ten CH, van Berkel TJ, Stassen FR, Kuiper J. Delivery of Chlamydia pneumoniae to the vessel wall aggravates atherosclerosis in LDLr−/− mice. Cardiovasc Res. 2006;14:280–288. doi: 10.1016/j.cardiores.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Aalto-Setala K, Laitinen K, Erkkila L, Leinonen M, Jauhiainen M, Ehnholm C, Tamminen M, Puolakkainen M, Penttila I, Saikku P. Chlamydia pneumoniae does not increase atherosclerosis in the aortic root of apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2001;14:578–584. doi: 10.1161/01.ATV.21.4.578. [DOI] [PubMed] [Google Scholar]
- Caligiuri G, Rottenberg M, Nicoletti A, Wigzell H, Hansson GK. Chlamydia pneumoniae infection does not induce or modify atherosclerosis in mice. Circulation. 2001;14:2834–2838. doi: 10.1161/01.CIR.103.23.2834. [DOI] [PubMed] [Google Scholar]
- Cao F, Castrillo A, Tontonoz P, Re F, Byrne GI. Chlamydia pneumoniae–induced macrophage foam cell formation is mediated by Toll-like receptor 2. Infect Immun. 2007;14:753–759. doi: 10.1128/IAI.01386-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes BK, Mahony JB. Chlamydia pneumoniae infection of human endothelial cells induces proliferation of smooth muscle cells via an endothelial cell-derived soluble factor(s) Infect Immun. 1999;14:2909–2915. doi: 10.1128/iai.67.6.2909-2915.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Yumoto H, Takahashi Y, Davey M, Gibson FC III, Genco CA. Pathogen-accelerated atherosclerosis occurs early after exposure and can be prevented via immunization. Infect Immun. 2006;14:1376–1380. doi: 10.1128/IAI.74.2.1376-1380.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi C, Papadopoulos G, Gudino CV, Weinberg EO, Barth KR, Madrigal AG, Chen Y, Ning H, LaValley M, Gibson FC III, Hamilton JA, Genco CA. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J Immunol. 2012;14:3681–3688. doi: 10.4049/jimmunol.1201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayada K, Yokota K, Hirai K, Fujimoto K, Kobayashi K, Ogawa H, Hatanaka K, Hirohata S, Yoshino T, Shoenfeld Y, Matsuura E, Oguma K. Regulation of cellular immunity prevents Helicobacter pylori-induced atherosclerosis. Lupus. 2009;14:1154–1168. doi: 10.1177/0961203309106600. [DOI] [PubMed] [Google Scholar]
- Kesavalu L, Lucas AR, Verma RK, Liu L, Dai E, Sampson E, Progulske-Fox A. Increased atherogenesis during Streptococcus mutans infection in ApoE-null mice. J Dent Res. 2012;14:255–260. doi: 10.1177/0022034511435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alber DG, Powell KL, Vallance P, Goodwin DA, Grahame-Clarke C. Herpesvirus infection accelerates atherosclerosis in the apolipoprotein E-deficient mouse. Circulation. 2000;14:779–785. doi: 10.1161/01.CIR.102.7.779. [DOI] [PubMed] [Google Scholar]
- Naghavi M, Wyde P, Litovsky S, Madjid M, Akhtar A, Naguib S, Siadaty MS, Sanati S, Casscells W. Influenza infection exerts prominent inflammatory and thrombotic effects on the atherosclerotic plaques of apolipoprotein E-deficient mice. Circulation. 2003;14:762–768. doi: 10.1161/01.CIR.0000048190.68071.2B. [DOI] [PubMed] [Google Scholar]
- Hsich E, Zhou YF, Paigen B, Johnson TM, Burnett MS, Epstein SE. Cytomegalovirus infection increases development of atherosclerosis in Apolipoprotein-E knockout mice. Atherosclerosis. 2001;14:23–28. doi: 10.1016/S0021-9150(00)00608-0. [DOI] [PubMed] [Google Scholar]
- Vliegen I, Duijvestijn A, Grauls G, Herngreen S, Bruggeman C, Stassen F. Cytomegalovirus infection aggravates atherogenesis in apoE knockout mice by both local and systemic immune activation. Microbes Infect. 2004;14:17–24. doi: 10.1016/j.micinf.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Doenhoff MJ, Stanley RG, Griffiths K, Jackson CL. An anti-atherogenic effect of Schistosoma mansoni infections in mice associated with a parasite-induced lowering of blood total cholesterol. Parasitology. 2002;14:415–421. doi: 10.1017/s0031182002002275. [DOI] [PubMed] [Google Scholar]
- Wright SD, Burton C, Hernandez M, Hassing H, Montenegro J, Mundt S, Patel S, Card DJ, Hermanowski-Vosatka A, Bergstrom JD, Sparrow CP, Detmers PA, Chao YS. Infectious agents are not necessary for murine atherogenesis. J Exp Med. 2000;14:1437–1442. doi: 10.1084/jem.191.8.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostos MA, Recalde D, Zakin MM, Scott-Algara D. Implication of natural killer T cells in atherosclerosis development during a LPS-induced chronic inflammation. FEBS Lett. 2002;14:23–29. doi: 10.1016/S0014-5793(02)02692-3. [DOI] [PubMed] [Google Scholar]
- Vliegen I, Herngreen SB, Grauls GE, Bruggeman CA, Stassen FR. Mouse cytomegalovirus antigenic immune stimulation is sufficient to aggravate atherosclerosis in hypercholesterolemic mice. Atherosclerosis. 2005;14:39–44. doi: 10.1016/j.atherosclerosis.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, Manalastas A, Hilfiker M, Pfister S, Schwerdel C, Riether C, Meyer U, Knuesel I. Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation. 2012;14:151. doi: 10.1186/1742-2094-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;14:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein SE, Zhu J, Najafi AH, Burnett MS. Insights into the role of infection in atherogenesis and in plaque rupture. Circulation. 2009;14:3133–3141. doi: 10.1161/CIRCULATIONAHA.109.849455. [DOI] [PubMed] [Google Scholar]
- McGuinness B, Passmore P. Can statins prevent or help treat Alzheimer’s disease? J Alzheimers Dis. 2010;14:925–933. doi: 10.3233/JAD-2010-091570. [DOI] [PubMed] [Google Scholar]
- Ballantyne CM, Raichlen JS, Nicholls SJ, Erbel R, Tardif JC, Brener SJ, Cain VA, Nissen SE. Effect of rosuvastatin therapy on coronary artery stenoses assessed by quantitative coronary angiography: a study to evaluate the effect of rosuvastatin on intravascular ultrasound-derived coronary atheroma burden. Circulation. 2008;14:2458–2466. doi: 10.1161/CIRCULATIONAHA.108.773747. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Nicholls SJ, Sipahi I, Libby P, Raichlen JS, Ballantyne CM, Davignon J, Erbel R, Fruchart JC, Tardif JC, Schoenhagen P, Crowe T, Cain V, Wolski K, Goormastic M, Tuzcu EM. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;14:1556–1565. doi: 10.1001/jama.295.13.jpc60002. [DOI] [PubMed] [Google Scholar]
- Sipahi I, Nicholls SJ, Tuzcu EM, Nissen SE. Coronary atherosclerosis can regress with very intensive statin therapy. Cleve Clin J Med. 2006;14:937–944. doi: 10.3949/ccjm.73.10.937. [DOI] [PubMed] [Google Scholar]
- Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;14:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;14:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem. 2007;14:1095–1104. doi: 10.1111/j.1471-4159.2007.04613.x. [DOI] [PubMed] [Google Scholar]
- Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang F, Hudspeth B, Hu S, Faull KF, Teter B, Cole GM, Frautschy SA. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther. 2008;14:196–208. doi: 10.1124/jpet.108.137455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed T, Gilani AH, Hosseinmardi N, Semnanian S, Enam SA, Fathollahi Y. Curcuminoids rescue long-term potentiation impaired by amyloid peptide in rat hippocampal slices. Synapse. 2011;14:572–582. doi: 10.1002/syn.20876. [DOI] [PubMed] [Google Scholar]
- Baum L, Lam CW, Cheung SK, Kwok T, Lui V, Tsoh J, Lam L, Leung V, Hui E, Ng C, Woo J, Chiu HF, Goggins WB, Zee BC, Cheng KF, Fong CY, Wong A, Mok H, Chow MS, Ho PC, Ip SP, Ho CS, Yu XW, Lai CY, Chan MH, Szeto S, Chan IH, Mok V. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol. 2008;14:110–113. doi: 10.1097/jcp.0b013e318160862c. [DOI] [PubMed] [Google Scholar]
- Olszanecki R, Jawien J, Gajda M, Mateuszuk L, Gebska A, Korabiowska M, Chlopicki S, Korbut R. Effect of curcumin on atherosclerosis in apoE/LDLR-double knockout mice. J Physiol Pharmacol. 2005;14:627–635. [PubMed] [Google Scholar]
- Shin SK, Ha TY, McGregor RA, Choi MS. Long-term curcumin administration protects against atherosclerosis via hepatic regulation of lipoprotein cholesterol metabolism. Mol Nutr Food Res. 2011;14:1829–1840. doi: 10.1002/mnfr.201100440. [DOI] [PubMed] [Google Scholar]
- Coban D, Milenkovic D, Chanet A, Khallou-Laschet J, Sabbe L, Palagani A, Vanden Berghe W, Mazur A, Morand C. Dietary curcumin inhibits atherosclerosis by affecting the expression of genes involved in leukocyte adhesion and transendothelial migration. Mol Nutr Food Res. 2012;14:1270–1281. doi: 10.1002/mnfr.201100818. [DOI] [PubMed] [Google Scholar]
- Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc Natl Acad Sci U S A. 2009;14:16764–16769. doi: 10.1073/pnas.0909142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Zhao H, Davies P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J Biol Chem. 2005;14:37377–37382. doi: 10.1074/jbc.M508246200. [DOI] [PubMed] [Google Scholar]
- Granzotto A, Zatta P. Resveratrol acts not through anti-aggregative pathways but mainly via its scavenging properties against Abeta and Abeta-metal complexes toxicity. PLoS One. 2011;14:e21565. doi: 10.1371/journal.pone.0021565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capiralla H, Vingtdeux V, Zhao H, Sankowski R, Al-Abed Y, Davies P, Marambaud P. Resveratrol mitigates lipopolysaccharide- and Abeta-mediated microglial inflammation by inhibiting the TLR4/NF-kappaB/STAT signaling cascade. J Neurochem. 2012;14:461–472. doi: 10.1111/j.1471-4159.2011.07594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TC, Lu KT, Wo YY, Wu YJ, Yang YL. Resveratrol protects rats from Abeta-induced neurotoxicity by the reduction of iNOS expression and lipid peroxidation. PLoS One. 2011;14:e29102. doi: 10.1371/journal.pone.0029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Pinto JT, Xu H, Chen HL, Beal MF, Gibson GE. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem Int. 2009;14:111–118. doi: 10.1016/j.neuint.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ho L, Zhao Z, Seror I, Humala N, Dickstein DL, Thiyagarajan M, Percival SS, Talcott ST, Pasinetti GM. Moderate consumption of Cabernet Sauvignon attenuates Abeta neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2006;14:2313–2320. doi: 10.1096/fj.06-6281com. [DOI] [PubMed] [Google Scholar]
- Patel KR, Scott E, Brown VA, Gescher AJ, Steward WP, Brown K. Clinical trials of resveratrol. Ann N Y Acad Sci. 2011;14:161–169. doi: 10.1111/j.1749-6632.2010.05853.x. [DOI] [PubMed] [Google Scholar]
- Fukao H, Ijiri Y, Miura M, Hashimoto M, Yamashita T, Fukunaga C, Oiwa K, Kawai Y, Suwa M, Yamamoto J. Effect of trans-resveratrol on the thrombogenicity and atherogenicity in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Blood Coagul Fibrinolysis. 2004;14:441–446. doi: 10.1097/00001721-200408000-00001. [DOI] [PubMed] [Google Scholar]
- Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;14:2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- Do GM, Kwon EY, Kim HJ, Jeon SM, Ha TY, Park T, Choi MS. Long-term effects of resveratrol supplementation on suppression of atherogenic lesion formation and cholesterol synthesis in apo E-deficient mice. Biochem Biophys Res Commun. 2008;14:55–59. doi: 10.1016/j.bbrc.2008.06.113. [DOI] [PubMed] [Google Scholar]
- Verschuren L, Wielinga PY, van DW, Tijani S, Toet K, van OB, Kooistra T, Kleemann R. A dietary mixture containing fish oil, resveratrol, lycopene, catechins, and vitamins E and C reduces atherosclerosis in transgenic mice. J Nutr. 2011;14:863–869. doi: 10.3945/jn.110.133751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zou J, Cao K, Hsieh TC, Huang Y, Wu JM. Dealcoholized red wine containing known amounts of resveratrol suppresses atherosclerosis in hypercholesterolemic rabbits without affecting plasma lipid levels. Int J Mol Med. 2005;14:533–540. [PubMed] [Google Scholar]
- Park CS, Lee YC, Kim JD, Kim HM, Kim CH. Inhibitory effects of Polygonum cuspidatum water extract (PCWE) and its component resveratrol [correction of rasveratrol] on acyl-coenzyme A-cholesterol acyltransferase activity for cholesteryl ester synthesis in HepG2 cells. Vascul Pharmacol. 2004;14:279–284. doi: 10.1016/j.vph.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Rong JX, Blachford C, Feig JE, Bander I, Mayne J, Kusunoki J, Miller C, Davis M, Wilson M, Dehn S, Thorp E, Tabas I, Taubman MB, Rudel LL, Fisher EA. ACAT inhibition reduces the progression of preexisting, advanced atherosclerotic mouse lesions without plaque or systemic toxicity. Arterioscler Thromb Vasc Biol. 2013;14:4–12. doi: 10.1161/ATVBAHA.112.252056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, Fujiwara Y, Hayashida A, Horlad H, Kato H, Rotinsulu H, Losung F, Mangindaan RE, de Voogd NJ, Takeya M, Tsukamoto S. Manzamine A, a marine-derived alkaloid, inhibits accumulation of cholesterol ester in macrophages and suppresses hyperlipidemia and atherosclerosis in vivo. Bioorg Med Chem. 2013;14:3831–3838. doi: 10.1016/j.bmc.2013.04.025. [DOI] [PubMed] [Google Scholar]
- Alegret M, Llaverias G, Silvestre JS. Acyl coenzyme A:cholesterol acyltransferase inhibitors as hypolipidemic and antiatherosclerotic drugs. Methods Find Exp Clin Pharmacol. 2004;14:563–586. doi: 10.1358/mf.2004.26.7.863738. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Konopka G, Pack-Chung E, Ingano LA, Berezovska O, Hyman BT, Chang TY, Tanzi RE, Kovacs DM. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat Cell Biol. 2001;14:905–912. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Peach C, Bhattacharyya R, Barren C, Pettingell W, Hutter-Paier B, Windisch M, Berezovska O, Kovacs DM. Inhibition of acyl-coenzyme A: cholesterol acyl transferase modulates amyloid precursor protein trafficking in the early secretory pathway. FASEB J. 2009;14:3819–3828. doi: 10.1096/fj.09-134999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen HJ, Havas D, Peach C, Barren C, Duller S, Xia W, Frosch MP, Hutter-Paier B, Windisch M, Kovacs DM. The acyl-coenzyme A: cholesterol acyltransferase inhibitor CI-1011 reverses diffuse brain amyloid pathology in aged amyloid precursor protein transgenic mice. J Neuropathol Exp Neurol. 2010;14:777–788. doi: 10.1097/NEN.0b013e3181e77ed9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SR, Chang CC, Dogbevia G, Bryleva EY, Bowen Z, Hasan MT, Chang TY. Acat1 knockdown gene therapy decreases amyloid-beta in a mouse model of Alzheimer’s disease. Mol Ther. 2013;14:1497–1506. doi: 10.1038/mt.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M, Rogers G, Peters J, Anderson R, Hoyle M, Miners A, Moxham T, Davis S, Thokala P, Wailoo A, Jeffreys M, Hyde C. The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of Technology Appraisal No. 111): a systematic review and economic model. Health Technol Assess. 2012;14:1–470. doi: 10.3310/hta16210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanaga K, Ichiki T, Miyazaki R, Takeda K, Hashimoto T, Matsuura H, Sunagawa K. Acetylcholinesterase inhibitors attenuate atherogenesis in apolipoprotein E-knockout mice. Atherosclerosis. 2010;14:52–58. doi: 10.1016/j.atherosclerosis.2010.07.027. [DOI] [PubMed] [Google Scholar]
- Koch MA, Waldmann H. Protein structure similarity clustering and natural product structure as guiding principles in drug discovery. Drug Discov Today. 2005;14:471–483. doi: 10.1016/S1359-6446(05)03419-7. [DOI] [PubMed] [Google Scholar]
- Benyamin B, Middelberg RP, Lind PA, Valle AM, Gordon S, Nyholt DR, Medland SE, Henders AK, Heath AC, Madden PA, Visscher PM, O'Connor DT, Montgomery GW, Martin NG, Whitfield JB. GWAS of butyrylcholinesterase activity identifies four novel loci, independent effects within BCHE and secondary associations with metabolic risk factors. Hum Mol Genet. 2011;14:4504–4514. doi: 10.1093/hmg/ddr375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan VK, Risacher SL, Nho K, Kim S, Swaminathan S, Shen L, Foroud TM, Hakonarson H, Huentelman MJ, Aisen PS, Petersen RC, Green RC, Jack CR, Koeppe RA, Jagust WJ, Weiner MW, Saykin AJ. APOE and BCHE as modulators of cerebral amyloid deposition: a florbetapir PET genome-wide association study. Mol Psychiatry. 2013;14:351–357. doi: 10.1038/mp.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelliah J, Smith JD, Fariss MW. Inhibition of cholinesterase activity by tetrahydroaminoacridine and the hemisuccinate esters of tocopherol and cholesterol. Biochim Biophys Acta. 1994;14:17–26. doi: 10.1016/0167-4838(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Czura CJ, Ivanova S. Mind over immunity. FASEB J. 2001;14:1575–1576. doi: 10.1096/fj.01-0148hyp. [DOI] [PubMed] [Google Scholar]
- Roher AE, Tyas SL, Maarouf CL, Daugs ID, Kokjohn TA, Emmerling MR, Garami Z, Belohlavek M, Sabbagh MN, Sue LI, Beach TG. Intracranial atherosclerosis as a contributing factor to Alzheimer’s disease dementia. Alzheimers Dement. 2011;14:436–444. doi: 10.1016/j.jalz.2010.08.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray M, Ruan J, Zhang W. Variations in the transcriptome of Alzheimer’s disease reveal molecular networks involved in cardiovascular diseases. Genome Biol. 2008;14:R148. doi: 10.1186/gb-2008-9-10-r148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos JS, Ramirez C, Sastre I, Bullido MJ, Valdivieso F. ApoE4 is more efficient than E3 in brain access by herpes simplex virus type 1. Neuroreport. 2003;14:1825–1827. doi: 10.1097/00001756-200310060-00013. [DOI] [PubMed] [Google Scholar]
- Nazzal D, Therville N, Yacoub-Youssef H, Garcia V, Thomsen M, Levade T, Segui B, Benoist H. Apolipoprotein E-deficient mice develop an anti-Chlamydophila pneumoniae T helper 2 response and resist vascular infection. J Infect Dis. 2010;14:782–790. doi: 10.1086/655700. [DOI] [PubMed] [Google Scholar]
- de BN, Netea MG, Demacker PN, Kullberg BJ, van der Meer JW, Stalenhoef AF. Apolipoprotein E-deficient mice have an impaired immune response to Klebsiella pneumoniae. Eur J Clin Invest. 2000;14:818–822. doi: 10.1046/j.1365-2362.2000.00715.x. [DOI] [PubMed] [Google Scholar]
- Roselaar SE, Daugherty A. Apolipoprotein E-deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J Lipid Res. 1998;14:1740–1743. [PubMed] [Google Scholar]
- Ghosh J, Das S, Guha R, Ghosh D, Naskar K, Das A, Roy S. Hyperlipidemia offers protection against Leishmania donovani infection: role of membrane cholesterol. J Lipid Res. 2012;14:2560–2572. doi: 10.1194/jlr.M026914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos JS, Ramirez C, Sastre I, Valdivieso F. Effect of apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J Virol. 2006;14:5383–5387. doi: 10.1128/JVI.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal LR, Fernandes LR, Pietra Pedroso VS, Santiago HC, Gazzinelli RT, Alvarez-Leite JI. Influence of low-density lipoprotein (LDL) receptor on lipid composition, inflammation and parasitism during Toxoplasma gondii infection. Microbes Infect. 2008;14:276–284. doi: 10.1016/j.micinf.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Burt TD, Agan BK, Marconi VC, He W, Kulkarni H, Mold JE, Cavrois M, Huang Y, Mahley RW, Dolan MJ, McCune JM, Ahuja SK. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc Natl Acad Sci U S A. 2008;14:8718–8723. doi: 10.1073/pnas.0803526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard HC, Wildt KL, Whittum-Hudson JA, Lai Z, Ager J, Hudson AP. The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb Pathog. 2005;14:19–26. doi: 10.1016/j.micpath.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Faragher EB, Todd JA, Koram KA, Riley EM, Itzhaki RF. Does apolipoprotein E polymorphism influence susceptibility to malaria? J Med Genet. 2003;14:348–351. doi: 10.1136/jmg.40.5.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathe R. Steroid and sterol 7-hydroxylation: Ancient pathways. Steroids. 2002;14:967–977. doi: 10.1016/S0039-128X(02)00044-2. [DOI] [PubMed] [Google Scholar]
- Morel DW, Edgerton ME, Warner GE, Johnson WJ, Phillips MC, Rothblat GH. Comparison of the intracellular metabolism and trafficking of 25-hydroxycholesterol and cholesterol in macrophages. J Lipid Res. 1996;14:2041–2051. [PubMed] [Google Scholar]
- Zhao C, Dahlman-Wright K. Liver X receptor in cholesterol metabolism. J Endocrinol. 2010;14:233–240. doi: 10.1677/JOE-09-0271. [DOI] [PubMed] [Google Scholar]
- Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A. 1999;14:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG. 27-hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J Biol Chem. 2001;14:38378–38387. doi: 10.1074/jbc.M105805200. [DOI] [PubMed] [Google Scholar]
- Nelson ER, DuSell CD, Wang X, Howe MK, Evans G, Michalek RD, Umetani M, Rathmell JC, Khosla S, Gesty-Palmer D, McDonnell DP. The oxysterol, 27-hydroxycholesterol, links cholesterol metabolism to bone homeostasis through its actions on the estrogen and liver X receptors. Endocrinology. 2011;14:4691–4705. doi: 10.1210/en.2011-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaya J, Aviram M, Mahmood S, Hayek T, Grenadir E, Hoffman A, Milo S. Selective distribution of oxysterols in atherosclerotic lesions and human plasma lipoproteins. Free Radic Res. 2001;14:485–497. doi: 10.1080/10715760100300431. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Jessup W. Oxysterols and atherosclerosis. Atherosclerosis. 1999;14:1–28. doi: 10.1016/S0021-9150(98)00196-8. [DOI] [PubMed] [Google Scholar]
- Heverin M, Bogdanovic N, Lutjohann D, Bayer T, Pikuleva I, Bretillon L, Diczfalusy U, Winblad B, Bjorkhem I. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J Lipid Res. 2004;14:186–193. doi: 10.1194/jlr.M300320-JLR200. [DOI] [PubMed] [Google Scholar]
- Shafaati M, Marutle A, Pettersson H, Lovgren-Sandblom A, Olin M, Pikuleva I, Winblad B, Nordberg A, Bjorkhem I. Marked accumulation of 27-hydroxycholesterol in the brains of Alzheimer’s patients with the Swedish APP 670/671 mutation. J Lipid Res. 2011;14:1004–1010. doi: 10.1194/jlr.M014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Newman K, Javitt NB. Alzheimer’s disease: brain desmosterol levels. J Alzheimers Dis. 2013;14:881–888. doi: 10.3233/JAD-2012-121453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt NB. Alzheimer’s disease: neuroprogesterone, epoxycholesterol, and ABC transporters as determinants of neurodesmosterol tissue levels and its role in amyloid protein processing. J Alzheimers Dis. 2013;14:441–450. doi: 10.3233/JAD-130044. [DOI] [PubMed] [Google Scholar]
- Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, Lacaze P, Watterson S, Griffiths SJ, Spann NJ, Meljon A, Talbot S, Krishnan K, Covey DF, Wenk MR, Craigon M, Ruzsics Z, Haas J, Angulo A, Griffiths WJ, Glass CK, Wang Y, Ghazal P. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity. 2013;14:106–118. doi: 10.1016/j.immuni.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diczfalusy U, Bjorkhem I. Still another activity by the highly promiscuous enzyme CYP3A4: 25-hydroxylation of cholesterol. J Lipid Res. 2011;14:1447–1449. doi: 10.1194/jlr.E017806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Bjorkhem I, Furster C, Wikvall K. 24-, 25- and 27-hydroxylation of cholesterol by a purified preparation of 27-hydroxylase from pig liver. Biochim Biophys Acta. 1993;14:177–182. doi: 10.1016/0005-2760(93)90094-P. [DOI] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A. 1999;14:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Kerr TA, Sakai J, Li WP, Russell DW. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem. 1998;14:34316–34327. doi: 10.1074/jbc.273.51.34316. [DOI] [PubMed] [Google Scholar]
- Diczfalusy U, Olofsson KE, Carlsson AM, Gong M, Golenbock DT, Rooyackers O, Flaring U, Bjorkbacka H. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J Lipid Res. 2009;14:2258–2264. doi: 10.1194/jlr.M900107-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Scott AL. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol. 2010;14:1081–1087. doi: 10.1189/jlb.0610318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Dana SE, Goldstein JL. Cholesterol ester formation in cultured human fibroblasts. Stimulation by oxygenated sterols. J Biol Chem. 1975;14:4025–4027. [PubMed] [Google Scholar]
- Cheng D, Chang CC, Qu X, Chang TY. Activation of acyl-coenzyme A: cholesterol acyltransferase by cholesterol or by oxysterol in a cell-free system. J Biol Chem. 1995;14:685–695. doi: 10.1074/jbc.270.2.685. [DOI] [PubMed] [Google Scholar]
- Gold ES, Ramsey SA, Sartain MJ, Selinummi J, Podolsky I, Rodriguez DJ, Moritz RL, Aderem A. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J Exp Med. 2012;14:807–817. doi: 10.1084/jem.20111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltl S, Patankar JV, Fauler G, Nusshold C, Ullen A, Eibinger G, Wintersperger A, Kratky D, Malle E, Sattler W. 25-Hydroxycholesterol regulates cholesterol homeostasis in the murine CATH.a neuronal cell line. Neurosci Lett. 2013;14:16–21. doi: 10.1016/j.neulet.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, Freiberg AN, Su L, Lee B, Cheng G. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity. 2013;14:92–105. doi: 10.1016/j.immuni.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N, Carlin AF, Spann NJ, Saijo K, Morello CS, McDonald JG, Romanoski CE, Maurya MR, Kaikkonen MU, Lam MT, Crotti A, Reichart D, Fox JN, Quehenberger O, Raetz CR, Sullards MC, Murphy RC, Merrill AH Jr, Brown HA, Dennis EA, Fahy E, Subramaniam S, Cavener DR, Spector DH, Russell DW, Glass CK. 25-Hydroxycholesterol activates the integrated stress response to reprogram transcription and translation in macrophages. J Biol Chem. 2013;14:35812–35823. doi: 10.1074/jbc.M113.519637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, Guerini D, Baumgarten BU, Roggo S, Wen B, Knochenmuss R, Noel S, Gessier F, Kelly LM, Vanek M, Laurent S, Preuss I, Miault C, Christen I, Karuna R, Li W, Koo DI, Suply T, Schmedt C, Peters EC, Falchetto R, Katopodis A, Spanka C, Roy MO, Detheux M. et al. Oxysterols direct immune cell migration via EBI2. Nature. 2011;14:524–527. doi: 10.1038/nature10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diczfalusy U. On the formation and possible biological role of 25-hydroxycholesterol. Biochimie. 2013;14:455–460. doi: 10.1016/j.biochi.2012.06.016. [DOI] [PubMed] [Google Scholar]
- Rose K, Allan A, Gauldie S, Stapleton G, Dobbie L, Dott K, Martin C, Wang L, Hedlund E, Seckl JR, Gustafsson JA, Lathe R. Neurosteroid hydroxylase CYP7B: vivid reporter activity in dentate gyrus of gene-targeted mice and abolition of a widespread pathway of steroid and oxysterol hydroxylation. J Biol Chem. 2001;14:23937–23944. doi: 10.1074/jbc.M011564200. [DOI] [PubMed] [Google Scholar]
- Yi T, Wang X, Kelly LM, An J, Xu Y, Sailer AW, Gustafsson JA, Russell DW, Cyster JG. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity. 2012;14:535–548. doi: 10.1016/j.immuni.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, van der Westhuyzen DR, Goldstein JL, Brown MS. Purification of oxysterol binding protein from hamster liver cytosol. J Biol Chem. 1989;14:9046–9052. [PubMed] [Google Scholar]
- Burgett AW, Poulsen TB, Wangkanont K, Anderson DR, Kikuchi C, Shimada K, Okubo S, Fortner KC, Mimaki Y, Kuroda M, Murphy JP, Schwalb DJ, Petrella EC, Cornella-Taracido I, Schirle M, Tallarico JA, Shair MD. Natural products reveal cancer cell dependence on oxysterol-binding proteins. Nat Chem Biol. 2011;14:639–647. doi: 10.1038/nchembio.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway ND, Dawson PA, Ho YK, Brown MS, Goldstein JL. Translocation of oxysterol binding protein to Golgi apparatus triggered by ligand binding. J Cell Biol. 1992;14:307–319. doi: 10.1083/jcb.116.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto A, Liu X, Robinson CA, Ridgway ND. Multisite phosphorylation of oxysterol-binding protein regulates sterol binding and activation of sphingomyelin synthesis. Mol Biol Cell. 2012;14:3624–3635. doi: 10.1091/mbc.E12-04-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Cordy JM, Chen CD, Guillily M, Suon S, Ray WJ, Seabrook GR, Abraham CR, Wolozin B. Oxysterol-binding protein-1 (OSBP1) modulates processing and trafficking of the amyloid precursor protein. Mol Neurodegener. 2008;14:5. doi: 10.1186/1750-1326-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Jauhiainen M, Hildebrand RB, van Willems DK, van Berkel TJ, Ehnholm C, Van EM, Olkkonen VM. Expression of human OSBP-related protein 1 L in macrophages enhances atherosclerotic lesion development in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2007;14:1618–1624. doi: 10.1161/ATVBAHA.107.144121. [DOI] [PubMed] [Google Scholar]
- Olkkonen VM. Macrophage oxysterols and their binding proteins: roles in atherosclerosis. Curr Opin Lipidol. 2012;14:462–470. doi: 10.1097/MOL.0b013e328356dba0. [DOI] [PubMed] [Google Scholar]
- Lappano R, Recchia AG, de Francesco EM, Angelone T, Cerra MC, Picard D, Maggiolini M. The cholesterol metabolite 25-hydroxycholesterol activates estrogen receptor alpha-mediated signaling in cancer cells and in cardiomyocytes. PLoS One. 2011;14:e16631. doi: 10.1371/journal.pone.0016631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods. 2013;14:259–264. doi: 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;14:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- IBC 50K CAD Consortium. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;14:e1002260. doi: 10.1371/journal.pgen.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papassotiropoulos A, Lambert JC, Wavrant-De Vrieze F, Wollmer MA, von der KH, Streffer JR, Maddalena A, Huynh KD, Wolleb S, Lutjohann D, Schneider B, Thal DR, Grimaldi LM, Tsolaki M, Kapaki E, Ravid R, Konietzko U, Hegi T, Pasch T, Jung H, Braak H, Amouyel P, Rogaev EI, Hardy J, Hock C, Nitsch RM. Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic Alzheimer’s disease. Neurodegener Dis. 2005;14:233–241. doi: 10.1159/000090362. [DOI] [PubMed] [Google Scholar]
- Laumet G, Chouraki V, Grenier-Boley B, Legry V, Heath S, Zelenika D, Fievet N, Hannequin D, Delepine M, Pasquier F, Hanon O, Brice A, Epelbaum J, Berr C, Dartigues JF, Tzourio C, Campion D, Lathrop M, Bertram L, Amouyel P, Lambert JC. Systematic analysis of candidate genes for Alzheimer’s disease in a French, genome-wide association study. J Alzheimers Dis. 2010;14:1181–1188. doi: 10.3233/JAD-2010-100126. [DOI] [PubMed] [Google Scholar]
- Holmes RS, Vandeberg JL, Cox LA. Genomics and proteomics of vertebrate cholesterol ester lipase (LIPA) and cholesterol 25-hydroxylase (CH25H) 3 Biotech. 2011;14:99–109. doi: 10.1007/s13205-011-0013-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Grabowski GA. Lysosomal acid lipase and atherosclerosis. Curr Opin Lipidol. 2004;14:539–544. doi: 10.1097/00041433-200410000-00007. [DOI] [PubMed] [Google Scholar]
- Sugiura H, Koarai A, Ichikawa T, Minakata Y, Matsunaga K, Hirano T, Akamatsu K, Yanagisawa S, Furusawa M, Uno Y, Yamasaki M, Satomi Y, Ichinose M. Increased 25-hydroxycholesterol concentrations in the lungs of patients with chronic obstructive pulmonary disease. Respirology. 2012;14:533–540. doi: 10.1111/j.1440-1843.2012.02136.x. [DOI] [PubMed] [Google Scholar]
- Matkovic Z, Miravitlles M. Chronic bronchial infection in COPD. Is there an infective phenotype? Respir Med. 2013;14:10–22. doi: 10.1016/j.rmed.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yu C, Liu J, Spencer TA, Chang CC, Chang TY. Cholesterol is superior to 7-ketocholesterol or 7 alpha-hydroxycholesterol as an allosteric activator for acyl-coenzyme A:cholesterol acyltransferase 1. J Biol Chem. 2003;14:11642–11647. doi: 10.1074/jbc.M211559200. [DOI] [PubMed] [Google Scholar]
- Miller SC, Melnykovych G. Regulation of cholesterol biosynthesis and esterification by 25-hydroxycholesterol in a macrophage-like cell line: uncoupling by progesterone. J Lipid Res. 1984;14:991–999. [PubMed] [Google Scholar]
- Chang TY, Chang CC, Bryleva E, Rogers MA, Murphy SR. Neuronal cholesterol esterification by ACAT1 in Alzheimer’s disease. IUBMB Life. 2010;14:261–267. doi: 10.1002/iub.305. [DOI] [PubMed] [Google Scholar]
- Lange Y, Ye J, Rigney M, Steck TL. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J Lipid Res. 1999;14:2264–2270. [PubMed] [Google Scholar]
- An S, Jang YS, Park JS, Kwon BM, Paik YK, Jeong TS. Inhibition of acyl-coenzyme A:cholesterol acyltransferase stimulates cholesterol efflux from macrophages and stimulates farnesoid X receptor in hepatocytes. Exp Mol Med. 2008;14:407–417. doi: 10.3858/emm.2008.40.4.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MP, Noble WJ, Olm V, Gaynor K, Casey E, LaFrancois J, Wang L, Duff K. Co-localization of cholesterol, apolipoprotein E and fibrillar Abeta in amyloid plaques. Brain Res Mol Brain Res. 2003;14:119–125. doi: 10.1016/S0169-328X(02)00647-2. [DOI] [PubMed] [Google Scholar]
- Nelson TJ, Alkon DL. Oxidation of cholesterol by amyloid precursor protein and beta-amyloid peptide. J Biol Chem. 2005;14:7377–7387. doi: 10.1074/jbc.M409071200. [DOI] [PubMed] [Google Scholar]
- Yoshimoto N, Tasaki M, Shimanouchi T, Umakoshi H, Kuboi R. Oxidation of cholesterol catalyzed by amyloid beta-peptide (Abeta)-Cu complex on lipid membrane. J Biosci Bioeng. 2005;14:455–459. doi: 10.1263/jbb.100.455. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Friedlich AL, Setchell KD, Nagano S, Opazo C, Cherny RA, Cherny RA, Barnham KJ, Wade JD, Melov S, Kovacs DM, Bush AI. Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. J Clin Invest. 2005;14:2556–2563. doi: 10.1172/JCI23610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del TK, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kovari E, Kukull WA, Leverenz JB, Love S, Mackenzie IR, Mann DM, Masliah E, McKee AC, Montine TJ, Morris JC, Schneider JA. et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;14:362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;14:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;14:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksler ME, Gouras G, Relkin NR, Szabo P. The immune system, amyloid-beta peptide, and Alzheimer’s disease. Immunol Rev. 2005;14:244–256. doi: 10.1111/j.0105-2896.2005.00264.x. [DOI] [PubMed] [Google Scholar]
- Finch CE, Sapolsky RM. The evolution of Alzheimer disease, the reproductive schedule, and apoE isoforms. Neurobiol Aging. 1999;14:407–428. doi: 10.1016/S0197-4580(99)00053-6. [DOI] [PubMed] [Google Scholar]
- Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;14:839–845. doi: 10.1093/oxfordjournals.aje.a010288. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I. Rediscovery of cerebrosterol. Lipids. 2007;14:5–14. doi: 10.1007/s11745-006-1003-2. [DOI] [PubMed] [Google Scholar]
- He X, Ong WY, Hua Q. Distribution of cholesterol 24-hydroxylase in the monkey brain. Neurosci Bull. 2010;14:197–204. doi: 10.1007/s12264-010-1224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolsch H, Lutjohann D, Ludwig M, Schulte A, Ptok U, Jessen F, von BK, Rao ML, Maier W, Heun R. Polymorphism in the cholesterol 24S-hydroxylase gene is associated with Alzheimer’s disease. Mol Psychiatry. 2002;14:899–902. doi: 10.1038/sj.mp.4001109. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, Huang DY, Pericak-Vance M, Schmechel D, Roses AD. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A. 1994;14:11183–11186. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, Virgin HW. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;14:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, Sidhu R, Onken MD, Harbour JW, Hagbi-Levi S, Chowers I, Edwards PA, Baldan A, Parks JS, Ory DS, Apte RS. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;14:549–561. doi: 10.1016/j.cmet.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;14:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]