

Abstract
Protein secretion, a key intercellular event for transducing cellular signals, is thought to be strictly regulated. However, secretion dynamics at the single-cell level have not yet been clarified because intercellular heterogeneity results in an averaging response from the bulk cell population. To address this issue, we developed a novel assay platform for real-time imaging of protein secretion at single-cell resolution by a sandwich immunoassay monitored by total internal reflection microscopy in sub-nanolitre-sized microwell arrays. Real-time secretion imaging on the platform at 1-min time intervals allowed successful detection of the heterogeneous onset time of nonclassical IL-1β secretion from monocytes after external stimulation. The platform also helped in elucidating the chronological relationship between loss of membrane integrity and IL-1β secretion. The study results indicate that this unique monitoring platform will serve as a new and powerful tool for analysing protein secretion dynamics with simultaneous monitoring of intracellular events by live-cell imaging.
Many secreted proteins have been identified as important functional mediators of intercellular communication for the purpose of initiating various cellular processes, including differentiation and migration1,2,3. Cytokines in particular are one of the best studied classes of secreted proteins with broad effects on immune responses4. For the proper functioning of the immune system, cytokine synthesis and secretion must be tightly regulated, both spatially and temporally5. However, recent investigations using single-cell analysis have shown that immune cells display highly heterogeneous levels of cytokine secretion, even in cells with apparently similar phenotypes6. Therefore, the relationship between heterogeneous cytokine secretion at the single-cell level and the maintenance of homeostasis of the immune system has become a primary subject of investigation in the field of immunology. To address this issue, a methodology is required that enables delineation of spatiotemporal heterogeneity of cytokine secretion at the single-cell level. We have particularly focused on cytokine induction processes that occur in single cells induced by external stimulation, specifically with regard to (1) cellular heterogeneity in protein secretion dynamics and (2) the chronological relationship between intracellular event(s) and protein secretion. However, the technology available for monitoring protein secretion from single cells remains in its infancy.
Several groups have reported population analysis of cytokine secretion from single cells by using antibody-based immunoassay applications. Love et al. generated a secretion profile for a large collection of single cells by using microengraving7 and succeeded in measuring the time course of cytokine secretion during T-lymphocyte maturation every 2 h for a period spanning several hours8. While these methods are efficient for their high throughput and/or the quantitative data generated, several challenges remain because of their inherent measurement limitations. In these methods, the accumulated cytokine molecules situated on a solid surface are labelled with a detection probe and are quantified after intensive wash steps, which are required to remove excess probe. Although this wash step, known as bound/free (B/F) separation, determines the signal/noise ratio for detection, this step also causes a lag between secretion and detection. Therefore, these methods cannot currently offer either a time interval of shorter than a few hours nor simultaneous real-time monitoring of a second intracellular variable (e.g. cell viability) over time.
Previously, our group and Salehi-Reyhani et al. respectively have successfully addressed this B/F separation issue in fluorescence immunoassays (FIAs) by taking advantage of near-field excitation in total internal reflection fluorescence microscopy (TIRFM)9,10. In these studies, target proteins in each single-cell lysate segmented by microwells were quantified by detecting formation of immunocomplexes on the microwell bottom. In the current study, we have developed a novel assay platform for real-time monitoring of live single-cell cytokine secretion (Fig. 1). Each single cell is deposited on a microfabricated-well array (MWA) chip, which restricts cell migration as well as compartmentalizes the secretory signals from individual cells. The anti-cytokine capture antibody immobilized on the microwell bottom immediately captures the cytokine secreted from a cell, which enables TIRFM-FIA to function in situ. An MWA chip of the platform has an open architecture at the top of each microwell to permit easy access for stimulus delivery and to maintain culture conditions similar to those of a bulk experiment.
Figure 1. Concept of the real-time single cell secretion assay platform.

The schematic illustrates the concept of the platform for the real-time single cell secretion assay. The platform works with micro-fabricated well-array chip on a fully automated fluorescence microscopy. The platform maintains the environment (temperature, concentration of CO2, and humidity) of the chip. The chip has an array of nanolitre-sized microwells with a glass bottom, into which individual cells were introduced separately. The well has open-ended structure; therefore, culture medium was exchanged constantly during the observation. The anti-cytokine capture antibody was immobilized on the well bottom, onto which secreted cytokine and fluorescently labelled detection antibody were bound to form a sandwich immunocomplex. Near-field excitation by total internal reflection enabled selective detection of the cytokine sandwich immunocomplex immediately following secretion without the requirement for wash steps.
We characterized our secretion assay platform using a MWA chip consisting of glass and polydimethylsiloxiane (PDMS) in a quantitative manner by model experiments, introducing minute amounts of cytokine into microwells to mimic the milieu of cytokine secretion from a single cell. The platform was applied to monitor cytokine secretion from human peripheral blood monocytes at 1-min time intervals. We examined the IL-1β secretion process while simultaneously observing membrane integrity to determine the intracellular processes that occur at the time of cytokine secretion, since the mechanism underlying the non-classical IL-1β secretion pathway currently remains unclear.
Results
Characterization of an assay platform for real-time secretion imaging in an MWA chip
In this study, we designed a real-time single-cell imaging platform to monitor the secretion processes over time. We fabricated a novel MWA chip consisting of a PDMS well wall and a glass well bottom (see Supplementary Fig. S1a online). An objective of numerical aperture 1.49 was used to avoid the stray light due to the higher refractive index of PDMS (n = 1.42, Sylgard®184, Dow Corning Co., http://www1.dowcorning.com/DataFiles/090007c8803bb6a1.pdf) than that of medium (n = 1.34; Supplementary Fig. S1b–d). To assess restriction of the horizontal movement of secreted cytokine molecules by the microwell structure, we compared the decrease of fluorescently labelled protein from an observation area for TIRFM with and without microwell structures. The microwell structure slowed the escape of fluorescent molecules by about 20-fold (see Supplementary Fig. S2).
Next, to assess the functionality of time-resolved FIA (even in the open MWA), we introduced a miniscule amount of cytokine into microwells by using a microinjector to mimic cytokine secretion from a single cell (Fig. 2a). Fluorescence signals began to increase immediately after pulsed injection of IL-1β (time = 0; Fig. 2b, c) and increased without apparent change in localization over time (Fig. 2b), indicating that most of the injected IL-1β was instantly captured by antibody on the MWA bottom before diffusion throughout the microwell. From a separate experiment, we found that the fluorescence signals after the injection of pre-formed IL-1β-detection antibody complexes increased 15 times faster than that after the injection of IL-1β alone, suggesting that the released IL-1β rapidly bound to the capture antibodies on the bottom surface of the MWA chip (Fig. S3a, b). Therefore, binding of the detection antibody to form sandwich immunocomplexes was likely the rate-limiting step for the increase in the fluorescence signals under the experimental conditions employed. Once the immunocomplex was formed, its dissociation occurred very slowly (dissociation constant koff < 2 × 10−6 s−1, Fig. S3c).
Figure 2. Performance evaluation of time-resolved FIA in the MWA on model experiments using a microinjector.

(a) Schema of the model experiment using pulsed injection of IL-1β via a microinjector. An increase in fluorescence signal was observed after pulse injecting 100 ng/mL of recombinant IL-1β into a microwell that was filled with the detection medium containing 30 nM fluorescently labelled detection antibody. (b) Representative images of developing fluorescence signals obtained from time-resolved FIA after introducing recombinant IL-1β into the microwell. Images were acquired once every second applying a 60-ms exposure time. The time elapsed after the pulsed injection is shown on each image. (c) Time course of the average intensity of the IL-1β signal in a microwell. Each dot denotes a measured value and solid lines denote fitted curves. Different colours denote different durations under a constant pressure of injection (1,200 Pa): 4 s (black), 2 s (green), 0.5 s (blue), and 0.1 s (cyan). Curve fitting was performed for every dataset from 0 to 5 min using the global parameter of time constant, τ, and local parameters of maximum intensity, I, and consequently τ was determined as 3 min.
The rising curves of the fluorescence signal obtained from IL-1β injection were fitted with a single exponential, especially during 5 min after injection, and the time constant was estimated to be 3 min (Fig. 2c). Using this parameter, we evaluated the accuracy for determination of the onset time by fitting a single exponential to the dataset quantifying the fluorescence increase obtained within an arbitrary time interval. The accuracy of computed onset time was determined within 0.1 min when using a dataset with 1-min time intervals (Fig. S4).
The increase in the amount of fluorescence signal depended upon the quantity of IL-1β injected (Fig. 2c). However, the absolute amount of secreted cytokine could not be determined because of the difficulties in controlling the local concentration of cytokines within an open-ended microwell to generate a standard curve. Additionally, immobilized cytokine captured by antibody on the MWA surface was considered to be part of the overall secreted cytokine, while the non-immobilized fraction diffused into the medium. The captured ratio would depend upon an uncontrollable variable, such as the height of the cytokine release point (which determines the probability of the cytokine encountering the capture antibody). Therefore, the assay platform developed in this study was best suited for detection of the onset of secretory molecule secretion from single cells at high time resolution (probably less than 1 min) while also providing semi-quantitative data on secreted molecules. In experiments performed using microwells closed with sealing oil, the platform could detect the signal from 2,000 molecules of IL-1β in a microwell (Supplementary Fig. S5).
Real-time monitoring of IL-6 secretion from single living monocytes within a 1-min time interval
As a proof-of-concept experiment, we assessed the performance of our assay platform for time-resolved observation using lipopolysaccharide (LPS)-stimulated human peripheral blood monocytes by simultaneous detection of cytokine secretion and live/dead signals. We confirmed that proinflammatory cytokines were detected in the culture supernatant of 1 × 105 monocytes stimulated with 1 μg/mL LPS (first priming; Supplementary Fig. S6). Among the detectable cytokines, we selected interleukin 6 (IL-6) as a typical cytokine, known to be released using classical pathways involving ER/Golgi trafficking11.
First, we examined the reactivity and viability of monocytes in the MWA from snapshot measurements of 2,500 microwells after LPS stimulation for 4 h. Of the 584 cells observed from 2,500 microwells, 23 cells displayed the IL-6 signal (4%). Calcein (+)/SYTOX (−) living cells accounted for 60% of the total cells, but 91% of IL-6–positive cells. Next, 40 microwells were scanned for 4 h for real-time IL-6 secretion imaging at 1-min time intervals. After three independent experiments, we could analyse 71 single cells, including 56 living cells; 7 individual cells were observed to secrete IL-6 (representative images are displayed in Fig. 3a and Supplementary movie 1). IL-6 signals increased gradually for over 1 h after stimulation (Fig. 3b) without change in the SYTOX signal (Fig. 3b), as observed in the displayed dead cell (Fig. 3c). All the remaining IL-6–secreting cells continued to survive during the observation period. These results demonstrated that the single-cell secretion assay platform enabled us to monitor physiological secretion of IL-6 from live monocytes.
Figure 3. Time-resolved monitoring of IL-6 secretion after a classical secretion pathway.

Calcein-charged human peripheral blood monocytes were introduced into the MWA chip. Signals of SYTOX, calcein and IL-6 secretion were observed with 1-min time intervals after administration of 1 μg/mL of LPS. (a) Representative images of multichannel microscopy. Morphological features of a human monocyte were monitored under diascopic illumination (DIA). The fluorescence signal of SYTOX-stained nuclei was magenta (SYTOX), that of calcein-stained cell bodies was green (Calcein), and that of secreted IL-6 was yellow (CF660R anti IL-6 Ab). Merged images of these three fluorescence signals are also displayed (Merged). Each image was generated at the described time point. Scale bar, 50 μm. Although cells floated soon after LPS stimulation, cells began to adhere to the microwell bottom at 1.5 h after LPS stimulation, with some exhibiting the IL-6 signal at 4 h after LPS stimulation. Although the fluorescence intensity of calcein per pixel was apparently decreased due to the cell deformation, it was distinct from a sharp drop of calcein signal observed for dead cells like in the bottom-right well. The cells that underwent cell death showed elevation of the SYTOX signal. (b), (c) Time course of the average intensity of IL-6 and SYTOX signals within microwells shown in Figure 3a: the top right (b) or the bottom right (c) microwell. (b) The IL-6 secretion signal gradually increased from 80 min after LPS stimulation without any change in SYTOX signal. (c) Typical example of a dead cell. Only the SYTOX signal was increased.
Simultaneous imaging of extracellular IL-1β secretion and plasma membrane integrity
The mechanism underlying IL-1β secretion remains poorly understood, although it is known that IL-1β is a key mediator of inflammation12,13,14. Several researchers have proposed various mechanisms for IL-1β secretion15,16; however, many details remain to be validated due to a lack of techniques for monitoring real-time secretion processes of IL-1β at single-cell resolution. Therefore, we monitored IL-1β secretion from individual monocytes at high temporal resolution in parallel with observation of the cellular physiological states at the time of secretion.
In this study, monocytes were costimulated with both LPS and adenosine triphosphate (ATP), because IL-1β release from monocytes is known to require a second signal (i.e. ATP) to activate the intracellular inflammasome in addition to priming with pathogen-associated molecular stimuli (i.e. LPS)17,18,19. Glycine was added to the culture medium during stimulation because glycine blocks cytolytic release of pro–IL-1β without affecting the secretion of mature IL-1β (Supplementary Fig. S6)15,20,21.
Before real-time monitoring, we analysed IL-1β secretion and the live/dead state by snapshot secretion measurements of single cells with calcein/SYTOX staining (Fig. 4a) in 2,500 microwells at 4 h after LPS/ATP stimulation (Supplementary Fig. S7). This snapshot measurement demonstrated that approximately 30% of single monocytes secreted IL-1β. Interestingly, 99% of these IL-1β–secreting cells lost the calcein signal and were stained with SYTOX. When lower LPS concentration (10 ng/mL) was tested, both the number of IL-1β-secreting cells and the amount of secreted IL-1β per monocytes were decreased while the percentage of dead cells was only slightly affected (Supplementary Table S1). The concomitant disappearance of calcein and the increase in SYTOX signal reflect the compromised status of the cell membrane. Therefore, these results suggested some degree of association between IL-1β secretion and the loss of cell membrane integrity, exhibiting a sharp contrast with IL-6 secretion from LPS-stimulated monocytes (in which the vast majority of IL-6 secreting cells remained calcein-positive).
Figure 4. Time-resolved monitoring of IL-1β secretion on the PDMS MWA chip.
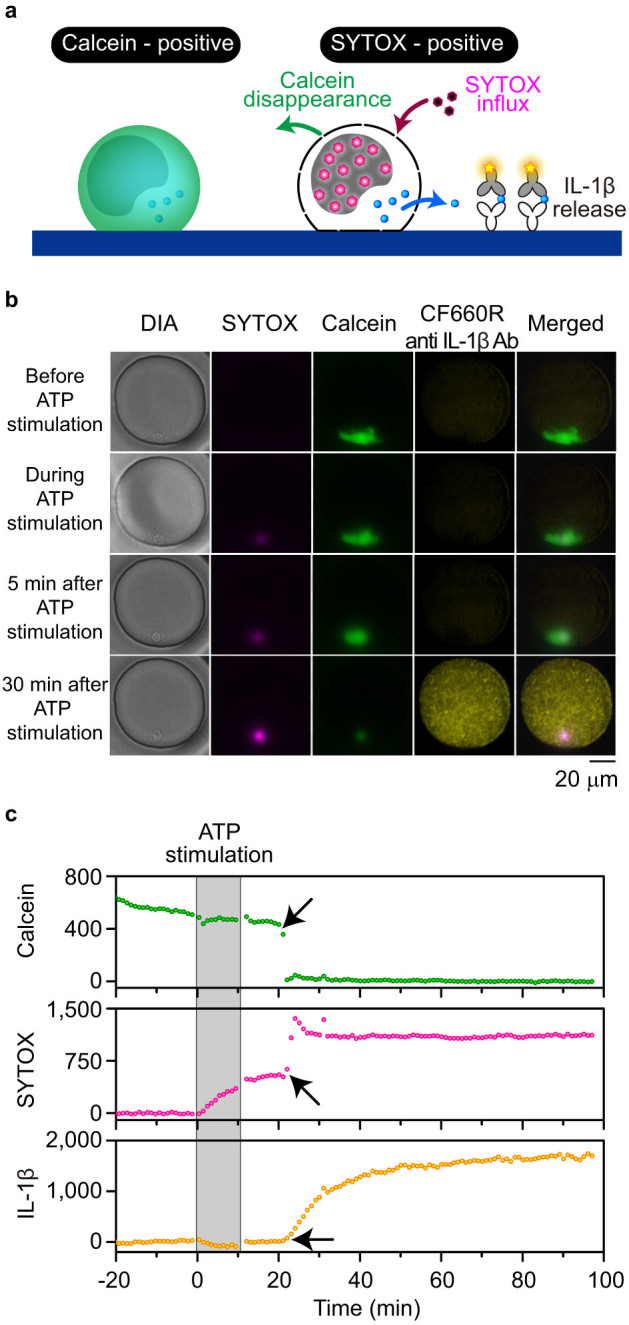
(a) Schematic of simultaneous monitoring of IL-1β secretion and cell membrane integrity using calcein and SYTOX staining. SYTOX influx and fluorescent calcein disappearance was observed due to compromised plasma membrane integrity. (b) Representative images of multichannel microscopy. Morphological features of a human monocyte were monitored under diascopic illumination (DIA). The fluorescence signal of SYTOX-stained nuclei was magenta (SYTOX), that of a calcein-stained cell bodies was green (Calcein), and that of secreted IL-1β was yellow (CF660R anti IL-1β Ab). Merged images of these three fluorescence signals are also displayed (Merged). Each image was obtained at the described period. Scale bar, 20 μm. (c) Example of the signal time course during time-resolved monitoring. Grey bands represent the period when the monocytes were exposed to ATP. Arrows represent the transition time of the respective signals.
We then performed real-time secretion imaging of IL-1β from monocytes, focusing on whether IL-1β secretion was preceded or followed by a loss of cellular membrane integrity. The experiments were performed twice, measuring 54 individual monocytes, 20 of which secreted detectable quantities of IL-1β. While all of these cells were calcein-positive before ATP stimulation, they lost their calcein signal and subsequently stained with SYTOX, consistent with the aforementioned snapshot observations (Supplementary movie 2 and Fig. 4b). Disappearance of the calcein signal occurred rapidly and was completed within a few minutes. By contrast, the observed increases in SYTOX signalling consisted of two phases: the first occurred gradually upon ATP stimulation and the second occurred abruptly at various moments following ATP stimulation. These two phases suggest that cell membrane permeability for SYTOX influx was altered through multiple stages.
Surprisingly, IL-1β secretion appeared to coincide with calcein disappearance and the second SYTOX influx (Supplementary movie 2 and Fig. 4c). The increase in the signal of IL-1β secretion occurred as a concave-down function, suggesting IL-1β was secreted in a burst release pattern (Supplementary Fig. S8). To evaluate the association between these events, the transition times were determined by curve fitting of the equation (1), (2), or (3) to the mean fluorescence intensities over time and were compared with one another (Supplementary Fig. S9). Although the response times after ATP stimulation were quite heterogeneous among cells, the transition time of calcein disappearance, SYTOX influx, and IL-1β release were quite similar in most cells (Fig. 5a and b). By focusing on the timeline of these events, lag times between SYTOX influx or IL-1β release and calcein disappearance were calculated. The SYTOX influx and the calcein disappearance (both resulting from membrane imperfections) occurred simultaneously or nearly simultaneously, and SYTOX influx was only slightly delayed following calcein disappearance (mean, 0.2 min; Fig. 5c). By contrast, IL-1β release occurred several minutes following calcein disappearance (mean, 2.0 min, except for 2 outliers; Fig. 5d), and its lag times were more variable than those of SYTOX influx. Two outliers exhibited extremely long delays (44.5 and 105.9 min) in IL-1β release after calcein disappearance. These results indicated that burst release of IL-1β was preceded by loss of cell membrane integrity.
Figure 5. High concordance of the three transition times observed in individual single cells.

(a), (b) Scatter plots of the signal transition times for SYTOX or IL-1β versus calcein. Signal transition occurred virtually simultaneously between calcein, SYTOX, and IL-1β. Each circle represents 20 individual monocytes. (d) Histograms of the lag times of SYTOX or IL-1β signal transition after the calcein disappearance. The lag times of SYTOX were distributed in a narrow range near 0, whereas those of IL-1β showed a wider distribution.
Discussion
In this study, we have developed a novel assay platform for real-time imaging of secretion at the single cell level at 1-min intervals. The dynamics of cytokine secretion against external stimuli have conventionally been investigated using a bulk population of cells with the same phenotype, based on the premise that these cells always display uniform responses. However, contrary to this premise, we have observed a wide distribution of onset time for IL-1β release triggered by ATP stimulation from individual human peripheral blood monocytes. Temporal heterogeneities at the single cell level have been demonstrated by many studies, but have been limited to intracellular processes, e.g. intracellular calcium elevation22. Our results indicated that extracellular reactions, such as protein secretion, were also chronologically heterogeneous at the single cell level. Furthermore, we successfully performed simultaneous measurement of cell membrane integrity and IL-1β release, indicating that our platform allowed for elucidation of the chronological relationship between intracellular process and the extracellular reaction at the single-cell level.
Imaging methods for secretion dynamics have been poorly developed for two primary reasons: First, secreted molecules disperse too rapidly in solution for efficient onsite monitoring. Second, a molecule of interest must be tagged by sensing moieties, but the tagging processes for molecular visualization are often accompanied with greater risks of generating artifacts, including functional modifications. Indeed, this latter point is the most serious limitation of live-cell imaging in general. The platform developed in this study permitted us to bypass such issues by immobilizing and labelling target molecules in the extracellular space. The detection strategy offers an advantage in its non-invasive monitoring of the physiological response of living cells, including clinical samples. A similar methodology with a label-free technique based upon nanoplasmonic imaging has been developed, although it is only applicable to up to three cells per experiment23. Sandwich FIA-based assay is expected to be a more sensitive and specific approach for small molecules like cytokines than the plasmonic approach, since the plasmonic signal is proportional to the molecular weight of the binding molecule.
Our platform uses MWAs that effectively trap floating cells as well as secreted cytokine molecules. This compartmentalization permitted integration of independently isolated single cells within a small area to increase the number of observable cells. More specifically, observation of a large number of cells made it feasible to perform statistical analyses on a small population of secreting monocytes. The open architecture of this device is well suited for cell manipulation using conventional tools, allowing for complex experimental arrangements. Additionally, the open architecture was beneficial for the maintenance of cellular physiology, because prolonged isolation within closed microwells may influence cellular conditions, e.g. oxygen starvation24. In this work, MWA chips were fabricated with PDMS, suitable for live cell imaging because of its biocompatibility25. PDMS offers low cost and fast processing in fabrication of the MWA chip. The only inconvenience was its higher refractive index than that of water, requiring a higher critical angle of the incident light for TIRFM. We resolved the issue by using an objective lens of high numerical aperture to achieve incidence angles greater than the critical angle for a glass/PDMS interface.
In our experiments, the rate of the apparent increase in the TIRF signal was 15 times slower than that of the apparent capture rate of the antigen onto the bottom surface of the MWA chip. This phenomenon was observed probably because (1) the difference in the local concentration of antibodies near the bottom surface and (2) decrease of the unbound antigen by diffusion followed pseudo first-order kinetics with a time constant of approximately 0.9 min (Supplementary Fig. S2). The increase in the concentration of fluorescent detection antibody accelerates the rate of the apparent increase in the TIRF signal while decreasing the detection sensitivity because of elevated background.
The 2,000-molecule detection limit for IL-1β is as low as that reported earlier6,8. The average rate of IL-1β secretion from a single monocyte calculated from bulk measurements (Supplementary Fig. S6) was approximately 40 molecules/min if all the monocytes continuously secreted IL-1β, implying that it takes more than 50 min to detect IL-1β secretion using our assay system. However, we observed sharp rise of IL-1β signals in some cells, which indicated the massive IL-1β secretion was in a transiently-released manner. This shows the power of our real-time imaging system, which allows the dynamic analysis with improved time resolution as well as analysis for temporal cell-to-cell variations in protein secretion. The concave-down increase of the IL-1β secretion signal also suggested a burst release, rather than continuous secretion of IL-1β. Use of the time constant for association of detection antibody, obtained from the results of a model experiment, enabled estimation of the onset time of a massive burst release of IL-1β with a time resolution of <1 min. The accuracy of onset time estimation was sufficient for identifying the relationship of biophysical phenomena on a minute-based time scale. Smaller time intervals of data acquisition improve the accuracy of curve fitting within a predetermined time window of 5 min, but simultaneously reduce the number of observable microwells during a single cycle of scanning. The nonequilibrium characteristics of the antigen-antibody interaction in the open structure made it difficult to establish a series of concentration standards, but were considered an acceptable trade-off for long-term monitoring of cellular activities.
In addition to previous studies15,16, our snapshot measurement of IL-1β secretion from monocytes stained with live/dead indicators demonstrated that most of the IL-1β–secreting cells were dead. These observations allow for several interpretations, because the chronological relationship between IL-1β release and the change in calcein/SYTOX staining remained unknown. However, our real-time monitoring experiment showed that IL-1β release always occurred after calcein/SYTOX transition. Since the monitored cells were prepared in the presence of glycine16, both the calcein/SYTOX transition and IL-1β release were likely caused by pore formation on the plasma membrane, and not by cytolysis16,17. Two phases of SYTOX influx were observed during and after ATP administration to monocytes, suggesting multiple cell-permeable processes, such as the ATP-dependent opening of a P2X7 pore allowing for the passage of molecules of up to 900 Da in size19,26 and large pore formation mediated by caspase 120. Moreover, our analysis of high temporal resolution uncovered a several-minute lag between calcein/SYTOX transition and IL-1β release. This lag time suggested that the calcein/SYTOX-permeable cells require another process to release IL-1β, a process that remains to be elucidated.
The cytokine secretion dynamics in monocytes described herein implies that cell-cell communication via cytokines also varies widely with regard to timing. Further studies are required to clarify the manner in which heterogeneous cell-cell communication affects the maintenance of homeostasis of the immune system under various complex physiological situations, such as T-helper subset differentiation27 and the switch from acute resolving to chronic persistent inflammation28. The establishment of the real-time secretion assay platform described in this study opens the way to addressing these issues through the monitoring of cytokine secretion dynamics in parallel with intracellular events at single-cell resolution.
Methods
Reagents
The DuoSet ELISA Development System was purchased from R&D Systems (Minneapolis, MN, USA), and the set of capture and detection antibodies was used for sandwich immunoassays for human IL-1β (DY201). Human IL-6 monoclonal antibody (clone 6708; MAB206) and human IL-6 biotinylated affinity-purified polyclonal antibody (BAF206) were also purchased from R&D Systems. Adenosine 5′-triphosphate disodium salt hydrate (ATP, A7699) and lipopolysaccharides from Escherichia coli 055:B5 (LPS, L4524) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Lipidure BL802, a water-soluble polymer of 2-methacryloyloxy ethyl phosphorylcholine, was purchased from NOF Corporation (Tokyo, Japan). Calcein AM (C3099) and SYTOX Blue nucleic acid stain (S11348) were purchased from Life Technologies (Carlsbad, CA, USA). CF660R streptavidin was purchased from Biotium (29040; Hayward, CA, USA). Dimethyl pimelimidate-2HCl (DMP, 21666) was purchased from Thermo Fisher Scientific (Rockford, IL, USA). Foetal bovine serum (FBS, s1560) was purchased from Biowest (Nuaillé, France).
Cell culture
The incubation of cells was performed in a CO2 incubator at 37°C in a humidified atmosphere with 5% CO2, unless otherwise indicated. For the isolation of human peripheral blood monocytes, 20 mL of venous blood was drawn from a healthy donor after obtaining institutional approval of the ethical committee of the Kyoto University Hospital, in accordance with Declaration of Helsinki. Monocytes from different donors were used for different experiments. The cell fraction was separated with Lymphoprep (Axis-Shield, Dundee, UK) according to the manufacturer's instructions. CD14+ monocytes were sequentially purified by MACS (Miltenyi Biotec, Bergisch Gladbach, Germany) with a negative selection reagent (i.e. monocyte isolation kit II) and a positive selection reagent (i.e. CD14 microbeads). The isolated monocytes were incubated overnight in RPMI medium containing 10% FBS.
Optical arrangement
All measurements were performed with a completely automated inverted microscope (ECLIPSE Ti-E; Nikon, Tokyo, Japan) equipped with a high NA 60× objective lens (TIRF 60 × H; NA, 1.49; Nikon). The microscope was customized to introduce external laser beams (635 nm; Radius 635-25; Coherent, Santa Clara, CA, USA) at the indicated incident angle to actualize TIRF illumination. The following sets of excitation (Ex) and emission (Em) filters and a dichroic mirror (DM) were used with a high-pressure xenon lamp: for SYTOX, Ex = FF01-448/20-25, Em = FF01-482/25-25, and DM = Di02-R442-25x36; and for calcein, Ex = FF02-472/30-25, Em = FF01-520/35-25, and DM = FF495-Di03-25x36. The following Ex and Em filters and DM were used with the 632 nm laser for the CF660R dye: Ex = FF02-628/40-25, Em = FF01-692/40-25, and DM = FF660-Di02-25x36. These optical filters were purchased from Semrock (Rochester, NY, USA). Each image was projected on an EM-CCD camera (ImagEM C9100-13; Hamamatsu Photonics K.K., Sizuoka, Japan) through a 0.7× lens (C-0.7x DXM Relay Lens; Nikon). A stage-top incubator (ONICS; Tokai Hit Co., Shizuoka, Japan) was used to control temperature, humidity, and gas concentration.
MWA chips
MWA chips were prepared from PDMS and microscopic-grade coverslips. The PDMS MWA chip was prepared as follows: A thin PDMS sheet, consisting of a 50 × 50 array of 50-μm or 70-μm through-holes with 100-μm centre-to-centre spacing and 80-μm thickness, was purchased from Fluidware Technologies (Saitama, Japan). The PDMS sheet and a glass slide were permanently bonded together after the contact surfaces between them were plasma-treated (SEDE-PFA; Meiwafosis, Tokyo, Japan). The bare glass surfaces that now functioned as the bottom of the microwells after bonding were aminated with Vecatabond Reagent (SP-1800; Vector Laboratories, Burlingame, CA, USA). The chip was mounted on a coverslip holder (Attofluor cell chamber, A7819; Life Technologies) adjacent to another PDMS block (Sylgard184; Dow Corning Toray, Tokyo, Japan) with an 8-mm diameter through-hole that served as a reservoir well. The interstices between the PDMS-glass chip and the PDMS block were filled with uncured PDMS. The integrated chip was baked at 130°C for 3 h.
A 100-μL mixture of capture antibodies (100 μg/mL) and dimethyl pimelimidate-2HCl (DMP; 7 mg/mL) was loaded onto the aminated glass surface to fix the capture antibody. The surface was blocked with monoethanolamine (0.1 M, pH 8.2) and Lipidure reagent (0.2% [w/v]), and stored at 4°C until use.
Preparation of detection medium
Detection antibody labelled with biotin was coupled with CF-labelled streptavidin at 1:10 molar ratios at room temperature in the dark for 3 h. Unoccupied sites on streptavidin were blocked with excess dPEG4-biotin acid (10199; Quanta BioDesign, Ltd., Powell, OH, USA). Unconjugated streptavidin and dPEG4-biotin were removed by ultrafiltration (Amicon Ultra-0.5, 100 kDa; Merck Millipore, Billerica, MA, USA). The detection media contained prepared CF-labelled detection antibody (30 nM), BSA (2.5% [w/v]), and the indicated combination of the following additives (with the final concentrations): SYTOX (0.8 μM), glycine (5 mM), LPS (1 μg/mL), and/or ATP (5 mM).
Handling and treatment of cells
Monocytes were stimulated with LPS for 3 h and incubated with 0.4 μM Calcein AM for 15 min before IL-1β secretion analysis on a PDMS MWA chip. The cells were washed with fresh medium and recovered from the bottom of 96-well plates by gentle pipetting. Approximately 2,000 cells were introduced into an MWA chip and allowed to settle by gravity for 10 min in a CO2 incubator. Next, the medium was replaced with the detection medium containing SYTOX, glycine, and LPS. The cells were first monitored for 20 min before ATP stimulation; after ATP administration, monitoring was resumed during the 10-min incubation period. The medium was then replaced with fresh detection medium containing SYTOX, glycine, and LPS. Measurements were then resumed and continued for 4 h.
Time-resolved monitoring of cytokine secretion from stimulated monocytes
Ten positions were selected in order to include as many single cell-containing microwells as possible. Monocytes in the selected position of microwells were monitored at 1-min intervals by multichannel microscopy, i.e. DIA images, epifluorescence images for calcein and SYTOX, and TIRF images for CF660R for cytokine secretion at controlled temperature and gas concentrations. The MFIs of each microwell at each time point were measured with the NIS Elements imaging software.
Transition time estimation of calcein, SYTOX and IL-1β signals obtained from LPS/ATP stimulated monocytes
The transition time of calcein disappearance, SYTOX influx, and IL-1β release were detected by data analysis software (Origin 8.6; OriginLab Co., Northampton, MA, USA) by using the following equations:
 |
for calcein disappearance,
 |
for SYTOX influx, and
 |
for IL-1β release, where I(t) represents intensity, I0 represents the background, m represents intensity drift, I1 represents amplitude, t0 represents transition time, and τ represents the time constant of exponential decay for each fluorescence signal. The subscripts c, s, and i of each parameter indicate calcein, SYTOX, and IL-1β labelling, respectively. The time constant of IL-1β (τi) was determined from a supplementary experiment with recombinant IL-1β (Fig. 2). The IL-1β secretion dataset was fitted by equation (3).
Author Contributions
Y.S., Y.H. and O.O. conceived the methods described; Y.S. and M.Y. set up the optical instruments; A.N., J.M. and S.S. contributed to the technical development of the MWA chip; Y.S. and N.S. prepared MWA chips; K.I., R.N. and T.H. performed and supervised clinical sample preparation; and Y.S. performed imaging experiments and analysis. The manuscript was prepared by Y.S., M.Y., and O.O. after discussion with all the authors.
Supplementary Material
movie 1
movie 2
Supplementary information
Acknowledgments
We thank Katsuyuki Shiroguchi (RIKEN) and Hirotsugu Oda (Kyoto Univ.) for critical reading of the manuscript. This work was partly supported by the Special Postdoctoral Researchers Program of RIKEN, by a Grant-in-Aid for Young Scientists (B) (no. 23770189, to Y.S.), a Grant-in-Aid for Scientific Basic Research (C) (no. 25440074, to Y.S.) and for Scientific Basic Research (S) (no. 23226010, to S.S.) from MEXT, Japan.
References
- Gnecchi M., Zhang Z. P., Ni A. G. & Dzau V. J. Paracrine Mechanisms in Adult Stem Cell Signaling and Therapy. Circ Res 103, 1204–1219 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander A. D. How Cells Know Where They Are. Science 339, 923–927 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stastna M. & Van Eyk J. E. Secreted proteins as a fundamental source for biomarker discovery. Proteomics 12, 722–735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg E. V. Cell lineage regulators in B and T cell development. Nat Immunol 8, 441–444 (2007). [DOI] [PubMed] [Google Scholar]
- Lacy P. & Stow J. L. Cytokine release from innate immune cells: association with diverse membrane trafficking pathways. Blood 118, 9–18 (2011). [DOI] [PubMed] [Google Scholar]
- Ma C. et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med 17, 738–743 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love J. C., Ronan J. L., Grotenbreg G. M., van der Veen A. G. & Ploegh H. L. A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat Biotechnol 24, 703–707 (2006). [DOI] [PubMed] [Google Scholar]
- Han Q., Bagheri N., Bradshaw E. M., Hafler D. A., Lauffenburger D. A. & Love J. C. Polyfunctional responses by human T cells result from sequential release of cytokines. P Natl Acad Sci USA 109, 1607–1612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasuga Y. et al. Single-cell chemical lysis method for analyses of intracellular molecules using an array of picoliter-scale microwells. Anal Chem 80, 9141–9149 (2008). [DOI] [PubMed] [Google Scholar]
- Salehi-Reyhani A. et al. A first step towards practical single cell proteomics: a microfluidic antibody capture chip with TIRF detection. Lab Chip 11, 1256–1261 (2011). [DOI] [PubMed] [Google Scholar]
- Stanley A. C. & Lacy P. Pathways for Cytokine Secretion. Physiology 25, 218–229 (2010). [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. Biologic basis for interleukin-1 in disease. Blood 87, 2095–2147 (1996). [PubMed] [Google Scholar]
- Fujisawa A. et al. Disease-associated mutations in CIAS1 induce cathepsin B-dependent rapid cell death of human THP-1 monocytic cells. Blood 109, 2903–2911 (2007). [DOI] [PubMed] [Google Scholar]
- Tanaka T. et al. Induced pluripotent stem cells from CINCA syndrome patients as a model for dissecting somatic mosaicism and drug discovery. Blood 120, 1299–1308 (2012). [DOI] [PubMed] [Google Scholar]
- Bergsbaken T., Fink S. L. & Cookson B. T. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7, 99–109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder C. Mechanisms of interleukin-1beta release. Immunobiology 214, 543–553 (2009). [DOI] [PubMed] [Google Scholar]
- Lopez-Castejon G. & Brough D. Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev 22, 189–195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006). [DOI] [PubMed] [Google Scholar]
- Ward J. R. et al. Temporal Interleukin-1 beta Secretion from Primary Human Peripheral Blood Monocytes by P2X7-independent and P2X7-dependent Mechanisms. Journal of Biological Chemistry 285, 23145–23156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink S. L. & Cookson B. T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8, 1812–1825 (2006). [DOI] [PubMed] [Google Scholar]
- Verhoef P. A., Kertesy S. B., Estacion M., Schilling W. P. & Dubyak G. R. Maitotoxin induces biphasic interleukin-1beta secretion and membrane blebbing in murine macrophages. Mol Pharmacol 66, 909–920 (2004). [DOI] [PubMed] [Google Scholar]
- Roy S. S. & Hajnoczly G. Calcium, mitochondria and apoptosis studied by fluorescence measurements. Methods 46, 213–223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael M. P., Christodoulides J. A., Delehanty J. B., Long J. P. & Byers J. M. Quantitative Imaging of Protein Secretions from Single Cells in Real Time. Biophys J 105, 602–608 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molter T. W. et al. A New Approach for Measuring Single-Cell Oxygen Consumption Rates. IEEE Trans Autom Sci Eng 5, 32–42 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M. C. & Marois Y. Hemocompatibility, biocompatibility, inflammatory and in vivo studies of primary reference materials low-density polyethylene and polydimethylsiloxane: A review. J Biomed Mater Res 58, 467–477 (2001). [DOI] [PubMed] [Google Scholar]
- Ferrari D. et al. The P2X(7) receptor: A key player in IL-1 processing and release. Journal of Immunology 176, 3877–3883 (2006). [DOI] [PubMed] [Google Scholar]
- Zhu J. F. & Paul W. E. Heterogeneity and plasticity of T helper cells. Cell Res 20, 4–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. & Ding A. H. Nonresolving Inflammation. Cell 140, 871–882 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
movie 1
movie 2
Supplementary information