

Abstract
Background
SCN2A is a gene that codes for the alpha subunit of voltage-gated, type II sodium channels, and is highly expressed in the brain. Sodium channel disruptions, such as mutations in SCN2A, may play an important role in psychiatric disorders. Recently, de novo SCN2A mutations in autism spectrum disorder (ASD) have been identified. The current study characterizes a de novo splice site mutation in SCN2A that alters mRNA and protein products.
Case presentation
We describe results from clinical and genetic characterizations of a seven-year-old boy with ASD. Psychiatric interview and gold standard autism diagnostic instruments (ADOS and ADI-R) were used to confirm ASD diagnosis, in addition to performing standardized cognitive and adaptive functioning assessments (Leiter-R and Vineland Adaptive Behavior Scale), and sensory reactivity assessments (Sensory Profile and Sensory Processing Scales). Genetic testing by whole exome sequencing revealed four de novo events, including a splice site mutation c.476 + 1G > A in SCN2A, a missense mutation (c.2263G > A) causing a p.V755I change in the TLE1 gene, and two synonymous mutations (c.2943A > G in the BUB1 gene, and c.1254 T > A in C10orf68 gene). The de novo SCN2A splice site mutation produced a stop codon 10 amino acids downstream, possibly resulting in a truncated protein and/or a nonsense-mediated mRNA decay. The participant met new DSM-5 criteria for ASD, presenting with social and communication impairment, repetitive behaviors, and sensory reactivity issues. The participant’s adaptive and cognitive skills fell in the low range of functioning.
Conclusion
This report indicates that a splice site mutation in SCN2A might be contributing to the risk of ASD. Describing the specific phenotype associated with SCN2A mutations might help to reduce heterogeneity seen in ASD.
Keywords: DSM-5, autism spectrum disorder, de novo SCN2A splice site mutation
Background
Autism spectrum disorders (ASDs) are a group of neurodevelopmental disorders characterized by social and communication impairments, repetitive behaviors, and sensory reactivity issues [1]. The etiology of ASD is genetically heterogeneous and de novo mutations and copy number variations account for 15-20% of ASD cases [2]. Recently, Sanders et al. [14] and Neale et al. [3] reported de novo SCN2A mutations (which codes for the alpha subunit of voltage-gated, type II sodium channel) in three cases with ASD, one of which [3] will be described in detail herein.
SCN2A is involved in generating action potentials in neurons and muscles and is highly expressed in the brain, specifically in neurons and glia [4]. A recent paper highlights the role of sodium channels in psychiatric disorders and neurological diseases [4]. Sodium-channel disruption, caused by mutations on SCN2A and related genes such as SCN1A or SCN9A, has been associated with ASD, intellectual disability, ataxia and increased sensitivity to pain [5-8]. SCN2A mutations have also been associated with various forms of epilepsy [9-12]. A case report documented seizures in a participant with SCN2A mutation, along with abnormal visual fixation and poor muscle tone [13]. However, not all patients with SCN2A mutations have been reported to have epilepsy [14,15].
In the current study, we characterize a de novo splice site mutation in SCN2A that alters mRNA. We used gold standard autism diagnostic instruments, structured clinical interview, standardized cognitive testing, and sensory reactivity assessments to probe for diagnostic status, cognitive and adaptive functioning, and sensory reactivity, including hyper-reactivity to stimuli in the environment. Describing phenotypes of SCN2A cases will eventually help to reduce the phenotypic heterogeneity seen in individuals with ASD.
Case presentation
Methods
The participant was ascertained as part of ongoing IRB-approved genetic research in ASD at the Seaver Autism Center for Research and Treatment at the Icahn School of Medicine at Mount Sinai. Genetic testing included chromosomal microarray analysis (CMA) (Agilent Human CGH 1 × 244A, Agilent Technologies, Santa Clara, CA) and whole exome sequencing (WES). Clinical assessments included: (1) neurological examination conducted by a pediatric neurologist; (2) psychiatric evaluation using the DSM [16] conducted by a board-certified child and adolescent psychiatrist; (3) the Autism Diagnostic Observation Schedule (ADOS) [17]; (4) the Autism Diagnostic Interview Revised (ADI-R); (5) cognitive testing using the Leiter International Performance Scale-Revised [18]; (6) adaptive ratings using the Vineland Adaptive Behavior Scales-II, Survey Edition [19]; and (7) tests of sensory reactivity, including the Caregiver Sensory Profile, a 125-item questionnaire [20] and the Sensory Processing Scales [21]. Magnetic Resonance Imaging (MRI) was conducted to check for structural brain abnormalities (Table 1).
Table 1.
Domains and measures used in the current study
| Domain | Measure |
|---|---|
| Genetic testing | |
| |
WES |
|
CMA | |
| Clinical assessment | |
| Neurological examination |
Clinical genetics; height, weight, dysmorphic features |
| Autism spectrum disorder (ASD) traits |
ADOS |
| |
ADI-R |
| Cognitive and adaptive behavior skills |
Leiter-R |
| |
Vineland Adaptive Behavior Scale |
| Sensory reactivity |
Sensory Profile |
| |
Sensory Over-Responsivity Scale |
| Brain development |
|
| Structural Imaging | MRI |
Abbreviations: CMA = chromosomal microarray analysis; WES = whole exome sequencing; ADOS = Autism Diagnostic Observation Schedule; ADI-R = Autism Diagnostic Interview Revised; MRI = Magnetic Resonance Imaging.
Genetic testing
Whole exome sequencing
High quality genomic DNA from peripheral blood of the participant and both parents were used for library preparation. A total of 3 ug DNA were sheared separately with a Covaris Acoustic Disruptor (E210) instrument set to time 250 sec, duty cycle 10, intensity 10 and 200 cycles per burst. The size distribution of fragments was centered about 200 bp and verified by Agilent Bioalayzer. NEBNext™ DNA Sample Prep Master Mix Set 1 (New England BioLabs) was used for creating the DNA library. During the adaptor ligation, a six-base sequence index was introduced into the adapter for multiplex sequencing on the Illumina platform.
NimbleGen Seq Cap EZ SR v2 kit (Roche NimbleGen) was used for whole exome library preparation using standard protocol. Prepared libraries were then sequenced with the Hiseq 2000 - V2.5 flowcells. The standard Illumina software was used for de-multiplexing.
Sequencing data was processed with Picard ( http://picard.sourceforge.net/), which utilizes base quality-score recalibration and BWA for mapping reads to hg19. Genetic variants were called using GATK and were annotated by AnnTools ( http://anntools.sourceforge.net/). Potential de novo mutation was defined as a heterozygous genotype in the participant and observed reference homozygote genotypes in both parents and in the absence of any other copy of the alternate allele. Each putative de novo event was validated in the participant and both parents using the Sanger sequencing method.
RT-PCR and cDNA sequencing in lymphoblastoid cell lines
Lymphoblastoid cell lines from the participant and parents were cultured and used to isolate total RNA using the RNeasy Mini kit (Qiagen). SuperScript® II reverse transcriptase and random primers (Life Technology) were used to generate cDNA from the RNA. Different pairs of primer from exon 2 to exon 6 were designed to amplify wildtype and mis-spliced cDNA.
Clinical assessments
Autism spectrum disorder (ASD) traits
The Autism Mental Status Exam (AMSE) was used to structure the direct observation of the subject during the clinical evaluation and was performed by a child and adolescent psychiatrist with expertise in ASD [22]. The ADOS, Module 1, was administered at age three and again at age seven. The ADI-R was conducted with the mother when the participant was three years old [23].
Cognitive and adaptive evaluations
The Leiter-R relies on nonverbal instructions for administration, requires minimal motor skills, and measures only nonverbal elements of cognitive functioning [18]. Ratio IQs were calculated using mental age estimates from the cognitive tests and used to provide an estimate of nonverbal IQ. The Vineland Adaptive Behavior Scale was used to measure communication, daily living skills, socialization and motor skills [19].
Sensory assessment
The Sensory Profile is a validated instrument, which was standardized using a sample of over 1000 typical developing children [20]. The Sensory Profile is a 125-item questionnaire that has identified sensory differences in over 90% of children and adults with ASD, compared to controls [24-31] and was used to query parents about the frequency (1 = always; 5 = never) of sensory behaviors such as, ‘avoids going barefoot, especially in sand or grass (item 3). The parents also filled out the Sensory Processing Scale (SP Scale) [21,32], which includes 60 items for Sensory Over-Responsivity (SOR) (These smells bother my child, e.g. soap) and 30 items for Sensory Under-Responsivity (Typically my child does not notice e.g. strong odors). Unlike other scales, the SP Scale reflects sensory processing across all sensory domains (tactile, visual, olfactory, auditory, vestibular, proprioception and gustatory). Parents complete the Inventories by scoring each item as a "1" if the behavior describes their child. Total scores are then computed for each subtype.
Results
Whole exome sequencing results
The participant first presented to the Seaver Autism Center for Research and Treatment at Mount Sinai for evaluation at the age of three. At that time, chromosome microarray was performed and no pathogenic alterations were detected. Follow-up testing using whole exome sequencing subsequently identified the SCN2A mutation, and the participant returned for another clinical assessment at age seven.
A total of four de novo events were identified by whole exome sequencing, including a splice site mutation c.476 + 1G > A in SCN2A, a missense mutation (c.2263G > A) causing a p.V755I change in the TLE1 gene, and two synonymous mutations (c.2943A > G in the BUB1 gene, and c.1254 T > A in C10orf68 gene). All of the four de novo mutations were heterozygous mutations and none have been reported in the dbSNP or other genomic databases. We validated each mutation and its de novo status by Sanger sequencing in the participant and parental DNA.
The SCN2A splice site mutation was in the donor site of intron 4 of the longest isoform (NM_021007) of SCN2A. This exon is conserved in two other isoforms (NM_001040142 and NM_001040143) of the gene as reported in the Refseq gene database.
Since the splice site mutation could affect mRNA splicing, we further examined cDNA of SCN2A in cell lines from the participant and parents (see Figure 1). cDNA sequencing showed both exon 3 and exon 4 were spliced out in the participant, while it is expressed normally in the parents. The mis-splicing caused 209 nt deletion in the mRNA and altered the reading frame of the mRNA starting with amino acid 90. It produced a stop codon 10 amino acids downstream, possibly resulting in a truncated protein and/or a nonsense-mediated mRNA decay.
Figure 1.
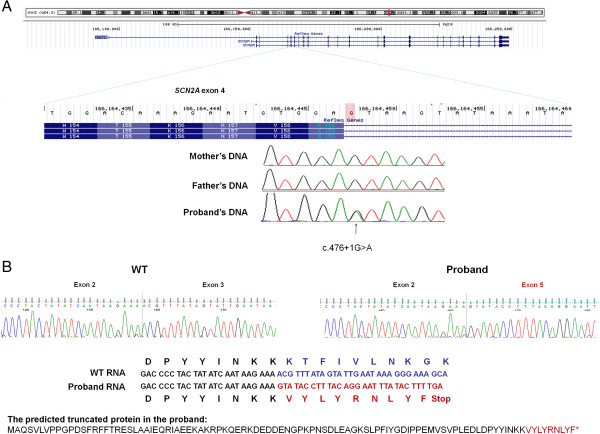
A de novo SCN2A splice mutation in a participant with ASD. (A) The de novo splice mutation c.476 + 1G > A is in the donor site of intron 4, which is conserved over three transcripts of SCN2A gene. (B) cDNA sequencing in the lymphoblastoid cell lines shows both exon 3 and exon 4 were spliced out in the participant, likely resulting in a truncated short protein.
Clinical assessments
Neurological and clinical genetic examinations
The participant’s height was 136 cm at age seven years and nine months (75th percentile), his weight was 32.8 kg (82nd percentile) and his head circumference was 52.2 cm (50th percentile). The participant showed some mild dysmorphic features, including eye asymmetry, fleshy earlobes, and a gap between front teeth. On a cardiovascular exam, the participant had regular heart rate and rhythm, and no murmur was apparent. Motor examination revealed mild hypotonia, but normal strength. Deep tendon reflexes and extensor signs were tested but difficult to elicit. There was no clonus. The participant exhibited a broad-based, abnormal gait with some waddling. He was unsteady standing or walking on uneven surfaces. However there were no abnormal movements. Cranial nerve examination was normal and visual fields were full. Sensory examination elicited symmetrical responses to touch and pain.
Psychiatric evaluation
The participant’s parents reported longstanding impairment in social and communicative functioning. His parents reported that his receptive language skills were more advanced than his expressive language. There was no report of delayed or immediate echolalia but there was report of intermittent vocal stereotypy. The participant did not exhibit the capacity for oral motor imitation skills. In the behavioral domain, the participant demonstrated repetitive, compulsive-like behaviors, including repetitive opening and closing of doors. Motor stereotypies were characterized by arm flapping. Encompassing pre-occupations were reported as specific pre-occupations with toys that emit noise.
The participant also had a low frustration tolerance and displayed intermittent tantrum behaviors, which in their severest form included banging his head with his arms and fists. With regard to adaptive functioning, the participant was not consistently bladder trained at night and was not bowel trained during the day. With regard to sensory symptoms, the participant had a notably high pain threshold, and he did not tolerate certain types of touch (e.g. getting a haircut). The participant was described as a picky eater.
As for medical history, the parents reported no history of seizures. However, in the first year of life there were frequent episodes of becoming "stone faced" and "limp". Pregnancy and birth were normal. Parents were both in their mid-thirties when the participant was born. There was no family history of ASD, language delay, intellectual disability or Attention Deficit/Hyperactivity Disorder. Family history was positive for Obsessive Compulsive Disorder.
Regarding developmental history, the participant had motor delays; he did not sit or crawl and only began walking at approximately two years of age. The participant had longstanding language impairment and had only begun using single words at age 4, after beginning Applied Behavioral Analysis (ABA). There was no report of loss of skills. As for educational history the participant was referred for early intervention services at 18 months due to concerns about motor and language delays at which time he began receiving occupational therapy (OT) and physical therapy (PT).
Autism spectrum disorder diagnostic test
Mental status exam
The participant showed significant social impairment and was unable to engage in joint attention or social reciprocity on examination. Although he was able to make fleeting eye contact, he was unable to point or follow pointing. Further, the participant showed motor stereotypies such as hand flapping and engaged in compulsive opening and closing of doors.
ADOS
Overall, the participant’s scores were consistent with a classification of autism in the areas of communication, reciprocal social interactions and repetitive behaviors, and in total at age three and at age seven. As for communication, at age seven the participant directed vocalizations towards the examiner infrequently and used mostly single words, along with an occasional phrase for the purpose of requesting (e.g. "Go bathroom"). The intonation of his speech tended either to be flat or to take on a "sing-song" quality. With respect to nonverbal communication, he did not point or use any other common gestures throughout the assessment (e.g., head nodding, shrugging).
As for the social domain, the participant demonstrated several emerging social skills, including the ability to respond to his name as well as bids for joint attention. However, eye contact and social responsiveness were inconsistent throughout the observation.
Regarding repetitive and stereotyped behaviors, the participant demonstrated sensory interests and hand mannerisms, such as touching the length of his index finger to his mouth and flapping his hands, respectively.
ADI-R
The participant met criteria for autism in all three diagnostic domains (Qualitative Abnormalities in Reciprocal Social Interaction, Qualitative Abnormalities in Communication, and Restricted, Repetitive and Stereotyped Patterns of Behavior) as reported by his mother.
According to his mother’s report, the participant was generally unresponsive to unfamiliar people, but he was responsive to familiar individuals, smiled, and displayed a range of appropriate facial expressions. Language was described as limited to two-word phrases, and beyond shaking his head or occasionally nodding, he did not use nonverbal gestures. Finally, the participant displayed excessive interest in books, prompting his parents to carry a book with them everywhere; he was reported to line up his toys, tended to focus on part of his toys rather than the whole toy, and was particularly drawn to visual reflections.
Cognitive and adaptive evaluations
Leiter international performance subscales
At age three years and nine months, the participant’s Full Scale IQ was estimated to be 53, which falls into the extremely low range of functioning. At age seven years and nine months, the estimated Full Scale IQ was 45, which falls in the extremely low range as well. This apparent decline in IQ between the ages of three and seven is unlikely to be related to loss of skills but instead due to the lack of appropriate gains as compared to typically developing peers. The participant had difficulty attending to tasks and could not complete all subtests. A relative strength was the participant’s ability to match basic visual-perceptual stimuli (see Table 2).
Table 2.
Leiter International Performance Scale scores at ages three and seven
| Subtest | Scaled score at age 3 years 9 month | Scales score at age 7 years and 9 month |
|---|---|---|
|
Visualization
|
|
|
|
Figure ground |
4 |
1 |
|
Matching |
5 |
1 |
|
Picture context |
4 |
- |
|
Paper folding |
- |
2 |
|
Form completion |
5 |
1 |
|
Reasoning
|
|
|
|
Classification |
2 |
- |
|
Sequential order |
3 |
5 |
|
Design analogies |
- |
5 |
| Repeated patterns | 4 | 1 |
Adaptive behavior
The participant had an overall adaptive behavior composite score and sub scores in the low to moderately low range (see Table 3).
Table 3.
Summary of Vineland Adaptive Behavior Scale results
| Vineland Adaptive Behavior Scale | SCN2A case | Compared to normative scores |
|---|---|---|
|
Adaptive behavior composite
|
64 |
1st percentile, low range |
|
Communication
|
35 |
8th percentile, low range |
|
Daily living skills
|
25 |
1st percentile, low range |
|
Socialization
|
30 |
3rd percentile, low range |
| Motor skills | 17 | 1st percentile, low range |
Sensory assessment
Sensory profile
The participant presented with sensory reactivity abnormalities as reported by his parents on the widely used Sensory Profile. Definite differences were reported (2 standard deviations above the mean) on oral, tactile, multisensory and vestibular processing. Probable differences (one standard deviation above from mean) were reported on auditory processing (see Table 4). In terms of sensory modulation, definite differences were shown on sensory processing related to endurance/tone, modulation of movement affecting activity level, and modulation of sensory input affecting emotional response. On the Factors of the Sensory Profile, the participant showed definite differences on all factors other than sensory sensitivity, which was in the typical range. Responses on all other factors including sensory seeking, poor registration, low endurance, fine motor, sedentary and distractibility showed definite differences as compared to typically developing children. Taken together the participant shows severe sensory reactivity issues.
Table 4.
Summary of the participant’s sensory profile scores
| Sensory processing | SCN2A case | Typical performance | Probable difference | Definite difference |
|---|---|---|---|---|
|
Oral sensory processing
|
25 |
55-48 |
47-45 |
44-11 |
|
Touch processing
|
59 |
90-73 |
72-65 |
64-18 |
|
Multisensory processing
|
19 |
35-27 |
26-24 |
23- 7 |
|
Vestibular processing
|
43 |
55-48 |
47-45 |
44-11 |
|
Visual processing
|
26 |
45-32 |
31-27 |
26-9 |
| Auditory processing | 26 | 40-30 | 29-26 | 25-8 |
The classifications of typical, probable and definite differences were based on the performance of children without disabilities (n = 1,037, see Dunn, 1997). Probable differences are characterized as being over 2 standard deviations away from norm and definite differences as being over 3 standard deviations away from norm.
Sensory over-responsivity inventory
The parents endorsed 35% of the items (21 out of 60) on the Sensory Over-Responsivity Scale (see Table 5). The participant’s score of 21 fell into the category of being sensory over-responsive. Specifically, tactile accessories (scarf, hat etc.), fuzzy or furry textures, wool clothes, washing or wiping face, cutting nails, having hair cut, and light stroking of the skin bother the participant. In terms of vision, brightly color material, fluorescent light, visually cluttered environments, and busy pictures bothered the participant. In terms of sound, small motor noises, restaurants and large gatherings bothered the participant. In addition, the parents endorsed 14 out of 31 items on the Sensory Under Responsivity Inventory. Parents reported that the participant responds less than others to bruises or cuts, hurting himself, dirtying himself, and bumping into things. In addition, the participant does not notice when his hands or face are messy/dirty, when he needs to use the toilet, or if an object cames close to his eyes.
Table 5.
Participants scores on the sensory over- responsivity scale
| Sensory processing | SCN2A score | Typical performance | Children with sensory processing difficulties |
|---|---|---|---|
| Sensory over-reactivity | 21 | 4 ± 4.7 | 18.5 ± 13.9 |
Scores for typically developing participant’s and participants with sensory difficulties were taken from Schoen, et al. 2008 [32].
Brain development
MRI
A brain MRI was performed using a Siemens Allegra 3 T head-only scanner. The research protocol on the diagnostic images showed no evidence of infra or supratentorial masses. The lateral ventricles appeared normal. No midline shift was appreciated and the ventricles and sulci were within normal limits. No white matter abnormalities were appreciated. The flow voids in the vessels appeared grossly within normal limits. The overall impression showed a normal research protocol of the brain.
Conclusions
The current study describes a 7-year-old boy with a de novo SCN2A splice site mutation (c.476 + 1G > A). Although this particular mutation has not been reported before, it could be considered as contributing to risk of ASD by causing abnormal gene splicing, leading to significantly shortened protein product and/or an abnormal message that is subject to nonsense-mediated mRNA decay. The participant also presented with three other de novo events, one missense mutation in TLE1 gene, and two synonymous mutations in BUB1 gene. While the four de novo mutations support the notion of multiple hits in ASD, the de novo SCN2A splice site mutation, a recently identified ASD gene, is likely to be driving the severity of symptoms. The participant presented with ASD as diagnosed using psychiatric evaluation and confirmed by the ADOS and ADI-R. The participant showed social and communication problems, repetitive behaviors and sensory over- and under-reactivity. Neurological and clinical genetic examinations revealed hypotonia, abnormal gait and fine motor coordination, and mild dysmorphic features. Further, the participant’s adaptive and cognitive skills fell in the low range of functioning. No structural brain abnormalities were evident on MRI, which is consistent with past reviews of participants with sodium channel disruptions [11].
Interestingly, the participant does not have any history of seizures. Participants with SCN2A mutations have been identified with seizures and various forms of epilepsy [15]. However, in the current case, no seizures were observed. Although electroencephalography was attempted, the participant’s sensory reactivity issues prevented any usable data from being recorded. The absence of seizures is consistent with a recent study by Sanders et al. (2012), which reported two de novo SCN2A cases that did not show any history of seizures [14]. However, in our study it was noted that there were frequent episodes of becoming "stone faced" and "limp" in the first year of life that may have been indicative of seizure activity. On the other hand, since SCN2A mutations are linked to various forms of epilepsy, a late onset cannot be ruled out either [33].
This participant demonstrated with significant sensory reactivity issues. Most sensory reactivity differences were reported on oral, tactile, multisensory, vestibular, and visual processing followed, less markedly, by auditory processing. The participant fell into the ‘sensation seeker’ and ‘poor registration’ category. Further, the participant seemed to be overwhelmed by some aspects of the environment such as touch (washing face, wool clothes), while being under-responsive to other aspects of the environment (bruises, cuts). Pain related perception is processed via nociceptors (e.g., unmyelinated C fibres) whereas light touch is processed by cutaneous receptors (e.g. Meissner’s corpuscles or Merkel discs) [34]. Examining sensory reactivity in cases with ASD and SCN2A mutations is important because comorbid sensory symptoms often compromise an individual’s and family’s quality of life [35]. Sensory over-reactivity has also been linked to high rates of anxiety in individuals with ASD [33,36,37].
Future research is needed to confirm the phenotypes observed in the current case report in more cases with SCN2A mutations. Identifying a robust phenotype for SCN2A cases will help to reduce the phenotypic heterogeneity seen in individuals with ASD.
Consent
Written informed consent was obtained from the parents of the patients.
Competing interests
The authors of this manuscript report no competing interests.
Authors’ contributions
JDB, AK, DG, ATW, DH and TT participated in the design of the study. LS, LB, ATW and SS coordinated the study and carried out the data collection and MRI acquisition. JC-L, DH and DG carried out the clinical assessments. GC carried out chromosomal microarray analysis and whole exome sequencing. TT drafted the manuscript and all authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Teresa Tavassoli, Email: teresa.tavassoli@mssm.edu.
Alexander Kolevzon, Email: alexander.kolevzon@mssm.edu.
A Ting Wang, Email: ting.wang@mssm.edu.
Jocelyn Curchack-Lichtin, Email: jocelyn.curchack-lichtin@mssm.edu.
Danielle Halpern, Email: danielle.halpern@mssm.edu.
Lily Schwartz, Email: lily.schwartz@mssm.edu.
Sarah Soffes, Email: sarah.soffes@mssm.edu.
Lauren Bush, Email: lauren.a.bush@mssm.edu.
David Grodberg, Email: david.grodberg@mssm.edu.
Guiqing Cai, Email: guiqing.cai@mssm.edu.
Joseph D Buxbaum, Email: joseph.buxbaum@mssm.edu.
Acknowledgments
We would like to thank the participant and his family for taking part in this research. Further we would like to thank Katherine Bellesheim, Silvia De Rubeis and Arthur Goldberg for their help and valuable discussions.
Funding
This work was supported by grants from the Beatrice and Samuel A. Seaver Foundation. TT received funding from the Wallace Research Foundation, the Seaver Foundation and the Autism Science Foundation during the period of this work. A.K. received research support from the NIH, the Autism Science Foundation, the Seaver Foundation, and Hofmann-LaRoche.
References
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5. Arlington, VA: American Psychiatric Publishing; 2013. [Google Scholar]
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;15(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;15(7397):242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbrici P, Camerino DC, Tricarico D. Major channels involved in neuropsychiatric disorders and therapeutic perspectives. Front Genet. 2013;15:76. doi: 10.3389/fgene.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, Reichert J, Buxbaum JD, Meisler MH. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry. 2003;15(2):186–194. doi: 10.1038/sj.mp.4001241. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Kaneda M, Sugawara T, Mazaki E, Okamura N, Montal M, Makita N, Tanaka M, Fukushima K, Fujiwara T, Inoue Y, Yamakawa K. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. 2004;15(11):2690–2698. doi: 10.1523/JNEUROSCI.3089-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JN, Boorman J. Voltage-gated sodium channel blockers; target validation and therapeutic potential. Curr Top Med Chem. 2005;15(6):529–537. doi: 10.2174/1568026054367584. [DOI] [PubMed] [Google Scholar]
- Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;15(8):2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YW, Yu MJ, Long YS, Qin B, He N, Meng H, Liu XR, Deng WY, Gao MM, Yi YH, Li BM, Liao WP. Mosaic SCN1A mutations in familial partial epilepsy with antecedent febrile seizures. Genes Brain Behav. 2012;15(2):170–176. doi: 10.1111/j.1601-183X.2011.00756.x. [DOI] [PubMed] [Google Scholar]
- Raymond G, Wohler E, Dinsmore C, Cox J, Johnston M, Batista D, Wang T. An interstitial duplication at 2q24.3 involving the SCN1A, SCN2A, SCN3A genes associated with infantile epilepsy. Am J Med Genet A. 2011;15(4):920–923. doi: 10.1002/ajmg.a.33929. [DOI] [PubMed] [Google Scholar]
- Goeggel Simonetti B, Rieubland C, Courage C, Strozzi S, Tschumi S, Gallati S, Lemke JR. Duplication of the sodium channel gene cluster on 2q24 in children with early onset epilepsy. Epilepsia. 2012;15(12):2128–2134. doi: 10.1111/j.1528-1167.2012.03676.x. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kato M, Osaka H, Yamashita S, Nakagawa E, Haginoya K, Tohyama J, Okuda M, Wada T, Shimakawa S, Imai K, Takeshita S, Ishiwata H, Lev D, Lerman-Sagie T, Cervantes-Barragán DE, Villarroel CE, Ohfu M, Writzl K, Gnidovec Strazisar B, Hirabayashi S, Chitayat D, Myles Reid D, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Hayasaka K, Matsumoto N, Saitsu H. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013;15(11):992–998. doi: 10.1212/WNL.0b013e3182a43e57. [DOI] [PubMed] [Google Scholar]
- Pereira S, Vieira JP, Barroca F, Roll P, Carvalhas R, Cau P, Sequeira S, Genton P, Szepetowski P. Severe epilepsy, retardation, and dysmorphic features with a 2q deletion including SCN1A and SCN2A. Neurology. 2004;15(1):191–192. doi: 10.1212/01.WNL.0000132844.20654.C1. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;15(7397):237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartnik M, Chun-Hui Tsai A, Xia Z, Cheung SW, Stankiewicz P. Disruption of the SCN2A and SCN3A genes in a patient with mental retardation, neurobehavioral and psychiatric abnormalities, and a history of infantile seizures. Clin Genet. 2011;15(2):191–195. doi: 10.1111/j.1399-0004.2010.01526.x. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook EH Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M. The Autism Diagnostic Observation Schedule-Generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;15(3):205–223. doi: 10.1023/A:1005592401947. [DOI] [PubMed] [Google Scholar]
- Roid GH, Miller LJ. Leiter International Performance Scale-Revised. Wood Dale, IL: Stoelting Co.; 1997. [Google Scholar]
- Sparrow SS, Cicchetti DV, Balla DA. Vineland-II Survey Forms Manual (Vineland Adaptive Behavior Scales) 2. Minneapolis, MN: AGS Publishing; 2005. [Google Scholar]
- Dunn W. Sensory Profile. San Antonio: The Psychological Corporation; 1999. [Google Scholar]
- Miller LJ, Schoen SA. The sensory processing scales inventory: Sensory Over-Responsivity (SenSI: SOR) Am J Occup Ther. 2012;15(4):393–406. doi: 10.5014/ajot.62.4.393. [DOI] [PubMed] [Google Scholar]
- Grodberg D, Weinger PM, Kolevzon A, Soorya L, Buxbaum JD. Brief report: the autism mental status examination: development of a brief autism-focused exam. J Autism Dev Disord. 2012;15(3):455–459. doi: 10.1007/s10803-011-1255-4. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;15(5):659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Kientz MA, Dunn W. A comparison of the performance of children with and without autism on the Sensory Profile. Am J Occup Ther. 1997;15(7):530–537. doi: 10.5014/ajot.51.7.530. [DOI] [PubMed] [Google Scholar]
- Dunn W, Myles BS, Orr S. Sensory processing issues associated with Asperger syndrome: a preliminary investigation. Am J Occup Ther. 2002;15(1):97–102. doi: 10.5014/ajot.56.1.97. [DOI] [PubMed] [Google Scholar]
- Watling RL, Deitz J, White O. Comparison of sensory profile scores of young children with and without autism spectrum disorders. Am J Occup Ther. 2001;15(4):416–423. doi: 10.5014/ajot.55.4.416. [DOI] [PubMed] [Google Scholar]
- Wiggins LD, Robins DL, Bakeman R, Adamson LB. Brief report: sensory abnormalities as distinguishing symptoms of autism spectrum disorders in young children. J Autism Dev Disord. 2009;15(7):1087–1091. doi: 10.1007/s10803-009-0711-x. [DOI] [PubMed] [Google Scholar]
- Crane L, Goddard L, Pring L. Sensory processing in adults with autism spectrum disorders. Autism. 2009;15(3):215–228. doi: 10.1177/1362361309103794. [DOI] [PubMed] [Google Scholar]
- Leekam SR, Nieto C, Libby SJ, Wing L, Gould J. Describing the sensory abnormalities of children and adults with autism. J Autism Dev Disord. 2007;15(5):894–910. doi: 10.1007/s10803-006-0218-7. [DOI] [PubMed] [Google Scholar]
- Kern JK, Trivedi MH, Grannemann BD, Garver CR, Johnson DG, Andrews AA, Savla JS, Mehta JA, Schroeder JL. Sensory correlations in autism. Autism. 2007;15(2):123–134. doi: 10.1177/1362361307075702. [DOI] [PubMed] [Google Scholar]
- Tomchek SD, Dunn W. Sensory processing in children with and without autism: a comparative study using the short sensory profile. Am J Occup Ther. 2007;15(2):190–200. doi: 10.5014/ajot.61.2.190. [DOI] [PubMed] [Google Scholar]
- Schoen SA, Miller LJ, Green KE. Pilot study of the Sensory Over-Responsivity Scales: assessment and inventory. Am J Occup Ther. 2008;15(4):393–406. doi: 10.5014/ajot.62.4.393. [DOI] [PubMed] [Google Scholar]
- Herlenius E, Heron SE, Grinton BE, Keay D, Scheffer IE, Mulley JC, Berkovic SF. SCN2A mutations and benign familial neonatal-infantile seizures: the phenotypic spectrum. Epilepsia. 2007;15(6):1138–1142. doi: 10.1111/j.1528-1167.2007.01049.x. [DOI] [PubMed] [Google Scholar]
- Barker RA, Barasi S, Neal MJ. Neuroscience at a Glance. 2. Oxford: Blackwell Publishing Ltd; 2003. [Google Scholar]
- Ben-Sasson A, Soto TW, Martinez-Pedraza F, Carter AS. Early sensory over-responsivity in toddlers with autism spectrum disorders as a predictor of family impairment and parenting stress. J Child Psychol Psychiatry. 2013;15(8):846–853. doi: 10.1111/jcpp.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane SJ, Reynolds S, Thacker L. Sensory over-responsivity and ADHD: differentiating using electrodermal response, cortisol and anxiety. Front Integr Neurosci. 2010;15(4):595–603. doi: 10.3389/fnint.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer B, Kinnealey M, Reed C, Herzberg G. Sensory modulation and affective disorders in children and adolescents with Asperger’s disorder. Am J Occup Ther. 2005;15(3):335–345. doi: 10.5014/ajot.59.3.335. [DOI] [PubMed] [Google Scholar]