

ABSTRACT
The resistance-nodulation-division (RND) family multidrug efflux system MexXY-OprM is a major determinant of aminoglycoside resistance in Pseudomonas aeruginosa, although the details of aminoglycoside recognition and export by MexY, the substrate-binding RND component of this efflux system, have not been elucidated. To identify regions/residues of MexY important for aminoglycoside resistance, plasmid-borne mexY was mutagenized and mutations that impaired MexY-promoted aminoglycoside (streptomycin) resistance were identified in a ΔmexY strain of P. aeruginosa. Sixty-one streptomycin-sensitive mexY mutants were recovered; among these, 7 unique mutations that yielded wild-type levels of MexY expression were identified. These mutations compromised resistance to additional aminoglycosides and to other antimicrobials and occurred in both the transmembrane and periplasmic regions of the protein. Mapping of the mutated residues onto a 3-dimensional structure of MexY modeled on Escherichia coli AcrB revealed that these tended to occur in regions implicated in general pump operation (transmembrane domain) and MexY trimer assembly (docking domain) and, thus, did not provide insights into aminoglycoside recognition. A region corresponding to a proximal binding pocket connected to a periplasm-linked cleft, part of a drug export pathway of AcrB, was identified in MexY and proposed to play a role in aminoglycoside recognition. To test this, selected residues (K79, D133, and Y613) within this pocket were mutagenized and the impact on aminoglycoside resistance was assessed. Mutations of D133 and Y613 compromised aminoglycoside resistance, while, surprisingly, the K79 mutation enhanced aminoglycoside resistance, confirming a role for this putative proximal binding pocket in aminoglycoside recognition and export.
IMPORTANCE
Bacterial RND pumps do not typically accommodate highly hydrophilic agents such as aminoglycosides, and it is unclear how those, such as MexY, which accommodate these unique substrates, do so. The results presented here indicate that aminoglycosides are likely not captured and exported by this RND pump component in a unique manner but rather utilize a previously defined export pathway that involves a proximal drug-binding pocket that is also implicated in the export of nonaminoglycosides. The observation, too, that a mutation in this pocket enhances MexY-mediated aminoglycoside resistance (K79A), an indication that it is not optimally designed to accommodate these agents, lends further support to earlier proposals that antimicrobials are not the intended pump substrates.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen that is frequently associated with life-threatening nosocomial infections in immunocompromised individuals, especially patients with cystic fibrosis (CF) (1). Intrinsically resistant to many clinically used antimicrobial agents and readily developing resistance during antimicrobial therapy (2), pseudomonal infections are often recalcitrant and difficult to manage (3). Among the major contributors to this intrinsic and acquired antimicrobial resistance are several multidrug efflux systems of the resistance-nodulation-division (RND) family (4). One of these, MexXY-OprM, is particularly noteworthy owing to its role in resistance to aminoglycosides (5, 6), which are uncommon RND pump substrates (4, 7), particularly in clinical P. aeruginosa isolates recovered from CF patients (8–11). MexXY proteins are encoded by a two-gene operon (5, 6), while OprM is encoded by the third gene of another multidrug efflux operon, mexAB-oprM (12). The mexXY operon is inducible by many of its antimicrobial substrates, specifically those, including the aminoglycosides, that target and disrupt the ribosome (13, 14).
The tripartite MexXY-OprM pump is comprised of a periplasmic membrane fusion protein (MexX), the inner-membrane (IM) drug/H+ antiporter (MexY; the RND component), and the outer-membrane (OM) channel (OprM) (15, 16). Crystal structures of the related AcrB protein from Escherichia coli indicate that RND components exist as asymmetric homotrimers with individual monomers, in a concerted fashion, adopting one of three conformations that represent different steps of the drug export process—access, binding, and extrusion (also known as loose, tight, and open, respectively) (17, 18). Individual monomers are proposed to cycle sequentially through these conformational states, as substrates first enter the pump (access state), bind (binding state), and are then extruded into the OM channel-forming constituent through an opening at the top of the RND component (extrusion state) in what has been termed a functionally rotating drug transport mechanism (17). RND monomers are comprised of a transmembrane (TM) region of 12 α-helices (TM1 to TM12) responsible for coupling the TM proton flux to pump operation, a periplasmic region divided into an IM-proximal porter domain of 4 β-strand-α-helix-β-strand subdomains (PC1, PC2, PN1, and PN2) involved in drug capture and extrusion, and an OM-proximal region formed by 2 β-sheet subdomains (DN and DC) that form a so-called docking domain (19). The polyspecific binding pocket of AcrB that is accessible in the binding/tight monomer (now called the deep [20] or distal [21] binding pocket) is well defined (17, 18, 22, 23), although more-recent studies have identified a second, so-called proximal binding pocket in the access/loose monomer (21). The proximal pocket is separated from the distal pocket by a switch-loop (20) or G-loop (23) that controls access to the distal pocket as monomers transition from the access state to the binding state (20). Two routes of drug entry into the binding pockets have been described, one via a cleft between PC1 and PC2 (the cleft pathway) (24–26) and the other involving the intermonomer vestibule found near the IM surface (27) and, possibly, a hydrophobic groove defined by TM8 and TM9 (the vestibule pathway) (25, 28). Recently, two distinct vestibule pathways, “a” and “b,” were identified in AcrB using molecular simulations and verified using site-directed mutagenesis (25). Evidence suggests that the cleft route is used by larger and, likely, hydrophilic substrates whereas the vestibule pathways accommodate lipophilic agents that are likely to partition into the IM (25).
While crystal structures, molecular simulations, and biochemical studies have provided evidence that many drugs, including those accommodated by MexY, use the aforementioned export pathways in AcrB, this RND pump does not accommodate/bind aminoglycosides (4, 23). As such, it is not clear that the heretofore-identified export pathways, while likely present in MexY, are used by aminoglycosides, which are much more hydrophilic than the typical RND/AcrB substrates (23). To identify regions/residues of MexY involved in the MexY-mediated aminoglycoside resistance and so provide insights into aminoglycoside accommodation by this RND pump, mexY was mutagenized and the impact on aminoglycoside resistance assessed. We report here that, in addition to mutations that likely compromise assembly and general pump operation, mutations in the MexY-equivalent proximal binding pocket impact aminoglycoside resistance, highlighting the probable involvement of the previously defined export pathway for other agents in aminoglycoside capture and extrusion by MexY.
RESULTS AND DISCUSSION
Isolation of MexY mutants exhibiting reduced aminoglycoside resistance.
To identify mutations in mexY that compromised MexY activity with respect to aminoglycoside resistance, a mexY-bearing pRK415 derivative, plasmid pCL10, was mutagenized and introduced into a P. aeruginosa PAO1 ΔmexY derivative, strain K3315. Elimination of the mexY gene in K3315 conferred susceptibility to known MexXY-OprM antimicrobial substrates, including aminoglycosides (Table 1), while the cloned wild-type (WT) mexY gene on plasmid pCL10 restored resistance (Table 1). As such, pCL10 derivatives carrying mexY mutations that compromise aminoglycoside resistance could be selected by their failure to restore MexYWT levels of aminoglycoside resistance. Thus, hydroxylamine-mutated pCL10-bearing K3315 (2,600 colonies) was screened for increased susceptibility to the aminoglycoside streptomycin. Of 61 colonies showing enhanced susceptibility to streptomycin, 7 unique single amino acid substitutions within the mexY gene that yield a wild-type level of MexY expression were uncovered (Fig. 1, top, lanes S16F to A960T; cf. WT lane). These mexY mutations (and not an additional mutation[s] elsewhere on pCL10) were subsequently confirmed to be responsible for the resistance defect by individually recloning the mutated mexY genes into pRK415 and assessing their impact on antimicrobial resistance and MexY production in strain K3315. Examination of the impact of the mexY mutations on susceptibility to additional MexXY-OprM antimicrobial substrates revealed a number of patterns, with some mutations (S16F, R184H, G216D, and A960T) conferring increased susceptibility to all tested antimicrobials and some (R166C, V339M, and P562S) increased susceptibility to a subset only. Initially, the V339M mutation appeared to specifically enhance susceptibility to aminoglycosides (including a related aminocyclitol, spectinomycin). Still, only one nonaminoglycoside (erythromycin) could be tested, since only agents capable of inducing mexXY could be assessed (while mexY genes present on pCL10 and its derivatives are constitutively expressed from the plasmid-borne lac promoter, expression of the chromosomally encoded MexX constituent requires drug-induced expression of the gene from its native promoter). Erythromycin and tetracycline are the only mexXY-inducing nonaminoglycoside/aminocyclitol MexY substrates (13) (while chloramphenicol induces mexXY expression [13], it does not appear to be a substrate for MexXY-OprM; Table 1), and since plasmid pCL10 carries a tetracycline resistance gene, the impact of MexY mutations on tetracycline resistance could not be assessed. To evaluate the impact of the MexYV339M mutation on resistance to additional antimicrobials, it was necessary to induce mexX expression with the nonsubstrate chloramphenicol. As seen in Table 1, chloramphenicol increased resistance to the quinolone norfloxacin and the β-lactam cefepime in strain K3315 expressing wild-type mexY but not in the same stain harboring a plasmid without an insertion or expressing the inactive MexYG216D variant (Table 1; see legend for MICs determined in the absence of chloramphenicol), consistent with chloramphenicol-promoted drug resistance being mediated by MexXY. Chloramphenicol-treated strain K3315 expressing MexYV339M showed an increase (relative to the vector-only control) in resistance to norfloxacin but not cefepime, indicating that this mutation compromised resistance to the β-lactam but not the quinolone. Thus, although the V339M mutation negatively impacts resistance to aminoglycosides, it does not do so selectively.
TABLE 1 .
Impact of MexY mutations on antimicrobial susceptibility of P. aeruginosaa
| Strain | Plasmid | MexY mutation |
MIC (μg/ml)b |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| STR | PAR | NEO | AMI | ERY | SPC | CAM | NORc | CEFd | |||
| K767 | None | WT | 32 | 256 | 32 | 2 | 512 | 512 | 64 | NDe | ND |
| K3315 | None | None | 2 | 8 | 4 | 0.5 | 64 | 64 | 64 | ND | ND |
| K3315 | pRK415 | None | 2 | 16 | 4 | 0.5 | 64 | 64 | 32 | 0.5 | 1 |
| K3315 | pCL10 | WT | 8 | 64 | 8 | 1 | 512 | 512 | 32 | 2 | 4 |
| K3315 | pCL14 | G216D | 2 | 16 | ND | ND | 64 | 64 | 32 | 0.5 | 1 |
| K3315 | pCL13 | R184H | 4 | 32 | ND | ND | 256 | 256 | 32 | ND | ND |
| K3315 | pCL11 | S16F | 4 | 32 | ND | ND | 256 | 256 | 32 | ND | ND |
| K3315 | pCL17 | A960T | 4 | 16 | ND | ND | 256 | 256 | 32 | ND | ND |
| K3315 | pCL15 | V339M | 4 | 16 | 8f | 1 | 512 | 256 | 32 | 2 | 1 |
| K3315 | pCL12 | R166C | 4 | 32 | ND | ND | 256 | 512 | 32 | 2 | 4 |
| K3315 | pCL16 | P562S | 8 | 32 | ND | ND | 256 | 512 | 32 | ND | ND |
| K3315 | pCL19 | D133A | 4 | 16 | 4 | 0.5 | 256 | 1024 | 32 | ND | ND |
| K3315 | pCL20 | D133S | 4 | 16 | 4 | 0.5 | 256 | 1024 | 32 | ND | ND |
| K3315 | pCL18 | K79A | 8 | 128 | 8g | 1 | 512 | 1024 | 32 | ND | ND |
| K3315 | pCL21 | Y613A | 4 | 16 | 4 | 0.5 | 512 | 256 | 32 | 2 | 2 |
The antimicrobial susceptibility of P. aeruginosa ΔmexY strain K3315 carrying the indicated plasmids expressing wild-type (WT) MexY or MexY derivatives with the indicated amino acid substitutions is reported. Results for WT strain K767 and plasmid-free K3315 are provided for comparison purposes.
MIC values lower than that measured for pCL10-carrying K3315 expressing WT MexY are bolded. MIC values higher than that measured for pCL10-carrying K3315 are italicized. STR, streptomycin; PAR, paromomycin; NEO, neomycin; AMI, amikacin; ERY, erythromycin; SPC, spectinomycin; CAM, chloramphenicol; NOR, norfloxacin; CEF, cefepime.
NOR MICs were determined in the presence of 8 µg/ml chloramphenicol. The NOR MIC for all strains in the absence of chloramphenicol was 0.5 µg/ml.
CEF MICs were determined in the presence of 8 µg/ml chloramphenicol. The CEF MIC determined in the absence of chloramphenicol was 1 (for pRK415 control and MexYG216D) or 2 (for MexYWT and its V339M, R166C, and Y613A variants) μg/ml.
ND, not determined.
At half the NEO MIC (4 µg/ml), MexYV339M-expressing K3315 grew reproducibly slower than K3315 expressing MexYWT (data not shown), indicating that the V339M mutation compromises neomycin resistance.
At half the NEO MIC (4 µg/ml), MexYK79A-expressing K3315 grew reproducibly faster than K3315 expressing MexYWT (data not shown), indicating that the K79A mutation enhances neomycin resistance.
FIG 1 .
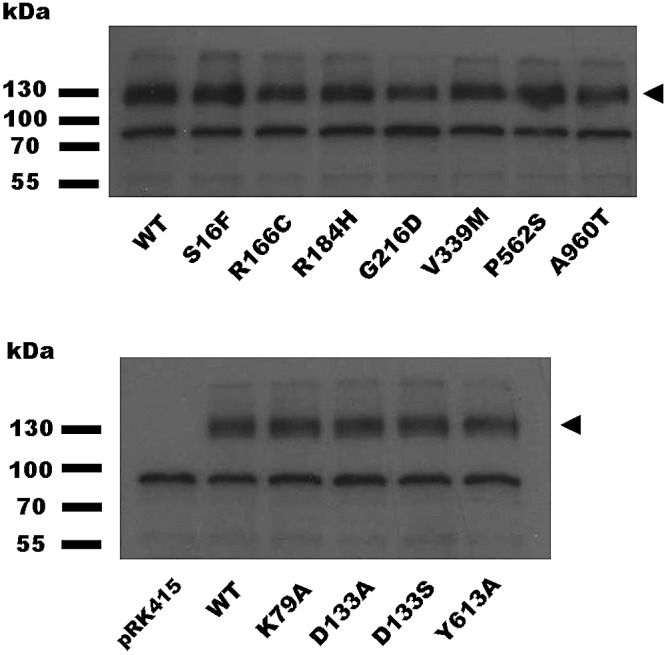
Expression of mutant MexY proteins in P. aeruginosa K3315. Whole-cell extracts of P. aeruginosa ΔmexY strain K3315 expressing plasmid pRK415-borne wild-type (WT) or mutant mexY genes (amino acid substitutions in MexY are indicated) were electrophoretically separated by SDS-PAGE, electroblotted, and developed with antibodies directed against the MexY protein. The MexY proteins, absent in strain K3315 harboring pRK415 without a cloned mexY gene (pRK415), are indicated by arrowheads. The migration positions of molecular mass markers are shown at left.
Noticeably, most of the identified mutations (except S16F and R166C) occur at residues that are conserved among a variety of RND family multidrug transporters, including those that accommodate aminoglycosides (MexY, AxyY, AmrB, AdeB, SmeZ, and AcrD) and those that do not (MexB, MexD, AcrB, and BpeB) (see Fig. 3). Only two aminoglycosides were assessed, however, since the cloned wild-type mexY gene restored aminoglycoside resistance in strain K3315 only partially (Table 1)—for some aminoglycosides (e.g., amikacin, neomycin), the wild-type gene provided such a modest increase in resistance (2-fold) that the impact of anything but a fully null mutation would not have been measurable. Why the plasmid-borne wild-type mexY gene failed to fully complement the mexY deficiency of strain K3315 with respect to aminoglycoside resistance while doing so for resistance to nonaminoglycosides is unclear. It is possible that the plasmid-encoded MexY is overproduced and so disrupts the cytoplasmic membrane, thereby sensitizing it to perturbation by aberrant polypeptides that are the eventual product of aminoglycoside action on the bacterial ribosome (29). In agreement with this explanation, several streptomycin-sensitive mutants lacking mutations in mexY but showing significantly increased MexY production as a result of vector backbone-borne mutations were also recovered in this study (data not shown).
FIG 3 .

Segments of amino acid sequence alignment of MexY and selected RND homologues in P. aeruginosa and other Gram-negative bacteria. The amino acid sequences of MexY and the indicated RND homologues were aligned using the multiple-sequence-alignment program T-coffee (55). Only those aligned sequences that encompassed the MexY residues whose mutation compromised drug resistance (indicated with an arrowhead and labeled) are shown. Residues that are conserved in other RND homologues are highlighted in bold lettering. Amino acid sequences were obtained from the NCBI Protein database (GenBank accession no. NP_250708 [MexY_P. aeruginosa PAO1], EGP45231 [AxyY_Achromobacter xylosoxidans AXX-A], YP_108402 [AmrB_Burkholderia pseudomallei K96243], CAJ77844 [AdeB_Acinetobacter baumannii AYE], YP_001972001 [SmeZ_Stenotrophomonas maltophilia K279a], NP_416965 [AcrD_Escherichia coli K-12 MG1655], NP_253288 [MexD_P. aeruginosa PAO1], NP_249117 [MexB_P. aeruginosa PAO1], NP_414995 [AcrB_E. coli K-12 MG1655], and YP_006275722 [BpeB_B. pseudomallei 1026b]). MexY, AxyY, AmrB, AdeB, SmeZ, and AcrD have been linked to aminoglycoside resistance.
Mapping of MexY mutations onto a three-dimensional model of MexY.
To better understand how the aforementioned mutations compromise MexY-mediated aminoglycoside resistance, we constructed a three-dimensional model of trimeric MexY based on the asymmetrical AcrB trimer structure that includes one monomer each in the access, binding, and extrusion conformations (PDB accession number 2HRT) (18). MexY and AcrB share an overall sequence identity of 51%, with an additional 19% of sequence similarity, rendering AcrB a suitable template for building a MexY structural model. The resultant asymmetric MexY trimer model (Fig. 2A) has an excellent geometric quality—95.0% of the modeled residues fall in the most favored regions of the Ramachandran plot, 4.2% in the additionally allowed region, and only 8 residues of 2,622 (0.3%) in the disallowed region, as validated by the PROCHECK (30) program. Mapping of the mexY mutations onto the MexY structure (mutations were mapped onto all three of the MexY monomers, although only the binding monomer is shown for illustrative purposes; Fig. 2B) revealed that mutations compromising resistance to the tested antimicrobials were scattered throughout the molecule, including within the outer-membrane-proximal docking (DN, DC), the periplasmic porter (PN, PC), and the transmembrane (TM) domains.
FIG 2 .

Mapping mutations impacting MexY-mediated antibiotic resistance on a 3-dimensional homology model of MexY. (A) An asymmetric trimer model of MexY constructed by homology modeling on the crystal structure of E. coli AcrB (PDB code 2HRT). Individual monomers are shown in space-fill formatting and are colored salmon red (loose/access conformation), gray (tight/binding conformation), and blue (open/extrusion conformation; mostly hidden behind the other 2 monomers). Structural details of the tight/binding monomer are shown in order to highlight the characteristic N- and C-terminal regions/subdomains of the docking domain (DN [yellow] and DC [purple], respectively), the porter domain (2 N-terminal [PN1, orange]/[PN2, pink] and 2 C-terminal [PC1, green]/[PC2, red] subdomains), and the transmembrane domain (transmembrane segments TM1-6 [light blue] and TM7-12 [dark blue], respectively) of RND transporters. The positions of the intermonomer vestibule and intramonomer cleft are also indicated. (B) Locations in the tight/binding MexY monomer of mutations (shown in space-fill formatting) that compromise MexY-mediated drug resistance. Relevant subdomains (as described for panel A) are highlighted in the corresponding colors. Structural models at left and right were rotated 50° counterclockwise and 100° clockwise, respectively, relative to the middle model in order to better illustrate the positions of various mutations. The dashed lines define the inner-membrane boundary. A cleft/opening, within which the putative proximal binding pocket occurs, is clearly seen in the structural model at left. A vestibule drug entry pathway is also highlighted in the leftmost structural model.
Docking domain mutations.
The G216D mutation, which had the biggest negative impact on MexY-mediated drug resistance (Table 1), maps to a protruding “thumb” structure (Fig. 2B, middle structural model) within the periplasmic DN subdomain that inserts into a “hole” in the docking domain of the neighboring protomer (Fig. 2A). This structure has previously been implicated in the trimerization of RND components (31, 32) with a mutation in the MexYG219-equivalent residue of P. aeruginosa MexB, G220, similarly compromising resistance to all antimicrobials (32) and mutations in the corresponding G217 (and neighboring) residue of E. coli AcrB compromising trimerization (31). Thus, MexYG216D is plausibly impaired in trimer assembly and this explains its inability to promote resistance to all tested antimicrobials. Similarly, the R184H mutation occurs within the hole into which the “thumb” structure of a neighboring protomer inserts and therefore likely compromises trimerization as well. While the increased susceptibility of MexYR184H-expressing P. aeruginosa to all tested antimicrobials is consistent with such a notion, the R184H mutation does not fully abrogate MexY-mediated resistance and so, unlike the G216D mutation, presumably does not fully block trimerization.
Periplasmic domain mutations.
The R166C mutation maps to the periplasmic PN2 subdomain of MexY (Fig. 2B, rightmost structural model) in a region that, in AcrB (28), contacts the PN1′ subdomain of a neighboring monomer. This PN2-PN1′ intermonomeric contact appears to be central to the “functionally rotating” transport mechanism of multidrug RND transporters, whereby adoption of a tight or binding conformation by one protomer sterically facilitates (though the PN2-PN1′ contact) the cycling of its adjacent monomer to an “open” drug extrusion conformation (28). Thus, the R166C mutation may interfere with this cycling process in MexY. Still, the R166C mutation does not impact resistance to all antimicrobials (spectinomycin, norfloxacin, and cefepime MICs are unaffected; Table 1) and does not fully compromise MexY-mediated resistance to those agents that are impacted. Hence, if this mutation is affecting MexY operation, the impact is only partial and has, for some as-yet-unknown reason, differential impacts on the export of substrate antimicrobials. It is possible that the R166C mutation is also indirectly affecting substrate recognition/accommodation and that this may be the reason behind its substrate-specific impact.
Transmembrane domain mutations.
Four mexY mutations map to the transmembrane domains, with 2 being cytoplasm proximal (S16F and A960T; Fig. 2B, middle and leftmost structural model, respectively) and 2 being periplasm proximal (V339M and P562S; Fig. 2B, middle and leftmost structural models, respectively). All but S16 are conserved among RND transporters (Fig. 3), implying that they somehow contribute to basic pump operation. Still, only the A960T (and S16F) mutations are associated with reduced resistance to all tested antimicrobials (Table 1), consistent with the cytoplasm-proximal region of MexY playing a conserved role in RND pump operation. How these mutations impact MexY function is currently unclear, although, given that conformational changes occur within the TM region of RND transporters as they cycle between the different monomer states during drug export (17, 18), likely mediated by proton movement though the RND constituent (33, 34), it may be that the productive proton-driven conformational changes in the TM region are compromised to some extent by these mutations. Despite their conservation among RND multidrug transporters, mutations at V339 (V339M) and P562 (P562S) have a substrate-specific impact on MexY function, suggesting that, unlike the cytoplasm-proximal TM mutations, they are not compromising general pump operation. Similarly situated mutations in MexB (V928M and M395I) also impacted resistance to only a subset of antimicrobial substrates (32), although the reasons are unclear. Interestingly, P562 occurs within a region of MexY that in the corresponding region of AcrB is variously at the extreme lower edge of the cleft that appears to provide access of periplasmic substrates to the proximal binding pocket (24) or at the upper reaches of a so-called vestibule pathway (Fig. 2B; left structural model), specifically, vestibule pathway b (25), that is suggested to accommodate lipophilic pump substrates that are likely to partition into the outer leaflet of the inner membrane (25, 28). Binding of the detergent and AcrB substrate dodecyl-β-d-maltoside to this region of AcrB has previously been demonstrated (20), and D566 in AcrB (which corresponds to the P562-adjacent E563 residue in MexY) appears to be involved in ciprofloxacin binding (26) and is accessible to the AcrB substrate Bodiyp FL maleimide (24). Thus, the region of MexY that encompasses P562 likely forms an entry pathway for certain MexXY-OprM substrate antimicrobials, explaining the selective impact of the P562S mutation on drug resistance. Given the proximity of MexYP562 to two entry pathways and the structure-altering nature of a P562S substitution, it is possible that both pathways are negatively impacted by this mutation. While one might expect the cleft pathway to be the route of choice for hydrophilic aminoglycosides, these agents do cross and therefore do interact with the IM, and given how little is known about this process, the possibility cannot be ruled out that these agents access the proximal binding pocket of MexY via the vestibule route.
Site-directed mutagenesis of a predicted substrate-binding pocket in MexY.
The observation that some mexY mutations showed substrate-specific effects on resistance that included aminoglycosides suggests that the latter agents are actual substrates of MexY. In an attempt to better elucidate the molecular details of aminoglycoside recognition by MexY, the protein was aligned with other aminoglycoside-accommodating RND pump components (e.g., AcrD of E. coli [35], AmrB of Burkholderia pseudomallei [36], AdeB of Acinetobacter baumannii [37], and AxyY of Achromobacter xylosoxidans [38]; Fig. 3), as well as several RND components that do not accommodate aminoglycosides (Fig. 3). The intent was to identify residues that were uniquely conserved in aminoglycoside-accommodating RND pump components and, so, potentially contribute to aminoglycoside binding and/or export. No residues were specifically conserved in the aminoglycoside-accommodating pumps that were examined, although 4 of 6 of these had a conserved aspartate residue (D133 in MexY; Fig. 3). Interestingly, D133 occurs within a region of MexY that corresponds structurally to the proximal multisite binding pocket of E. coli AcrB (21) (see Fig. 2B). Moreover, the D133-equivalent residue in AcrB, S134 (Fig. 3), is one of the 4 key residues that coordinate the proximal binding pocket of this RND pump (21) and its mutation compromises AcrB-mediated drug resistance (21). MexYD133 was, therefore, considered a candidate residue involved in aminoglycoside recognition/binding. The amino acid sequence alignment of RND proteins also revealed a number of residues that were uniquely present in a subset (e.g., MexY, AmrB, and AxyY) of the aminoglycoside-accommodating RND pumps. AmrB (66% identity) and AxyY (70% identity) exhibit the highest degree of homology to MexY among all the aminoglycoside-accommodating RND proteins (identity with the remainder is ≤50%), and while this may simply reflect a closer evolutionary relationship between these RND proteins (phylogenetic analysis shows that these protein cluster; data not shown), it may also be that there are multiple ways for RND pump components to recognize/accommodate aminoglycosides and that these three share a mechanism. Intriguingly, one of these conserved residues, Y613 (Fig. 3), whose unique presence in these three proteins was confirmed in a more extensive comparison involving all “confirmed to be resistance-mediating” RND proteins from P. aeruginosa, E. coli, and B. pseudomallei (22 proteins in total; data not shown), was also identified in the vicinity of the proposed proximal binding pocket of MexY (Fig. 2B), on the tip of the switch-loop (20) that separates the proximal and distal binding pockets. Mutations of the switch-loop in AcrB have been shown to compromise AcrB-mediated resistance to erythromycin and doxorubicin, apparently owing to mutation-driven structural changes in the loop interfering with the binding of these larger substrates to the proximal binding pocket (20, 21). MexYY613 was, thus, possibly involved in aminoglycoside binding as well. Finally, a lysine residue unique to MexY (K79) was identified within the putative MexY proximal binding pocket (Fig. 2B) and a role for it in aminoglycoside binding was also proposed. The chosen candidate residues were individually mutated to alanine on plasmid pCL10, and the impact on MexY-mediated antimicrobial resistance of P. aeruginosa strain K3315 was assessed. MexYD133 was also mutated to serine, which is the residue at this position in E. coli AcrB and the one which has been implicated in erythromycin binding (21). It was reasoned that, should D133 contribute to substrate binding and MexY-mediated antimicrobial resistance, then the change to the AcrB-equivalent serine might specifically compromise aminoglycoside binding/resistance but retain erythromycin binding/resistance, while the alanine substitution would compromise binding of/resistance to both.
All site-directed mutation-bearing mexY genes yielded wild-type levels of MexY protein (Fig. 1, bottom). As shown in Table 1 (bottom), both mutations of D133 reduced resistance to all agents tested, with the exception of spectinomycin, where an increase in resistance was noted. These observations suggest that the mutationally altered binding pocket is better able to accommodate spectinomycin while concomitantly being less able to accommodate the other substrates, confirming a role for the putative MexY proximal binding pocket and D133 specifically in substrate (including, importantly, aminoglycoside) recognition and MexY-mediated antimicrobial resistance. Interestingly, the Y613A mutation compromised resistance to the aminoglycoside/aminocyclitol agents but not erythromycin (Table 1), initially suggesting that it might specifically impact the former agents only. In cells treated with chloramphenicol to induce chromosomal mexX, however, this mutation also compromised cefepime resistance but not norfloxacin resistance (Table 1). These data are consistent with the loop functioning directly in the binding/recognition of these agents or, more likely, the Y613A mutation impacting the disposition of the switch-loop, sterically hindering access of these agents to the proximal binding pocket, as proposed, for example, for the switch-loop mutations in E. coli AcrB (20, 21). This further supports the idea of the putative binding pocket of MexY playing a significant role in substrate (including aminoglycoside) recognition/accommodation. In contrast to the results reported above, mutation of MexYK79 failed to compromise resistance to any agent tested and, in fact, enhanced resistance to several agents, including a number of aminoglycosides and spectinomycin (Table 1). The reason for this is unclear, though this result highlights once more the significance of this putative proximal binding pocket of MexY in recognition of antimicrobials, including aminoglycoside. The fact that native MexY retains a residue that is less than optimal for drug accommodation/resistance gives further evidence that antimicrobials are not the intended substrates for this RND efflux system.
The observation that mutations in regions of MexY that correspond to a previously defined drug export pathway(s) in AcrB (vestibule/cleft drug entry pathway and proximal drug binding pocket) impact resistance to aminoglycosides is consistent with these unusual hydrophilic RND pump substrates using a common drug export pathway versus one unique to these agents. Thus, the failure of AcrB and other RND pumps to accommodate aminoglycosides presumably stems simply from their lacking the necessary binding constituents in their entry/export pathways to accommodate aminoglycosides.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are described in Table 2. Bacterial cells were cultured in Luria broth (L-broth) and on Luria agar (L-agar), with antibiotics as necessary, at 37°C. Plasmid pEX18Tc and its derivatives were maintained or selected in Escherichia coli with 10 µg/ml tetracycline. Plasmid pRK415 and its derivatives were maintained or selected with 10 (in E. coli) or 50 (in Pseudomonas aeruginosa) μg/ml tetracycline.
TABLE 2 .
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| E. coli strains | ||
| DH5α | φ80dlacZΔM15 endA1 recA1 hsdR17 (rK− mK+) supE44 thi-1 gyrA96 relA1 F− Δ(lacZYA-argF) U169 | 49 |
| S17-1 | thi pro hsdR recA Tra+ | 50 |
| P. aeruginosa strains | ||
| K767 | PAO1 prototroph wild type | 51 |
| K1525 | K767ΔmexXY | 52 |
| K3315 | K767ΔmexY | This study |
| Plasmids | ||
| pEX18Tc | Broad-host-range gene replacement vector; sacB Tcr | 53 |
| pCL8 | pEX18Tc::ΔmexY | This study |
| pCL9 | pEX18Tc::mexY | This study |
| pRK415 | P. aeruginosa-E. coli shuttle cloning vector; Tcr | 54 |
| pCL10 | pRK415::mexYWT | This study |
| pCL11 | pRK415::mexYS16F | This study |
| pCL12 | pRK415::mexYR166C | This study |
| pCL13 | pRK415::mexYR184H | This study |
| pCL14 | pRK415::mexYG216D | This study |
| pCL15 | pRK415::mexYV339M | This study |
| pCL16 | pRK415::mexYP562S | This study |
| pCL17 | pRK415::mexYA960T | This study |
| pCL18 | pRK415::mexYK79A | This study |
| pCL19 | pRK415::mexYD133A | This study |
| pCL20 | pRK415::mexYD133S | This study |
| pCL21 | pRK415::mexYY613A | This study |
Tcr, tetracycline resistance; WT, wild type.
DNA methods.
Standard protocols were used for restriction endonuclease digestion, ligation, transformation, and agarose gel electrophoresis, as described by Sambrook and Russell (39). Plasmid and chromosomal DNA was prepared as described before (40). DNA fragments used for cloning were extracted from agarose gels using a Wizard SV gel and PCR cleanup system (Fisher Scientific, Ltd., Nepean, Canada). CaCl2-competent E. coli (41) and electrocompetent P. aeruginosa (42) cells were prepared as previously described. Oligonucleotide (PCR primer) synthesis was carried out by Integrated DNA Technologies (Coralville, IA). Nucleotide sequencing was carried out by ACGT Corp. (Toronto, Canada) using universal and custom primers (Table 3).
TABLE 3 .
Oligonucleotides used in this study
| Primer | Oligonucleotide sequence (5′ → 3′)a | Source |
|---|---|---|
| mexYUP-F | ATTAAGAGCTCTACCGCCAGGCTG | This study |
| mexYUP-R | GCTGCATCTAGATCGTAGCGTTCTC | This study |
| mexYDN-F | AGTCGATCTAGATGCCCCTAGCGAAAC | This study |
| mexYDN-R | GCAGACAAGCTTCTGGCCGACTATC | This study |
| mexYscreen-F | TGTTCCGCAATCCGCATC | This study |
| mexYscreen-R | GCGTAGCCGATCATCTGTC | This study |
| mexY-3138-F | GGCTCGAAGCTTATGGCTCGTTTCTTC | This study |
| mexY-3138-R | ATCTAGGATCCTCAGGCTTGCTCCGTG | This study |
| mexY-58-F | TTATACAAGCTTCGACGCCCCCTCACCGCT | This study |
| mexY-58-R | TGGCGGTCATTTGGTTGACC | This study |
| mexYseq-F1 | GCCTGAAGATCGTCGAGTC | This study |
| mexYseq-F2 | CAAGCTGACCTCGATGAACCT | This study |
| mexYseq-F3 | CGATCAACGTGCTGACCATGT | This study |
| mexYseq-R4 | CAGTCCCTGCATCAATTGCT | This study |
| mexYseq-R5 | ATCTCGTCCATGCTCACGC | This study |
| mexY-K79A-F | GGCCTGCTCTACACCGCGGCCACCAGCAGCAC | This study |
| mexY-K79A-R | GTGCTGCTGGTGGCCGCGGTGTAGAGCAGGCC | This study |
| mexY-D133A-F | TGGAGAAGGCGGCGGCCAGCATCCAGCTGAT | This study |
| mexY-D133A-R | ATCAGCTGGATGCTGGCCGCCGCCTTCTCCA | This study |
| mexY-D133S-F | TGGAGAAGGCGGCGTCCAGCATCCAGCTGAT | This study |
| mexY-D133S-R | ATCAGCTGGATGCTGGACGCCGCCTTCTCCA | This study |
| mexY-Y613A-F | GGCGGCTTCAGCCTGGCCGGCGACGGCACCAG | This study |
| mexY-Y613A-R | CTGGTGCCGTCGCCGGCCAGGCTGAAGCCGCC | This study |
Restriction endonuclease cleavage sites are underlined. Site-directed mutations are bolded and italicized.
Construction of a mexY deletion strain of P. aeruginosa.
To introduce an in-frame deletion of mexY into wild-type P. aeruginosa PAO1 strain K767, a pEX18Tc-based deletion construct was first prepared by sequentially cloning PCR-amplified 1-kb DNA fragments corresponding to the regions upstream and downstream of mexY into pEX18Tc. The mexY upstream and downstream regions were individually amplified using primers mexYUP-F and mexYUP-R and primers mexYDN-F and mexYDN-R, respectively. Amplification was achieved in a 50-µl reaction mixture formulated as described before (40) and heated for 3 min at 98°C, followed by 35 cycles of 0.5 min at 98°C, 0.5 min at 68°C, and 0.5 min at 72°C, before finishing with 10 min at 72°C. The resultant ΔmexY deletion vector, pCL8, was mobilized into P. aeruginosa strain K767 from E. coli S17-1 (40), and tetracycline-resistant transconjugants were recovered and patched onto sucrose as described before (40). Sucrose-resistant colonies were then screened for chromosomal deletion of mexY using colony PCR (43) with 2.5 U Taq polymerase in 10% (vol/vol) dimethyl sulfoxide (DMSO). Colony PCR was carried out using primers mexYscreen-F and mexYscreen-R at a 0.6 µM final concentration and deoxynucleoside triphosphates (dNTPs) at a 0.2 mM final concentration in a 10-µl reaction volume, with the mixture heated for 3 min at 95°C, followed by 35 cycles of 0.5 min at 95°C, 0.5 min at 59.1°C, and 4 min at 72°C, before finishing with 10 min at 72°C.
Cloning of mexY.
Plasmid pCL10, a pRK415 derivative carrying the mexY gene and 58 bp of upstream sequence, was constructed using a two-step cloning approach. First, the mexY gene (coding sequence only) was PCR amplified from the chromosome of K767 using primers mexY-3138-F and mexY-3138-R and cloned into pEX18Tc as a HindIII-BamHI-restricted fragment. The PCR conditions were as described above for generation of the mexY deletion fragments but using instead an annealing temperature of 70°C and an extension time of 100 s. The 58-bp upstream sequence (carrying the ribosome-binding site) was amplified from the chromosome of K767 also as described above but with a 30-s extension time, using primers mexY-58-F and MexY-58-R, and cloned into the aforementioned mexY-carrying pEX18Tc derivative as a HindIII-SalI-restricted fragment to create plasmid pCL9. The mexY gene together with its 58 bp of upstream sequence was then excised from pCL9 as a HindIII-BamHI fragment and cloned into pRK415 to yield pCL10.
Hydroxylamine mutagenesis of mexY.
Ten micrograms of mexY-carrying pCL10 was mutagenized with hydroxylamine hydrochloride (460 mM) for 30 min at 70°C as previously described (44). Eighty microliters of the 500-µl reaction mixture was added to 720 µl of prechilled 100 mM Tris-HCl (pH 8.0)–10 mM EDTA to stop the mutagenesis reaction, and plasmid DNA was subsequently recovered in a final volume of 30 µl. E. coli S17-1 was transformed with 4 µl of hydroxylamine-treated plasmid DNA and allowed to recover in L-broth (1 ml) at 37°C before being diluted with 9 ml of L-broth containing 10 µg/ml of tetracycline and cultured overnight at 37°C with shaking. The mutagenized pCL10 was then mobilized into the mexY-deficient P. aeruginosa K3315 strain from the overnight culture of E. coli S17-1 as described before (40) except that the mating mixture on the L-agar plate was incubated for 4 h at 37°C. P. aeruginosa K3315 carrying mutated plasmid-borne mexY was recovered on L-agar plates containing tetracycline (50 µg/ml) and chloramphenicol (5 µg/ml, to counterselect the E. coli S17-1) and screened for aminoglycoside susceptibility (lack of growth after 24 h on L-agar plates containing streptomycin [15 µg/ml]) as an indicator of impaired MexY function. Streptomycin-sensitive transformants were screened for production of MexY using MexY-specific antibodies in Western immunoblot analyses of whole-cell extracts prepared from overnight cultures as described below. Plasmid DNA was then prepared from streptomycin-sensitive transformants producing wild-type levels of MexY, and the mexY genes were sequenced using primers mexYseq-F1, mexYseq-F2, mexYseq-F3, mexYseq-R4, and mexYseq-R5 (Table 3). Mutations in mexY were identified following sequence alignment performed with the wild-type gene using DNAMAN (ver. 7) software.
Site-directed mutagenesis of mexY.
To introduce specific mutations into mexY, a site-directed mutagenesis single-primer PCR protocol was employed, with minor modifications (45). Briefly, 2 single-primer amplifications for each mutagenesis were carried out using the mutagenic primer pairs mexY-K79A-F and mexY-K79A-R (for mexYK79A), mexY-D133A-F and mexY-D133A-R (for mexYD133A), mexY-D133S-F and mexY-D133S-R (for mexYD133S), and mexY-Y613A-F and mexY-Y613A-R (for mexYY613A) (Table 3) with each at a 0.6 µM final concentration in a 50-µl reaction mixture containing 25 ng of mexY-encoding plasmid pCL9, 1 U of Phusion DNA polymerase, 1× Phusion HF buffer, 5% (vol/vol) DMSO, and dNTPs at a 0.2 mM final concentration. The mixtures were heated for 3 min at 98°C, followed by 25 cycles of 0.5 min at 98°C, 0.5 min at 72°C, and 4.75 min at 72°C, before finishing with 10 min at 72°C. Ten microliters each of the 2 PCR products generated for a given mexY mutation were then mixed and heated at 95°C, followed by a slow stepwise cooling to 37°C to promote reannealing of the denatured plasmid templates and PCR products (45), which were then digested with DpnI for 1 h at 37°C. Following transformation of E. coli DH5α with the DpnI-treated DNAs, plasmid DNA was recovered from transformants and the mexY gene sequenced to identify plasmids carrying the desired mutation. The mutated mexY genes were excised on BamHI-HindIII restriction fragments and cloned into pRK415 for mobilization into P. aeruginosa K3315. Production of the mutant mexY gene products was again assessed following immunoblotting of whole-cell extracts.
Antibiotic susceptibility assay.
The susceptibility of P. aeruginosa to antimicrobial agents was assessed using the 2-fold serial microtiter broth dilution method described previously (46), with an inoculum of approximately 5 × 105 cells per ml. MICs were recorded as the lowest concentrations of antibiotic inhibiting visible growth after 18 h of incubation at 37°C. In some experiments, chloramphenicol was included in the growth medium at one-fourth the MIC (8 µg/ml). In some instances, growth of the bacterium was assessed continuously over the period of the assay using a previously described methodology (40).
Cell extracts and Western immunoblotting.
Whole-cell protein extracts of P. aeruginosa prepared as described previously (47) were separated using SDS-PAGE and electroblotted and screened with MexY-specific antibodies (48) using a previously described protocol (32).
Protein modeling.
The MexY homology model was generated using ESyPred3D Web Server 1.0 (http://webapps.fundp.ac.be/esypred/) following alignment and threading of the MexY sequence with or against the three individual monomers of the E. coli AcrB trimer (PDB accession number 2HRT). ESyPred3D is an automated homology-modeling program that uses the MODELLER modeling package to build the final three-dimensional structure. The geometry of the final MexY model was validated using PROCHECK (30), and the model was rendered for viewing using PyMol (PyMol Molecular Graphics System, version 1.5.0.4 Schrödinger; http://www.pymol.org).
ACKNOWLEDGMENTS
This work was funded by operating grants from Cystic Fibrosis Canada and Canadian Institutes of Health Research (to K.P.).
We thank Z. Jia for providing the MexY model, N. Gotoh for providing the anti-MexY antibodies, and R. Hurowitz for assisting with plasmid extractions.
Footnotes
Citation Lau CHF, Hughes D, Poole K. 2014. MexY-promoted aminoglycoside resistance in Pseudomonas aeruginosa: involvement of a putative proximal binding pocket in aminoglycoside recognition. mBio 5(2):e01068-14. doi:10.1128/mBio.01068-14.
REFERENCES
- 1. Lyczak JB, Cannon CL, Pier GB. 2002. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15:194–222. 10.1128/CMR.15.2.194-222.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2:65. 10.3389/fmicb.2011.00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cantón R, Cobos N, De Gracia J, Baquero F, Honorato J, Gartner S, Alvarez A, Salcedo A, Oliver A, García-Quetglas E, Spanish Consensus Group for Antimicrobial Therapy. in the Cystic Fibrosis Patient 2005. Antimicrobial therapy for pulmonary pathogenic colonisation and infection by Pseudomonas aeruginosa in cystic fibrosis patients. Clin. Microbiol. Infect. 11:690–703. 10.1111/j.1469-0691.2005.01217.x [DOI] [PubMed] [Google Scholar]
- 4. Poole K. 2005. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 56:20–51. 10.1093/jac/dki171 [DOI] [PubMed] [Google Scholar]
- 5. Westbrock-Wadman S, Sherman DR, Hickey MJ, Coulter SN, Zhu YQ, Warrener P, Nguyen LY, Shawar RM, Folger KR, Stover CK. 1999. Characterization of a Pseudomonas aeruginosa efflux pump contributing to aminoglycoside impermeability. Antimicrob. Agents Chemother. 43:2975–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aires JR, Köhler T, Nikaido H, Plésiat P. 1999. Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 43:2624–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morita Y, Tomida J, Kawamura Y. 2012. MexXY multidrug efflux system of Pseudomonas aeruginosa. Front. Microbiol. 3:408. 10.3389/fmicb.2012.00408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sobel ML, McKay GA, Poole K. 2003. Contribution of the MexXY multidrug transporter to aminoglycoside resistance in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 47:3202–3207. 10.1128/AAC.47.10.3202-3207.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Islam S, Oh H, Jalal S, Karpati F, Ciofu O, Høiby N, Wretlind B. 2009. Chromosomal mechanisms of aminoglycoside resistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Clin. Microbiol. Infect. 15:60–66. 10.1111/j.1469-0691.2008.02097.x [DOI] [PubMed] [Google Scholar]
- 10. Vogne C, Aires JR, Bailly C, Hocquet D, Plésiat P. 2004. Role of the multidrug efflux system MexXY in the emergence of moderate resistance to aminoglycosides among Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob. Agents Chemother. 48:1676–1680. 10.1128/AAC.48.5.1676-1680.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Henrichfreise B, Wiegand I, Pfister W, Wiedemann B. 2007. Resistance mechanisms of multiresistant Pseudomonas aeruginosa strains from Germany and correlation with hypermutation. Antimicrob. Agents Chemother. 51:4062–4070. 10.1128/AAC.00148-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mine T, Morita Y, Kataoka A, Mizushima T, Tsuchiya T. 1999. Expression in Escherichia coli of a new multidrug efflux pump, MexXY, from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43:415–417. 10.1093/jac/43.3.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morita Y, Sobel ML, Poole K. 2006. Antibiotic inducibility of the MexXY multidrug efflux system of Pseudomonas aeruginosa: involvement of the antibiotic-inducible PA5471 gene product. J. Bacteriol. 188:1847–1855. 10.1128/JB.188.5.1847-1855.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jeannot K, Sobel ML, El Garch F, Poole K, Plésiat P. 2005. Induction of the MexXY efflux pump in Pseudomonas aeruginosa is dependent on drug-ribosome interaction. J. Bacteriol. 187:5341–5346. 10.1128/JB.187.15.5341-5346.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nikaido H, Takatsuka Y. 2009. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta 1794:769–781. 10.1016/j.bbapap.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blair JM, Piddock LJ. 2009. Structure, function and inhibition of RND efflux pumps in gram-negative bacteria: an update. Curr. Opin. Microbiol. 12:512–519. 10.1016/j.mib.2009.07.003 [DOI] [PubMed] [Google Scholar]
- 17. Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. 2006. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443:173–179. 10.1038/nature05076 [DOI] [PubMed] [Google Scholar]
- 18. Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. 2006. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313:1295–1298. 10.1126/science.1131542 [DOI] [PubMed] [Google Scholar]
- 19. Murakami S, Nakashima R, Yamashita E, Yamaguchi A. 2002. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419:587–593. 10.1038/nature01050 [DOI] [PubMed] [Google Scholar]
- 20. Eicher T, Cha HJ, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grütter MG, Diederichs K, Pos KM. 2012. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. U. S. A. 109:5687–5692. 10.1073/pnas.1114944109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. 2011. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480:565–569 [DOI] [PubMed] [Google Scholar]
- 22. Bohnert JA, Schuster S, Seeger MA, Fähnrich E, Pos KM, Kern WV. 2008. Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J. Bacteriol. 190:8225–8229. 10.1128/JB.00912-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vargiu AV, Nikaido H. 2012. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 109:20637–20642. 10.1073/pnas.1218348109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Husain F, Nikaido H. 2010. Substrate path in the AcrB multidrug efflux pump of Escherichia coli. Mol. Microbiol. 78:320–330. 10.1111/j.1365-2958.2010.07330.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yao XQ, Kimura N, Murakami S, Takada S. 2013. Drug uptake pathways of multidrug transporter AcrB studied by molecular simulations and site-directed mutagenesis experiments. J. Am. Chem. Soc. 135:7474–7485. 10.1021/ja310548h [DOI] [PubMed] [Google Scholar]
- 26. Yu EW, Aires JR, McDermott G, Nikaido H. 2005. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J. Bacteriol. 187:6804–6815. 10.1128/JB.187.19.6804-6815.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Husain F, Bikhchandani M, Nikaido H. 2011. Vestibules are part of the substrate path in the multidrug efflux transporter AcrB of Escherichia coli. J. Bacteriol. 193:5847–5849. 10.1128/JB.05759-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pos KM. 2009. Drug transport mechanism of the AcrB efflux pump. Biochim. Biophys. Acta 1794:782–793. 10.1016/j.bbapap.2008.12.015 [DOI] [PubMed] [Google Scholar]
- 29. Davis BD, Chen LL, Tai PC. 1986. Misread protein creates membrane channels: an essential step in the bactericidal action of aminoglycosides. Proc. Natl. Acad. Sci. U. S. A. 83:6164–6168. 10.1073/pnas.83.16.6164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. 10.1107/S0021889892009944 [DOI] [Google Scholar]
- 31. Fang J, Yu L, Wu M, Wei Y. 2013. Dissecting the function of a protruding loop in AcrB trimerization. J. Biomol. Struct. Dyn. 31:385–392. 10.1080/07391102.2012.703065 [DOI] [PubMed] [Google Scholar]
- 32. Middlemiss JK, Poole K. 2004. Differential impact of MexB mutations on substrate selectivity of the MexAB-OprM multidrug efflux pump of Pseudomonas aeruginosa. J. Bacteriol. 186:1258–1269. 10.1128/JB.186.5.1258-1269.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamane T, Murakami S, Ikeguchi M. 2013. Functional rotation induced by alternating protonation states in the multidrug transporter AcrB: all-atom molecular dynamics simulations. Biochemistry 52:7648–7658. 10.1021/bi400119v [DOI] [PubMed] [Google Scholar]
- 34. Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. 2006. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J. Bacteriol. 188:7290–7296. 10.1128/JB.00684-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosenberg EY, Ma D, Nikaido H. 2000. AcrD of Escherichia coli is an aminoglycoside efflux pump. J. Bacteriol. 182:1754–1756. 10.1128/JB.182.6.1754-1756.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moore RA, DeShazer D, Reckseidler S, Weissman A, Woods DE. 1999. Efflux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei. Antimicrob. Agents Chemother. 43:465–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Magnet S, Courvalin P, Lambert T. 2001. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 45:3375–3380. 10.1128/AAC.45.12.3375-3380.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bador J, Amoureux L, Blanc E, Neuwirth C. 2013. Innate aminoglycoside resistance of Achromobacter xylosoxidans is due to AxyXY-OprZ, an RND-type multidrug efflux pump. Antimicrob. Agents Chemother. 57:603–605. 10.1128/AAC.01243-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 40. Lau CH, Fraud S, Jones M, Peterson SN, Poole K. 2012. Reduced expression of the rplU-rpmA ribosomal protein operon in mexXY-expressing pan-aminoglycoside-resistant mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 56:5171–5179. 10.1128/AAC.00846-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Inoue H, Nojima H, Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28. 10.1016/0378-1119(90)90336-P [DOI] [PubMed] [Google Scholar]
- 42. Choi KH, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64:391–397. 10.1016/j.mimet.2005.06.001 [DOI] [PubMed] [Google Scholar]
- 43. Sheu DS, Wang YT, Lee CY. 2000. Rapid detection of polyhydroxyalkanoate-accumulating bacteria isolated from the environment by colony PCR. Microbiology 146:2019–2025 [DOI] [PubMed] [Google Scholar]
- 44. Garinot-Schneider C, Pommer AJ, Moore GR, Kleanthous C, James R. 1996. Identification of putative active-site residues in the DNase domain of colicin E9 by random mutagenesis. J. Mol. Biol. 260:731–742. 10.1006/jmbi.1996.0433 [DOI] [PubMed] [Google Scholar]
- 45. Edelheit O, Hanukoglu A, Hanukoglu I. 2009. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9:61. 10.1186/1472-6750-9-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jo JT, Brinkman FS, Hancock RE. 2003. Aminoglycoside efflux in Pseudomonas aeruginosa: involvement of novel outer membrane proteins. Antimicrob. Agents Chemother. 47:1101–1111. 10.1128/AAC.47.3.1101-1111.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kilburn L, Poole K, Meyer JM, Neshat S. 1998. Insertion mutagenesis of the ferric pyoverdine receptor FpvA of Pseudomonas aeruginosa: identification of permissive sites and a region important for ligand binding. J. Bacteriol. 180:6753–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hocquet D, Vogne C, El Garch F, Vejux A, Gotoh N, Lee A, Lomovskaya O, Plésiat P. 2003. MexXY-OprM efflux pump is necessary for adaptive resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 47:1371–1375. 10.1128/AAC.47.4.1371-1375.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 1992. Short protocols in molecular biology, 2nd ed. John Wiley & Sons, New York, NY. [Google Scholar]
- 50. Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Nat. Biotechnol. 1:784–791. 10.1038/nbt1183-784 [DOI] [Google Scholar]
- 51. Masuda N, Ohya S. 1992. Cross-resistance to meropenem, cephems, and quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 36:1847–1851. 10.1128/AAC.36.9.1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. De Kievit TR, Parkins MD, Gillis RJ, Srikumar R, Ceri H, Poole K, Iglewski BH, Storey DG. 2001. Multidrug efflux pumps: expression patterns and contribution to antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 45:1761–1770. 10.1128/AAC.45.6.1761-1770.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. 10.1016/S0378-1119(98)00130-9 [DOI] [PubMed] [Google Scholar]
- 54. Keen NT, Tamaki S, Kobayashi D, Trollinger D. 1988. Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70:191–197. 10.1016/0378-1119(88)90117-5 [DOI] [PubMed] [Google Scholar]
- 55. Notredame C, Higgins DG, Heringa J. 2000. T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]