

Abstract
Loss-of-function mutations in the KRIT1 gene (CCM1) have been associated with the pathogenesis of cerebral cavernous malformations (CCM), a major cerebrovascular disease. However, KRIT1 functions and CCM pathogenetic mechanisms remain incompletely understood. Indeed, recent experiments in animal models have clearly demonstrated that the homozygous loss of KRIT1 is not sufficient to induce CCM lesions, suggesting that additional factors are necessary to cause CCM disease. Previously, we found that KRIT1 is involved in the maintenance of the intracellular reactive oxygen species (ROS) homeostasis to prevent ROS-induced cellular dysfunctions, including a reduced ability to maintain a quiescent state. Here, we show that KRIT1 loss of function leads to enhanced expression and phosphorylation of the redox-sensitive transcription factor c-Jun, as well as induction of its downstream target COX-2, in both cellular models and human CCM tissues. Furthermore, we demonstrate that c-Jun upregulation can be reversed by either KRIT1 re-expression or ROS scavenging, whereas KRIT1 overexpression prevents forced upregulation of c-Jun induced by oxidative stimuli. Taken together with the reported role of c-Jun in vascular dysfunctions triggered by oxidative stress, our findings shed new light on the molecular mechanisms underlying KRIT1 function and CCM pathogenesis.
Abbreviations: CCM, cerebral cavernous malformation; MEF, mouse embryonic fibroblast; ROS, reactive oxygen species; siRNA, short interfering RNA; RT-qPCR, real-time quantitative PCR; NAC, N-acetylcysteine; HUVEC, human umbilical vein endothelial cells
Keywords: Cerebral cavernous malformations, KRIT1, Reactive oxygen species, Cellular antioxidant defense mechanisms, c-Jun, COX-2, Free radicals
Graphical abstract
Highlights
-
•
KRIT1 loss of function leads to the upregulation of c-Jun.
-
•
c-Jun upregulation occurs in human cerebral cavernous malformation (CCM) lesions.
-
•
KRIT1 loss-dependent c-Jun upregulation can be reversed by ROS scavenging.
-
•
KRIT1 overexpression prevents forced upregulation of c-Jun induced by oxidative stimuli.
-
•
A novel mechanism for CCM pathogenesis is suggested.
Cerebral cavernous malformation (CCM) is a major cerebrovascular disease with a prevalence of 0.3–0.5% in the general population. It is characterized by closely clustered, abnormally dilated, and leaky capillary channels (caverns) surrounded by a thick, segmental layered basal membrane, and may cause serious clinical symptoms, including recurrent headaches, neurological deficits, seizures, stroke, and fatal intracerebral hemorrhage [1]. However, only approximately 30% of people with CCM lesions will eventually develop clinical symptoms, which usually occurs between the 2nd and 5th decades of life.
Although advances have been made toward understanding the natural history and molecular basis of CCM disease, a complete understanding of the pathogenic mechanisms is still a research challenge for defining pharmacological therapies and prognostic factors [1–4]. Indeed, to date there are no direct therapeutic approaches for CCM disease, other than the surgical removal of accessible lesions in patients with recurrent hemorrhage or intractable seizures. In particular, novel pharmacological strategies are required for preventing the de novo formation of CCM lesions in susceptible individuals and the progression of the disease.
CCM is a disease of proven genetic origin (OMIM 116860) that may arise sporadically or be inherited as an autosomal dominant condition with incomplete penetrance and variable expressivity. Sporadic cases usually present with a single lesion on cerebral MRI, whereas the familial form is characterized by the presence of multiple and evolutive lesions.
Genetic studies have so far identified three genes whose mutation causes CCM: KRIT1 (CCM1), MGC4607 (CCM2), and PDCD10 (CCM3) [5]. Comprehensive studies in cellular and animal models have revealed a major role for these genes in blood vessel formation and quiescence maintenance, showing a causal link between their loss-of-function mutations and the hyperactivation of the RhoA GTPase, which destabilizes endothelial barrier function leading to increased vascular permeability [6–8], and suggesting a potential therapy for CCM based on RhoA signaling inhibitors, including statins and fasudil [6,9,10]. However, whereas the mechanisms underlying Rho activation in CCM lesions have yet to be clearly defined, the mechanisms of CCM pathogenesis remain incompletely understood and still fundamental challenges for basic and translational research [2,3]. In fact, recent experiments in conditional knockout mouse models have clearly demonstrated that the homozygous loss of CCM genes is not sufficient to induce CCM lesions, suggesting that additional factors, possibly specific to the neurovascular microenvironment, are necessary to cause CCM disease [11]. Deeper insights into the mechanisms by which CCM proteins affect vascular functions are therefore urgently needed ultimately to provide better treatment options [3].
Previously, we found that KRIT1 is involved in the maintenance of the intracellular reactive oxygen species (ROS) homeostasis to prevent ROS-mediated cellular dysfunctions via an antioxidant pathway involving superoxide dismutase 2 (SOD2), the major cellular antioxidant enzyme, and the transcriptional factor forkhead box protein O1 (FoxO1), a master regulator of cell responses to oxidative stress and a modulator of SOD2 levels. Moreover, we demonstrated that the role of KRIT1 in preventing the accumulation of intracellular ROS facilitates the downregulation of cyclin D1 expression required for cell transition from proliferative growth to quiescence [12].
To test whether the inverse relationship between KRIT1 and ROS levels could affect sensitive intracellular targets involved in the control of important biological functions, including vascular permeability and angiogenesis, we analyzed gene-regulatory proteins known to be targets of oxidative stimuli as well as being involved in the modulation of endothelial barrier function, including c-Jun, a major redox-sensitive component of the transcription factor Activator Protein 1 (AP-1), which mediates endothelial cell gene responses to oxidants [13,14]. Indeed, whereas there is clear evidence that ROS can act as intracellular second messengers that trigger c-Jun expression and activity [13,15–19], it is known that enhanced ROS levels and c-Jun activation have a critical role in the induction of endothelial dysfunction and vascular permeability [17,20–26].
The experimental outcomes showed that KRIT1 loss of function is associated with the upregulation of c-Jun in both cellular models and human CCM tissue samples. Conversely, either KRIT1 re-expression or ROS scavenging reversed the upregulation of c-Jun induced by KRIT1 loss. Furthermore, c-Jun upregulation was correlated with the induction of cyclooxygenase-2 (COX-2), a c-Jun downstream target involved in oxidative stress and inflammatory and angiogenic responses [27–29], whereas KRIT1 overexpression prevented forced upregulation of c-Jun induced by exogenous oxidative stimuli.
Taken together, these results demonstrate that KRIT1 controls c-Jun expression through a mechanism involving the modulation of ROS homeostasis, suggesting that KRIT1 function may exert a protective role in CCM pathogenesis by preventing c-Jun-dependent induction of endothelial dysfunction and vascular permeability triggered by oxidative insults.
Material and methods
Cell culture, transfection, and gene silencing
KRIT1−/− (K−/−) and KRIT1+/+ (K+/+) mouse embryonic fibroblast (MEF) cell lines were established from KRIT1−/− and KRIT1+/+ E8.5 mouse embryos, respectively, whereas KRIT1 9/6 (K9/6) MEFs were obtained by infecting KRIT1−/− cells with a lentiviral vector encoding KRIT1 (Lv-KRIT1) [12]. MEF and HeLa cells were cultured at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, and 100 U/ml penicillin/streptomycin. Human Umbilical Vein Endothelial cells (HUVECs) were cultivated in M199 supplemented with 10% FCS, heparin, and bovine brain extract and maintained at 37 °C and 5% CO2.
HeLa cells were transiently transfected with a pEGFP-KRIT1A construct or pEGFP empty vector as control with FuGENE 6 transfection reagent (Roche) according to the manufacturer's instructions. At 48 h after transfection, cells were used for Western blotting analysis [30].
The expression of KRIT1 in HeLa cells was silenced by the RNA interference (RNAi) technology using two distinct short interfering double-stranded RNA oligomers (siRNAs), Silencer Validated Nos. 15655 (siK655) and 15469 (siK469) siRNAs (Ambion), corresponding to exon 12 and exon 9 sequences (GenBank Accession No. NM_194455), respectively. The BLOCK-iT Alexa Fluor red fluorescent oligo (Invitrogen) was used for determination of efficiency of siRNA transfection as well as RNAi negative control along with the Silencer Negative Control 1 siRNA (Ambion) (siNC). Cells were reverse transfected with 30 nM KRIT1-specific or negative control siRNAs using the Amaxa HUVEC Nucleofector kit and electroporation device (Lonza) according to the optimized manufacturer's reverse transfection protocol. Briefly, cells were harvested by trypsinization and cell density was determined using the Countess automated cell counter (Invitrogen). Cells (5×105 per sample) were pelleted, resuspended in 100 µl of supplemented HUVEC Nucleofector solution, combined with the appropriate dilution of siRNAs, electroporated using the U-001 Nucleofector program, and seeded into six-well plates containing complete culture medium. Forty-eight to seventy-two hours posttransfection, cells were lysed and analyzed by real-time quantitative PCR (RT-qPCR) and Western blotting.
Real-time quantitative PCR
DNA-free RNA was obtained by purification from cell monolayers using the PureLink RNA Mini Kit and DNase I treatment (Invitrogen) and used for cDNA synthesis with the High-Capacity cDNA reverse transcription kit (Invitrogen) according to the manufacturer's instructions. To quantify transcript expression levels, an optimal TaqMan real-time PCR assay was designed for each target transcript using the ProbeFinder software (version 2.45) of the Universal Probe Library from Roche. TaqMan gene expression assays were performed in triplicate on MicroAmp 96-well optical plates using a 7300 real-time PCR system (Applied Biosystems). Reactions were carried out in 25 μl, containing 8 μl diluted (1:10) cDNA, 12.5 μl 2× qPCR Master Mix (Invitrogen), 0.2 μl each primer (20 μM; Sigma), 0.2 μl probe (10 μM; Roche), and 3.9 μl H2O, using the following parameters: 50 °C for 2 min, 95 °C for 2 min, and 45 cycles of 90 °C for 15 s and 60 °C for 30 s. The amounts of the target gene expressed in a sample were normalized to the amounts of internal normalization controls, including the endogenous “housekeeping” 18S rRNA and glyceraldehyde-3-phosphate dehydrogenase transcripts. All TaqMan PCR data were collected using the Sequence Detector software (SDS version 1.3.1; Applied Biosystems).
Western blotting analysis
Cells grown in complete culture medium were either mock-treated or treated with either hydrogen peroxide (H2O2; 0.1 mM) or the ROS scavenger N-acetylcysteine (NAC; 20 mM) in complete medium at 37 °C for various time periods. Cells were then lysed and total cell lysates were analyzed by Western blotting as previously described [31]. Briefly, cell lysates containing equal amounts of total proteins (~50 µg) were separated by either 10 or 12% SDS–PAGE and electroblotted onto Hybond-C transfer membrane (Amersham). The blots were blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS) containing 0.1% Tween 20 for 1 h at 42 °C and incubated with appropriate dilutions of primary antibodies overnight at 4 °C and subsequently with horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 h at room temperature. Proteins were then visualized by an enhanced chemiluminescence detection system (Millipore).
The following antibodies were used: rabbit polyclonal antibodies (pAbs) against KRIT1 (K2 pAb; [12]), c-Jun (H-79:sc-1694; Santa Cruz Biotechnology), JNK (sc-571; Santa Cruz Biotechnology), green fluorescent protein (GFP; G1544; Sigma); mouse monoclonal antibodies (mAbs) against α-tubulin (T5168; Sigma), phospho-c-Jun (P-c-Jun) (KM-1:sc-822; Santa Cruz Biotechnology), phospho-JNK (P-JNK) (sc-6254; Santa Cruz Biotechnology), COX-2 (610203; BD Transduction Laboratories), and vinculin [65]. Primary antibodies were detected using affinity-purified HRP-conjugated secondary antibodies (Sigma).
To compare the density of protein bands of interest and test for significant differences between samples in Western blotting experiments, blots were scanned and quantified using ImageJ software (http://rsb.info.nih.gov/ij/index.html). Band optical density values (mean±SD) were expressed as relative protein level units and plotted in representative histograms showed in figures.
Fluorescence microscopy
For immunofluorescence microscopy analyses, cells were fixed in 3.7% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 in TBS for 1 min, incubated with 1% BSA in TBS for 1 h, and stained with primary antibodies and Alexa Fluor 488 (Invitrogen) secondary antibodies for 1 h each at room temperature. Cells were then counterstained with a blue fluorescent nuclear dye (DAPI) and mounted on microscope slides with ProLongH Gold antifade reagent (Molecular Probes, Invitrogen) before imaging. Confocal microscopy imaging was performed on a Leica TCS_SP5 confocal microscope (Leica Microsystems) using a 40x objective. Instrument parameters for sequential image acquisition, including pinhole diameter, laser intensity, exposure time, PMT gain and offset, were set and held constant to minimize autofluorescence and for comparison between samples.
Immunohistochemistry
Histological samples of surgically resected CCM specimens fixed with Carnoy's fluid (methanol:chloroform:acetic acid, 6:3:1) and embedded in paraffin were retrieved from files of the Surgical Pathology Unit of Turin's Pediatric Hospital (OIRM—Ospedale Infantile Regina Margherita). At the time of neurosurgery the patient (or parent or legal representative if a minor) was asked by the neurosurgeon for an informed consent for genetic analysis of the known CCM genes, including CCM1 (KRIT1), and for scientific use of residual materials according to Institutional Rules defined by OIRM ethics committee.
Specifically, histological samples of two surgically resected CCM specimens from distinct KRIT1 loss-of-function mutation carriers were available, including carriers of either a frameshift (c.1254delA, 419fs436X) or an aberrant splicing (c.1255-4delGTA) mutation.
Histological serial sections (4 µm thick) of paraffin-embedded CCM specimens were obtained and processed by a two-step immunohistochemical staining technique (DAKO EnVision+ System, HRP). Briefly, histological sections were deparaffinized, rehydrated, and subjected to three cycles of 5 min at boiling temperature in citrate buffer (0.01 M, pH 6.0) for antigen retrieval. Endogenous peroxidase activity was blocked by incubation with H2O2 0.3% in methanol for 15 min, and nonspecific binding was prevented by blocking with normal goat serum (ab7481; Abcam). Thereafter, the sections were incubated with either rabbit polyclonal or mouse monoclonal primary antibodies, including a pAb for c-Jun (H-79:sc-1694; Santa Cruz Biotechnology) and mAbs for P-c-Jun (KM-1:sc-822; Santa Cruz Biotechnology) and COX-2 (610203; BD Transduction Laboratories), diluted 1:200 in 0.01 M phosphate-buffered saline containing 0.1% BSA and 0.01% sodium azide or negative control reagent, followed by incubation with an HRP-labeled polymer conjugated to secondary antibodies, using two sequential 30-min incubations at room temperature. Labeling was then visualized by a 5- to 10-min incubation with 3,3′-diaminobenzidine+H2O2 substrate–chromogen, which results in a brown-colored precipitate at the antigen site. The sections were subsequently counterstained with hematoxylin.
Statistics
Data are expressed as the mean ± SD. Statistical analyses were performed by one-way analysis of variance followed by Bonferroni correction. P<0.05 was used as the threshold for statistically significant differences. The results shown in the figures are representative of at least three independent experiments.
Results
KRIT1 regulates c-Jun expression
Previously we showed that KRIT1 loss is associated with an increase in intracellular ROS levels as well as with ROS-mediated cellular dysfunctions, including a reduced ability to maintain a quiescent state [12].
A growing body of evidence suggests that the cellular response to unbalanced ROS overproduction and detoxification is primarily regulated at the level of transcription. Indeed, posttranslational modification of redox-sensitive transcription factors may provide a mechanism by which cells sense these redox changes [18].
To further characterize the functional significance of KRIT1 involvement in the maintenance of the intracellular ROS homeostasis, we analyzed the effects of KRIT1 loss on the expression of c-Jun, a redox-sensitive transcription factor known to be involved in the modulation of endothelial barrier function and angiogenesis [13,18,21,22,25,26].
As a first approach, we performed RT-qPCR and Western blotting analysis of c-Jun mRNA and protein expression levels in KRIT1−/− (K−/−) and wild-type (K+/+) MEFs, established from KRIT1−/− and KRIT1+/+ E8.5 mouse embryos, respectively, as well as in KRIT1−/− MEFs re-expressing KRIT1 (K9/6) [12].
The outcomes of these experiments showed that c-Jun expression was significantly higher in K−/− than in K+/+ and K9/6 cells at both the mRNA (Fig. 1A) and the protein (Fig. 1B) levels, suggesting that KRIT1 loss leads to c-Jun upregulation.
Fig. 1.

KRIT1 regulates c-Jun expression — KRIT1 knockout and re-expression approach. KRIT1−/− (K−/−) and wild-type (K+/+) MEFs and KRIT1−/− MEFs re-expressing KRIT1 (K9/6) were grown to confluence under standard conditions and analyzed by (A) RT-qPCR, (B, C) Western blotting, and (D) immunofluorescence as described under Material and methods. (A) RT-qPCR analysis of c-Jun mRNA expression levels. The amount of each target mRNA expressed in a sample was analyzed in triplicate using appropriate TaqMan gene expression assays (Roche) and normalized to the amounts of internal normalization control transcripts (18S rRNA). Results are expressed as relative mRNA level units referred to the average value obtained for the K−/− samples and represent the mean (±SD) of n ≥ 3 independent RT-qPCR experiments. ***P≤0.001 versus K−/− cells. Notice that c-Jun mRNA levels are significantly higher in K−/− compared to K+/+ and K9/6 MEFs. (B) Representative Western blot analysis of the relative c-Jun, phospho-c-Jun, and KRIT1 expression levels. Tubulin (α-Tub) was used as loading control. Notice that both c-Jun and phospho-c-Jun levels are significantly higher in K−/− compared to K+/+ and K9/6 MEFs. An inverse correlation between c-Jun/phospho-c-Jun and KRIT1 protein levels is also evident. (C) Histograms showing quantitative results of Western blot analysis of the relative c-Jun, phospho-c-Jun, and KRIT1 expression levels. Optical density values are expressed as relative protein level units referred to the average value obtained for the K−/− samples and represent the mean (±SD) of n ≥ 3 independent Western blotting experiments. **P ≤ 0.01 and ***P ≤ 0.001 versus K−/− cells. Notice that differences in phospho-c-Jun levels are correlated with differences in total c-Jun levels. (D) Confocal microscopy analysis of phospho-c-Jun levels and subcellular localization in K−/− and K9/6 MEF cells. Phospho-c-Jun and nuclei were visualized with anti-phospho-c-Jun mAb coupled to Alexa Fluor 488 secondary antibody and DAPI dye, respectively. Notice that phospho-c-Jun is correctly localized to the nucleus in cells lacking KRIT1 (K−/−) and shows enhanced levels compared to K9/6. Scale bar, 15 μm.
To evaluate whether this effect was associated with c-Jun activating phosphorylation and efficient import into the nucleus, we examined phospho-c-Jun levels and subcellular localization. Western blotting analysis of whole-cell extracts with a mAb specific for the active, phosphorylated form of c-Jun (at Ser-63 and Ser-73) (P-c-Jun) showed that phospho-c-Jun levels were always correlated with total c-Jun levels, being significantly higher in K−/− than in K+/+ and K9/6 MEF cells (Fig. 1B, P-c-Jun, and 1C). Furthermore, fluorescence microscopy analysis of K−/− and K9/6 MEF cells with the anti-phospho-c-Jun mAb confirmed that phospho-c-Jun levels were higher in K−/− than in K9/6 MEF cells (Fig. 1D, images a, b) and showed a correct nuclear localization in cells lacking KRIT1 (Fig. 1D, images a, c, e).
Remarkably, an inverse correlation between KRIT1 and c-Jun expression/phosphorylation levels was also observed (Figs. 1A–C), suggesting that KRIT1 plays a role in controlling c-Jun expression and activity.
To further assess this evidence, we used other cell types, including epithelial and endothelial cells, and modulated the expression of KRIT1 by two additional and complementary approaches, such as knockdown and overexpression approaches.
KRIT1 knockdown was performed in HeLa cells (Figs. 2A–E) and HUVECs (Fig. 2F) using two distinct KRIT1-specific siRNAs (siK655 and siK469), which induced a significant decrease in KRIT1 expression at both mRNA (Fig. 2A) and protein (Figs. 2C–F) levels. Notably, as detected by RT-qPCR and Western blotting assays, the siRNA-mediated knockdown of KRIT1 resulted in a significant upregulation of c-Jun mRNA (Fig. 2B) and protein (Figs. 2C–F) expression levels, supporting the evidence that KRIT1 downregulation causes the upregulation of c-Jun. Notably, the upregulated levels of c-Jun were again correlated with corresponding enhanced levels of the active, phosphorylated form of c-Jun (Fig. 2D).
Fig. 2.

KRIT1 regulates c-Jun expression — KRIT1 downregulation (siRNA) approach. (A–E) HeLa cells and (F) HUVECs were mock-transfected or transfected with either a KRIT1-specific siRNA (siK655 or siK469) or a negative control siRNA (siNC). 48 h posttransfection, cells were lysed and analyzed by (A, B) RT-qPCR and (C, D, F) Western blotting to assess c-Jun and KRIT1 mRNA and protein levels, respectively. 18S rRNA and tubulin (α-Tub) were used as endogenous controls for RT-qPCR normalization and Western blotting loading, respectively. Notice that the siRNA-mediated knockdown of KRIT1 results in the upregulation of c-Jun and phospho-c-Jun expression levels. Histograms show quantitative results of (A, B) RT-qPCR and (E, F) Western blot analysis of the relative c-Jun and KRIT1 mRNA and protein expression levels, respectively. mRNA levels (A, B) and optical density values of Western blot bands (E, F) are expressed as relative level units referred to the average value obtained for the mock- (E) or siNC-transfected (F) cells and represent the mean (±SD) of n = 3 independent experiments. ***P ≤ 0.001 versus mock-transfected cells.
Taken together, the KRIT1 knockout and knockdown approaches demonstrate that KRIT1 loss/downregulation is associated with a significant upregulation of c-Jun/phospho-c-Jun levels. Conversely, the forced re-expression of KRIT1 in KRIT1−/− MEF cells to levels higher than wild-type cells (Fig. 1B, compare KRIT1 levels in K9/6 and K+/+ MEFs) caused a significant downregulation of c-Jun/phospho-c-Jun expression at both the protein (Fig. 1B) and the mRNA (Fig. 1A) levels, suggesting a dose-dependent inverse relationship between KRIT1 and c-Jun levels.
To provide further support to the existence of this inverse relationship, we induced KRIT1 overexpression in HeLa cells via transient transfection with a GFP-tagged KRIT1 construct [30]. Consistent with the outcomes of the alternative KRIT1 knockout and knockdown approaches described above, this third complementary approach showed that the forced upregulation of KRIT1 leads to a strong downregulation of c-Jun protein levels (Figs. 3A and B), clearly demonstrating that KRIT1 is able to keep c-Jun expression under strict control.
Fig. 3.

KRIT1 regulates c-Jun expression — KRIT1 overexpression approach. HeLa cells were mock-transfected or transiently transfected with a GFP-tagged KRIT1A construct. 48 h posttransfection, cells were lysed and analyzed by Western blotting with anti-c-Jun (c-Jun) and anti-GFP (GFP-KRIT1 and GFP) antibodies. Tubulin (α-Tub) was used as loading control. Notice that KRIT1 overexpression in HeLa cells results in the downregulation of c-Jun protein levels. Histograms show quantitative results of Western blotting analysis of the relative c-Jun and KRIT1 expression levels. Band optical density values are expressed as relative protein level units referred to the average value obtained for the mock-transfected cells and represent the mean (±SD) of n=3 independent Western blotting experiments. ***P≤0.001 versus mock-transfected cells.
All together, these results suggest that KRIT1 plays a dose-dependent role in limiting c-Jun expression and activity.
ROS scavenging reverses the upregulation of c-Jun expression/phosphorylation caused by KRIT1 loss
There is clear evidence that ROS trigger c-Jun activity by both inducing c-Jun expression and activating phosphorylation [13,18].
To test whether the c-Jun upregulation observed in KRIT1−/− MEF cells was attributable to the previously reported KRIT1 loss-dependent enhanced steady-state levels of intracellular ROS [12], we analyzed both c-Jun expression and phosphorylation in K−/−, K+/+, and K9/6 MEF cells after cell treatment with the antioxidant NAC, which was previously demonstrated to be effective at reducing the levels of ROS in K−/− cells near to the levels of K9/6 cells and rescuing KRIT1 loss-dependent ROS-mediated molecular and cellular dysfunctions, including the upregulation of cyclin D1 and the reduced cell ability to maintain a quiescent state [12]. The outcomes of these experiments showed that treatment of KRIT1−/− cells with NAC led to a significant reduction in both c-Jun expression and phosphorylation compared with relative levels in untreated cells (Figs. 4A–D), indicating that the enhanced c-Jun expression and phosphorylation associated with KRIT1 loss is a redox-sensitive phenomenon. Furthermore, the reduced levels of c-Jun expression and phosphorylation observed in KRIT1−/− cells upon NAC treatment were close to levels observed in untreated wild-type cells (Figs. 4A and B), suggesting that the enhanced c-Jun expression and phosphorylation associated with KRIT1 loss may indeed be largely reversed by antioxidants.
Fig. 4.

ROS scavenging overcomes the upregulation of c-Jun expression/phosphorylation caused by KRIT1 loss. KRIT1−/− (K−/−), wild-type (K+/+), and KRIT1−/− re-expressing KRIT1 (K9/6) MEFs grown to confluence were either mock-treated or treated with the ROS scavenging agent NAC (20 mM in complete medium) for 120 min at 37 °C. The cells were then lysed and analyzed by Western blotting with either (A) c-Jun (c-Jun) or (B) phospho-c-Jun (P-c-Jun) antibodies. Vinculin was used as loading control. Notice that c-Jun expression and phosphorylation levels in KRIT1−/− cells treated with the ROS scavenger NAC (K−/− NAC) are significantly reduced compared with untreated KRIT1−/− cells and close to the levels of untreated wild-type cells (K+/+). (C, D) Histograms showing quantitative results of Western blot analysis of c-Jun and phospho-c-Jun expression levels. Band optical density values are expressed as relative protein level units referred to the average value obtained for untreated K−/− cells and represent the mean (±SD) of n = 3 independent Western blotting experiments. **P≤0.01 versus untreated K−/− cells.
KRIT1 overexpression prevents forced upregulation of c-Jun induced by oxidative stimuli
There is strong evidence that oxidative stress due to either exogenous oxidants or the unbalanced overproduction and detoxification of intracellular ROS, including superoxide anion (O2•−) and H2O2, leads to an increase in c-Jun expression and transcriptional activity [15,16,18,19,32–35].
On the other hand, we showed previously that KRIT1 prevents oxidative stress-mediated cellular dysfunctions by limiting the accumulation of intracellular ROS in a dose-dependent manner [36].
In this light, we hypothesized that the expression of KRIT1 could prevent the increase in c-Jun expression levels triggered by exogenous oxidative stimuli. To test this hypothesis, c-Jun protein levels were assayed in K−/−, K+/+, and K9/6 MEF cells either mock-treated or treated with H2O2.
Consistent with the above reported finding that KRIT1 dose-dependently regulates c-Jun steady-state levels, these levels were inversely proportional to KRIT1 expression levels in untreated MEFs (Fig. 5A, lanes 1, 3, and 5; 5B, and 5C). However, whereas c-Jun was significantly upregulated upon H2O2 treatment in both K−/− and K+/+ MEFs, confirming that oxidative stimuli induce c-Jun upregulation [18], this did not occur in K9/6 MEFs (Fig. 5A, lanes 2, 4, 6, and 5B), indicating that KRIT1 overexpression prevents forced upregulation of c-Jun induced by oxidative stimuli and further suggesting that KRIT1 plays a role in protecting cells against exogenous oxidative insults.
Fig. 5.

KRIT1 overexpression prevents forced upregulation of c-Jun induced by oxidative stimuli. (A, D) KRIT1−/− (K−/−) and wild-type (K+/+) MEFs and KRIT1−/− re-expressing KRIT1 (K9/6) MEFs grown to confluence were either mock-treated or treated with H2O2 (0.1 mM in complete medium) for 60 min at 37 °C. The cells were then lysed and analyzed by Western blotting with anti-c-Jun (c-Jun) and anti-KRIT1 (KRIT1) antibodies. Tubulin (α-Tub) was used as loading control. Notice that KRIT1 overexpression prevents forced upregulation of c-Jun induced by oxidative stimuli. (B, C, E) Histograms show quantitative results of Western blot analysis of the relative c-Jun (B), KRIT1 (C), and P-c-Jun (E) expression levels. Band optical density values are expressed as relative protein level units referred to the average value obtained for untreated K−/− cells and represent the mean (±SD) of n=3 independent Western blot experiments. ***P≤0.001 versus untreated K−/− cells.
Furthermore, whereas previous FACS analysis demonstrated that KRIT1 overexpression prevents ROS enhancement in response to cell treatment with either inorganic or organic oxidants, including H2O2 and tert-butylhydroperoxide [12], Western blotting assays showed that phospho-c-Jun levels were again correlated with total c-Jun levels (Figs. 5A–E). Intriguingly, a slight downregulation of KRIT1 protein levels was also observed upon H2O2 treatment in both wild-type (K+/+) and KRIT1-overexpressing (K9/6) MEF cells (Figs. 5A, C, and D), which deserve future investigation.
c-Jun expression/phosphorylation is enhanced in CCM lesions from KRIT1 loss-of-function mutation carriers
CCM lesions are characterized by altered blood–brain barrier function and increased vessel permeability due to the weakening of endothelial cell–cell junctions [6–8]. Interestingly, there is clear evidence that c-Jun upregulation is linked to the induction of endothelial dysfunction and vascular permeability [21,22,25,26]. In this light, we hypothesized that the KRIT1 loss-of-function-dependent upregulation of c-Jun expression and phosphorylation observed in cellular models could also occur in vivo. To address this hypothesis, we performed immunohistochemical analysis of c-Jun expression and phosphorylation levels in surgically resected human CCM specimens from patients carrying a KRIT1 loss-of-function mutation. Notably, the results of these experiments showed a significant positive staining for both total c-Jun and phospho-c-Jun in endothelial cells lining the lumen of CCM lesions compared with perilesional normal vessels, with up to 90% of positive cells in the most abnormally dilated vessels (Fig. 6), clearly demonstrating that the upregulation of c-Jun caused by KRIT1 loss-of-function occurs also in vivo and suggesting a potential relationship with CCM disease.
Fig. 6.

c-Jun expression and phosphorylation are enhanced in CCM lesions from KRIT1 loss-of-function mutation carriers. Histological sections (4 µm) of paraffin-embedded CCM surgical specimens, deriving from a KRIT1 loss-of-function mutation carrier, were processed by a two-step immunohistochemical staining technique (DAKO EnVision+ System, HRP) with c-Jun and phospho-c-Jun antibodies to assess (A, B) c-Jun and (C, D) phospho-c-Jun expression in perilesional and CCM lesion vessels. Notice that many endothelial cells lining the lumen (l) of CCM vessels (B, D) were positive for c-Jun (B) and phospho-c-Jun (D), whereas neither c-Jun- (A) nor phospho-c-Jun- (C) positive staining was detected in perilesional normal vessels (A, C). Original magnification: 20×.
KRIT1 loss of function induces a ROS-dependent activation of JNK
There is evidence that the role of oxidants and oxidative stress in enhancing c-Jun expression and transcriptional activity is mediated, at least in part, by the c-Jun NH2-terminal kinase (JNK), a major upstream regulator of c-Jun. Indeed, oxidative stress induces phosphorylation and activation of JNK, facilitating its entry into the nucleus. Nuclear JNK phosphorylates c-Jun at the serine 63 and 73 regulatory sites within the N-terminal transactivation domain, enhancing its transcriptional activities [13,18].
To test whether c-Jun upregulation induced by KRIT1 loss of function was correlated with the activation of JNK, we performed Western blotting analysis of cell extracts from K−/− and K9/6 MEF cells using a monoclonal antibody specific for the active, phosphorylated form of JNK (at Thr-183 and Tyr-185) (P-JNK). The outcomes of these experiments showed that phospho-JNK levels were significantly higher in K−/− compared to K9/6 MEF cells (Fig. 7A, P-JNK), thus paralleling the enhanced P-c-Jun levels (Fig. 7A, P-c-Jun, and previous figures); in contrast, the levels of total JNK were not significantly varied (Fig. 7A, JNK).
Fig. 7.

KRIT1 loss of function induces a ROS-dependent activation of JNK. KRIT1−/− (K−/−) and KRIT1−/− re-expressing KRIT1 (K9/6) MEFs grown to confluence were (A) left untreated or (B) either mock-treated or treated with the ROS-scavenging agent NAC (20 mM in complete medium) for 120 min at 37 °C. The cells were then lysed and analyzed by Western blotting as described under Material and methods. The phosphorylated JNK and total JNK were probed using anti-phospho-JNK (Thr-183/Tyr-185) antibody and anti-JNK antibody and compared to the relative P-c-Jun and KRIT1 expression levels. Total JNK and tubulin (α-Tub) served as loading controls. (A) Notice that JNK phosphorylation is significantly higher in K−/− compared to K9/6 MEFs and correlated with P-c-Jun levels. An inverse correlation between P-JNK and KRIT1 protein levels is also evident. (B) Notice that P-JNK levels in K−/− MEFs treated with the ROS scavenger NAC (K−/− NAC+) are significantly reduced compared with untreated K−/− cells (K−/− NAC−) and close to the levels of untreated K9/6 MEFs (K9/6 NAC−). A direct correlation between P-JNK and P-c-Jun and an inverse correlation between P-JNK and KRIT1 levels are also evident.
To test whether the activating phosphorylation of JNK observed in KRIT1−/− MEF cells was ROS-dependent, we then analyzed P-JNK levels upon cell treatment with the antioxidant NAC. Indeed, cell treatment with NAC was effective at reducing both JNK and c-Jun phosphorylation (Fig. 7B), indicating that KRIT1 loss of function induces a ROS-dependent activation of JNK and suggesting that this activation plays an upstream regulatory role in mediating the ROS-dependent upregulation of c-Jun. However, cell treatment with an inhibitor of JNK (SP600125) rescued only partially the ROS-dependent upregulation of c-Jun induced by KRIT1 loss (data not shown), suggesting that additional regulatory factors acting upstream of c-Jun are probably involved.
KRIT1 loss of function induces downstream targets of c-Jun
Activation of c-Jun promotes induction of both proliferative and proinflammatory gene products. Notably, we previously found that KRIT1 loss of function leads to a ROS-mediated upregulation of cyclin D1 [12,37], a major c-Jun target gene involved in cell cycle progression through the G1 phase [12,37], suggesting a plausible involvement of c-Jun in the reduced cell ability to maintain a quiescent state caused by KRIT1 loss. To further extend the potential functional significance of the inverse relationship between KRIT1 expression and c-Jun expression and its oxidative stress-induced activation, we then tested c-Jun target genes known to be involved in proinflammatory responses, including COX-2, a major oxidative stress biomarker and inflammatory mediator involved in angiogenesis and vascular dysfunction [27–29].
As detected by RT-qPCR and Western blotting assays, COX-2 expression was significantly higher in K−/− compared to K+/+ and K9/6 cells, at both the mRNA (Fig. 8A) and the protein (Fig. 8B) levels, suggesting that KRIT1 loss leads to COX-2 upregulation. Moreover, COX-2 protein levels were directly correlated with P-c-Jun levels (Fig. 8B), suggesting a potential relationship. Furthermore, immunohistochemical analysis of COX-2 expression in surgically resected human CCM specimens from patients carrying a KRIT1 loss-of-function mutation showed a significant positive staining in endothelial cells lining the lumen of CCM lesions, with most cells showing a prevalent COX-2 perinuclear/nuclear localization (Fig. 8C), suggesting that the upregulation of COX-2 caused by KRIT1 loss of function may occur also in vivo and pointing to a potential relationship with CCM disease.
Fig. 8.

KRIT1 loss of function causes the upregulation of COX-2. (A, B) KRIT1−/− (K−/−), wild-type (K+/+), and KRIT1-/- re-expressing KRIT1 (K9/6) MEFs were grown to confluence under standard conditions and analyzed by (A) RT-qPCR and (B) Western blotting. (A) RT-qPCR analysis. COX-2 mRNA expression levels were analyzed in triplicate using appropriate TaqMan gene expression assays (Roche) and normalized to the amounts of internal normalization control transcripts (18S rRNA). Results are expressed as relative mRNA level units referred to the average value obtained for the K−/− samples and represent the mean (±SD) of n=3 independent RT-qPCR experiments. ***P≤0.001 versus K−/− cells. (B) Representative Western blot analysis. COX-2 levels in cell lysates were analyzed by Western blotting with an anti-COX-2 mAb and compared to the relative P-c-Jun and KRIT1 levels. Tubulin (α-Tub) was used as loading control. Notice that KRIT1 loss of function reproduced in K−/− cells caused a significant upregulation of COX-2 expression at both mRNA and protein levels, which was completely rescued by the re-expression of KRIT1 (K9/6). It is also evident that COX-2 protein levels are directly and inversely correlated with P-c-Jun and KRIT1 protein levels, respectively. (C) Immunohistochemical analysis of paraffin-embedded human cerebral cavernous malformations with anti-COX-2 antibodies. Cavernous malformation tissue was collected from a CCM1 mutation carrier with familial disease at the time of surgical resection under an approved institutional review board protocol. Notice that many endothelial cells lining the lumen (l) of CCM vessels were positive for COX-2.
Whereas further studies based on c-Jun dominant negative inhibition and RNAi-mediated knockdown are required to verify the putative direct relationship between the KRIT1 loss-dependent upregulation of c-Jun and its target genes, including but not limited to cyclin D1 and COX2, the findings that KRIT1 plays a role in regulating distinct proteins involved in oxidative stress responses open novel avenues for future investigations aimed at better defining the molecular mechanisms of CCM pathogenesis.
Discussion
KRIT1 loss-of-function mutations have been clearly associated with the pathogenesis of CCM, a major cerebrovascular disease characterized by abnormally enlarged and leaky capillaries that predispose to seizures, focal neurological deficits, and fatal intracerebral hemorrhage. In particular, comprehensive analysis of the KRIT1 gene in CCM patients has suggested that KRIT1 functions need to be severely impaired for pathogenesis, whereas studies in cellular and animal models have demonstrated that KRIT1 deficiency leads to the major molecular and cellular hallmarks of CCM disease, including destabilization of endothelial cell–cell junctions, reduced ability of the cell to maintain a quiescent state, and increased vascular permeability and angiogenic potential [6,12,38–42]. However, recent experiments in animal models have shown that loss of function of CCM genes, including KRIT1 (CCM1), is not sufficient to induce CCM lesions, suggesting that additional triggers occurring locally at neurovascular units, including microenvironmental stress factors and brain injuries, are necessary to promote the onset and progression of CCM disease [3,11].
Among the microenvironmental stress events that might account for a sort of environmental second hit, triggering CCM lesion formation in sensitive vascular areas of CCM mutation carriers, there is oxidative stress. This may occur as a consequence of an endogenous imbalance between the production of ROS and the ability of cellular antioxidant mechanisms to readily prevent excessive ROS accumulation, maintaining a physiological equilibrium, as well as exogenous oxidative insults, including cell exposure to xenobiotics or ionizing radiations [36]. Furthermore, pro-oxidant factors may be released locally after inflammatory responses, impaired neurovascular coupling, and ischemia–reperfusion events [20]. Remarkably, there is now a wealth of evidence indicating that oxidative stress is indeed a major cause of vascular remodeling and neurovascular unit dysfunction associated with cerebrovascular diseases [20,23,43]. In particular, oxidative stress has been clearly implicated in all the major molecular and cellular dysfunctions related to CCM diseases, including destabilization of endothelial cell–cell junctions, reduced cell ability to maintain a quiescent state, and increased vascular permeability and angiogenic activity [24,36,44], suggesting that it might represent a significant additive factor involved in the initiation and progression of CCM disease.
Consistently, we previously reported that KRIT1 is involved in the maintenance of intracellular ROS homeostasis to prevent ROS-mediated cell dysfunctions through an antioxidant pathway involving SOD2, the major cellular antioxidant enzyme, and the transcriptional factor FoxO1, a master regulator of cell responses to oxidative stress and a modulator of SOD2 levels, raising the hypothesis that CCM lesions may result from an impaired oxidative stress defense in microvascular districts of genetically predisposed subjects and opening new therapeutic perspectives [12,45].
These original results and hypothesis are now further supported and expanded by novel findings showing that KRIT1 loss of function causes the upregulation of c-Jun in both cellular models and human CCM tissue samples, as well as that this upregulation can be reversed by either KRIT1 re-expression or ROS scavenging with antioxidant compounds, including NAC, suggesting that KRIT1 controls c-Jun expression through the regulation of intracellular ROS homeostasis.
c-Jun, a prominent member of the AP-1 transcription family, has been implicated in the regulation of a wide range of biological processes including development, differentiation, transformation, and apoptosis [14]. It is now clear that c-Jun activity is closely associated with a steady elevation of its expression through an autocrine and feed-forward transcriptional mechanism, as well as with the phosphorylation of two serine residues, Ser-63 and Ser-73, located within the N-terminal transcription activation domain, whereas its effects on cellular responses depend strongly on the context of other regulatory influences that the cell is receiving [14]. Both c-Jun expression and phosphorylation are highly induced in response to environmental cues, including mitogenic stimuli and various stresses [14,46].
According to our findings, there is clear evidence that c-Jun is highly upregulated in response to either oxidants or oxidative stress and associated with vascular dysfunctions, including vascular remodeling and inflammation, and enhanced vascular permeability [13,15–19,47]. Conversely, there is also evidence that increased cellular glutathione levels by NAC inhibit the expression and transcriptional activity of c-Jun triggered by numerous stimuli [18], including the binding of c-Jun to the AP-1 element within the VEGF promoter [48].
Notably, the finding that KRIT1 high expression levels prevent forced upregulation of c-Jun induced by oxidative stimuli suggests that the dose-dependent inverse relationship between KRIT1 and c-Jun levels may play an important role in protecting cells against exogenous oxidative insults. Accordingly, previously reported findings showed that KRIT1 overexpression prevents ROS enhancement in response to cell treatment with either inorganic or organic oxidants, including H2O2 and tert-butyl hydroperoxide, conferring resistance to exogenous oxidative challenge-induced DNA damage and apoptotic response [12].
Moreover, our novel findings are reinforced by complementary results showing that KRIT1 loss-of-function-dependent and redox-sensitive upregulation of c-Jun correlates with the upregulation of both upstream regulators and downstream targets of c-Jun. In particular, we show that the upregulation of c-Jun is accompanied by a redox-sensitive activating phosphorylation of JNK, a major c-Jun upstream regulator known to be activated by oxidants and oxidative stress [13,18]. Accordingly and intriguingly, an enhanced phosphorylation of JNK has been previously observed in CCM2-depleted cells [6–8], suggesting that it might represent a general consequence of the deficiency of any CCM protein.
In addition to JNK, multiple signaling molecules have been proposed as potential upstream regulators for ROS-mediated induction of c-Jun expression and activity, including extracellular signal-regulated kinases, p38 mitogen-activated protein kinases, phospholipase A2, arachidonic acid, lipoxygenase, and protein kinase C, as well as tyrosine kinases and tyrosine and serine/threonine phosphatases [13,15–19]. Accordingly, cell treatment with an inhibitor of JNK (SP600125) rescued only partially the ROS-dependent upregulation of c-Jun induced by KRIT1 loss, suggesting that additional regulatory factors acting upstream of c-Jun are likely to be involved. Further studies based on pharmacologic inhibition and RNAi-mediated knockdown of distinct c-Jun upstream regulators are therefore required to gain further insights into the probably complex redox-sensitive molecular machinery that links KRIT1 to the ROS-mediated activation of c-Jun.
On the other hand, previous results showed that KRIT1 loss-of-function leads to a ROS-mediated upregulation of cyclin D1 [12,37], a major c-Jun target gene involved in cell cycle progression [12,37], suggesting a potential involvement of c-Jun in the reduced cell ability to maintain a quiescent state caused by KRIT1 loss [12,37]. In addition, here we report that the upregulation of c-Jun is paralleled by the induction of COX-2, a major c-Jun target gene [49], as well as a major oxidative stress biomarker and inflammatory mediator involved in vascular dysfunction [27–29], raising the possibility that KRIT1 loss of function might be implicated in synergistic oxidative stress and inflammatory responses. Consistently, a close relationship between oxidative stress and local inflammation has been clearly established and shown to underlie vascular disease of diverse etiology [43,50]. Furthermore, whereas a recent report shows that mice heterozygous for the deletion of the KRIT1 gene (KRIT1+/−) exhibit an enhanced sensitivity to inflammatory stimuli [38], there is evidence that an inflammatory response occurs in CCM lesions [51,52], supporting a potential role for inflammatory processes in the pathogenesis of CCM disease. On the other hand, whereas evidence in animal models suggests that neoangiogenic events are necessary to cause CCM disease [11], a key molecular link connecting oxidative stress and angiogenesis has been also established, with oxidative stress having potential primacy [53]. Indeed, reciprocity of inflammation, oxidative stress, and neovascularization is emerging as an important mechanism underlying numerous biological processes [53], raising the possibility that these mechanisms play key sequential and synergistic roles in CCM pathogenesis.
Consistently, an increased ROS production has been associated with endothelial dysfunction and onset of the CCM-related disease hereditary hemorrhagic telangiectasia [54,55].
In this light, the evidence that the expression levels of c-Jun, including its phosphorylated, active form, and COX-2 are enhanced in endothelial cells lining CCM lesions from KRIT1 loss-of-function mutation carriers suggests that the upregulation of these proteins may actively contribute to the pathogenesis of CCM disease by acting synergistically with local microenvironmental factors affecting endothelial function and vascular permeability, including locally released pro-oxidant, inflammatory, and angiogenic factors.
Future focused research is required to address this possibility, as well as to assess whether COX-2 is activated as a consequence of the KRIT1 loss-of-function-dependent and ROS-mediated upregulation of c-Jun. Indeed, although it is clearly established that c-Jun activation is upstream of COX-2 expression, there is also evidence that COX-2 can be induced by other redox-sensitive transcription factors, including NF-κB, and may act both upstream and downstream of ROS signaling [27–29]. Nonetheless, our findings open a promising novel research avenue and provide a useful framework for paving the way toward a better understanding of the molecular events underlying the pathogenesis of CCM disease.
Remarkably, and consistent with our finding and hypothesis, c-Jun upregulation has been linked with pathological angiogenesis and microvascular diseases in humans, whereas its inhibition has been shown to be effective as a rescue therapy for these diseases [21,22,25,26,34,56]. In particular, it has been demonstrated that targeting of c-Jun inhibits microvascular endothelial cell proliferation, migration, invasion, tubule formation, and endothelial cell production of matrix metalloproteinase-2 (MMP-2) in vitro [25,26], and suppresses VEGF-induced neovascularization, MMP-2 production, and vascular permeability and inflammation in vivo [21,25,26].
In this light, it is noteworthy that an upregulation of c-Jun downstream targets involved in cell cycle progression and extracellular matrix remodeling, including cyclin D1 and MMP-2 [25,37], has been previously observed in cellular models of CCM [12] and human CCM lesions [57], respectively. On the other hand, our recently reported findings related to a role for TGF-β in CCM patients [58] might also be connected to and downstream of deregulation of oxidative stress and c-Jun activity. Indeed, whereas there is clear evidence that ROS can stimulate the activation of the TGF-β pathway with important consequences on cellular functions [59,60], it has been demonstrated that this ROS-mediated regulation is dependent on AP-1 transcriptional activity [60].
Conclusions
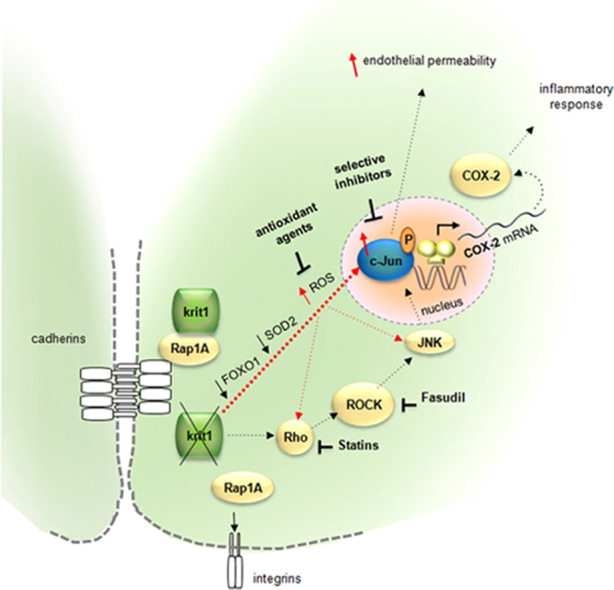
Taken together with the above observations, our experimental results demonstrating that KRIT1 controls c-Jun expression and phosphorylation through a mechanism involving the modulation of ROS homeostasis suggest an important role for ROS and the redox-sensitive transcription factor c-Jun in CCM pathogenesis, and point to a model mechanism whereby the dose of KRIT1 may be relevant in preventing c-Jun-dependent induction of vascular dysfunctions triggered by oxidative stress (Fig. 9).
Fig. 9.

Schematic model representing the inverse relationship between KRIT1 and c-Jun expression levels and its putative functional significance. See text for details.
Importantly, our findings provide also an alternative explanation to the suggested effectiveness of fasudil and statins as potential therapy for CCM disease [6,9,10]. Indeed, whereas it has been demonstrated that the Rho GTPase pathway can be directly activated by ROS [61], there is clear evidence that both fasudil and statins exert powerful intracellular antioxidant activities in endothelial cells, including the inhibition of superoxide production and the improvement of both ROS scavenging and NO bioavailability [62–64].
Future progress toward this novel perspective should pave the way for the development of novel, safe, and effective therapeutic strategies for prevention and treatment of CCM disease.
Acknowledgments
We are grateful to Alessandro Morina, Valentina Cutano, Rosalia Canzoneri, Silvia Gianoglio, Debora Monteleone, Elena Tamagno, and Eva Baldini, for helping in some experiments, and Alfredo Brusco, Carlo Arduino (Medical Genetics Laboratory, A.O. Città della Salute e della Scienza di Torino), and Cristiana Marchese (Department Area Diagnostica Patologico-Clinica, Azienda Ospedaliera Ordine Mauriziano) for help with genetic analyses. We also wish to gratefully acknowledge Alessandra De Luca for creating and drawing the model shown in graphical abstract and Fig. 9, and clinical and basic research units of the Italian Research Network for Cerebral Cavernous Malformation (http://www.ccmitalia.unito.it) and the Associazione Italiana Angiomi Cavernosi (http://www.ccmitalia.unito.it/aiac) for supporting the study with biological materials and helpful discussion. This work was supported by grants from the University of Torino (Local Research Funding 2012/SFR), MIUR (PRIN2008BP25KN to S.F.R.), and Fondazione Telethon (GGP06222 to S.F.R.).
Authors' Contributions
S.F.R., L.G., and E.D.L. conceived and designed the experiments. L.G., E.D.L., S.B., E.T., and M.G. performed the experiments. L.G., E.D.L., M.F., and S.F.R. analyzed the data. M.F., L.T., A.M., and F.B. contributed reagents/materials/analysis tools. S.F.R., L.G., and E.D.L. wrote the paper.
References
- 1.Cavalcanti D.D., Kalani M.Y., Martirosyan N.L., Eales J., Spetzler R.F., Preul M.C. Cerebral cavernous malformations: from genes to proteins to disease. J. Neurosurg. 2012;116:122–132. doi: 10.3171/2011.8.JNS101241. [DOI] [PubMed] [Google Scholar]
- 2.Bacigaluppi S., Retta S.F., Pileggi S., Fontanella M., Goitre L., Tassi L., La Camera A., Citterio A., Patrosso M.C., Tredici G., Penco S. Genetic and cellular basis of cerebral cavernous malformations: implications for clinical management. Clin. Genet. 2013;83:7–14. doi: 10.1111/j.1399-0004.2012.01892.x. [DOI] [PubMed] [Google Scholar]
- 3.Fischer A., Zalvide J., Faurobert E., Albiges-Rizo C., Tournier-Lasserve E. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol. Med. 2013;19:302–308. doi: 10.1016/j.molmed.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Storkebaum E., Quaegebeur A., Vikkula M., Carmeliet P. Cerebrovascular disorders: molecular insights and therapeutic opportunities. Nat. Neurosci. 2011;14:1390–1397. doi: 10.1038/nn.2947. [DOI] [PubMed] [Google Scholar]
- 5.Riant F., Cecillon M., Saugier-Veber P., Tournier-Lasserve E. CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics. 2013;14:133–141. doi: 10.1007/s10048-013-0362-0. [DOI] [PubMed] [Google Scholar]
- 6.Stockton R.A., Shenkar R., Awad I.A., Ginsberg M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010;207:881–896. doi: 10.1084/jem.20091258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitehead K.J., Chan A.C., Navankasattusas S., Koh W., London N.R., Ling J., Mayo A.H., Drakos S.G., Jones C.A., Zhu W., Marchuk D.A., Davis G.E., Li D.Y. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009;15:177–184. doi: 10.1038/nm.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng X., Xu C., Di Lorenzo A., Kleaveland B., Zou Z., Seiler C., Chen M., Cheng L., Xiao J., He J., Pack M.A., Sessa W.C., Kahn M.L. CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J. Clin. Invest. 2010;120:2795–2804. doi: 10.1172/JCI39679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li D.Y., Whitehead K.J. Evaluating strategies for the treatment of cerebral cavernous malformations. Stroke. 2010;41:S92–94. doi: 10.1161/STROKEAHA.110.594929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonald D.A., Shi C., Shenkar R., Stockton R.A., Liu F., Ginsberg M.H., Marchuk D.A., Awad I.A. Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease. Stroke. 2012;43:571–574. doi: 10.1161/STROKEAHA.111.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boulday G., Rudini N., Maddaluno L., Blecon A., Arnould M., Gaudric A., Chapon F., Adams R.H., Dejana E., Tournier-Lasserve E. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J. Exp. Med. 2011;208:1835–1847. doi: 10.1084/jem.20110571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goitre L., Balzac F., Degani S., Degan P., Marchi S., Pinton P., Retta S.F. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS One. 2010;5:e11786. doi: 10.1371/journal.pone.0011786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunsch C., Medford R.M. Oxidative stress as a regulator of gene expression in the vasculature. Circ. Res. 1999;85:753–766. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 14.Meng Q., Xia Y. c-Jun, at the crossroad of the signaling network. Protein Cell. 2011;2:889–898. doi: 10.1007/s13238-011-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beiqing L., Chen M., Whisler R.L. Sublethal levels of oxidative stress stimulate transcriptional activation of c-jun and suppress IL-2 promoter activation in Jurkat T cells. J. Immunol. 1996;157:160–169. [PubMed] [Google Scholar]
- 16.Lee S.F., Huang Y.T., Wu W.S., Lin J.K. Induction of c-jun protooncogene expression by hydrogen peroxide through hydroxyl radical generation and p60SRC tyrosine kinase activation. Free Radic. Biol. Med. 1996;21:437–448. doi: 10.1016/0891-5849(96)00040-8. [DOI] [PubMed] [Google Scholar]
- 17.Lum H., Roebuck K.A. Oxidant stress and endothelial cell dysfunction. Am. J. Physiol. Cell Physiol. 2001;280:C719–741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 18.Marshall H.E., Merchant K., Stamler J.S. Nitrosation and oxidation in the regulation of gene expression. FASEB J. 2000;14:1889–1900. doi: 10.1096/fj.00.011rev. [DOI] [PubMed] [Google Scholar]
- 19.Pinkus R., Weiner L.M., Daniel V. Role of oxidants and antioxidants in the induction of AP-1, NF-kappaB, and glutathione S-transferase gene expression. J. Biol. Chem. 1996;271:13422–13429. doi: 10.1074/jbc.271.23.13422. [DOI] [PubMed] [Google Scholar]
- 20.Chrissobolis S., Miller A.A., Drummond G.R., Kemp-Harper B.K., Sobey C.G. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front. Biosci. 2011;16:1733–1745. doi: 10.2741/3816. [DOI] [PubMed] [Google Scholar]
- 21.Fahmy R.G., Waldman A., Zhang G., Mitchell A., Tedla N., Cai H., Geczy C.R., Chesterman C.N., Perry M., Khachigian L.M. Suppression of vascular permeability and inflammation by targeting of the transcription factor c-Jun. Nat. Biotechnol. 2006;24:856–863. doi: 10.1038/nbt1225. [DOI] [PubMed] [Google Scholar]
- 22.Folkman J. Angiogenesis and c-Jun. J. Natl. Cancer Inst. 2004;96:644. doi: 10.1093/jnci/djh148. [DOI] [PubMed] [Google Scholar]
- 23.Fraser P.A. The role of free radical generation in increasing cerebrovascular permeability. Free Radic. Biol. Med. 2011;51:967–977. doi: 10.1016/j.freeradbiomed.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signaling. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang G., Fahmy R.G., diGirolamo N., Khachigian L.M. JUN siRNA regulates matrix metalloproteinase-2 expression, microvascular endothelial growth and retinal neovascularisation. J. Cell Sci. 2006;119:3219–3226. doi: 10.1242/jcs.03059. [DOI] [PubMed] [Google Scholar]
- 26.Zhang G.X., Kimura S., Nishiyama A., Shokoji T., Rahman M., Abe Y. ROS during the acute phase of Ang II hypertension participates in cardiovascular MAPK activation but not vasoconstriction. Hypertension. 2004;43:117–124. doi: 10.1161/01.HYP.0000105110.12667.F8. [DOI] [PubMed] [Google Scholar]
- 27.Hsieh H.L., Lin C.C., Chan H.J., Yang C.M., c-Src-dependent E.G.F. receptor transactivation contributes to ET-1-induced COX-2 expression in brain microvascular endothelial cells. J. Neuroinflammation. 2012;9:152. doi: 10.1186/1742-2094-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishikawa T., Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signaling. 2007;9:343–353. doi: 10.1089/ars.2006.1458. [DOI] [PubMed] [Google Scholar]
- 29.Ushio-Fukai M., Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266:37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francalanci F., Avolio M., De Luca E., Longo D., Menchise V., Guazzi P., Sgro F., Marino M., Goitre L., Balzac F., Trabalzini L., Retta S.F. Structural and functional differences between KRIT1A and KRIT1B isoforms: a framework for understanding CCM pathogenesis. Exp. Cell Res. 2009;315:285–303. doi: 10.1016/j.yexcr.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Balzac F., Avolio M., Degani S., Kaverina I., Torti M., Silengo L., Small J.V., Retta S.F. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the crossroads between cadherin and integrin function. J. Cell Sci. 2005;118:4765–4783. doi: 10.1242/jcs.02584. [DOI] [PubMed] [Google Scholar]
- 32.Aharoni-Simon M., Reifen R., Tirosh O. ROS-production-mediated activation of AP-1 but not NFkappaB inhibits glutamate-induced HT4 neuronal cell death. Antioxid. Redox Signaling. 2006;8:1339–1349. doi: 10.1089/ars.2006.8.1339. [DOI] [PubMed] [Google Scholar]
- 33.Datta R., Hallahan D.E., Kharbanda S.M., Rubin E., Sherman M.L., Huberman E., Weichselbaum R.R., Kufe D.W. Involvement of reactive oxygen intermediates in the induction of c-jun gene transcription by ionizing radiation. Biochemistry. 1992;31:8300–8306. doi: 10.1021/bi00150a025. [DOI] [PubMed] [Google Scholar]
- 34.Deng Z., Sui G., Rosa P.M., Zhao W. Radiation-induced c-Jun activation depends on MEK1–ERK1/2 signaling pathway in microglial cells. PLoS One. 2012;7:e36739. doi: 10.1371/journal.pone.0036739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li D.W., Spector A. Hydrogen peroxide-induced expression of the proto-oncogenes, c-jun, c-fos and c-myc in rabbit lens epithelial cells. Mol. Cell. Biochem. 1997;173:59–69. doi: 10.1023/a:1006828402225. [DOI] [PubMed] [Google Scholar]
- 36.Goitre L., Pergolizzi B., Ferro E., Trabalzini L., Retta S.F. Molecular crosstalk between integrins and cadherins: do reactive oxygen species set the talk? J. Signal Transduction. 2012;2012:807682. doi: 10.1155/2012/807682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wisdom R., Johnson R.S., Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999;18:188–197. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corr M., Lerman I., Keubel J.M., Ronacher L., Misra R., Lund F., Sarelius I.H., Glading A.J. Decreased Krev interaction-trapped 1 expression leads to increased vascular permeability and modifies inflammatory responses in vivo. Arterioscler. Thromb. Vasc. Biol. 2012;32:2702–2710. doi: 10.1161/ATVBAHA.112.300115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glading A., Han J., Stockton R.A., Ginsberg M.H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 2007;179:247–254. doi: 10.1083/jcb.200705175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lampugnani M.G., Orsenigo F., Rudini N., Maddaluno L., Boulday G., Chapon F., Dejana E. CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J. Cell Sci. 2010;123:1073–1080. doi: 10.1242/jcs.059329. [DOI] [PubMed] [Google Scholar]
- 41.Whitehead K.J., Plummer N.W., Adams J.A., Marchuk D.A., Li D.Y. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437–1448. doi: 10.1242/dev.01036. [DOI] [PubMed] [Google Scholar]
- 42.Wustehube J., Bartol A., Liebler S.S., Brutsch R., Zhu Y., Felbor U., Sure U., Augustin H.G., Fischer A. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc. Natl. Acad. Sci. USA. 2010;107:12640–12645. doi: 10.1073/pnas.1000132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faraci F.M. Protecting against vascular disease in brain. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H1566–1582. doi: 10.1152/ajpheart.01310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukai T., Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guazzi P., Goitre L., Ferro E., Cutano V., Martino C., Trabalzini L., Retta S.F. Identification of the Kelch family protein Nd1-L as a novel molecular interactor of KRIT1. PLoS One. 2012;7:e44705. doi: 10.1371/journal.pone.0044705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leppa S., Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- 47.Rojas A., Figueroa H., Re L., Morales M.A. Oxidative stress at the vascular wall: mechanistic and pharmacological aspects. Arch. Med. Res. 2006;37:436–448. doi: 10.1016/j.arcmed.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 48.Tsai C.H., Chiang Y.C., Chen H.T., Huang P.H., Hsu H.C., Tang C.H. High glucose induces vascular endothelial growth factor production in human synovial fibroblasts through reactive oxygen species generation. Biochim. Biophys. Acta. 2013;1830:2649–2658. doi: 10.1016/j.bbagen.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 49.Yamaguchi K., Lantowski A., Dannenberg A.J., Subbaramaiah K. Histone deacetylase inhibitors suppress the induction of c-Jun and its target genes including COX-2. J. Biol. Chem. 2005;280:32569–32577. doi: 10.1074/jbc.M503201200. [DOI] [PubMed] [Google Scholar]
- 50.Basuroy S., Bhattacharya S., Leffler C.W., Parfenova H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2009;296:C422–432. doi: 10.1152/ajpcell.00381.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi C., Shenkar R., Du H., Duckworth E., Raja H., Batjer H.H., Awad I.A. Immune response in human cerebral cavernous malformations. Stroke. 2009;40:1659–1665. doi: 10.1161/STROKEAHA.108.538769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shenkar R., Shi C., Check I.J., Lipton H.L., Awad I.A. Concepts and hypotheses: inflammatory hypothesis in the pathogenesis of cerebral cavernous malformations. Neurosurgery. 2007;61:693–702. doi: 10.1227/01.NEU.0000298897.38979.07. (discussion 702-693) [DOI] [PubMed] [Google Scholar]
- 53.West X.Z., Malinin N.L., Merkulova A.A., Tischenko M., Kerr B.A., Borden E.C., Podrez E.A., Salomon R.G., Byzova T.V. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–976. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Belik J., Jerkic M., McIntyre B.A., Pan J., Leen J., Yu L.X., Henkelman R.M., Toporsian M., Letarte M. Age-dependent endothelial nitric oxide synthase uncoupling in pulmonary arteries of endoglin heterozygous mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;297:L1170–1178. doi: 10.1152/ajplung.00168.2009. [DOI] [PubMed] [Google Scholar]
- 55.Jerkic M., Kabir M.G., Davies A., Yu L.X., McIntyre B.A., Husain N.W., Enomoto M., Sotov V., Husain M., Henkelman M., Belik J., Letarte M. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc. Res. 2011;92:375–384. doi: 10.1093/cvr/cvr232. [DOI] [PubMed] [Google Scholar]
- 56.Vleugel M.M., Greijer A.E., Bos R., van der Wall E., van Diest P.J. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol. 2006;37:668–674. doi: 10.1016/j.humpath.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 57.Fujimura M., Watanabe M., Shimizu H., Tominaga T. Expression of matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinase (TIMP) in cerebral cavernous malformations: immunohistochemical analysis of MMP-2, -9 and TIMP-2. Acta Neurochir. (Wien) 2007;149:179–183. doi: 10.1007/s00701-006-0929-8. (discussion 183) [DOI] [PubMed] [Google Scholar]
- 58.Maddaluno L., Rudini N., Cuttano R., Bravi L., Giampietro C., Corada M., Ferrarini L., Orsenigo F., Papa E., Boulday G., Tournier-Lasserve E., Chapon F., Richichi C., Retta S.F., Lampugnani M.G., Dejana E. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 2013;498:492–496. doi: 10.1038/nature12207. [DOI] [PubMed] [Google Scholar]
- 59.Fukawa T., Kajiya H., Ozeki S., Ikebe T., Okabe K. Reactive oxygen species stimulates epithelial mesenchymal transition in normal human epidermal keratinocytes via TGF-beta secretion. Exp. Cell Res. 2012;318:1926–1932. doi: 10.1016/j.yexcr.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 60.Gonzalez-Ramos M., Mora I., de Frutos S., Garesse R., Rodriguez-Puyol M., Olmos G., Rodriguez-Puyol D. Intracellular redox equilibrium is essential for the constitutive expression of AP-1 dependent genes in resting cells: studies on TGF-beta1 regulation. Int. J. Biochem. Cell Biol. 2012;44:963–971. doi: 10.1016/j.biocel.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 61.Aghajanian A., Wittchen E.S., Campbell S.L., Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adam O., Laufs U. Antioxidative effects of statins. Arch. Toxicol. 2008;82:885–892. doi: 10.1007/s00204-008-0344-4. [DOI] [PubMed] [Google Scholar]
- 63.Kuhlmann C.R., Gerigk M., Bender B., Closhen D., Lessmann V., Luhmann H.J. Fluvastatin prevents glutamate-induced blood–brain-barrier disruption in vitro. Life Sci. 2008;82:1281–1287. doi: 10.1016/j.lfs.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 64.Ma Z., Zhang J., Ji E., Cao G., Li G., Chu L. Rho kinase inhibition by fasudil exerts antioxidant effects in hypercholesterolemic rats. Clin. Exp. Pharmacol. Physiol. 2011;38:688–694. doi: 10.1111/j.1440-1681.2011.05561.x. [DOI] [PubMed] [Google Scholar]
- 65.Retta S.F., Barry S.T., Critchley D.R., Defilippi P., Silengo L., Tarone G. Focal adhesion and stress fiber formation is regulated by tyrosine phosphatase activity. Exp Cell Res. 1996;229:307–317. doi: 10.1006/excr.1996.0376. [DOI] [PubMed] [Google Scholar]