

Abstract
Importance of the field:
Many promising targeted agents and combination therapies are being investigated for brain cancer. However, the results from recent clinical trials have been disappointing. A better understanding of the disposition of drug in the brain early in drug development would facilitate appropriate channeling of new drugs into brain cancer clinical trials.
Areas covered in this review:
Barriers to successful drug activity against brain cancer and issues affecting intratumoral drug concentrations are reviewed. The use of the microdialysis technique for extracellular fluid (ECF) sampling and its application to drug distribution studies in brain are reviewed using published literature from 1995 to the present. The benefits and limitations of microdialysis for performing neuorpharmacokinetic (nPK) and neuropharmacodynamic (nPD) studies are discussed.
What the reader will gain:
The reader will gain an appreciation of the challenges involved in identifying agents likely to have efficacy in brain cancer, an understanding of the general principles of microdialysis, and the power and limitations of using this technique in early drug development for brain cancer therapies.
Take home message:
A major factor preventing efficacy of anti-brain cancer drugs is limited access to tumor. Intracerebral microdialysis allows sampling of drug in the brain ECF. The resulting nPK/nPD data can aid in the rational selection of drugs for investigation in brain tumor clinical trials.
Keywords: BBB, brain cancer, clinical trial, drug delivery, microdialysis, pharmacodynamics, pharmacokinetics, tumor
1. Introduction
Since the 1940s when nitrogen mustard was initially recognized as effective at inducing remission in some acute forms of leukemia, there has been a concerted effort to identify drugs that will halt cancer. The fruits of this collective effort are roughly 125 anticancer drugs that have been approved by the Food and Drug Administration (FDA) for the treatment of various forms of cancer. In addition to traditional cytotoxic chemotherapy agents, there has been a surge in the number of targeted agents in development for clinical use. These new agents inhibit signaling pathways that are critical for tumor growth. Despite the addition of these therapies to the anticancer armamentarium, cure or even long-term disease stabilization remains elusive for most advanced cancers, a reflection of the complexity of developing effective treatments for cancer.
Brain cancers, such as glioblastoma (GBM), serve as an excellent example of the challenges involved in successful drug development for aggressive cancers. GBM is the most common type of primary brain cancer in adults (representing roughly 60% of all primary brain malignancies) [1]. It is a particularly devastating cancer affecting people in the prime of their professional and personal lives, often causing substantial disability and near universal mortality, with a median overall survival of only 15 months [2]. In children, brain tumors are the second most common type of cancer [1]. Although GBM is relatively uncommon in children, it is equally fatal. Moreover, treatment of brain cancers in children presents additional challenges due to the sensitivity of the developing brain to radiation-associated injury [3]. Better anticancer drug therapies are urgently needed for the treatment of GBM and primary brain cancers.
An increasing number of chemotherapies that either interrupt cell division or target pathways specifically implicated in the tumorigenesis or survival of gliomas are undergoing active investigation [4,5]. Some of these agents have been adopted from research and discovery in other cancers (e.g., erlotinib for lung cancer; bevacizumab for colon cancer) while others, such as temozolomide (TMZ), have undergone focused development for gliomas [2,6]. Unfortunately, despite sound scientific rationale and promising preclinical results, the majority of agents studied in GBM have demonstrated only modest clinical benefit [4,5,7]. Hence, the field of neuro-oncology remains challenged to efficiently translate preclinical knowledge about the molecular, genetic and physiochemical properties of GBM into rational selection of the agent(s) most likely to yield clinical benefit in patients.
1.1 Glioblastoma resistance to chemotherapy
Intrinsic resistance to therapy is one of the major factors limiting successful treatment of gliomas. GBM represents a heterogeneous population of rapidly dividing and non-dividing cells with variable regions of hypoxia, increased perfusion due to neo-angiogenesis and infiltrating cancer cells admixed with normal brain [7-9]. Hence, there is no single, defined target or local environment for anticancer agents in GBM. Even in controlled preclinical assays, GBM cells are relatively chemotherapy-resistant across various in vitro assays. [10,11]. The etiology for the relative drug-resistance in GBM is not clearly understood, but recent investigations have suggested that cancer stem cells (defined as cells that are independently tumorigenic) may be a major factor [12,13].
In addition to cell-based mechanisms contributing to GBM chemoresistance, the microenvironment in which cancer cells live plays a role in drug resistance [14]. For example, elevated glutathione levels enhance cellular resistance to several alkylating agents [15]. Similarly, a key regulator protein in tumor stroma (heat shock protein-90) appears to protect gliomas from cell death [16]. Hence, feedback loops among glioma cells, microglia, capillaries and other stromal cells may contribute to treatment resistance [14,17].
Importantly, the local tumor environment also contributes to treatment resistance by limiting access of the drug to the cancer cells. Irregular vasculature and increased interstitial pressure result in poor delivery of molecules via the capillary network [14,18-20]. Failure to achieve the minimal drug concentration in tumor for the required period of time to produce the desired effect (i.e., growth arrest or apoptosis) may result in an apparent insensitivity to drug or development of secondary resistance [14,20]. In addition, variable blood perfusion can contribute to intratumoral hypoxia and acidity that may change the behavior of cancer cells (i.e., decreased proliferation), their response to chemotherapy or the activity of drugs in vivo.
In summary, GBM cells appear to be intrinsically chemoresistant; however, several factors including the cell lines, incubation times and drug dosing influence the in vitro sensitivity reported [21,22]. Once in vitro efficacy is established against GBM, access to tumor at the required concentration, for the required time, is the next critical hurdle. This cannot be inferred from preclinical efficacy studies or serum pharmacokinetics. Hence, the addition of in vivo assessments of drug concentration in tumor is recommended to gather information about a drug’s ability to cross the blood brain barrier (BBB) and achieve adequate concentration for effect.
1.2 BBB and drug concentrations
A major obstacle to successful pharmacologic management of all brain cancers is the presence of the BBB (Figure 1). The BBB is a physical and chemical barrier existing at the level of brain capillary endothelial cells that effectively restricts the diffusion of most substances into the brain. It is charged, composed predominantly of lipids, has tight junctions between endothelial cells and a continuous basement membrane reinforced by astrocytic foot processes. These features contribute to preventing polar or large molecules from getting into brain tissue unless they are carried by designated transporters such as those for glucose and select amino acids [23-27]. Typically, drugs gain access to the brain only if they have the appropriate properties of being lipophilic, nonpolar, low protein binding, low in molecular mass or are carried by the transporter pumps [24,26,28,29]. Most anticancer drugs are unable to cross an intact BBB because they lack these properties or are substrates for effusion transporters such as the ATP-binding cassette (ABC) transporters [30-33]. For example, anthracyclines, vinca alkaloids and taxanes as well as some of the newer receptor tyrosine kinase inhibitors, such as gefitinib and dasatinib, are substrates of ABC transporters that rapidly eliminate these agents from the brain [24,30,31]. In the setting of brain cancer, inflammation, ischemia and neovascularization contribute to development of leaky capillaries in the brain. These capillaries are fenestrated and do not have tight junctions, permitting even hydrophilic chemotherapies to pass through this partially disrupted BBB [34]. However, the amount of drug that actually reaches the tumor is variable and unpredictable [20,35,36].
Figure 1. Schematic of the BBB.
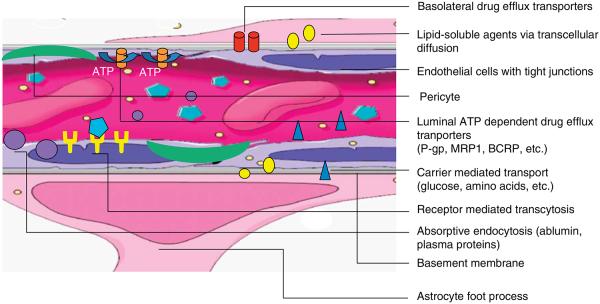
Cartoon depiction of the major components that contribute to the BBB and the pathways by which agents may access brain across the intact BBB. The major anatomical components of the BBB are: the endothelial cells with tight junctions and a specialized basement membrane, the reinforcing pericytes and the astrocytic foot processes. Transport mechanisms may include transcellular diffusion of un-bound, non-polar, lipid soluble agents (yellow circles), receptor-mediated transcytosis (turquoise hexagons), carrier-mediated transport (blue triangles) and absorptive endocytosis (purple circles). Even after successful transport across the BBB into brain via one of these mechanisms, agents may be extruded via ATP- and non-ATP-dependent drug efflux transporters (orange and red cylinders).
The CNS has several compartments and drug concentration cannot necessarily be extrapolated from one compartment to another [26,37]. The concentration of a drug in the cerebral spinal fluid (CSF) does not necessarily reflect its concentration in brain or brain tumor [37]. One explanation for this is that the choroid plexus is not within the BBB and it serves as a major site for direct transportation of compounds from systemic circulation into CSF. The CSF is also often not subject to the same inflammatory factors or vascular-mediated changes as brain in the setting of cancer; CSF white blood cell and protein levels (typical markers of inflammation) may be entirely normal with GBM [38]. Therefore, detecting a drug in the CSF is not an absolute indication that the drug is also present, or present in the same concentration, in the brain or brain tumor. Similarly, plasma pharmacokinetic (PK) studies (the core of early drug investigation) do not predict brain concentrations. Even for the most widely used chemotherapies in neuro-oncology, the brain AUC is often a fraction of the plasma PK [39,40]. In summary, a complex collection of normal and pathologic features create variable access to brain for most chemotherapies, and direct measurement may be required to understand intratumoral drug disposition.
2. Body: intratumoral pharmacokinetics and pharmacodynamics, importance in clinical drug development
2.1 Relationship between tumor concentration and efficacy
Across many medical disciplines, failure to achieve sufficient levels of certain drugs within the tissue of interest, for an adequate period of time, results in poor clinical outcome [41]. Unfortunately, there are very few studies exploring the relationship between drug concentration in brain cancer and clinical outcome [42-44]. The best examples regarding this relationship exist for erlotinib, an EGFR inhibitor, in primary and secondary brain cancers. A dose–concentration relationship was established for erlotinib in the CSF of patients with metastatic disease to the brain [42,43]. However, only one study has investigated tissue concentrations, local effect on EGFR phosphorylation and clinical outcome in brain cancer [44,45]. In this study, patients who received erlotinib 150 mg/day did not achieve adequate intratumoral levels, did not have phosphorylation of EGFR and the final conclusion of the Phase II trial was no significant clinical benefit [44,45]. The converse, achieving high intratumoral tissue concentration without significant clinical efficacy, has also been demonstrated with other therapies in brain cancer [46,47]. All of these studies investigated tumor concentration via analysis of tumor homogenate which can be vulnerable to artifact due to inadvertent measurement of intravascular drug [44].
The relationship between tissue concentration and efficacy has been explored with microdialysis in some solid tumors with agents such as cisplatin [48] and topotecan [49,50] and data suggest that an intratumoral dose–response relationship exists. To date, this relationship has been best documented in the infectious diseases literature where there are clear associations among drug concentrations achieved in the ECF of a tissue, the minimum inhibitory concentration required for efficacy of an antimicrobial therapy and the clinical outcome [51-56]. Cumulatively, the results from brain cancer, systemic cancers and infectious disease studies suggest that clinical outcome may be related to concentrations achieved in the tissue ECF.
Currently, intratumoral drug concentration assessment is limited to tissue sampling, microdialysis or imaging. Both tissue sampling and microdialysis are invasive; however, microdialysis has the advantage of allowing repeat measurements over time and specific sampling of the ECF space. Non-invasive measures such as magnetic resonance-based techniques and positron emission technologies are in development but have not yet been validated and are costly and inefficient for clinical investigation. Although similarly cumbersome, microdialysis is clinically feasible, increasingly cost efficient and reliable for assessing tumor neuorpharmacokinetic (nPK) data in preclinical and clinical settings. Indeed, it has been endorsed by a task force led by the American Association of Pharmaceutical Scientists and the FDA to improve the efficiency and accuracy of preclinical and early clinical development of new agents by identifying drugs likely to be effective at the site of disease [57]. Hence, there is strong scientific and regulatory support for the application of microdialysis for nPK and neuropharmacodynamic (nPD) studies.
2.2 Principles of microdialysis
Microdialysis is a well-validated technique for continuously sampling the concentration of a drug or biomolecule (i.e., glucose, lactate and neurotransmitters) in the ECF of a body tissue. This method, first developed in the 1970s, consists of placing a catheter that contains a semi-permeable membrane at its tip into a body tissue [58]. Perfusion fluid, similar in composition to the ECF, is slowly and continuously pumped through the inlet tubing of the microdialysis catheter at low flow rates ranging from 0.1 to 5 μl/min. The diffusion of molecules occurs in both directions across the semi-permeable membrane as the ECF equilibrates with the perfusion fluid (Figure 2). The dialysate (solution that exits the probe) is comprised of small organic molecules, water and inorganic ions that passively diffuse down their concentration gradients [59,60]. Drugs or other molecules of interest (such as neurotransmitters) may also diffuse from brain ECF to the dialysate. Hence, this technique can be applied to assess disposition of molecules in the ECF in both disease states and under normal physiologic conditions [61]. A microvial collects dialysate from the outlet tubing of the catheter and is changed at regular intervals (chosen based on volume desired, solute of interest, analytical technique, etc.) and the dialysate is sent for analysis.
Figure 2. Schematic of microdialysis catheter membrane.

Schematic depiction of the probe portion of a microdialysis catheter (not drawn to scale). The semi-permeable membrane at the tip of the probe allows diffusion of fluid (perfusate out and ECF in) along concentration gradients. Perfusate enters the central portion of the probe under a constant flow rate (i.e., 1 μl/min). It then moves to the outer compartment of the probe where it is admixed with the ECF and the solute under investigation (grey drops) is collected in the outlet tubing.
ECF: Extracellular fluid.
The ultimate goal of microdialysis is to obtain an accurate representation of the composition of the ECF in tissue in vivo. There are several variables that the investigator can manipulate to accomplish this. A low flow rate for the perfusion fluid is used to maximize the relative recovery from the ECF. The low flow allows time for movement of molecules from ECF to dialysate [59,60,62]. The trade off is that small volumes of dialysate (in the microliter range) are collected in each sample. Highly sensitive analytical methods such as HPLC and tandem mass spectrometry are then needed to detect minute concentrations of drug [63,64].
Low flow rates have the additional advantage of no net gain or loss of fluid across the membrane. This ‘volume-neutral’ aspect of microdialysis makes it a particularly appealing technique for scenarios such as sampling brain where infusion of large volumes of fluid may not be well tolerated or where there are limitations on how much blood sampling can be performed such as in pediatric PK studies and in small animal preclinical studies [57,59]. Importantly, the lack of large fluid shifts over the membrane ensures that fluid exchange does not artificially alter the disposition of the drug in the ECF [65].
2.3 Relative recovery methods for estimating the true drug concentration in tissue
The dialysate collected with microdialysis catheters contains a representative proportion of the molecule or unbound drug that is in the tissue ECF. The concentration of free drug in the dialysate is not the true concentration in the tissue. This is because full equilibration does not take place across the dialysis membrane when there is a continuous flow of perfusion fluid (required to move the dialysate through the catheter tubing) [60]. Hence, only a fraction of the drug in the ECF is present in the dialysate. This fraction is referred to as the relative recovery and is defined as the ratio of drug concentration in dialysate compared to the actual concentration of drug in tissue at the sampling site. Relative recovery is influenced by various factors such as flow rate, membrane pore size and length, properties of the drug or biomolecule of interest, and the approach used to estimate recovery [60,66]. Slower perfusion rates and longer dialysis membranes produce a higher relative recovery [60,61]. Several other factors including the type of probe used, the tissue under investigation, and the perfusion fluid and anylate of interest influence the assessment of drug concentrations [59,60,62]. Drugs with high lipophilicity or polarity may have low recovery rates or unreliable recovery over time. Although there are no clear guidelines regarding minimal accepted rate of recovery, the key factors that influence the interpretation of nPK data corrected for recovery are the reproducibility of recovery results and the sensitivity of the analytical method.
2.3.1 In vitro relative recovery
In vitro assessments of recovery determine if a drug or compound can be recovered from aqueous solution via a microdialysis probe. It is a test of feasibility for applying microdialysis to measure a given compound. In this setting, a known amount of drug is put into an aqueous solution, the catheter is allowed to equilibrate and serial samples are collected. This process can be repeated using various flow rates to determine the flow rate that results in the maximum recovery of the compound of interest. If there is no adequate recovery in the in vitro system, it is unlikely that there will be adequate recovery in tissue studies. For example, lipophilic drugs, such as docetaxel, tend to bind to the microdialysis tubing resulting in decreased drug recovered in the dialysate [67,68]. In vitro microdialysis studies allow assessment of the extent to which this may be happening for a given drug and what techniques can be applied to optimize recovery. An in vitro study is required as a first step to assess the feasibility of measuring a given compound via microdialysis.
2.3.2 In vivo relative recovery
Although in vitro relative recovery studies are important for understanding the relationship between experimental factors and drug recovery for a given agent, these studies represent a comparatively simple system that can overestimate the percent recovery. When in vitro relative recovery is used as a correction factor for subsequent in vivo results, the overestimated recovery can lead to an underestimation of the true concentration of drug in tissue. This is because in vitro studies cannot account for many factors that influence in vivo disposition of drug such as tortuosity of the tissue, blood flow, intratumoral pressure gradients and the rate of drug clearance from the ECF [60,66,69]. Therefore, it is more reliable to calibrate for in vivo drug studies with in vivo relative recovery methods that take into account the influence of the many tissue-related factors. Several reviews [62,66,69,70] provide detailed discussions regarding commonly applied in vivo methods for determining relative recovery, such as the no-net-flux method [71], the dynamic no-net flux method [72] and the retrodialysis method [73].
Retrodialysis is the most direct and simple method for in vivo catheter calibration. This method is based on the assumption that the loss of drug across the semi-permeable membrane into the interstitium is equivalent to its recovery. In other words, the percent of in vivo drug lost into the ECF is used as a measure of the in vivo recovery. After placement of the microdialysis probe into tissue, a known amount of the drug (or calibrator agent chemically similar to the drug of interest) is perfused through the catheter into tissue. After a period of equilibration, the dialysate is sampled and the recovery is calculated as a ratio of the difference between the perfusate and dialysate concentrations and the known perfusate concentration [61,66,73]. The probe is then perfused without drug (or calibrator) and after an appropriate washout period, the investigational drug is given systemically and microdialysis collection in the brain ECF is initiated. The raw ECF concentrations measured from the dialysate after systemic drug may then be adjusted using the relative recovery of the drug. A theoretical limitation of this approach is that the factors influencing recovery are dynamic and a single time point assessment, either at the start or conclusion of microdialysis studies, reflects the state of recovery only at that sampling time. A practical clinical limitation is that drugs available only in pill form cannot be used as a calibrator for retrodialysis as they are not sterile and cannot be reliably solubilized for delivery via perfusate. Despite these limitations, in general, retrodialysis is the optimal approach for accounting for the many in vivo factors that influence the measurement of drug in the ECF. In most cases, it is helpful to report both the raw concentrations measured and the values corrected based on the calculated relative recovery. This allows the reader the opportunity to assess the full data set for maximal interpretation given that relative recovery is an estimation in itself.
2.4 Preclinical experience with using microdialysis to perform pharmacokinetic studies of chemotherapy agents
Compared to traditional in vivo intratumoral PK studies that use multiple animals at multiple time points to determine a single CNS concentration–time profile, microdialysis is an animal-sparing, data-rich technique that allows for measurement of drug levels at many time points in the same animal, thus also avoiding inter-animal variability [60,62]. Serial sample collections can be done while animals are awake and freely moving, without the interference of an anesthetic or physiological processes. Microdialysis has been used in animal models to evaluate various chemotherapies within tumor as well as to explore the physiologic changes in neurotransmitters in and around tumor [64,74-85]. These studies have recently been reviewed in detail [61,63,66,86].
Knowing the concentration–time profiles of chemotherapies in tumor can aid in choosing a drug with favorable properties for efficacy testing in cancer patients and can help with optimizing dosing schedules [61,66,69]. Preclinical microdialysis studies can also explore interactions between agents such as cytotoxic and antiangiogenesis agents that may not be feasible in the clinical setting due to the patient risks [82-84].
2.5 Clinical experience with microdialysis to perform pharmacokinetic assessments of chemotherapy
Microdialysis can be performed in almost any human tissue, such as skeletal muscle, skin, lung, liver, brain and myocardium, as well as blood and CSF. Clinical PK microdialysis studies have been carried out with several traditional cytotoxic chemotherapies in patients with easily accessible tumors [65], such as breast cancer [87-89], melanoma [90] and oral cancers [91]. Several of these earlier studies have been expertly reviewed by Zhou and Gallo 2005 [66] and we, therefore, highlight only clinical microdialysis studies of anticancer agents that have been performed in brain.
Microdialysis was originally introduced as a technique for measuring concentrations of neurotransmitters in the brains of lab animals [92]. It has played an important role in assessing changes in levels of neurotransmitters and brain biochemistry in a variety of clinical scenarios [58]. Intracerebral microdialysis has mainly been applied to the study of head trauma [93-98], intracerebral hemorrhage [99-104] and epilepsy [105]. In these settings, microdialysis catheters are used to monitor metabolic changes (i.e., varying levels of lactate, glucose and glutamate) in the ECF in order to predict clinical outcome and to evaluate the effects of therapeutic interventions. More recently, intracerebral microdialysis has been applied for performing nPK assessments of drugs. When intracerebral microdialysis is used to determine drug nPK, concurrent systemic PK assessments are recommended to control for interpatient variability in drug metabolism.
There are two microdialysis catheters that are commercially available for intracerebral use in humans (CMA Microdialysis, Solna, Sweden). One of these catheters has a molecular mass (MM) cutoff semi-permeable membrane of 20,000 Daltons and can consistently recover a drug or biomolecule that has an MM of < 5000 Daltons (Figure 3). It has received FDA clearance for cerebral tissue monitoring. The other catheter has an MM cutoff membrane of 100,000 Daltons and is suitable for recovery of macromolecules, such as cytokines, chemokines and growth factors that have an MM of < 25,000 Daltons. This high MM cut-off membrane catheter is CE-marked in Europe, but the manufacturer has not applied for FDA approval.
Figure 3. Clinical translational paradigm for microdialysis in neuro-oncology.

Patients who plan to undergo surgery may be eligible for participation in an investigation of disposition of a drug in brain via microdialysis. Catheters are placed in residual tumor at the time of clinically planned biopsy or resection (panel 1). The commercially available catheter and pump for clinical use (panel 2). The CT image shows the radio-opaque tip of the catheter. After a period of postoperative recovery, the patient is given the drug of interest systemically (i.v. or p.o.) and the ECF samples are collected from the microdialysis catheter for the appropriate amount of time based on the drug PK. The catheter is then removed at the bedside.
ECF: Extracellular fluid; i.v.: Intravenous; PK: Pharmacokinetics; p.o.: By mouth.
Intracerebral microdialysis using the catheter with the lower MM cutoff membrane has been performed in hundreds of patients with acute brain injuries. Poca et al. [106] has published the largest series to date of microdialysis catheter use in neurointensive care units. They found that among the 97 patients studied, there were no catheter-associated infections and only 3% of patients had small, clinically insignificant bleeding (≤ 1 ml) around the catheter seen on CT scan. The catheters are smaller in caliber than those typically used for performing ventriculostomies or monitoring intracranial pressure. With the use of image guidance, they can be safely placed in brain interstitium and targeted to specific regions of brain. Each catheter has a gold tip that makes it visible on a CT scan to allow confirmation of correct positioning (Figure 3).
More than one catheter can be used at the same time to measure concentrations of a drug in different areas of the brain [107]. Sample collections can be done while patients are awake and mobile. These catheters can safely be left in patients for > 2 weeks [108], although catheter performance may deteriorate with prolonged use due to possible development of gliosis at the membrane tip or clogging of the catheter [109,110]. This can either reduce recovery or lead to outright catheter failure. In two recent clinical studies of microdialysis in brain tumors, catheter failure rates ranged from 25 to 28% [35,107]. It is possible that catheters may be particularly prone to this in the setting of tumors where the ECF is often highly proteinaceous.
Microdialysis for assessing drug levels in the human brain remains a strictly investigational endeavor. Brain microdialysis studies were initially done in epilepsy patients [111-113] to determine CNS levels of anticonvulsants. The first study to use microdialysis to measure drug in brain tumor assessed levels of the antibiotic rifampin in the brains of eight patients who were undergoing craniotomies for tumor resection [114]. They showed that there are regional differences in rifampin concentrations between tumor and surrounding normal brain and that assessment of the ECF drug concentration via microdialysis (versus tissue studies) provided the most reliable and reproducible nPK data [114].
Bergenheim et al. [107] assessed the uptake and elimination of p-boronophenylalanine (BPA) in four patients with GBM. Up to three catheters were placed in each patient’s brain: normal brain parenchyma, tumor and peritumoral tissue. Microdialysis catheters remained in place for ∼ 1 week. All patients tolerated the microdialysis well and were able to move freely during collection of the dialysate samples. Through the use of this sampling technique, the investigators were able to demonstrate within the same patient that there are variations in the temporal nPK of BPA within different areas of the brain.
Another study investigated high-dose methotrexate (12 g/m2) in patients with recurrent high-grade gliomas [35] in whom catheters were placed in either contrast enhancing or non-enhancing residual tumor tissue at the time of clinically indicated surgery. Digital fusion of the non-contrast brain CT scan (radio-opaque catheter tip) with the brain MRI (indicating regions of BBB disruption) allowed precise catheter localization. Methotrexate levels were considerably higher in contrast enhancing regions of brain (AUC ecf/AUC plasma 28 – 31%) compared with non-enhancing brain (AUC ecf/AUC plasma 3.2 – 9.4%). These results confirmed that there is variable delivery of systemic MTX to regions of tumor, with the lowest concentrations in non-contrast enhancing tumor and suggested that the technique is useful for investigating the influence of BBB integrity on drug disposition. This study also showed that brain microdialysis can be performed in the course of clinically indicated brain surgery without interruption of peri-operative care.
Portnow et al. [40] measured levels of TMZ in non-enhancing peritumoral tissue of brain tumor patients who were undergoing surgical resection. TMZ is part of the standard treatment for patients with newly-diagnosed GBM. It is given daily during focal brain radiation therapy and continued as a monotherapy after the radiation [2]. When used concurrently with radiation therapy, patients are typically instructed to take TMZ 1 h prior to the radiation. This recommendation is based on data from Phase I studies of TMZ [115,116] in patients with solid tumors where it was determined that peak levels in the blood occurred ∼ 1 h after ingestion. Portnow et al. found that, in contrast to plasma, peak levels of TMZ in brain interstitium occurred between 1.2 and 3.4 h after an oral dose, with the 2 – 3 h period representing a time of highest TMZ concentration [40]. TMZ levels in the brain at 1 h were as much as seven-fold lower than the maximum levels achieved between 2 and 3 h. This suggests that dosing of TMZ relative to radiation therapy might be further optimized based on nPK data.
In addition to traditional cytotoxic agents, many targeted therapies, such as tyrosine kinase inhibitors, proteasome inhibitors, mammalian target of rapamycin (mTOR) inhibitors and anti-angiogenic agents, are now available [4,5]. Intracerebral microdialysis is a technique that can be applied for screening these new agents for potential use in Phase I and II brain tumor clinical trials by directly determining if these drugs can cross the BBB in vivo in brain cancer patients.
For example, before committing time and patient resources to a phase II study of a new agent for brain cancer when it is not known how well the drug gets into brain, a translational intracerebral microdialysis study could be performed using a relatively small number of patients. One potential study design is to place one or more microdialysis catheters in tumor or peritumoral tissue at the time a patient undergoes a craniotomy or brain biopsy (Figure 3). Approximately 24 h after the surgery, when the patient has recovered from the acute effects of surgery, retrodialysis may be performed with a calibrator. The catheter would then be perfused with artificial CSF until the calibrator is no longer detectable in dialysate (‘washout period’). At this point, the drug of interest is given systemically (preferably with the proposed phase II dosing) and serial dialysate samples are collected over a period determined by the half-life of the drug or its metabolites. For a drug with a long half-life, it is preferable to perform microdialysis after multiple doses have been administered and the drug has reached steady-state.
The nPK parameters measured in the ECF often include the peak concentration (Cmax), time to Cmax, AUC and time to elimination. These values can be compared to those simultaneously obtained in plasma after systemic drug administration to provide important information about the relationship between systemic and brain PK, the optimal timing for dosing and, ultimately, the likelihood that the agent can reach the required time:concentration ratio in brain. If no drug is detected in brain ECF dialysate samples, this argues against advancing the drug to clinical trials for brain tumors. If the drug is detected in the brain at concentrations similar to free-drug concentrations in plasma or at levels sufficient for efficacy based on preclinical studies, then it is reasonable to move forward with efficacy testing. Finally, microdialysis-derived nPK data may guide further dose-escalation studies before embarking on an efficacy trial.
2.6 Limitations of microdialysis for brain studies
There are several potential limitations associated with the microdialysis technique (Table 1). Theoretically, the trauma to brain tissue that inevitably occurs with placement of an intracerebral microdialysis catheter could result in disruption of the BBB, thereby overestimating the amount of drug that gets into the brain [117,118]. However, other literature suggests there is rapid normalization of the BBB after placement and that allowing equilibration may be sufficient to minimize the artifact that can be associated with iatrogenic BBB disruption [62,119,120]. Moreover, this concern is probably not as relevant for nPK studies in patients with brain cancer where the BBB is often already disrupted.
Table 1.
Considerations for the use of microdialysis for neuro-pharmacokinetic studies in patients with brain tumors.
| Advantages | Disadvantages |
|---|---|
| Near real-time measurement of unbound drug near site of action in the brain; distinction between extracellular space and other compartments |
Invasive technique |
| Simultaneous measurement of drug pharmacokinetics in brain and plasma contributes to decreased interpatient variability |
Requires hospitalization |
| CNS concentration–time profiles of drugs can be determined using a relatively small number of patients |
Expensive, particularly if hospitalization is prolonged beyond clinically indicated durations (i.e., prolonged dialysate collections) |
| Serial sample collections can be done while patients are awake and in the course of standard clinical care |
Catheter calibration is needed to estimate the true drug concentration in tissue |
| Safe with low rates of bleeding or infections (comparable to other intracerebral monitoring techniques) |
Sensitive analytical methods are needed to detect drug concentrations in the dialysate samples |
| More than one catheter can be placed at one time, enabling concurrent measurement in tumor core, peritumoral tissue and normal brain |
In vitro testing is required to determine if the drug of interest is a candidate for microdialysis; many new targeted therapies and large proteins will not be good candidates |
| Accuracy of catheter placement can be confirmed with imaging studies |
Accurate placement of catheter into regions of tumor is challenging |
The major advantages and limitations of the microdialysis technique for clinical translational studies in neuro-oncology.
Logistically, placement of microdialysis catheters into brain tumors can be challenging due to: i) variable intraoperative visualization of residual gliomas at the resection margin, ii) brain shift during and after surgery that can interfere with the accuracy of targeted placement and iii) the relatively short length and flexibility of the catheter can inhibit precise placement in deeper regions. The risks associated with the catheters are very low as discussed above; however, they do include a small chance of hemorrhage or infection. Long-term studies are challenging due to decrease in catheter performance over time and the need for study subjects to remain hospitalized while the dialysate samples are collected. Hence, most nPK studies are functionally limited to a few days. As discussed above, catheter calibration can be an imperfect estimate of in vivo relative recovery, often resulting in an underestimation of the true drug concentration in a tissue. Hence, it is recommended to report both actual and corrected nPK values. New targeted agents are more likely to be lipophilic or very large, thus, limiting the usefulness of microdialysis for performing nPK assessments of these drugs (Table 1). Finally, intracerebral microdialysis is labor intensive with a number of opportunities for error. It requires an experienced team of pharmacologists, neurosurgeons, neuro-oncologists and nurses to successfully complete the study and obtain meaningful data.
2.7 Future directions
In addition to assessing the nPK of chemotherapies, microdialysis catheters may potentially be used as a means for studying whether the drug reaches its target within brain at concentrations high enough to produce a desired pharmacodynamic effect. The advantage of using the catheter with the 100,000 Dalton MM cutoff semi-permeable membrane is that it has the ability to measure chemotherapy concentrations and macromolecules that may change in response to treatment. This catheter can be more challenging to use than the 20,000 Dalton catheter because the larger pore size can lead to hyperfiltration, resulting in a net loss of fluid into tissue. Some investigators have found that using perfusion fluid containing dextran or albumin for increasing its colloid osmotic pressure balances transmembrane fluid flow. Experience is growing with applying the high MM cutoff catheter to measure changes in cytokines, chemokines, growth factors and other proteins in the brain [66,121-127]. An investigation in brain tumor patients using this catheter to measure changes in intracerebral levels of cytokines, such as VEGF and IL-1β, in response to treatment with a systemically administered mTOR inhibitor is currently underway [128].
In addition, complex pre-clinical nPK studies have been pursued with microdialysis to understand the impact of concurrent therapies on drug delivery to tumor [82,83,129]. Such studies are particularly important with the recent pairing of antiangiogenesis therapies with standard cytotoxic chemotherapies for treating brain cancer [130]. Microdialysis is also being applied to investigate the intratumoral PK of various compounds in the setting of ABC transporter inhibitors and with various doses of radiation. As the complexity of therapies continues to increase, microdialysis may be a useful tool for distinguishing the net effect of multiple therapies on intratumoral nPK and nPD.
2.8 Conclusion
Drug delivery to brain cancer is inherently complex. Poor drug delivery to tumor contributes to the disappointing results of clinical trials in brain cancer. Microdialysis is a powerful research tool that enables serial sampling of drug levels in brain with simultaneous assessment of plasma PK. Such data provide temporal resolution of drug disposition in brain tissue, insights about the variability between brain and plasma PK, and information about optimal dosing for the desired intratumoral result. Intracerebral microdialysis is a safe and feasible technique for measuring intratumoral concentrations and experience is growing with using this technique to perform nPK and nPD studies in humans. With training and experience, microdialysis can be incorporated into early drug development of new therapies for brain tumors to assist in the rational selection of agents to advance to efficacy studies and establish dosing regimens.
3. Expert opinion
Preclinical drug studies focus on the efficacy of an agent against a given cancer cell line using an optimal drug concentration and time exposure [21,22]. In vivo animal studies assess toxicity and efficacy at doses found to achieve drug concentrations that were successful in vitro. Similarly, the first study performed in patients is often a dose-finding study in which the maximum tolerated dose is defined based on toxicity and tolerability. Plasma PK is often obtained in these early trials to ensure that concentrations thought to be effective for cell kill preclinically are achieved with acceptable tolerability. However, plasma PK does not predict intratumoral PK, the site of action for most drugs. Preclinical and clinical studies have demonstrated that tumor drug concentration is rarely predicted by plasma PK [35,37,44,46,47] and that for some agents drug concentration in tumor is correlated with treatment response [44,49,50,66,91]. In brain cancers, both the tumor characteristics and the physiological properties of the BBB conspire to limit access of many chemotherapy agents to the tumor. Indeed, poor access of drug to tumor is cited as a contributing factor to the disappointing results of most chemotherapies for GBM [24,26,39,108]. Despite this, intratumoral drug concentrations are only rarely investigated in patients.
As the cost of drug development continues to rise and there are more agents available for investigation, it is important that drugs only proceed to efficacy studies after their intratumoral disposition is understood. If chemotherapy cannot reach the target cells in adequate concentration, there is little hope of clinical efficacy, and further studies to maximize drug delivery to tumor are required. This relatively simple step of confirming drug access to tumor cells may reduce the attrition rate of agents that look effective in preclinical studies, but have disappointing results in patients.
Various techniques have been applied for this purpose in brain cancer patients. Tissue sampling at the time of a planned tumor resection is popular and does assess tumor versus plasma PK [44,46,47]. However, this provides only a single time point and drug concentration is reported from tissue homogenate rather than the individual pharmacological compartments. Finally, the location of tissue sampled is often not available, making assessment of variable regions of tumor difficult. Imaging studies have been developed to track drug disposition in tumor, but these are early in development for clinical application and cumbersome to be applied for general drug discovery due to cost and the need to develop specific drug ligands. That said, imaging studies are non-invasive and can potentially assess both delivery and effect at the site of disease and may be more feasible if they can be developed to assess target pathways rather than individual agents.
Microdialysis is a minimally invasive technique that allows assessment of intratumoral PK. The catheter allows sampling of the ECF within tumor to measure the time–concentration curve. This is important as concentrations in the ECF are thought to most accurately reflect the availability of drug to tumor cells for many clinically relevant cancer therapies. It has been used to effectively study drug delivery in breast cancer, melanoma, colon cancer and brain cancer preclinically and has demonstrated correlations between intratumoral drug concentration and efficacy in vivo [64,74-82,84,85,88,90,131,132]. Although studied extensively in preclinical cancer models, microdialysis for intratumor drug studies may have even greater applicability in early clinical trials. There are commercially available catheters for use in humans and at least three clinical studies in brain cancer patients have shown the ability to assess intratumoral disposition of drug safely in patients [35,40,107].
Increased use of microdialysis to identify agents that have adequate access to tumor for efficacy in the brain may reduce the frequency of negative clinical trials in which agents with unknown tumor access are tested. Microdialysis can also be applied to assess the interactions amongst drugs given in combination or the optimal dosing strategy for multimodality treatments. Understanding the PK relationship of combination therapies is crucial for optimal timing and ordering of the various therapies in a regimen. This is particularly timely with the increasing practice of combining cytotoxic therapies, anti-angiogenesis therapies and radiation therapy in brain cancer patients [82-84,129]. Each of these interventions may have unique effects on the BBB and the local tumor environment influencing intratumoral PK. Finally, the larger bore microdialysis catheters are being used with increasing frequency and may provide insight about the local effect of drugs within brain tumors such that in a single study, one may be able to use microdialysis to assess the intratumoral drug nPK and nPD [66,121-127].
However, microdialysis does have some limitations that must be considered. It is invasive, can only reliably measure drugs with favorable properties for the technique (low lipophilicity, MM of < 5000 Daltons, low protein binding, etc.) and requires sensitive analytic techniques for each anylate of interest. It also has a spatial limitation, sampling only in the immediate area where the catheter is placed. Finally, it can be expensive and time consuming. Experience and skill with the technique, as well as accurate calculation of the relative recovery, are required for optimal results. Despite these limitations, for many anti-cancer agents, microdialysis is a feasible and highly informative approach for determining PK in tumor. Ultimately, this technology may be able to assess if drugs also produce their desired effect at their target. Applying microdialysis to early drug development may contribute to more time- and cost-efficient development of therapeutics for brain cancer.
Article highlights.
Poor drug delivery to tumor is one of the factors contributing to drug resistance in brain cancer.
Measuring intratumoral distribution of drug can assist with the selection of agents or optimal dosing and schedule for efficacy testing.
Microdialysis is a powerful research tool for obtaining both neuorpharmacokinetic and neuropharmacodynamic data in preclinical and clinical studies.
There are several advantages and disadvantages to intracerebral microdialysis that have to be considered when using this technique in early drug development.
This box summarizes key points contained in the article.
Acknowledgments
Declaration of interest
J Blakeley’s and J Portnow’s work has been supported by John Hopkins University and the City of Hope Cancer Center. J Blakeley is also supported by GlaxoSmithKine, the National Cancer Institute (NCI) and the Cancer Therapy Evaluation Program (CTEP). J Portnow has also received funding from the NCI and the California Institute of Regenerative Medicine.
Bibliography
- 1. CBTRUS Statistical Report: primary brain and central nervous system tumors diagnosed in eighteen states in 2002 – 2006. Published by the central brain tumor registry of the United States, Hinsdale, IL. 2009 – 2010. Available from: www.cbtrus.org.
- 2.Stupp R, Mason WP, van den Bent MJ, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Merchant TE, Pollack IF, Loeffler JS. Brain tumors across the age spectrum: biology, therapy, and late effects. Semin Radiat Oncol. 2010;20(1):58–66. doi: 10.1016/j.semradonc.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minniti G, Muni R, Lanzetta G, et al. Chemotherapy for glioblastoma: current treatment and future perspectives for cytotoxic and targeted agents. Anticancer Res. 2009;29(12):5171–84. [PubMed] [Google Scholar]
- 5.Omuro AM. Exploring multi-targeting strategies for the treatment of gliomas. Curr Opin Investig Drugs. 2008;9(12):1287–95. [PubMed] [Google Scholar]
- 6.Yung WK, Albright RE, Olson J, et al. A Phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83(5):588–93. doi: 10.1054/bjoc.2000.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reardon DA, Wen PY. Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist. 2006;11(2):152–64. doi: 10.1634/theoncologist.11-2-152. [DOI] [PubMed] [Google Scholar]
- 8.Dunbar E, Yachnis AT. Glioma diagnosis: immunohistochemistry and beyond. Adv Anat Pathol. 2010;17(3):187–201. doi: 10.1097/PAP.0b013e3181d98cd9. [DOI] [PubMed] [Google Scholar]
- 9.Burger PC, Heinz ER, Shibata T, Kleihues P. Topographic anatomy and CT correlations in the untreated glioblastoma multiforme. J Neurosurg. 1988;68(5):698–704. doi: 10.3171/jns.1988.68.5.0698. [DOI] [PubMed] [Google Scholar]
- 10.Salmaggi A, Boiardi A, Gelati M, et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia. 2006;54(8):850–60. doi: 10.1002/glia.20414. [DOI] [PubMed] [Google Scholar]
- 11.Leuraud P, Taillandier L, Medioni J, et al. Distinct responses of xenografted gliomas to different alkylating agents are related to histology and genetic alterations. Cancer Res. 2004;64(13):4648–53. doi: 10.1158/0008-5472.CAN-03-3429. [DOI] [PubMed] [Google Scholar]
- 12.Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–35. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chua C, Zaiden N, Chong KH, et al. Characterization of a side population of astrocytoma cells in response to temozolomide. J Neurosurg. 2008;109(5):856–66. doi: 10.3171/JNS/2008/109/11/0856. [DOI] [PubMed] [Google Scholar]
- 14.Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–54. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 15.Colvin OM, Friedman HS, Gamcsik MP, et al. Role of glutathione in cellular resistance to alkylating agents. Adv Enzyme Regul. 1993;33:19–26. doi: 10.1016/0065-2571(93)90006-y. [DOI] [PubMed] [Google Scholar]
- 16.Siegelin MD, Plescia J, Raskett CM, et al. Global targeting of subcellular heat shock protein-90 networks for therapy of glioblastoma. Mol Cancer Ther. 2010;9(6):1638–46. doi: 10.1158/1535-7163.MCT-10-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99(21):1583–93. doi: 10.1093/jnci/djm187. [DOI] [PubMed] [Google Scholar]
- 18.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 19.Netti PA, Baxter LT, Boucher Y, et al. Time-dependent behavior of interstitial fluid pressure in solid tumors: implications for drug delivery. Cancer Res. 1995;55(22):5451–8. [PubMed] [Google Scholar]
- 20.Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271(1):58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 21.Zhang M, Aguilera D, Das C, et al. Measuring cytotoxicity: a new perspective on LC50. Anticancer Res. 2007;27(1A):35–8. [PubMed] [Google Scholar]
- 22.Wolff JE, Trilling T, Molenkamp G, et al. Chemosensitivity of glioma cells in vitro: a meta analysis. J Cancer Res Clin Oncol. 1999;125(8-9):481–6. doi: 10.1007/s004320050305. [DOI] [PubMed] [Google Scholar]
- 23.Blakeley J. Drug delivery to brain tumors. Curr Neurol Neurosci Rep. 2008;8(3):235–41. doi: 10.1007/s11910-008-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cecchelli R, Berezowski V, Lundquist S, et al. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6(8):650–61. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- 25.Jones AR, Shusta EV. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24(9):1759–71. doi: 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groothuis DR. The blood-brain and blood-tumor barriers: a review of strategies for increasing drug delivery. Neuro Oncol. 2000;2(1):45–59. doi: 10.1093/neuonc/2.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muldoon LL, Pagel MA, Kroll RA, et al. A physiological barrier distal to the anatomic blood-brain barrier in a model of transvascular delivery. AJNR Am J Neuroradiol. 1999;20(2):217–22. [PMC free article] [PubMed] [Google Scholar]
- 28.Colgan OC, Collins NT, Ferguson G, et al. Influence of basolateral condition on the regulation of brain microvascular endothelial tight junction properties and barrier function. Brain Res. 2007;1193:84–92. doi: 10.1016/j.brainres.2007.11.072. [DOI] [PubMed] [Google Scholar]
- 29.Szakacs G, Paterson JK, Ludwig JA, et al. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219–34. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 30.Agarwal S, Sane R, Gallardo JL, et al. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334(1):147–55. doi: 10.1124/jpet.110.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Agarwal S, Shaik NM, et al. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther. 2009;330(3):956–63. doi: 10.1124/jpet.109.154781. [DOI] [PubMed] [Google Scholar]
- 32.Decleves X, Bihorel S, Debray M, et al. ABC transporters and the accumulation of imatinib and its active metabolite CGP74588 in rat C6 glioma cells. Pharmacol Res. 2008;57(3):214–22. doi: 10.1016/j.phrs.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Nies AT, Jedlitschky G, Konig J, et al. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience. 2004;129(2):349–60. doi: 10.1016/j.neuroscience.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 34.Warnke PC, Kopitzki K, Timmer J, Ostertag CB. Capillary physiology of human medulloblastoma: impact on chemotherapy. Cancer. 2006;107(9):2223–7. doi: 10.1002/cncr.22212. [DOI] [PubMed] [Google Scholar]
- 35.Blakeley JO, Olson J, Grossman SA, et al. New Approaches to Brain Tumor Therapy (NABTT) Consortium. Effect of blood brain barrier permeability in recurrent high grade gliomas on the intratumoral pharmacokinetics of methotrexate: a microdialysis study. J Neurooncol. 2009;91(1):51–8. doi: 10.1007/s11060-008-9678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zunkeler B, Carson RE, Olson J, et al. Quantification and pharmacokinetics of blood-brain barrier disruption in humans. J Neurosurg. 1996;85(6):1056–65. doi: 10.3171/jns.1996.85.6.1056. [DOI] [PubMed] [Google Scholar]
- 37.de Lange EC, Danhof M. Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin Pharmacokinet. 2002;41(10):691–703. doi: 10.2165/00003088-200241100-00001. [DOI] [PubMed] [Google Scholar]
- 38.Blakeley JO, Laterra J. Neoplastic and paraneoplastic disorders. In: Irani D, editor. Cerebrospinal fluid in clinical practice. Vol. 1. Saunders Elsevier; Philadephia, PA: 2009. p. 233. [Google Scholar]
- 39.Jacobs S, McCully CL, Murphy RF, et al. Extracellular fluid concentrations of cisplatin, carboplatin, and oxaliplatin in brain, muscle, and blood measured using microdialysis in nonhuman primates. Cancer Chemother Pharmacol. 2010;65(5):817–24. doi: 10.1007/s00280-009-1085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Portnow J, Badie B, Chen M, et al. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: potential implications for the current approach to chemoradiation. Clin Cancer Res. 2009;15(22):7092–8. doi: 10.1158/1078-0432.CCR-09-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lovich MA, Edelman ER. Tissue concentration of heparin, not administered dose, correlates with the biological response of injured arteries in vivo. Proc Natl Acad Sci USA. 1999;96(20):11111–16. doi: 10.1073/pnas.96.20.11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clarke JL, Pao W, Wu N, et al. High dose weekly erlotinib achieves therapeutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancer. J Neurooncol. 2010;99(2):283–6. doi: 10.1007/s11060-010-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Togashi Y, Masago K, Fukudo M, et al. Cerebrospinal fluid concentration of erlotinib and its active metabolite OSI-420 in patients with central nervous system metastases of non-small cell lung cancer. J Thorac Oncol. 2010;5(7):950–5. doi: 10.1097/JTO.0b013e3181e2138b. [DOI] [PubMed] [Google Scholar]
- 44.Lassman AB, Rossi MR, Raizer JJ, et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res. 2005;11(21):7841–50. doi: 10.1158/1078-0432.CCR-05-0421. [DOI] [PubMed] [Google Scholar]
- 45.Raizer JJ, Abrey LE, Lassman AB, et al. North American Brain Tumor Consortium. A Phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12(1):95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holdhoff M, Supko JG, Gallia GL, et al. Intratumoral concentrations of imatinib after oral administration in patients with glioblastoma multiforme. J Neurooncol. 2010;97(2):241–5. doi: 10.1007/s11060-009-0008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Razis E, Selviaridis P, Labropoulos S, et al. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clin Cancer Res. 2009;15(19):6258–66. doi: 10.1158/1078-0432.CCR-08-1867. [DOI] [PubMed] [Google Scholar]
- 48.Zamboni WC, Gervais AC, Egorin MJ, et al. Inter- and intratumoral disposition of platinum in solid tumors after administration of cisplatin. Clin Cancer Res. 2002;8(9):2992–9. [PubMed] [Google Scholar]
- 49.Zamboni WC, Houghton PJ, Hulstein JL, et al. Relationship between tumor extracellular fluid exposure to topotecan and tumor response in human neuroblastoma xenograft and cell lines. Cancer Chemother Pharmacol. 1999;43(4):269–76. doi: 10.1007/s002800050894. [DOI] [PubMed] [Google Scholar]
- 50.Zamboni WC, Stewart CF, Cheshire PJ, et al. Studies of the efficacy and pharmacology of irinotecan against human colon tumor xenograft models. Clin Cancer Res. 1998;4(3):743–53. [PubMed] [Google Scholar]
- 51.Hutschala D, Kinstner C, Skhirtladze K, et al. The impact of perioperative atelectasis on antibiotic penetration into lung tissue: an in vivo microdialysis study. Intensive Care Med. 2008;34(10):1827–34. doi: 10.1007/s00134-008-1122-8. [DOI] [PubMed] [Google Scholar]
- 52.Karjagin J, Lefeuvre S, Oselin K, et al. Pharmacokinetics of meropenem determined by microdialysis in the peritoneal fluid of patients with severe peritonitis associated with septic shock. Clin Pharmacol Ther. 2008;83(3):452–9. doi: 10.1038/sj.clpt.6100312. [DOI] [PubMed] [Google Scholar]
- 53.Zeitlinger BS, Zeitlinger M, Leitner I, et al. Clinical scoring system for the prediction of target site penetration of antimicrobials in patients with sepsis. Clin Pharmacokinet. 2007;46(1):75–83. doi: 10.2165/00003088-200746010-00004. [DOI] [PubMed] [Google Scholar]
- 54.Bellmann R, Kuchling G, Dehghanyar P, et al. Tissue pharmacokinetics of levofloxacin in human soft tissue infections. Br J Clin Pharmacol. 2004;57(5):563–8. doi: 10.1111/j.1365-2125.2004.02059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tomaselli F, Maier A, Matzi V, et al. Penetration of meropenem into pneumonic human lung tissue as measured by in vivo microdialysis. Antimicrob Agents Chemother. 2004;48(6):2228–32. doi: 10.1128/AAC.48.6.2228-2232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Presant CA, Wolf W, Waluch V, et al. Association of intratumoral pharmacokinetics of fluorouracil with clinical response. Lancet. 1994;343(8907):1184–7. doi: 10.1016/s0140-6736(94)92399-x. [DOI] [PubMed] [Google Scholar]
- 57.Chaurasia CS, Muller M, Bashaw ED, et al. AAPS-FDA workshop white paper: microdialysis principles, application and regulatory perspectives. Pharm Res. 2007;24(5):1014–25. doi: 10.1007/s11095-006-9206-z. [DOI] [PubMed] [Google Scholar]
- 58.Ungerstedt U. Microdialysis–principles and applications for studies in animals and man. J Intern Med. 1991;230(4):365–73. doi: 10.1111/j.1365-2796.1991.tb00459.x. [DOI] [PubMed] [Google Scholar]
- 59.de Lange EC, de Boer BA, Breimer DD. Microdialysis for pharmacokinetic analysis of drug transport to the brain. Adv Drug Deliv Rev. 1999;36(2-3):211–27. doi: 10.1016/s0169-409x(98)00089-1. [DOI] [PubMed] [Google Scholar]
- 60.de Lange EC, Danhof M, de Boer AG, Breimer DD. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood-brain barrier. Brain Res Brain Res Rev. 1997;25(1):27–49. doi: 10.1016/s0165-0173(97)00014-3. [DOI] [PubMed] [Google Scholar]
- 61.Brunner M, Derendorf H. Clinical microdialysis: current applications and potential use in drug development. Trends Anal Chem. 2006;25(7):674–80. [Google Scholar]
- 62.Elmquist WF, Sawchuk RJ. Application of microdialysis in pharmacokinetic studies. Pharm Res. 1997;14(3):267–88. doi: 10.1023/a:1012081501464. [DOI] [PubMed] [Google Scholar]
- 63.Muller M. Microdialysis in clinical drug delivery studies. Adv Drug Deliv Rev. 2000;45(2-3):255–69. doi: 10.1016/s0169-409x(00)00113-7. [DOI] [PubMed] [Google Scholar]
- 64.de Lange EC, de Vries JD, Zurcher C, et al. The use of intracerebral microdialysis for the determination of pharmacokinetic profiles of anticancer drugs in tumor-bearing rat brain. Pharm Res. 1995;12(12):1924–31. doi: 10.1023/a:1016239822287. [DOI] [PubMed] [Google Scholar]
- 65.Konings IR, Engels FK, Sleijfer S, et al. Application of prolonged microdialysis sampling in carboplatin-treated cancer patients. Cancer Chemother Pharmacol. 2009;64(3):509–16. doi: 10.1007/s00280-008-0898-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Q, Gallo JM. In vivo microdialysis for PK and PD studies of anticancer drugs. AAPS J. 2005;7(3):E659–67. doi: 10.1208/aapsj070366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loos WJ, Zamboni WC, Engels FK, et al. Pitfalls of the application of microdialysis in clinical oncology: controversial findings with docetaxel. J Pharm Biomed Anal. 2007;45(2):288–94. doi: 10.1016/j.jpba.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 68.Schuck VJ, Rinas I, Derendorf H. In vitro microdialysis sampling of docetaxel. J Pharm Biomed Anal. 2004;36(4):807–13. doi: 10.1016/j.jpba.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 69.Chu J, Gallo JM. Application of microdialysis to characterize drug disposition in tumors. Adv Drug Deliv Rev. 2000;45(2-3):243–53. doi: 10.1016/s0169-409x(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 70.Kehr J. A survey on quantitative microdialysis: theoretical models and practical implications. J Neurosci Methods. 1993;48(3):251–61. doi: 10.1016/0165-0270(93)90096-a. [DOI] [PubMed] [Google Scholar]
- 71.Lonnroth P, Jansson PA, Smith U. A microdialysis method allowing characterization of intercellular water space in humans. Am J Physiol. 1987;253(2 Pt 1):E228–31. doi: 10.1152/ajpendo.1987.253.2.E228. [DOI] [PubMed] [Google Scholar]
- 72.Olson RJ, Justice JB., Jr Quantitative microdialysis under transient conditions. Anal Chem. 1993;65(8):1017–22. doi: 10.1021/ac00056a012. [DOI] [PubMed] [Google Scholar]
- 73.Wang Y, Wong SL, Sawchuk RJ. Microdialysis calibration using retrodialysis and zero-net flux: application to a study of the distribution of zidovudine to rabbit cerebrospinal fluid and thalamus. Pharm Res. 1993;10(10):1411–19. doi: 10.1023/a:1018906821725. [DOI] [PubMed] [Google Scholar]
- 74.Carcaboso AM, Elmeliegy MA, Shen J, et al. Tyrosine kinase inhibitor gefitinib enhances topotecan penetration of gliomas. Cancer Res. 2010;70(11):4499–508. doi: 10.1158/0008-5472.CAN-09-4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jacobs S, McCully CL, Murphy RF, et al. Extracellular fluid concentrations of cisplatin, carboplatin, and oxaliplatin in brain, muscle, and blood measured using microdialysis in nonhuman primates. Cancer Chemother Pharmacol. 2010;65(5):817–24. doi: 10.1007/s00280-009-1085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sani SN, Henry K, Bohlke M, et al. The effects of drug transporter inhibitors on the pharmacokinetics and tissue distribution of methotrexate in normal and tumor-bearing mice: a microdialysis study. Cancer Chemother Pharmacol. 2010;66(1):159–69. doi: 10.1007/s00280-009-1146-y. [DOI] [PubMed] [Google Scholar]
- 77.Apparaju SK, Gudelsky GA, Desai PB. Pharmacokinetics of gemcitabine in tumor and non-tumor extracellular fluid of brain: an in vivo assessment in rats employing intracerebral microdialysis. Cancer Chemother Pharmacol. 2008;61(2):223–9. doi: 10.1007/s00280-007-0464-1. [DOI] [PubMed] [Google Scholar]
- 78.Zhou Q, Guo P, Kruh GD, et al. Predicting human tumor drug concentrations from a preclinical pharmacokinetic model of temozolomide brain disposition. Clin Cancer Res. 2007;13(14):4271–9. doi: 10.1158/1078-0432.CCR-07-0658. [DOI] [PubMed] [Google Scholar]
- 79.Zamboni WC, Gervais AC, Egorin MJ, et al. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother Pharmacol. 2004;53(4):329–36. doi: 10.1007/s00280-003-0719-4. [DOI] [PubMed] [Google Scholar]
- 80.Dukic SF, Kaltenbach ML, Heurtaux T, et al. Influence of C6 and CNS1 brain tumors on methotrexate pharmacokinetics in plasma and brain tissue. J Neurooncol. 2004;67(1-2):131–8. doi: 10.1023/b:neon.0000021820.12444.4c. [DOI] [PubMed] [Google Scholar]
- 81.Johansen MJ, Thapar N, Newman RA, Madden T. Use of microdialysis to study platinum anticancer agent pharmacokinetics in preclinical models. J Exp Ther Oncol. 2002;2(3):163–73. doi: 10.1046/j.1359-4117.2002.01019.x. [DOI] [PubMed] [Google Scholar]
- 82.Ma J, Li S, Reed K, et al. Pharmacodynamic-mediated effects of the angiogenesis inhibitor SU5416 on the tumor disposition of temozolomide in subcutaneous and intracerebral glioma xenograft models. J Pharmacol Exp Ther. 2003;305(3):833–9. doi: 10.1124/jpet.102.048587. [DOI] [PubMed] [Google Scholar]
- 83.Ma J, Pulfer S, Li S, et al. Pharmacodynamic-mediated reduction of temozolomide tumor concentrations by the angiogenesis inhibitor TNP-470. Cancer Res. 2001;61(14):5491–8. [PubMed] [Google Scholar]
- 84.Devineni D, Klein-Szanto A, Gallo JM. Uptake of temozolomide in a rat glioma model in the presence and absence of the angiogenesis inhibitor TNP-470. Cancer Res. 1996;56(9):1983–7. [PubMed] [Google Scholar]
- 85.Devineni D, Klein-Szanto A, Gallo JM. In vivo microdialysis to characterize drug transport in brain tumors: analysis of methotrexate uptake in rat glioma-2 (RG-2)-bearing rats. Cancer Chemother Pharmacol. 1996;38(6):499–507. doi: 10.1007/s002800050518. [DOI] [PubMed] [Google Scholar]
- 86.Brunner M, Muller M. Microdialysis: an in vivo approach for measuring drug delivery in oncology. Eur J Clin Pharmacol. 2002;58(4):227–34. doi: 10.1007/s00228-002-0475-0. [DOI] [PubMed] [Google Scholar]
- 87.Hunz M, Jetter A, Warm M, et al. Plasma and tissue pharmacokinetics of epirubicin and Paclitaxel in patients receiving neoadjuvant chemotherapy for locally advanced primary breast cancer. Clin Pharmacol Ther. 2007;81(5):659–68. doi: 10.1038/sj.clpt.6100067. [DOI] [PubMed] [Google Scholar]
- 88.Mader RM, Schrolnberger C, Rizovski B, et al. Penetration of capecitabine and its metabolites into malignant and healthy tissues of patients with advanced breast cancer. Br J Cancer. 2003;88(5):782–7. doi: 10.1038/sj.bjc.6600809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Muller M, Mader RM, Steiner B, et al. 5-Fluorouracil kinetics in the interstitial tumor space: clinical response in breast cancer patients. Cancer Res. 1997;57(13):2598–601. [PubMed] [Google Scholar]
- 90.Joukhadar C, Klein N, Mader RM, et al. Penetration of dacarbazine and its active metabolite 5-aminoimidazole-4-carboxamide into cutaneous metastases of human malignant melanoma. Cancer. 2001;92(8):2190–6. doi: 10.1002/1097-0142(20011015)92:8<2190::aid-cncr1562>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 91.Tegeder I, Brautigam L, Seegel M, et al. Cisplatin tumor concentrations after intra-arterial cisplatin infusion or embolization in patients with oral cancer. Clin Pharmacol Ther. 2003;73(5):417–26. doi: 10.1016/s0009-9236(03)00008-0. [DOI] [PubMed] [Google Scholar]
- 92.Benveniste H. Brain microdialysis. J Neurochem. 1989;52(6):1667–79. doi: 10.1111/j.1471-4159.1989.tb07243.x. [DOI] [PubMed] [Google Scholar]
- 93.Salci K, Nilsson P, Howells T, et al. Intracerebral microdialysis and intracranial compliance monitoring of patients with traumatic brain injury. J Clin Monit Comput. 2006;20(1):25–31. doi: 10.1007/s10877-006-2864-x. [DOI] [PubMed] [Google Scholar]
- 94.Vespa P, Boonyaputthikul R, McArthur DL, et al. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit Care Med. 2006;34(3):850–6. doi: 10.1097/01.CCM.0000201875.12245.6F. [DOI] [PubMed] [Google Scholar]
- 95.Chan TV, Ng SC, Lam JM, et al. Monitoring of autoregulation using intracerebral microdialysis in patients with severe head injury. Acta Neurochir Suppl. 2005;95:113–16. doi: 10.1007/3-211-32318-x_24. [DOI] [PubMed] [Google Scholar]
- 96.Vespa P, Bergsneider M, Hattori N, et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005;25(6):763–74. doi: 10.1038/sj.jcbfm.9600073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vespa PM, McArthur D, O’Phelan K, et al. Persistently low extracellular glucose correlates with poor outcome 6 months after human traumatic brain injury despite a lack of increased lactate: a microdialysis study. J Cereb Blood Flow Metab. 2003;23(7):865–77. doi: 10.1097/01.WCB.0000076701.45782.EF. [DOI] [PubMed] [Google Scholar]
- 98.Goodman JC, Valadka AB, Gopinath SP, et al. Extracellular lactate and glucose alterations in the brain after head injury measured by microdialysis. Crit Care Med. 1999;27(9):1965–73. doi: 10.1097/00003246-199909000-00041. [DOI] [PubMed] [Google Scholar]
- 99.Wang E, Ho CL, Lee KK, et al. Changes in brain biochemistry and oxygenation in the zone surrounding primary intracerebral hemorrhage. Acta Neurochir Suppl. 2008;102:293–7. doi: 10.1007/978-3-211-85578-2_55. [DOI] [PubMed] [Google Scholar]
- 100.Miller CM, Vespa PM, McArthur DL, et al. Frameless stereotactic aspiration and thrombolysis of deep intracerebral hemorrhage is associated with reduced levels of extracellular cerebral glutamate and unchanged lactate pyruvate ratios. Neurocrit Care. 2007;6(1):22–9. doi: 10.1385/NCC:6:1:22. [DOI] [PubMed] [Google Scholar]
- 101.Nilsson OG, Polito A, Saveland H, et al. Are primary supratentorial intracerebral hemorrhages surrounded by a biochemical penumbra? A microdialysis study. Neurosurgery. 2006;59(3):521–8. doi: 10.1227/01.NEU.0000227521.58701.E5. discussion 521-8. [DOI] [PubMed] [Google Scholar]
- 102.Sarrafzadeh AS, Sakowitz OW, Kiening KL, et al. Bedside microdialysis: a tool to monitor cerebral metabolism in subarachnoid hemorrhage patients? Crit Care Med. 2002;30(5):1062–70. doi: 10.1097/00003246-200205000-00018. [DOI] [PubMed] [Google Scholar]
- 103.Kett-White R, Hutchinson PJ, Al-Rawi PG, et al. Adverse cerebral events detected after subarachnoid hemorrhage using brain oxygen and microdialysis probes. Neurosurgery. 2002;50(6):1213–21. doi: 10.1097/00006123-200206000-00008. discussion 1221-2. [DOI] [PubMed] [Google Scholar]
- 104.Staub F, Graf R, Gabel P, et al. Multiple interstitial substances measured by microdialysis in patients with subarachnoid hemorrhage. Neurosurgery. 2000;47(5):1106–15. doi: 10.1097/00006123-200011000-00016. discussion 1115-16. [DOI] [PubMed] [Google Scholar]
- 105.During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341(8861):1607–10. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- 106.Poca MA, Sahuquillo J, Vilalta A, et al. Percutaneous implantation of cerebral microdialysis catheters by twist-drill craniostomy in neurocritical patients: description of the technique and results of a feasibility study in 97 patients. J Neurotrauma. 2006;23(10):1510–17. doi: 10.1089/neu.2006.23.1510. [DOI] [PubMed] [Google Scholar]
- 107.Bergenheim AT, Capala J, Roslin M, Henriksson R. Distribution of BPA and metabolic assessment in glioblastoma patients during BNCT treatment: a microdialysis study. J Neurooncol. 2005;71(3):287–93. doi: 10.1007/s11060-004-1724-0. [DOI] [PubMed] [Google Scholar]
- 108.Benjamin RK, Hochberg FH, Fox E, et al. Review of microdialysis in brain tumors, from concept to application: first annual Carolyn Frye-Halloran symposium. Neuro Oncol. 2004;6(1):65–74. doi: 10.1215/S1152851703000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ruggeri M, Zoli M, Grimaldi R, et al. Aspects of neural plasticity in the central nervous system-III. Methodological studies on the microdialysis technique. Neurochem Int. 1990;16(4):427–35. doi: 10.1016/0197-0186(90)90004-d. [DOI] [PubMed] [Google Scholar]
- 110.Benveniste H, Diemer NH. Cellular reactions to implantation of a microdialysis tube in the rat hippocampus. Acta Neuropathol. 1987;74(3):234–8. doi: 10.1007/BF00688186. [DOI] [PubMed] [Google Scholar]
- 111.Lindberger M, Tomson T, Lars S. Microdialysis sampling of carbamazepine, phenytoin and phenobarbital in subcutaneous extracellular fluid and subdural cerebrospinal fluid in humans: an in vitro and in vivo study of adsorption to the sampling device. Pharmacol Toxicol. 2002;91(4):158–65. doi: 10.1034/j.1600-0773.2002.910402.x. [DOI] [PubMed] [Google Scholar]
- 112.Scheyer RD, During MJ, Hochholzer JM, et al. Phenytoin concentrations in the human brain: an in vivo microdialysis study. Epilepsy Res. 1994;18(3):227–32. doi: 10.1016/0920-1211(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 113.Hau P, Fabel K, Baumgart U, et al. Pegylated liposomal doxorubicin-efficacy in patients with recurrent high-grade glioma. Cancer. 2004;100(6):1199–207. doi: 10.1002/cncr.20073. [DOI] [PubMed] [Google Scholar]
- 114.Mindermann T, Zimmerli W, Gratzl O. Rifampin concentrations in various compartments of the human brain: a novel method for determining drug levels in the cerebral extracellular space. Antimicrob Agents Chemother. 1998;42(10):2626–9. doi: 10.1128/aac.42.10.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dhodapkar M, Rubin J, Reid JM, et al. Phase I trial of temozolomide (NSC 362856) in patients with advanced cancer. Clin Cancer Res. 1997;3(7):1093–100. [PubMed] [Google Scholar]
- 116.Reid JM, Stevens DC, Rubin J, Ames MM. Pharmacokinetics of 3-methyl-(triazen-1-yl)imidazole-4-carboximide following administration of temozolomide to patients with advanced cancer. Clin Cancer Res. 1997;3(12 Pt 1):2393–8. [PubMed] [Google Scholar]
- 117.Morgan ME, Singhal D, Anderson BD. Quantitative assessment of blood-brain barrier damage during microdialysis. J Pharmacol Exp Ther. 1996;277(2):1167–76. [PubMed] [Google Scholar]
- 118.Groothuis DR, Ward S, Schlageter KE, et al. Changes in blood-brain barrier permeability associated with insertion of brain cannulas and microdialysis probes. Brain Res. 1998;803(1-2):218–30. doi: 10.1016/s0006-8993(98)00572-1. [DOI] [PubMed] [Google Scholar]
- 119.Terasaki T, Deguchi Y, Kasama Y, et al. Determination of in vivo steady-state unbound drug concentration in the brain interstitial fluid by microdialysis. Int J Pharmaceut. 1992;81:143–52. [Google Scholar]
- 120.Tossman U, Ungerstedt U. Microdialysis in the study of extracellular levels of amino acids in the rat brain. Acta Physiol Scand. 1986;128(1):9–14. doi: 10.1111/j.1748-1716.1986.tb07943.x. [DOI] [PubMed] [Google Scholar]
- 121.Marklund N, Blennow K, Zetterberg H, et al. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg. 2009;110(6):1227–37. doi: 10.3171/2008.9.JNS08584. [DOI] [PubMed] [Google Scholar]
- 122.Mellergard P, Aneman O, Sjogren F, et al. Changes in extracellular concentrations of some cytokines, chemokines, and neurotrophic factors after insertion of intracerebral microdialysis catheters in neurosurgical patients. Neurosurgery. 2008;62(1):151–7. doi: 10.1227/01.NEU.0000311072.33615.3A. discussion 157-8. [DOI] [PubMed] [Google Scholar]
- 123.Hillman J, Aneman O, Persson M, et al. Variations in the response of interleukins in neurosurgical intensive care patients monitored using intracerebral microdialysis. J Neurosurg. 2007;106(5):820–5. doi: 10.3171/jns.2007.106.5.820. [DOI] [PubMed] [Google Scholar]
- 124.Waelgaard L, Pharo A, Tonnessen TI, Mollnes TE. Microdialysis for monitoring inflammation: efficient recovery of cytokines and anaphylotoxins provided optimal catheter pore size and fluid velocity conditions. Scand J Immunol. 2006;64(3):345–52. doi: 10.1111/j.1365-3083.2006.01826.x. [DOI] [PubMed] [Google Scholar]
- 125.Clough GF. Microdialysis of large molecules. AAPS J. 2005;7(3):E686–92. doi: 10.1208/aapsj070369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hillman J, Aneman O, Anderson C, et al. A microdialysis technique for routine measurement of macromolecules in the injured human brain. Neurosurgery. 2005;56(6):1264–8. doi: 10.1227/01.neu.0000159711.93592.8d. discussion 1268-70. [DOI] [PubMed] [Google Scholar]
- 127.Hutchinson PJ, O’Connell MT, Nortje J, et al. Cerebral microdialysis methodology–evaluation of 20 kDa and 100 kDa catheters. Physiol Meas. 2005;26(4):423–8. doi: 10.1088/0967-3334/26/4/008. [DOI] [PubMed] [Google Scholar]
- 128.Badie B, Chen M, Synold T, Portnow J. Assessment of the peritumoral micro-environment following craniotomy using intracerebral microdialysis [abstract #558] Neuro Oncol. 2009;11(5):690. [Google Scholar]
- 129.Zhou Q, Gallo JM. Differential effect of sunitinib on the distribution of temozolomide in an orthotopic glioma model. Neuro Oncol. 2009;11(3):301–10. doi: 10.1215/15228517-2008-088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–40. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 131.Hunz M, Jetter A, Warm M, et al. Plasma and tissue pharmacokinetics of epirubicin and Paclitaxel in patients receiving neoadjuvant chemotherapy for locally advanced primary breast cancer. Clin Pharmacol Ther. 2007;81(5):659–68. doi: 10.1038/sj.clpt.6100067. [DOI] [PubMed] [Google Scholar]
- 132.Thompson JF, Siebert GA, Anissimov YG, et al. Microdialysis and response during regional chemotherapy by isolated limb infusion of melphalan for limb malignancies. Br J Cancer. 2001;85(2):157–65. doi: 10.1054/bjoc.2001.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]