

Abstract
Despite significant progress in the treatment of breast cancer particularly through the use of targeted therapy, relapse and chemo-resistance remain a major hindrance to the fight to minimize the burden of the disease. It is becoming increasingly clear that a rare subpopulation of cells known as cancer stem cells (CSC), able to be generated through epithelial to mesenchymal transition (EMT) and capable of tumor initiation and self-renewal, contributes to treatment resistance and metastases. This means that a more effective therapy should target both the chemoresistant CSCs and the proliferating epithelial cells that give rise to them in order to reverse EMT and attenuate their conversion to CSCs. Here, we demonstrate a novel function of AXL in acting upstream to induce EMT in normal and immortalized human mammary epithelial cells in an apparent positive feedback loop mechanism and regulate breast CSC (BCSC) self-renewal and chemoresistance. Downregulation of AXL using MP470 (amuvatinib) reversed EMT in mesenchymal normal human mammary epithelial cells and murine BCSCs attenuating self-renewal and restored chemosensitivity of the BCSCs. AXL expression was also found to be associated with expression of stem cell genes, regulation of metastases genes, increased tumorigenicity, and was important for BCSC invasion and migration. Inactivation of AXL also led to downregulation of NFκB pathway and reduced tumor formation in vivo. Together, our data suggest that targeted therapy against AXL, in combination with systemic therapies, has the potential to improve response to anti-cancer therapies and to reduce breast cancer recurrence and metastases.
Keywords: AXL, EMT, CSCs, BCSCs, Tyrosine kinase
Introduction
Most breast cancer deaths result from disease relapse with invasion and metastases. Accumulated evidence suggest that breast cancer stem cells (BCSCs), which show resistance to chemotherapy and radiation, drive tumor initiation and disease recurrence.1–3 Recent evidence also suggests that breast cancer stem cells can be generated by epithelial to mesenchymal transition (EMT), a dedifferentiation program characterized by loss of epithelial characteristics and adhesion proteins and the acquisition of mesenchymal traits and invasive properties.4–8 EMT involves coordinated molecular, biochemical and cellular change resulting in the loss of cell-cell adhesion, apical-basolateral polarity, and epithelial markers, for acquisition of motility, spindle-cell shape, and mesenchymal markers. Epithelial cells that undergo EMT lose their epithelial cell characteristics marked by downregulation of E-cadherin in adherens junctions, occludins and claudins in tight junctions to acquire a mesenchymal phenotype characterized by upregulation of mesenchymal proteins vimentin, N-cadherin, fibronectin as well as activation of transcription factors including Snail, Slug, ZEB1, Zeb2 and Twist.8–10 EMT can be induced in vitro by treatment of epithelial cells with inducers such as EGF, HGF, PDGF or TGF-β, matrix metalloproteinases, or through overexpression of Snail or Twist.9, 11
AXL is a member of the TAM (Tyro3, Axl, Mer) family of receptor tyrosine kinases (RTK), originally identified as a transforming gene in cells of chronic myelogenous leukemia (CML) patients. AXL is activated through several mechanisms, including binding of its ligand, growth arrest specific 6 (Gas6), and extracellular domain-mediated dimerization or crosstalk with HER2/neu.12–14 AXL is overexpressed in a wide variety of human cancers with significant correlation with tumor stage in breast cancer patients and plays a role in cancer progression and metastases.15–17 Activation of AXL regulates a number of signal transduction pathways including NF-κB, STAT, Akt and MAP kinases. AXL is upregulated by EMT induction and has been shown to mediate acquired resistance in lapatinib-resistant, HER2/neu-positive and ER-positive BT474 cells.18, 19 However, it has not been shown whether AXL is sufficient to directly induce EMT or to activate the breast cancer stem cell phenotype. In this study, we show that AXL is constitutively activated in BCSCs and induces EMT by regulating the expression of EMT markers such as E- and N-Cadherin, Snail and Slug. We also show that downregulation of AXL using the tyrosine kinase inhibitor MP470 (Amuvatinib) reverses EMT in both human and mouse mesechymal cells which have been induced to undergo EMT, reduces the tumorigenicity of BCSC, downregulates NFκB pathway and restores sensitivity of the BCSC to chemotherapy. We also demonstrate that AXL is associated with expression of stem cell marker genes, regulates metastases genes and plays a role in BCSC migration and invasion.
Results
AXL is unregulated and ligand-independently activated in breast cancer stem cells
Previous studies have shown that EMT-induced regulation of AXL plays an essential role in breast metastases and correlates with poor patient survival.18 We have previously reported the generation of BCSCs through immune induced EMT4 and have also observed upregulation of AXL expression in the mesenchymal breast cancer stem cells (Figure 1A). Here, we assessed whether AXL plays a direct role in EMT induction or regulates the function of BCSCs. To determine the activation status of AXL in ANV BCSCs, we measured its phosphorylation at Ser 744 which showed that AXL is activated in ANVs but not in MMC cells. To determine if AXL activation in ANVs was dependent on GAS6 binding, we treated ANV5 and MMC cells with increasing doses of Gas6. Immunoblotting of Akt phosphorylation, a downstream target of the AXL pathway, showed no change in Akt phosphorylation and therefore no increase in AXL activation upon Gas6 stimulation (Figure 1B). In agreement with these results, treating ANV5 cell with increasing doses of Gas6 showed no change in the levels of activated AXL (Figure 1C). Furthermore expression of Axl and Gas6 mRNAs showed that whereas AXL expression is upregulated in ANV BCSCs, Gas6 expression was downregulated, confirming that constitutive activation of AXL is independent of Gas6 binding 9 (Figure 1D).
Figure 1.

AXL is upregulated and ligand-independently activated in breast cancer stem cells. A–B, Immunoblot showing expression of AXL in mouse BCSCs (ANVs) and human breast cancer cell lines. C, Immunoblot of phospho-Akt, Akt, Axl in ANVs and MMC treated with or without AXL ligand Gas6. β-actin is shown as a loading control. D, Treatment of ANV5 cells with increasing doses of Gas6 shows no effect on the activation status of AXL. E, RT-PCR results of endogenous AXL and Gas6 expression in ANV and MMC cells. GAPDH is a RNA loading control. The results of the experiments in this figure were repeated two times with similar results.
AXL overexpression induces EMT in human normal and immortalized mammary epithelial cells
EMT induction has been shown to regulate AXL expression.18 We observed an increase in AXL expression in mesenchymal cell lines MMCTT and ANV5 cells compared to parental epithelial MMC cells and MMCTTE epithelial cells derived through mesenchymal to epithelial transition (MET) of MMCTT cells (Figure 2A, upper panel). We also observed increased AXL expression in MCF10ATT cells generated from MCF10A treated with TGFβ and TNF-α (Figure 2A, lower panel). To test whether AXL plays a role in EMT induction, we assessed the effect of AXL overexpression on expression of EMT markers. The results showed that AXL overexpression in HMLE cells.5, 20 strongly downregulated E-Cadherin expression while the expression of mesenchymal markers such as N-Cadherin, Snail and Slug were upregulated (Figure 2B). A similar result was observed by forced expression of AXL in normal human mammary epithelial cell line MCF10A (Figure 2C). Consistent with the above results, silencing of AXL using viral mediated shRNA transfection led to upregulated expression of N-cadherin, Vimentin, fibronectin and Snail whiles E-cadherin expression was downregulated (Figure 2D). Comparable results were found when AXL expression was depleted in ETTM cells (Figure S1A).21 Change in cell morphology consistent with MET was also observed in ANV5 and ETTM cells (Figure S1B–C). These results demonstrated a novel role of AXL in EMT induction and suggest a potential feedback regulation since AXL is in turn regulated by EMT.
Figure 2.

AXL overexpression regulates expression of EMT markers. A, Immunoblot analysis showing the effect of TGFβ/TNFα-mediated EMT induction on AXL expression in MMC. MMCTT are MMCs treated with TGFβ and TNFα MMCTTE are MMCTT mesenchymal cells that reverted back to epithelial cells following treatment withdrawal. ANV5 are shown as control. Also shown is AXL expression in MCF-10A following treatment with TGF-β. B–C, Immunoblot analyses of EMT marker expression in HMLE and MCF10A. Overexpression of AXL was induced in HMLE and MCF10A using two separate expression vectors, pCi-AXL1 and pCi-AXL2. D–E, Silencing of AXL in ANV5 and ETTM cells reverses mesenchymal phenotype of ANV5 cells characterized by increased E-cadherin expression but decreased expression of AXL, N-cadherin, Vimentin, Fibronectin, Zeb1 and Snail. Control cells expressed a scrambled shRNA. The results of the experiments in this figure were repeated independently two times with similar results.
Inactivation of AXL by MP470 reverses epithelial to mesenchymal transition
To further explore the role of AXL in EMT process, we assessed changes in gene expression profile upon AXL inactivation using AXL inhibitor MP470 (Amuvatinib). MP470 is a structure-based multi-targeted RTK inhibitor that targets mutant c-Kit, PDGFRα and AXL. MP470 in combination with Erlotinib targets the HER family/PI3K/Akt pathway and inhibits tumor growth in prostate cancer and sensitizes glioblastoma cells to chemotherapy.22, 23 First, transient EMT was induced in MCF10A cells by TGFβ and TNFα treatment21 to generate mesenchymal MCF10ATT cells followed by treatment with varying doses of MP470 or DMSO. Pathway analysis using DMSO-treated control cells as a reference showed differential regulation of EMT markers in mesenchymal MCF10ATT cells, shown by downregulation of mesenchymal markers Foxc2, Snai1, FGFBP1 and BMP7 and upregulation of MMP9 and Oclnb (Figure 3A). Immunoblotting of protein expression revealed dose-dependent upregulation of E-Cadherin, β-Catenin and downregulation of N-Cadherin, phospho-AXL, and Snail expression (Figure 3C). Since MP470 is known as a c-MET inhibitor we also evaluated the impact of the inhibitor on levels of both c-MET and its activated form phosphorylated at tyrosine 1002. As shown in Figure 3C, c-MET was expressed, albeit weakly in MCF10ATT cells and activation is not detectable. Furthermore, in parallel with upregulation of epithelial markers and downregulation of mesenchymal markers, c-MET levels increased of which a small fraction remained activated despite exposure to MP470. Downregulation of MET in epithelial cells should drive an EMT phenotype and therefore would not be expected to be activated or expressed in the mesenchymal MCF10ATT cells.
Figure 3.

Inactivation of AXL by MP470 reverses epithelial to mesenchymal transition. A, RT-PCR gene expression analysis of TGFβ and TNFα-treated MCF10A cells or B, ANV5s treated with varying concentrations of MP470. C, Immunoblot analyses of lysates from TGFβ/TNFα –treated MCF10A cells and D, ANV5 cells treated with varying amounts of MP470 for 72 hours. E, Immunoblot analyses of lysates from TGFβ/TNFα–treated MCF10A cells treated with varying doses of imatinib. The results of the experiments in this figure were repeated independently two times with similar results.
We also assessed the effect of AXL inhibition on mesenchymal phenotype of ANV5 cells. Gene expression analysis of mRNA isolated from the ANV5 cells previously treated with the AXL inhibitor showed differential regulation of EMT genes (Figure 3B). Consistent with these results, immunoblotting of EMT markers showed downregulation of Snail, N-Cadherin, phosphoAKT as well as upregulation of E-cadherin and β-catenin in response to treatment with MP470 (Figure 3D). The use of MP470 to inhibit AXL activity also resulted in downregulation of AXL expression. This could be due to a feedback mechanism whereby the reversal of EMT caused by AXL inactivation in turn drives down AXL expression since EMT also functions as an upstream regulator of AXL expression (Figure 2A, lower panel). It also possible that through a similar mechanism, inactivation of AXL pathway represses pathways that maintain AXL expression since the pathway is no longer activated. Finally, since MP470 also targets PDGFRα and c-Met, we assessed the contribution of PDGFRα and c-Met downregulation on EMT by treating ANV5 cells with imatinib mesylate, an inhibitor of PDGFR and c-Met, and found no change in N-Cadherin or E-Cadherin expression by immunoblotting (Figure 3E), suggesting that the observed effect of MP470 on EMT reversal was due to a combination of AXL activity inhibition and downregulation. Together, these results show that downregulation of AXL using MP470 reverses EMT and supports the finding that AXL plays a direct role in EMT induction.
Inactivation of AXL downregulates NFKB pathway
To elucidate the mechanism of AXL regulation of EMT induction, we focused on NFκB pathway since this pathway acts downstream of Akt pathway and is also a downstream target of AXL activation.24, 25 To assess the effect of AXL on NFκβ activation, we determined the expression of components of the NFκB pathway in ANV5 cells treated with varying doses of MP470 for 72 hours. Immunoblotting showed downregulation of NFκB-p65, NFκB-p50 and IκKα as well as upregulation of IκBα kinases (Figure 4A). To confirm that AXL downregulates NFκB pathway in our cells, we performed NFκB luciferase reporter assay using viral-mediated transduction of NFκB reporter vector. ANV5 cells transduced with NFκB reporter viral particles were treated with increasing doses of MP470 or TNFα and assayed for the expression of the luciferase gene. Compared to the controls, MP470 treated cells showed substantial downregulation of NFκB pathway whereas activation was increased by TNFα (Figure 4B). To confirm these results, a similar experiment was performed using anti-AXL antibodies. As expected, cells treated with the anti-AXL antibody downregulated NFκB pathway activation in dose-dependent manner, suggesting that NFκB is a downstream target of AXL activation (Figure 4C). Figure 4D confirms downregulation of both AXL expression and activity upon antibody treatment and therefore underscores a direct role of AXL in regulating of the NFκB pathway. To show further proof that NFκB pathway mediates the function of AXL in driving EMT, MCF10ATT cells were treated with the proteasome inhibitor MG132, a potent inhibitor of NFκB and the effect on expression of EMT genes analyzed.26 The results showed that treatment with MG132 led to increased expression of BMP2, MMP3, KRT14, IGFBP4, OCLN and SERPINE1 as well as downregulation of ZEB2, COL5A2, AHNAK, SPARC, WNT5A and CDH2 (Figure 4E) indicating that NFκB pathway may mediate the role of AXL in regulating EMT phenotype.
Figure 4.

Inactivation of AXL downregulates NFκβ pathway. A. Immunoblot analyses of expression of p65, p50 IkBα, IKKα in ANV5 treated with varying levels of AXL inhibitor, MP470, for 72 hrs. B–C, Mean (± SEM, N=4) NFκB-driven luciferase reporter activity levels in ANV5 cells treated with increasing doses of MP470 (B) or AXL antibody (C). RLU = relative luminescence units (RLU). D, Treatment of ANV5 cells with AXL antibody resulted in downregulation of AXL expression and activity. E, Inhibition of NFκB in MCF10ATT using MG132 drives downregulation of epithelial markers and upregulation of mesenchymal genes. The experiments were repeated independently at least two times with similar results.
AXL expression correlates with increased tumorigenicity in breast cancer stem cells
AXL plays a critical role in tumor growth, metastasis, and is associated with poor prognosis in breast, lung and brain cancers.15–17 AXL was found to be aberrantly expressed in breast primary tumors.17 To determine whether AXL expression is associated with increased tumorigenicity, we stained and sorted ANV5 cells into AXL-positive and AXL-negative populations and implanted into syngeneic mice using unsorted ANV5 cells as a control. The results showed that ANV5 cells expressing AXL were more tumorigenic compared to AXL-negative population or unsorted ANV5 cells (Figure 5A–B). To validate these results, we treated ANV5 cells with MP470 in culture to effectively target the BCSCs before injection into mice. The results showed that inactivation of AXL reduced the tumorigenicity of the BCSCs in a dose-dependent manner (Figure 5C). We also generated stable AXL knockdown and control ANV5 cells for in vivo tumor formation in mice which confirmed the observation that downregulation of AXL negatively impacted the tumorigenic capacity of the BCSCs (Figure 5D). These results suggest that AXL also mediates the tumorigenicity of BCSCs and therefore could serve as a target for modulating breast cancer.
Figure 5.

AXL expression correlates with increased tumorigenicity in BCSCs. A, AXL staining dot plots used to sort ANV into AXL positive and AXL negative populations. B, Mean (± SEM, n=5) tumor volumes over time in mice injected with either AXL+, AXL− or unsorted ANV5s. Curves for AXL5- and unsorted ANV5s are superimposed because the two cell populations formed tumors to the same extent. C, Mean (± SEM, n=5) tumor volumes over time in mice that received MP470- or control-treated ANV5s. D, Mean (± SEM, n=5) tumor volumes over time mice injected with AXL knockdown or control ANV5 cells. The experiments were repeated at least two times independently with similar results. Statistical significance was determined by two-way ANOVA. *p<0.05; ***p<0.001. While all lines have error bars, some errors are small and obscured by the symbols.
AXL is important for BCSC motility and associates with expression of metastases and stem cell genes
We then assessed the role of AXL in mesenchymal BCSC migration and invasion. Using stable AXL knockdown and control ANV5 cells, we found that downregulation of AXL reduced the migration and invasion of AXL depleted ANV5 cells compared to control cells (Figure 6A–B). To determine whether AXL expression was associated with expression of stem cell marker genes, we performed gene expression analyses of AXL-positive cells compared to AXL-negative cells and found increased expression of several stemness genes that were associated with AXL expression including Isl1, Cdc2a and Bglap1 (Figure 6C). Despite that, other stemness genes such as Smad9, S100b, Mme, and Col1a1 appeared to be down regulated. The effect of AXL on expression of metastases genes was also assessed by gene expression analysis of AXL knockdown cells which also identified several genes to be differentially regulated upon AXL downregulation (Figure 6D).
Figure 6.

AXL is important for BCSC motility and regulates expression of metastases and stem cell genes. A–B, Shown are photomicrographs and graphs of the mean (±SEM, n=10) number of migrating (A) or invading (B) ANV5 cells that had stable AXL shRNA expression (ANV5-AXL shRNA/E1) compared to control cells (ANV5-PLK01) that received a control vector. The experiments in this figure were repeated independently at least two times with similar results. C, Mean fold difference plot of expression of stem cell genes in AXL+ ANV5 cells relative to AXL− ANV5 cells. D, Mean fold difference plot of gene expression of metastases genes, in stable AXL knockdown stable cells relative to vector control cells.
MP470 inhibition of AXL decreases self-renewal of BCSCs and restores sensitivity to chemotherapy
Two important functions of BCSCs are their self-renewal capacity and resistance to chemotherapy. BCSCs are believed to utilize a variety of mechanisms to evade eradication by conventional therapy such as chemotherapy, hormonal therapy and radiation.27–30 To elucidate the role of AXL in BCSC self-renewal, ANV5 cells were treated with MP470 and then transferred to mammosphere media for anchorage independent growth. Treatment with AXL inhibitor impacted the ability of the cells to form mammospheres compared to control treated cells (Figure 7A). A quantification of the number of mammospheres formed is shown in Figure 7B. We next assessed the effect of AXL inhibition on chemoresistance of the stem cells. We compared ANV5 cells previously treated with DMSO or MP470 followed by treatment with Salinomycin and assessed cells proliferation and viability by analyzing the slope of their growth curves. Compared to control treated cells, MP470 treated cells showed increased sensitivity to chemotherapy (Figure 7C). Similarly, we determined the effect of Etoposide and Paclitaxel on control and AXL inhibited ANV5 cells and observed that AXL depleted cells were more sensitive to chemotherapy compared to control cells (Figure 7D–E). These results together demonstrate that AXL is not only important for BSCS self-renewal but also mediates the cells’ resistance to chemotherapy, and therefore properly targeting this pathway could significantly block BCSC function and arrest disease progression.
Figure 7.
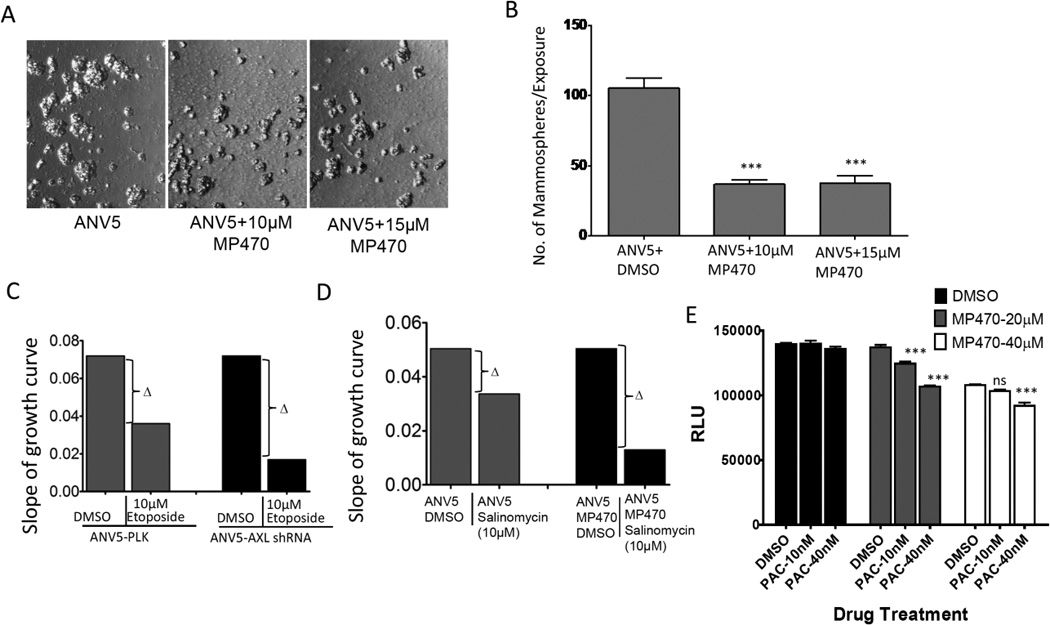
MP470 inhibition of AXL decreases self-renewal of BCSCs and restores sensitivity to chemotherapy. A, Photomicrograph of mammospheres generated from ANV5 cells treated with varying concentration of MP470 or only DMSO (ANV5). B, Bar graphs showing the mean (± SEM, N=8) number of the mammospheres formed by different treatments described above. C, Chemosensitivity graphs are shown for stable AXL-knockdown and control ANV5 cells were treated with Etoposide. Each bar is the slope of growth tracing as measured by impedance. Note that the change in slope is greater in the AXL-knockdown cells as compared to control ANV5-PLK cells. D, Chemosensitivity graphs of DMSO control or MP470-pretreated (20 µM) ANV5 cells subsequently exposed to Salinomycin. E, Shown are the mean (±SEM, n=6) relative luminescence units (RLU) of Control or MP470-pre-treated ANV5 cells subsequently treated with varying doses of paclitaxel. p-values were derived using one-way ANOVA within treatments. ***p<0.001; ns, not significant. The experiments in this figure were repeated independently at least three times with similar results.
Discussion
In spite of recent improvements in the breast cancer management, such as the use of targeted therapy which has led to improved patient survival and reduced mortality, recurrence of breast cancer due to resistance to conventional hormonal and chemotherapy remains a serious clinical problem. Recent studies have shown that BCSCs, characterized by low expression of CD24 and high expression of CD44 surface proteins, appear to mediate resistance to chemotherapy. Although neoplastic transformation of putative tissue stem cells could give rise to BCSCs, recent studies from several laboratories including ours suggest that induction of EMT can generate mesenchymal cells with stem cell-like properties.4, 5, 28–30 A review of the literature on EMT and BCSCs also show that EMT phenotype in breast cancer cell lines correlates with CD44+/CD24− phenotype.31 It therefore follows that identification of pathways and targets that are critical to EMT and regulate CSC function could lead to the development of potent targeted therapy to manage specific cancers.
The goal of this study was to delineate the role of the receptor tyrosine kinase AXL in EMT induction and regulation of BCSC function. Using AXL overexpression, RNAi mediated inactivation and receptor tyrosine kinase inhibitor MP470, we have shown that AXL is constitutively activated in BCSCs and induce EMT by regulating expression of EMT markers such as E- and N-Cadherin, Snail and Slug. We also demonstrated that downregulation of AXL using its inhibitor MP470 reverses EMT in mesenchymal MCF10A and ANV5 cells, reduces the tumorigenicity of BCSCs, downregulates NFκB pathway and restores sensitivity of BCSCs to chemotherapy. The results show that AXL expression is associated with stem cell marker genes, regulates metastases genes and is important for BCSC motility. AXL has been found to be overexpressed in a number of carcinomas and malignancies including ovary, brain, lung, colon, breast, kidney, melanoma and osteosarcoma.15, 17, 32–38 AXL expression also correlated with reduced overall survival and recurrence-free survival in pancreatic ductal adenocarcinomas. Its expression in primary breast cancers also independently predicted reduced overall survival with substantial increased expression in metastases lesions.18, 39 Gjerdrum and colleagues recently showed that AXL expression was upregulated during EMT, meaning that AXL is a downstream target of EMT.18 Vimentin, a marker of EMT, has been shown to regulate AXL expression and also mediate slug induced EMT.40 Our results extended these findings and identified a novel function of AXL as an upstream regulator of EMT induction. Furthermore, downregulation of AXL using a small molecule inhibitor MP470 reversed mesenchymal phenotype of BCSCs, suggesting a possible feedback loop mechanism of regulation of EMT. Although EMT inducers and AXL regulators such as Zeb1, Twist and Snail have shown association with poor prognosis of breast cancer, no correlation was found between AXL expression and E-cadherin in the study by Gjerdrum and colleagues despite both being downstream targets of EMT.18, 41, 42 It is however not clear whether a homogenous patient sample was used in that study which could affect the ability to detect any such association.
AXL has been shown to play an important role in invasion and cell survival in breast cancer, malignant melanoma, glioma, and liver cancers. Downregulation of AXL in breast cancer or pancreatic ductal carcinomas using siRNAs or Src/Abl inhibitor, bosutinib abolished Gas6-dependent AXL activation of Akt resulting in decreased cell invasion, motility, and survival of the breast and pancreatic cancers.17, 34, 39, 43, 44 A potent small-molecule AXL inhibitor R428 also blocked AXL functions in Akt phosphorylation, cell invasion and cancer metastases.45 AXL expression is also retained in metastases with strong correlation with breast cancer mortality, suggesting that the expression of the kinase could be directly linked to metastasis progression.18 Our results also show that AXL expression in BCSCs is associated with differential expression of metastases genes and is important for BCSC migration and invasion, which corroborates findings of similar roles of AXL in other cancers. We also established that downregulation of AXL using MP470 reduced the tumorigenicity of cancer cells and restored the sensitivity of BCSC to chemotherapy suggesting an important role of AXL in chemo-resistance, particularly in cancer stem cells. This is consistent with studies showing that inactivation of AXL using monoclonal antibodies and RNAi interference attenuated tumor growth, reduced cell survival and enhanced the effect of anti-cancer therapies.39, 46
In conclusion, we have shown that AXL is constitutively activated in BCSCs and induce EMT through direct regulation of expression of EMT markers such as E- and N-Cadherin, Snail and Slug. Furthermore, downregulation of AXL using its inhibitor MP470 reversed EMT in mesenchymal MCF10A and ANV5 cells, reduces the tumor growth, downregulates NFκB pathway and restores sensitivity of the BCSC to chemotherapy. We also demonstrate that AXL expression associated with stem cell gene, regulates metastases genes and is important for BCSC motility. These results demonstrate the potential of AXL as an important therapeutic target capable of targeting not only the bulk of epithelial cells of the tumor but also embedded CSCs responsible for resistance chemotherapy and recurrence. This also means that a combinatorial therapeutic approach to blocking EMT induction of epithelial cells and preventing CSC survival, chemoresistance, and tumorigenicity could be a better approach than strategies targeting the epithelial tumor cells or CSCs alone.
Materials and methods
Cell culture and reagents
ANV5 cells is a neu antigen-loss variants (ANV cells) with stem cell-like properties derived from mouse mammary carcinoma cell line (MMC), generated and established from a spontaneous tumor of a neu-tg mouse as previously described.4 The MMC, ANVs and their derivatives ETTM, MCTTE were generated and validated as previously described4, 21 and maintained in RPMI supplemented with 10% FBS, Glutamate, and antibiotics. HMLE, an immortalized human mammary epithelial cell line generated in Dr. Robert Weinberg’s laboratory were generous gift from Dr. Jing Yang (UCSD). All cells were immediately recovered and expanded in their respective media. MCF10A and HMLE cells were maintained in Dulbecco's modified Eagle's medium (DMEM)/F12 medium containing 5% horse serum, epidermal growth factor (EGF), insulin, hydrocortisone, and antibiotics, except that HMLE media included MEGM in 1:1 ratio to DMEM/F12. ATCC conducted authentication on the above cell lines through short tandem repeat profiling, karyotyping, and cytochrome c oxidase I testing. Test for bacterial and fungal contamination was carried out by ATCC by using current United States Pharmacopeia methods for viral testing adhering to United States Code of Federal Regulation (9 CFR 113.53) guidelines, mycoplasma testing via direct culture and Hoechst DNA staining and Limulus amoebocyte lysate assay to measure endotoxin values. HMLE cells were validated in Dr. Weinbergs’ laboratory. Prior to being used in experiments, all cell lines were treated with Plasmocin (InvivoGen) for two weeks to prevent mycoplasma contamination. Transient mesenchymal MCF10A cells were generated by treatment of MCF10A cells with 100ng/ml of TGFβ and 50ng/ml of TNFα for 7 days. TGFβ and TNFα were purchased from R&D System (St Paul, MN). AXL inhibitor, MP470, was purchased from Selleck Chemicals (Houston, TX).
RNAi mediated gene silencing
Bacterial stocks of shRNA clones were purchased from Invitrogen and Sigma-Aldrich. Vector DNA was extracted using HiSpeed Plasmid purification kit (Qiagen, Valencia, CA). 293TN cells were then transfected with shRNA vector clones mixed with viral package vectors pMD2 and psPAX2 using Lipofectamine 2000 transfection reagent (Invitrogen). After 48 hours, culture media containing viral particles was mixed with polybrene and centrifuged at 10,000 rpm to precipitate and concentrate the viral particles.
RNA Isolation, RT-PCR and qPCR
Total RNA was purified from ANV5 and MCF10A cells using RNeasy Plus kit (Qiagen, Valencia, CA). The quantity of the RNA and purity were determined using a NanoDrop ND-1000. AXL, Gas6 and GAPDH primers were designed with PrimerQuest and synthesized by IDT (Coralville, IA). Reverse transcription-PCR (RT-PCR) was performed using SuperScript One-Step RT-PCR with Platinum Taq (Invitrogen, Carlsbad, CA) using 500 ng of RNA in a Bio-Rad MyCycler. PCR samples were analyzed on 1.5% agarose gels and imaged on a Gel Doc XR (Bio-Rad, Hercules, CA). First strand cDNA for qPCR was synthesized using the RT2 First Strand cDNA Kit (Qiagen, Valencia, CA). Gene expression and signaling pathway analyses were done using RT2 Profiler PCR Array qPCR kit and detected with the RT2 SYBR Green qPCR Master Mix (SABioscience) according to the manufacturer's protocol and run on ABI 7900HT with standard 96 block (Applied Biosystems, Carlsbad, CA). Analysis of the gene expression arrays was performed using the manufacturer’s online analysis tool (http://www.sabiosciences.com/pcr/arrayanalysis.php) and normalized to housekeeping genes. Differential expression is measured as fold expression relative to the untreated cells.
Immunoblot analysis
Cell lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer and quantified using the Biorad BCA protein assay. Equal amounts of protein from each sample were resolved by SDS-PAGE gel, transferred onto PVDF membranes, blocked with 5% nonfat milk in TBST and incubated with primary antibody at room temperature for 4hrs or overnight at 4°C. After incubation with appropriate horseradish peroxidase-conjugated secondary antibodies in blocking buffer, protein expression was detected using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL). Primary polyclonal antibody to AXL, E-cadherin, N-Cadherin, β-catenin, Her2, Zeb1, Fibrinectin and β-actin-HRP, and secondary antibodies were purchased from Santa Cruz (Santa Cruz, CA). Monoclonal antibody to Akt, phospho-Akt(S473), Snail, Slug, MET, p-MET, Vimentin and PDGRα was purchased from Cell signaling (Cell signaling, Danvers, MA). Phopho-AXL was obtained from R&D Systems (R&D Systems, Minneapolis, MN). Anti–β-actin was used as loading control.
Mammosphere formation assay and cell imaging
For mammosphere formation, ANV5 and MP470-treated ANV5 cells were maintained in 4 mls DMEM/F12 media with 1:50 B27 (Invitrogen), 20 ng/mL EGF, 20 ng/mL, 10 µg/mL insulin, penicillin, streptomycin, and amphotericin B in 12-well plates at a density of 5,000 cells/mL as described previously.47 Additional 0.5mLs of media were added every 3 days for 15 days. The number of mammospheres formed were observed and counted under a Leica DC 200 microscope (Leica Microsystems, Bannockburn, IL). Images of spheroids and adherent cells were obtained with a Leica DC 200 microscope (Leica Microsystems) and Fujifilm FinePix 6800 Zoom camera (Fujifilm, Valhalla, NY).
Flow cytometry and cell sorting
Cell surface expression of AXL on ANV5 cells was determined by flow cytometry analysis. AXL-positive and negative ANV5 cells were sorted on a BD FACSVantage Cell Sorter (Becton Dickinson, Franklin Lakes, NJ). Cells were stained with primary antibodies at 4°C for 30 min, followed by secondary antibody staining for 30 min. The cells were then fixed with washing buffer containing 0.5% formaldehyde and run on a BD FACScan flow cytometer (Becton Dickinson). Flow data were analyzed using WinMDI ver 2.8 software (http://en.bio-soft.net/other/WinMDI.html). Secondary antibody FITC goat anti-rabbit IgG was purchased from Jackson ImmunoResearch Laboratories.
In vivo tumorigenicity
For in vivo tumor formation, female neu-tg mice on the FVB/N background were maintained as a colony and used in the experiment according to institutional animal care and use (IACUC) policy. About 100,000 of DMSO or MP470 treated ANV5 cells as well as stable control or AXL shRNA stable cells were injected subcutaneously into the mice and tumor size measured until the mice were sacrificed. Tumors were measured every other day with vernier calipers, and volumes were calculated as the product of length × width × height × 0.5236.
In vitro cell migration and invasion assay
Migration and invasion of control or AXL depleted ANV5 cells were assessed using transwells (24-well inserts; pore size, 8 µm; BD Biosciences). About 5 × 104 cells suspended in serum free medium were plated in the top chamber of the transwell and medium supplemented with serum was used as a chemoattractant in the lower chamber. The invasion assay was conducted as described for migration assay using 1.5 × 105 cells and matrigel-coated membrane (24-well insert, pore size, 8 µm; BD Biosciences). After 24 hrs of incubation, cells remaining on top of the membrane and those that did not migrate were removed using a cotton swab. Migrated cells on the lower surface of the membrane were stained with Hema 3 Stain (Fisher Scientific, Pittsburgh, PA), photographed, and counted.
Chemotherapy and cytotoxicity assays
Chemo-sensitivity assays were performed using the Xcelligence unit (Roche, Indianapolis, IN) as previously described21 or the CellTiter-Glo assay (Promega). DMSO or MP470 treated ANV5 cells were counted and seeded in xCelligence E-plates (Roche, Indianapolis, IN). The xCelligence plates measure impedance in real time generating a growth. Faster growth leads to steeper growth curve slopes. After 18 hrs, MP470 treated and control cells were then treated with 10µM salinomycin and the slopes of the growth curves calculated. Stable AXL knockdown ANV5 cells (ANV5-AXL shRNA) and control ANV5 cells (PLK-ANV5) growing in E-plates were also treated with 10µM and 20µM Etoposide and the growth of the cells recorded in real time followed by determination of slope. For CellTiter-Glo assay, ANV5 cells were treated with indicated concentrations of MP470 for 48hrs followed by paclitaxel treatment for another 48hrs. The number of metabolically active cells was determined by measuring the amount of ATP produced after cell lysis which was measured in relative luminescence units (RLU). The drugs Etoposide and Salinomycin were purchased from LC Laboratories (Woburn, MA).
NFκB Luciferase reporter Assay
Lentiviral stocks of NFκB reporter vectors (Qiagen, Valencia, CA) were transduced into ANV5 cells for 24 hrs and then treated with varying doses (5µM, 10µm and 15µM) of MP470 or treated with AXL antibody (1 to 6ug/ml). TNFα was added as a positive control. After 6 hrs of incubation, equal volume (100µl) of Bright-Glow luciferase substrate (Promega, Madison, WI) was added to each well and analyzed on a Victor 3 fluorometer. NFκB pathway activation was determined by the luciferase activity measured in relative luciferase units (RLU).
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 4.00 for Windows, GraphPad Software, San Diego California USA, (www.graphpad.com). Two-tailed Student's t-test, the Mann-Whitney Test, One-way analysis of variance or the Two-way ANOVA test was performed followed by the Bonferroni post test to determine statistically significant difference. p < 0.05 was considered as significant.
Supplementary Material
Acknowledgments
This work was supported by a generous gift from Martha and Bruce Atwater (KLK); Howard Temin Award, K01-CA100764 (KLK); R01-CA122086 (DCR); and the Mayo Clinic Breast Cancer Specialized Program of Research Excellence Award, P50-CA116201 (JNI). This publication was also supported by NIH/NCRR CTSA Grant Number UL1 RR024150. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The authors acknowledge the strong support of the Mayo Clinic Comprehensive Cancer Center for providing access to core facilities.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Han JS, Crowe DL. Tumor initiating cancer stem cells from human breast cancer cell lines. Int J Oncol. 2009;34:1449–1453. [PubMed] [Google Scholar]
- 2.Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells--old concepts, new insights. Cell Death Differ. 2008;15:947–958. doi: 10.1038/cdd.2008.20. [DOI] [PubMed] [Google Scholar]
- 3.Vicente-Duenas C, Cobaleda C, Perez-Losada J, Sanchez-Garcia I. The evolution of cancer modeling: the shadow of stem cells. Dis Model Mech. 2010;3:149–155. doi: 10.1242/dmm.002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santisteban M, et al. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69:2887–2895. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radisky DC. Epithelial-mesenchymal transition. J Cell Sci. 2005;118:4325–4326. doi: 10.1242/jcs.02552. [DOI] [PubMed] [Google Scholar]
- 7.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 10.Cowin P, Welch DR. Breast cancer progression: controversies and consensus in the molecular mechanisms of metastasis and EMT. J Mammary Gland Biol Neoplasia. 2007;12:99–102. doi: 10.1007/s10911-007-9041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Bryan JP, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11:5016–5031. doi: 10.1128/mcb.11.10.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bose R, et al. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc Natl Acad Sci U S A. 2006;103:9773–9778. doi: 10.1073/pnas.0603948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hafizi S, Dahlback B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. Febs J. 2006;273:5231–5244. doi: 10.1111/j.1742-4658.2006.05529.x. [DOI] [PubMed] [Google Scholar]
- 15.Hutterer M, et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin Cancer Res. 2008;14:130–138. doi: 10.1158/1078-0432.CCR-07-0862. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, et al. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene. 2009;28:3442–3455. doi: 10.1038/onc.2009.212. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YX, et al. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008;68:1905–1915. doi: 10.1158/0008-5472.CAN-07-2661. [DOI] [PubMed] [Google Scholar]
- 18.Gjerdrum C, et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107:1124–1129. doi: 10.1073/pnas.0909333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69:6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 20.Gupta PB, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGF{beta}/TNF{alpha}-Mediated Epithelial-Mesenchymal Transition Generates Breast Cancer Stem Cells with a Claudin-Low Phenotype. Cancer Res. 2011;71:4707–4719. doi: 10.1158/0008-5472.CAN-10-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi W, et al. MP470, a novel receptor tyrosine kinase inhibitor, in combination with Erlotinib inhibits the HER family/PI3K/Akt pathway and tumor growth in prostate cancer. BMC Cancer. 2009;9:142. doi: 10.1186/1471-2407-9-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welsh JW, Mahadevan D, Ellsworth R, Cooke L, Bearss D, Stea B. The c-Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. Radiat Oncol. 2009;4:69. doi: 10.1186/1748-717X-4-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng F, Liu L, Chin PC, D'Mello SR. Akt is a downstream target of NF-kappa B. J Biol Chem. 2002;277:29674–29680. doi: 10.1074/jbc.M112464200. [DOI] [PubMed] [Google Scholar]
- 25.Tai KY, Shieh YS, Lee CS, Shiah SG, Wu CW. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene. 2008;27:4044–4055. doi: 10.1038/onc.2008.57. [DOI] [PubMed] [Google Scholar]
- 26.Chen W, Li Z, Bai L, Lin Y. NF-kappaB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front Biosci. 2011;16:1172–1185. doi: 10.2741/3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shipitsin M, Polyak K. The cancer stem cell hypothesis: in search of definitions, markers, and relevance. Lab Invest. 2008;88:459–463. doi: 10.1038/labinvest.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wicha MS. Cancer stem cells and metastasis: lethal seeds. Clin Cancer Res. 2006;12:5606–5607. doi: 10.1158/1078-0432.CCR-06-1537. [DOI] [PubMed] [Google Scholar]
- 29.Gangemi R, et al. Cancer stem cells: a new paradigm for understanding tumor growth and progression and drug resistance. Curr Med Chem. 2009;16:1688–1703. doi: 10.2174/092986709788186147. [DOI] [PubMed] [Google Scholar]
- 30.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 31.Blick T, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/−) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:235–252. doi: 10.1007/s10911-010-9175-z. [DOI] [PubMed] [Google Scholar]
- 32.Rankin EB, et al. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010;70:7570–7579. doi: 10.1158/0008-5472.CAN-10-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakano T, et al. Biological properties and gene expression associated with metastatic potential of human osteosarcoma. Clin Exp Metastasis. 2003;20:665–674. doi: 10.1023/a:1027355610603. [DOI] [PubMed] [Google Scholar]
- 34.Vajkoczy P, et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A. 2006;103:5799–5804. doi: 10.1073/pnas.0510923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shieh YS, et al. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia. 2005;7:1058–1064. doi: 10.1593/neo.05640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung BI, Malkowicz SB, Nguyen TB, Libertino JA, McGarvey TW. Expression of the proto-oncogene Axl in renal cell carcinoma. DNA Cell Biol. 2003;22:533–540. doi: 10.1089/10445490360708946. [DOI] [PubMed] [Google Scholar]
- 37.van Ginkel PR, et al. Expression of the receptor tyrosine kinase Axl promotes ocular melanoma cell survival. Cancer Res. 2004;64:128–134. doi: 10.1158/0008-5472.can-03-0245. [DOI] [PubMed] [Google Scholar]
- 38.Craven RJ, et al. Receptor tyrosine kinases expressed in metastatic colon cancer. Int J Cancer. 1995;60:791–797. doi: 10.1002/ijc.2910600611. [DOI] [PubMed] [Google Scholar]
- 39.Song X, et al. Overexpression of receptor tyrosine kinase Axl promotes tumor cell invasion and survival in pancreatic ductal adenocarcinoma. Cancer. 117:734–743. doi: 10.1002/cncr.25483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vuoriluoto K, et al. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30:1436–1448. doi: 10.1038/onc.2010.509. [DOI] [PubMed] [Google Scholar]
- 41.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 42.Yang J, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Koorstra JB, et al. The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol Ther. 2009;8:618–626. doi: 10.4161/cbt.8.7.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee WP, Liao Y, Robinson D, Kung HJ, Liu ET, Hung MC. Axl-gas6 interaction counteracts E1A-mediated cell growth suppression and proapoptotic activity. Mol Cell Biol. 1999;19:8075–8082. doi: 10.1128/mcb.19.12.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holland SJ, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70:1544–1554. doi: 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]
- 46.Keating AK, et al. Inhibition of Mer and Axl receptor tyrosine kinases in astrocytoma cells leads to increased apoptosis and improved chemosensitivity. Mol Cancer Ther. 2010;9:1298–1307. doi: 10.1158/1535-7163.MCT-09-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grimshaw MJ, et al. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res. 2008;10:R52. doi: 10.1186/bcr2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.