

Abstract
Insulin controls nutrient and metabolic homeostasis via the IRS–PI3K–AKT signaling cascade that targets FOXO1 and mTOR. Mitochondria, as the prime metabolic platform, malfunction during insulin resistance in metabolic diseases. However, the molecular link between insulin resistance and mitochondrial dysfunction remains undefined. Here we review recent studies on insulin action and the mechanistic association with mitochondrial metabolism. These studies suggest that insulin signaling underpins mitochondrial electron transport chain integrity and activity by suppressing FOXO1/HMOX1 and maintaining the NAD+/NADH ratio, the mediator of the SIRT1/PGC1α pathway for mitochondrial biogenesis and function. Mitochondria generate moderately reactive oxygen species (ROS) and enhance insulin sensitivity upon redox regulation of protein tyrosine phosphatase and insulin receptor. However, chronic exposure to high ROS levels could alter mitochondrial function and thereby cause insulin resistance.
Insulin signaling and insulin receptor substrates
The insulin signaling system coordinates systemic growth and development with peripheral and central nutrient homeostasis, fertility and lifespan [1,2]. At the molecular level, insulin regulates many pathways including the stimulation of protein synthesis (in muscle and liver), lipid synthesis and storage (liver and adipose tissue), glycolysis and glucose storage (muscle and liver), and the inhibition of ketogenesis and gluconeogenesis (liver) [3].
The insulin receptor substrate (IRS) proteins integrate the activated membrane-bound insulin receptor (IR) kinases to downstream adapter proteins and enzymes. Four IRS-proteins have been identified in rodents, but only three (IRS1, IRS2 and IRS4) are expressed in human: IRS4 is largely restricted to the hypothalamus and thymus [4,5]. IRS1 and IRS2 are expressed in brain, muscle, heart, adipocyte, liver, kidney, ovary and mammary gland where they contribute to a broad array of physiologic functions including pancreatic β-cell growth and function, central nutrient sensing, neurodegeneration, cancer progression and lifespan [6]. Although the role of each of these protein phosphorylation sites merits attention, work with transgenic mice reveals that most if not all insulin/IGF responses – especially those that are associated with somatic growth, carbohydrate and lipid metabolism – are initiated through IRS1 and IRS2 [7].
IRS-proteins are targeted by the specific binding of their N-terminal phosphotyrosine binding (PTB) domain to a phosphorylated NPXY-motif in the cytoplasmic juxtamembrane region of the insulin or IGF1 receptors (Figure 1). The pleckstrin homology (PH) domain of the IRS-proteins is structurally similar but functionally distinct from the PTB-domain [8]. Deletion of the PH domain changes the signaling potential of IRS1 and IRS2 [9]. Although the PH domain can be interchanged among the IRS-proteins without noticeable loss of bioactivity, substitutions with heterologous PH-domains – βARK (beta adrenergic receptor kinase), phospholipase Cγ, or spectrin – inhibit IRS1 function [10]. Several proteins can interact with the PH domains of IRS1 and/or IRS2, including the Lon protease, myeloblast protein, and nucleolin [10]. Each protein contains an acidic motif that interacts with the PH domain of IRS-2, but only the acidic motif in nucleolin binds to IRS1: synthetic peptides based on the acidic motif in Lon protease and myeloblast protein inhibit the binding of nucleolin to the PH domain of IRS2 but not to the PH domain of IRS1 [10].
Figure 1.
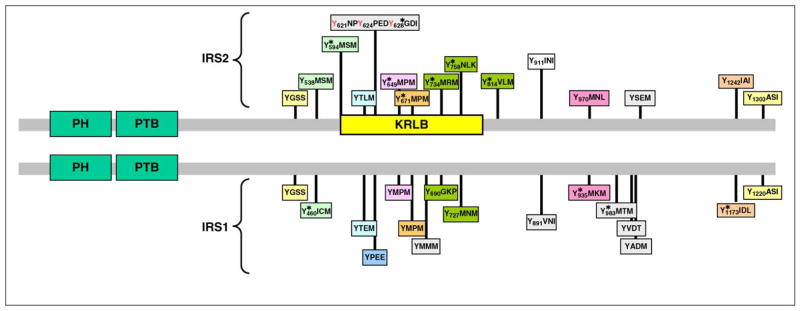
Comparison of IRS1 and IRS2 protein sequences, including the relative location of the amino-terminal pleckstrin homology (PH) and phosphotyrosine binding (PTB) domains, and numerous known (*, phosphorylation sites revealed by MS/MS) or expected tyrosine phosphorylation sites. The amino acid sequences surrounding tyrosine sites are shown, and motifs conserved between IRS1 and IRS2 are coded with a similar background color. The kinase regulatory loop-binding (KRLB) domain in IRS2 is indicated by a yellow box, which includes the tyrosine residue that binds in the ATP binding pocket (Y621).
The C termini of IRS1 and IRS2 are poorly conserved, but they both contain over twenty recognizable tyrosine phosphorylation motifs in similar positions that can bind signaling molecules (Figure 1). The activated IR kinase phosphorylates tyrosine residues within specific amino acid motifs, including the YMXM, YVNI and YIDL motifs [11–14]. IRS2 utilizes an additional motif located between amino acid residues 591 and 786 – especially Tyr624 and Tyr628 – to interact with the activated insulin receptor [15,16]. This binding region in IRS2 was originally called the kinase regulatory-loop binding (KRLB) domain because tris-phosphorylation of the insulin receptor activation (A)-loop was required to observe the interaction [15]. Because autophosphorylation moves the A-loop out of the catalytic site, the functional part of the KRLB-domain –residues 620–634 in murine IRS2 – can fit into the catalytic site [17]. This interaction aligns Tyr628 of IRS2 for phosphorylation; however, it also inserts Tyr621 into the ATP binding pocket, and this might attenuate signaling by blocking ATP access to the catalytic site. By contrast, this interaction could promote signaling by opening the catalytic site before tris-autophosphorylation. For unknown reasons, the KRLB-motif does not bind to the IGF1R, and this might explain signaling differences between IR and IGF1R, as well as the receptor hybrids [17].
The IRS→PI3K→AKT→mTOR cascade
One of the best-studied and most important signaling cascades activated by insulin involves the production of phosphatidylinositol lipids by the class 1A phosphotidylinositide 3-kinase (PI3K). The type 1A PI3K is a dimer composed of a catalytic subunit – either p110α, p110β or p110δ – and one of five regulatory subunit isoforms encoded by three different genes – Pik3r1, Pik3r2, and Pik3r3 (Figure 2) [18–20]. PI3K is activated when its SH2-domains in the regulatory subunit (p85) are occupied by phosphorylated YXXM-motifs in IRS-proteins [21]. The p85α regulatory protein in PI3K binds directly to and enhances the lipid phosphatase PTEN activity in addition to promoting the stabilization and localization of p110-PI3K activity, revealing its dual regulatory role in maintaining the balance of PI3K/PTEN signaling [22]. Partial loss of the regulatory subunits increases insulin sensitivity, and this appears to be related to diminished negative feedback to the IRS-proteins [23]. By contrast, complete disruption of hepatic Pik3r1 and Pik3r2 markedly reduces insulin-stimulated PI3K activity and PIP3 accumulation – at least in part by destabilizing the catalytic subunits – and this dysregulates glucose and lipid homeostasis, hepatic size and function [24]. Thus, PI3K activity is crucial for insulin’s actions.
Figure 2.

Insulin and insulin-like signaling cascade. Two main branches propagate signals generated via the IRS-proteins: PI3K→PDK1→AKT and GRB2/SOS→RAS kinase cascades. Activation of the receptors for insulin and IGF-1 results in tyrosine phosphorylation of the IRS-proteins, which bind PI3K and GRB2/SOS. The GRB2/SOS complex promotes GDP/GTP exchange on p21ras, which activates the RAS→RAF→MEK→ERK1/2 cascade. Activated ERK stimulates transcriptional activity by direct phosphorylation of elk1 and by indirect phosphorylation of fos through p90rsk. The activation of PI3K by IRS-protein recruitment produces PI3,4P2 and PI3,4,5P3 (antagonized by the action of PTEN or SHIP2), which recruit PDK1 and AKT to the plasma membrane. AKT is activated via phosphorylation at T308 by PDK1 and at S473 by mTOR in complex with rictor. The mTOR kinase is activated by RhebGTP, which accumulates upon inhibition of the GAP activity of the TSC1–TSC2 complex following PKB-mediated phosphorylation of TSC2. mTOR is also activated by AKT-mediated PRAS40 phosphorylation. The S6K is primed through mTOR-mediated phosphorylation for activation by PDK1. AKT phosphorylates many cellular proteins, and this inactivates PGC1α, p21kip, GSK3β, BAD and AS160, and activates PDE3b and eNOS. mTOR also promote cleavage and activation of SREBP1c (Clv’d SREBP1C), which stimulates the expression of genes needed for lipid synthesis. AKT-mediated phosphorylation of forkhead proteins, including FOXO1, results in their sequestration in the cytoplasm, which inhibits their influence upon transcriptional activity. Insulin stimulates protein synthesis by altering the intrinsic activity or binding properties of key translation initiation and elongation factors (eIFs and eEFs, respectively) as well as crucial ribosomal proteins. Components of the translation machinery that are targets of insulin regulation include eIF2B, eIF4E, eEF1, eEF2 and the S6 ribosomal protein [117]. TNFα activates JNK which can phosphorylate IRS1, inhibiting its interaction with the insulin receptor and its subsequent tyrosine phosphorylation. IRS2 expression is promoted by nuclear FOXO, which increases IRS2 concentration in the fasted liver.
PI-3,4,5-P3 produced by PI3K recruits several Ser/Thr kinases to the plasma membrane, including PDK1 (3′-phosphoinosite-dependent protein kinase-1) and AKT, where AKT is activated when its Thr308 is phosphorylated by PDK1 (Figure 2). AKT can phosphorylate several substrates relevant to insulin-like signaling: GSK3α/β (blocks inhibition of glycogen synthesis), AS160 (promotes GLUT4 translocation), the BAD·–BCL2 heterodimer (inhibits apoptosis), the FOXO (forkhead box O) transcription factors (regulate gene expression in liver, β cells and the hypothalamus), p21CIP1 and p27KIP1 (block cell cycle inhibition), eNOS (stimulates NO synthesis and vasodilatation), PDE3b (hydrolyzes cAMP), and TORC1. Of the three AKT isoforms (AKT1, AKT2 and AKT3), AKT2 was found to participate in metabolic regulation [18,25–28]. Thus, AKT below refers to AKT2 unless specified elsewhere.
To integrate cell growth and metabolism the TOR signaling complexes (TORC1 and TORC2) sense growth factor signals, energy status, oxygen availability and amino acid concentrations [29]. TORC1 is composed of the mammalian target of rapamycin (mTOR), mLST8, raptor and deptor, and the TORC1 complex is coupled to insulin signaling by AKT-mediated inhibition of TSC2 – a GTPase that inactivates RHEB (RAS homolog enriched in brain) [30,31]. AKT phosphorylates TSC2 and inhibits its GTPase to allow RHEB to accumulate in the GTP-bound form, and this activates mTORC1. FKBP38 (FK506 binding protein 8) also binds to mTORC1 and inhibits mTOR until RHEB–GTP binds to FKBP38 to reverse inhibition of the kinase [32–34]. This complex regulatory cascade is augmented by other pathways including the direct inhibition of the mTOR catalytic site by PRAS40 (the proline-rich AKT substrate of 40 kDa) until AKT-mediated phosphorylation of PRAS40 reverses the inhibition [18]. The mTORC2 complex is also composed of mTOR and mLST8 but, instead of raptor, this rapamycin-insensitive complex contains rictor and mSIN1 and is mainly regulated by nutrients [29]. The downstream effectors of mTORC1 – p70S6K and 4E-BP1 – control protein synthesis and cell growth (Figure 2) [18,35,36]. Disruption of the S6k1 gene in mice causes glucose intolerance owing to reduced size of pancreatic islet β cells [37].
Recent work suggests that TORC1 also plays an important role in lipid biosynthesis by promoting the cleavage and activation of SREBP1 [30]. Cleaved SREBP1 is a transcription factor that promotes the expression of diverse genes with important roles in lipid synthesis, including FASN (fatty acid synthase), GPAT (glycerol-3-phosphate acyltransferases), ACLY (ATP citrate lyase), ACC (acetyl-CoA carboxylase), SCD1 (stearoyl-CoA desaturase 1), and GK (glucokinase) (Figure 2) [30]. PI3K and AKT were found to be required for gluconeogenesis and lipogenesis, but mTORC1 was only required for lipogenesis via inducing SREBP-1c activity [38]. This established mTORC1 as an essential component at the point of divergence in the insulin signaling pathway that leads to selective insulin resistance – where insulin fails to block glucose production (gluconeogenesis) but continues to promote fatty acid synthesis (lipogenesis). TORC1 also inhibits insulin signaling by feedback inhibition of IRS1 through TOR and p70S6K-mediated serine phosphorylation [39]. Moreover, hepatic SREBP-1c expression inhibits TFE3/FOXO-mediated IRS2 expression, and this could be explained by competition between overlapping binding sites for SREBPs (SRE) and FOXO1 (IRE) [40]. Upregulation of IRS2 expression by TFE3/FOXO and downregulation by SREBP-1c parallels the switch from glycogenolysis and gluconeogenesis during fasting to lipogenesis following feeding. An imbalance in this reciprocal regulation could ultimately contribute to the pathophysiological effects of overnutrition, leading to the development of the metabolic syndrome and diabetes. More work is needed to establish whether this dysregulation contributes significantly to the pathophysiology of type 2 diabetes.
The IRS→AKT→FOXO cascade and hepatic lipid homeostasis
AKT phosphorylates the FOXO transcription factors –FOXO1, FOXO3 and FOXO4 – which control the expression of hundreds of genes, including several that mediate gluconeogenesis, lipid metabolism and stress resistance [41–44]. Earlier studies suggest IRS1 and IRS2 play distinct roles in glucose and lipid metabolism. IRS2 apparently controls gluconeogenesis by inhibiting FOXO1 and CREB-binding protein (CBP), whereas IRS1 controls lipid oxidation alongside IRS2 by regulating FOXA2 [45]. However, recent evidence from genetic mouse models show that both IRS1 and IRS2 are strong inhibitors of FOXO1, through AKT-mediated phosphorylation [42,46]. Moreover, the IRS1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis, because nutrient-sensitive transcripts are normally expressed in the liver of IRS2 knockout mice (LKO2-mice) but are significantly dysregulated in liver lacking IRS1 (LKO1-mice) [46]. High-fat diet (HFD) treatment decreases tyrosine phosphorylation of IRS2 in LKO1-mice but increases the postprandial tyrosine phosphorylation of IRS1 in LKO2-mice as hyperglycemia develops [47]. Moreover, a set of key gluconeogenic and lipogenic genes is markedly dysregulated in IRS1 and IRS2 double knockout (DKO-mice), which show severe glucose intolerance and impaired lipid metabolism [42,46]. Deletion of hepatic IRS1 and IRS2, and obesity-induced hepatic insulin resistance, hyperactivate FOXO1 that in turn induces HMOX1, the enzyme that consumes heme and disrupts the integrity of the mitochondrial electron transport chain (ETC) [41]. As a result, mitochondrial oxidative and phosphorylation activities are impaired, such as ATP generation and fatty acid oxidation [41]. Therefore, the IRS→AKT→FOXO cascade is required for nutrient and metabolic homeostasis in the liver.
Insulin resistance is associated with mitochondrial dysfunction
The characteristic of insulin resistance is the failure of insulin to suppress hepatic glucose production or stimulate glucose uptake by peripheral tissues, and this in turn causes hyperglycemia, hyperinsulinemia and dyslipidemia [1,2,7]. The dysregulation of glucose and lipid metabolism can induce a cohort of systemic disorders, including obesity, cardiovascular disease and hypertension, infertility, neurodegeneration, and type 2 diabetes when pancreatic β cells fail to secrete sufficient insulin to compensate for peripheral insulin resistance [48].
Systemic glucose and lipid metabolism converge in mitochondria that generate the majority of cellular energy (ATP) by coupling the tricarboxylic acid cycle (TCA) cycle with oxidative phosphorylation (OXPHOS) (Figure 3) [41,49–51]. Acetyl-CoA generated from glycolysis (glucose) and fatty acid β-oxidation (lipid) enters the TCA cycle in the mitochondrial matrix, in which the substrates are oxidized with the formation of CO2, NADH and FADH2. The electrons from NADH and FADH2 are taken up by complexes I and II, respectively, and passed to complex III and IV through ubiquinone (Q) and cytochrome c (C) (Figure 3). At complex IV, molecular oxygen accepts the electrons and is then converted into water. During this redox process, complexes I, II and IV pump protons from the matrix into the mitochondrial intermembrane space (IMS), and this generates an electrochemical gradient (membrane potential) and drives ATP generation through complex V (Figure 3). Mitochondrial metabolism is responsible for the major energy supply to vital cell functions including the maintenance of transmembrane ion gradients, protein synthesis, and vesicular transport [50]. Particularly, ATP and other mitochondrial factors accomplish the coupling of glucose metabolism to insulin secretion in the pancreatic β cell [50].
Figure 3.
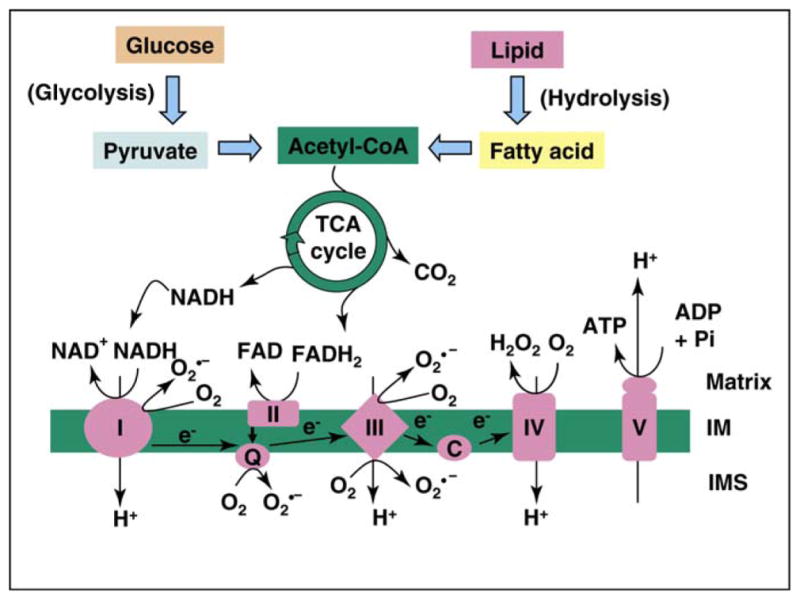
Mitochondria integrate glucose and lipid metabolism for energy generation. Glycolysis (glucose metabolism) and fatty acid oxidation (from lipid) generate acetyl-CoA, the substrate for tricarboxylic acid (TCA) cycle that oxidize the substrates into carbon dioxide and reducing products NADH and FADH2. NADH and FADH2 donate electrons to complexes I and II, respectively, of the respiration chain that generate an electrochemical gradient (membrane potential) by pumping protons in matrix across the inner membrane (IM) into the intermembrane space (IMS). The electrochemical gradient drives ATP generation at complex V and pumps protons back into the matrix. Moderately reactive oxygen species (such as O2•−) can be generated as a second messenger under physiological conditions, but a significant ROS burst can occur during mitochondrial hyperpolarization.
Mitochondrial function is impaired during insulin resistance that progresses to metabolic disease [52–54]. The muscles of patients with type 2 diabetes contain fewer mitochondria than those of age-matched insulin-sensitive individuals [55]. Nuclear magnetic resonance spectroscopy studies on individuals with insulin resistance showed marked reduction of mitochondrial oxidative and phosphorylation activity in their muscle and liver tissues [52,53]. Moreover, mitochondrial function is impaired in subjects with age-associated insulin resistance, indicated by reduced insulin-stimulated muscle glucose uptake and/or metabolism and an approximately 40% reduction in mitochondrial OXPHOS activity [56]. In contrast to insulin stimulation of mitochondrial protein synthesis and oxidative capacity in healthy subjects [57,58], sustained insulin infusion failed to promote the expression of mRNA transcripts encoding key mitochondrial proteins in the skeletal muscle of diabetic patients [57]. In line with dysregulated OXPHOS gene expression in the insulin-resistant liver [59], insulin signaling was recently shown to underpin ETC integrity and activity by suppressing FOXO1/HMOX1 and maintaining the NAD+/NADH ratio, the mediator of SIRT1/PGC1α pathway for mitochondrial biogenesis and function [41,49]. These findings strongly suggest that insulin signaling is required for normal mitochondrial function in metabolism [41,49,60–62].
A noteworthy observation in insulin-resistant elderly individuals is the significantly higher levels of triglycerides in both muscle and liver [56]. These data imply that insulin resistance might arise from defects in mitochondrial fatty acid oxidation, and that the deficiency increases intracellular fatty acid metabolites (fatty-acyl-CoA and diacylglyerol) that can disrupt insulin signaling through activation of stress-sensitive kinases such as PKC, IKK and JNK. These stress-sensitive kinases can phosphorylate and inactivate IRS1 and IRS2 [52]. However, a growing body of evidence teaches against this concept [54,63–64]. In particular, endurance-trained athletes have elevated levels of intramuscular triglycerides but they are highly insulin sensitive [65]. Although aerobic exercise can substantially increase mitochondrial capacity, it fails to improve insulin sensitivity [65–66]. In fact, excess lipid availability is found to increase mitochondrial fatty acid oxidative capacity in the muscle of rodents [67], and a HFD causes insulin resistance despite an increase in muscle mitochondria [68].
The controversy can be attributed to the complexity and difference of experimental conditions used in these studies. First, during nutrient deprivation (such as in fasting and endurance exercise) or in diabetic conditions, a metabolic switch from carbonate to lipid occurs in skeletal muscle [63,69]. Thus, this could possibly explain why increased levels of intramuscular triglycerides and fatty acid metabolites are observed in diabetic patients and endurance-trained athletes [52,56,65]. Secondly, physiological adaptation of mitochondria to energy demand and fuel influx must be taken into consideration in interpreting these data. The increased energy demand during aerobic exercise and an elevated fuel influx due to HFD could both induce an adaptive increase in mitochondrial biogenesis and function in muscle so that the nutrients are efficiently combusted for energy generation [67–68]. Third, short-term and chronic insults could produce different results [70–71]. Although HFD could increase mitochondrial number and activity due to a prompt adaptive response, oversupply of fuel can over-ride mitochondrial compensation. In this case, lipid accumulation and insulin resistance might not be due to mitochondrial dysfunction, but instead to the imbalance of fuel influx and consumption capacity of healthy mitochondria [67–68]. However, a chronic increase in fuel influx can induce mitochondrial hyperpolarization that causes ROS overproduction and mitochondrial damage (Figure 3) [71,72]. Regardless, it is suggested that mitochondrial abnormalities do not precede the onset of insulin resistance [71]. Together, the cause–effect relationship between mitochondrial dysfunction and insulin resistance requires further evidence.
Insulin signaling regulates mitochondrial metabolism
Given that insulin signaling is required for mitochondrial DNA and protein synthesis and potently stimulates mitochondrial oxidative capacity and ATP production [57,72], we reason that impaired insulin action can dysregulate mitochondrial function. This concept has been increasingly corroborated in different tissues. In β cells, the mitochondrion forms a tethering complex with glucokinase (GK) and the pro-apoptotic protein, BAD(S) [60]. However, mitochondria in β cells from patients with type 2 diabetes exhibited attenuated function and reduced BAD(S), GK and protein kinase A in the complex [60]. In line with this, β-cell-specific insulin receptor knockout (betaIRKO) mice show a similar phenotype, and re-expression of insulin receptors in betaIRKO cells partially restored the stoichiometry of the complex and mitochondrial function, suggesting that insulin signaling regulates mitochondrial function in β cells [60].
In cardiac muscle, activation of PI3K increases myocardial fatty acid oxidation capacity, whereas impaired PI3K signaling leads to cardiac mitochondrial dysfunction and prevents mitochondrial adaptations in response to physiological hypertrophic stimuli [62]. In mice with cardiomyocyte-specific deletion of IRS1 (CIRS1KO), IRS2 (CIRS2KO), or of IRS1 + IRS2 (CIRS12KO), there was a striking decrease in ADP-stimulated mitochondrial oxygen consumption and ATP synthesis, concomitant with a coordinate downregulation of OXPHOS genes. In line with this, deletion of myocardial insulin receptor (IR) and IGF1 receptor (IGF1R), whose signaling cascades converge at IRS proteins, resulted in downregulation of genes of the ETC and of mitochondrial fatty acid β-oxidation in the heart [73]. These findings reveal a crucial role for IRS-mediated signaling in the regulation of mitochondrial gene and protein expression, and for mitochondrial function in cardiomyocytes [61,73].
Mitochondrial biogenesis is controlled by the transcription co-activator PGC1α (peroxisome proliferator-activated receptor γ coactivator-1α) [74]. The AKT→FOXO1 cascade was shown to suppress PGC1α expression [75], and mitochondrial production and activity in liver and skeletal muscle (gastrocnemius) were increased in mice with insulin deficiency or resistance [76,77]. By contrast, however, PGC1α expression and PGC1α-responsive genes involved in OXPHOS are coordinately downregulated in skeletal muscle of subjects with diabetes or insulin resistance [78,79]. Moreover, mitochondrial production and activity were dampened regardless of increased PGC1α protein expression in insulin-resistant liver [41,49]. This controversy reflects the complexity of the molecular link between insulin signaling and mitochondrial metabolism. It should be noted, however, that different experiment models and conditions were used in those studies and could account for the varying results, especially when studies were carried out at different stages of the pathogenic progression of diabetes or insulin resistance [70,71]. In addition, PGC1α protein undergoes post-translational modification, such as reversible acetylation/deacetylation that suppresses/promotes its activity [41,80–83]. Thus, the post-translational regulation of PGC1α must be considered in addition to its total protein/gene expression level.
Using hepatic IRS1/IRS2 double knockout (DKO) mice, a possible molecular mechanism by which insulin regulates mitochondrial function in the liver was recently established [41]. The DKO mice develop insulin resistance and systemic hyperglycemia due to a substantial blockade of insulin signaling [42,46]. Hepatic insulin resistance hyperactivates hepatic FOXO1, which increases the expression of hundreds of genes including heme oxygenase 1 (HMOX1), the enzyme that breaks down heme into biliverdin (BV), Fe(III) and CO2 (Figure 4) [41,49]. This causes defects in the mitochondrial ETC by depleting heme, because the latter is the essential cofactor that facilitates electron transport and ensures the expression, stability and function of ETC components [84]. Consequently, NADH oxidation is impaired and the NAD+/NADH ratio decreases, which blunts the SIRT1→PGC1α pathway of mitochondrial biogenesis (Figure 4) [41,49,80–83]. The ETC defects can impair fatty acid oxidation rate due to the reduced concentration of NAD+ – the essential cofactor required for acyl-CoA dehydrogenase [85]. Mild lipid accumulation is present in the DKO liver, presumably because of the impaired fatty acid oxidation and reduced mitochondrial number [41,49]. Ablation of FOXO1 (gene knockout) or HMOX1 (siRNA knockdown) restores the SIRT1→PGC1α pathway for mitochondrial biogenesis, and rescues mitochondrial function (ETC activity, NAD+/NADH ratio and fatty acid oxidation rate), with normalization of hepatic triglyceride concentrations [41,46,49]. This mechanism can explain how failure of insulin action might impair mitochondrial function through the redox node composed of HMOX1 and the redox couple NAD+/NADH.
Figure 4.

Mechanistic association of insulin signaling with mitochondrial function. Insulin elicits the IR→IRS→PI3K→AKT signaling cascade and inhibits the transcriptional factor FOXO1 under normal conditions. During insulin resistance, including genetic deletion of IRS1 and IRS2, or physiological challenge of obesity, FOXO1 is hyperactivated and induces HMOX1. HMOX1 oxidizes heme to biliverdin (BV) and free Fe3+. Because heme is essential for the function and stability of electron transport proteins, insulin resistance impairs the ETC activity that is essential for NADH oxidation. Consequently, NAD+ levels decrease and the NAD+/NADH ratio increases, and this can inhibit the activity of the NAD+-dependent deacetylase SIRT1. Therefore, mitochondrial function and biogenesis are impaired under insulin-resistant conditions owing to the relative inactivity of SIRT1. Moreover, mitochondria can generate ROS (e.g., O2•− and H2O2) as a second messenger to regulate IR-mediated signaling cascade. ROS functions by oxidizing the β chain of the insulin receptor to facilitate its autophosphorylation (activation) or through oxidative modification of protein tyrosine phosphatases, especially PTP1B and PTEN, which leads to hyperphosphorylation of the insulin receptor and IRS1/2, and increased activity of the PI 3-kinase. Suppression of mitochondrial ROS causes insulin resistance, whereas knockout of the ROS-scavenger enzyme improves insulin responsiveness [104–105,118].
Mitochondrial regulation of insulin action
The mitochondrial respiration chain takes electrons from NADH and FADH2 that are produced during glucose and lipid oxidation and generates the electrochemical gradient (membrane potential) that drives ATP generation to meet cell energy demand (Figure 3) [41,50–51]. Thus, functional mitochondria are responsible for the finely tuned redox couple NAD+/NADH ratio, which has been shown to regulate insulin action through SIRT1 [80–83]. In particular, small molecules that reduce the Michaelis constant of SIRT1 for NAD+ have been used to improve systemic glucose homeostasis greatly in insulin-resistant mouse models [80–83], presumably by suppressing FOXO1 [86,87] and enhancing IRS2 signaling [88]. SIRT1 inhibition increased acetylation and decreased phosphorylation of IRS2, suggesting that SIRT1 can enhance insulin signaling in part by deacetylating IRS2 [88].
Single-electron reduction of molecular oxygen at complexes I and III generates superoxide (O2•−) that is converted into hydrogen peroxide (H2O2) (Figure 3) [89]. Although chronic exposure to high level of ROS can induce mitochondrial alteration [71] and possibly insulin resistance [90,91], accumulated evidence suggests that modest ROS generation in cells during insulin stimulation plays an integral role in the tyrosine phosphorylation for IR activation [92–94]. Early studies implicated NADPH oxidase (Nox) in the ROS elevation [92,94]. Recently, it was found that mitochondrial respiration could account significantly for the insulin-induced H2O2 production and tyrosine phosphorylation of the insulin receptor [94], and an H2O2 scavenger (N-acetylcysteine) could prevent both insulin-stimulated H2O2 generation and tyrosine phosphorylation of the insulin receptor. Inhibition of mitochondrial respiration-mediated H2O2 production by a potent chemical uncoupler of mitochondria (FCCP) diminishes both insulin-induced H2O2 and phosphorylation of the insulin receptor. Moreover, in neurons the respiratory substrate succinate substantially promotes insulin receptor phosphorylation [94]. These results reveal a functional relationship between mitochondrial respiration and insulin receptor autophosphorylation, which reveals how redox balance can modulate directly insulin sensitivity (Figure 4) [92].
Consistent with the treatment of cells with antioxidants reducing insulin responsiveness, mild oxidative conditions cause a decrease in insulin receptor β-chain sulfhydryl groups and greatly enhance the induction of insulin-stimulated tyrosine autophosphorylation [95]. Structural data indicate that the ATP binding site is blocked by the dephosphorylated A-loop in the insulin receptor β subunit. However, the conversion of any of the four cysteine residues (Cys1056, Cys1138, Cys1234, and Cys1245) into sulfenic acid leads to structural changes that bring Tyr1158 into close contact with Asp1083 and render the catalytic site accessible to ATP for tyrosine phosphorylation [96]. Thus redox priming can promote autophosphorylation and insulin receptor signal, even in the absence of insulin.
In addition to the role of receptor kinases, tyrosine phosphorylation of the proteins in IRS-branch signaling pathway is regulated by phosphatases (e.g. PTP1B and PTEN) (Figure 4) [97,98]. In line with the increased PTP1B expression in insulin resistant human subjects [99], mice lacking PTP1B are insulin sensitive and do not develop diet-induced obesity, whereas re-expression of PTP1B decreases insulin sensitivity [100,101]. PTPase activity is generally sensitive to the redox state in cells because the catalytic center consists of a readily oxidized 11-residue signature sequence (I/V)HCXAGXXR(S/T/G) [102]. The cysteine residue forms a cysteinyl-phosphate intermediate to catalyze the hydrolysis of protein-phosphotyrosine, which is inhibited by oxidation of the catalytic thiol by ROS [103,104]. Conversely, use of antioxidants to counteract the ROS effect can diminish exercise-induced insulin sensitivity [105].
PTPase activity undergoes a universal mechanism of reversible redox modulation of the active-site cysteine residues into Cys-SOH, glutathiolylated Cys, a disulfide bond with the neighboring Cys, or a sulfenyl-amide intermediate [102–104]. PTEN contains an N-terminal phosphatase domain with specificity toward both phospha-tidylinositol-3,4,5-trisphosphate (PI-3,4,5-P3) and to a lesser degree tyrosine-phosphorylated proteins. Being a lipid phosphatase, PTEN can reduce PI-3,4,5-P3 concentrations and suppress Akt phosphorylation/activation, which activates FOXO1 and increases glucose production in the liver [42,46]. However, PTP1B can directly dephosphorylate tyrosine residues in the insulin receptor and the IRS-proteins to block the insulin-signaling cascade [106–108]. Therefore, the dephosphorylation of IR and IRS-1 by PTP1B can modulate insulin receptor and IRS protein activities that contribute to insulin resistance (Figure 4). To this end, systemic and tissue-specific knockout of PTP1B is beneficial to enhance insulin signaling and ameliorate metabolic syndromes in obese and diabetic rodents [108–110].
The oxidation of PTEN by H2O2 results in a disulfide bond between Cys124 and Cys71 that blocks the active site, and this increases PIP3 concentration and activates downstream signaling events through the PI3K→Akt cascade [111–113]. Similarly, hyperglycemia-induced H2O2 in adipocytes substantially reduces PTP1B activity and increases the phosphorylation of the insulin receptor, IRS1, and Akt in response to insulin [114]. In particular, a chemical-induced mitochondrial ROS burst suppresses PTEN and greatly stimulates the PI3K signaling pathway, which can promote hepatocyte steatosis [115]. In a recent study, mice lacking Gpx1 (glutathione peroxidase 1), the key ROS scavenger, showed increased insulin sensitivity and were protected from HFD-induced metabolic syndrome [104]. The underlying mechanism is a significant elevation of ROS in muscle that leads to enhanced oxidation/inactivation of PTEN, thus improving PI3K/Akt signaling and glucose uptake in muscle (Figure 4). However, administration of the antioxidant N-acetylcysteine diminishes the phenotype in Gpx1-deficient mice. Consistent with this, supplementation with a combination of vitamin C and vitamin E abolishes exercise-induced ROS and diminishes the health-promoting effects (i.e. enhanced insulin sensitivity) of physical exercise in humans [105].
Summary and Perspectives
Insulin activates IR kinase and propagates two main branches of signaling via the IRS-proteins: the PI3K→PDK1→AKT and GRB2/SOS→RAS kinase cascades. Both of the branches of IRS signals are shown to regulate mitochondrial function [41,49,60–62]. Impaired insulin action deregulates systemic glucose and lipid metabolism, and leads to hyperglycemia and dyslipidemia in diabetic patients. It is established that under insulin resistance, insulin fails to block FOXO1 that can induce gluconeogenesis via PEPCK and G6P, but an outstanding question remaining to be addressed is how insulin resistance promotes dyslipidemia in the liver. Recent evidence that insulin resistance causes mitochondrial dysfunction and impairs fatty acid oxidation can at least in part account for this [41,49].
Insulin signaling has been established to underpin normal mitochondrial function in metabolism of the liver, skeletal and cardiac muscles as well as in pancreatic β cells. Impaired insulin action can cause abnormalities in both mitochondrial biogenesis and functions through the FOXO1–HMOX1–SIRT1–PGC1α axis. On the other hand, mitochondria produce ROS that serve as a second messenger and enhance insulin sensitivity via oxidative modification of the insulin receptor and inactivation of PTEN and PTP1B. As such, mitochondrial ROS and persistent oxidative challenge are suggested to induce systemic adaptations, during which both insulin action and antioxidant capacity are improved [104–105,116]. However, other evidence shows that hyperglycemia or obesity-induced ROS activates stress-sensitive kinases (e.g. JNK and IKK), and promotes insulin resistance in rodent animal models and humans [90–91]. Thus, the ‘threshold’ at which ROS switches from an enhancer to suppressor of insulin sensitivity must be defined before strategies can be developed for clinically treating metabolic syndrome.
Acknowledgments
This project was supported by US National Institutes of Health grants DK38712, DK55326 (M.F.W.), and an American Diabetes Association Mentor-Based Postdoctoral Fellowship 7-08-MN-63 (M.F.W. and Z.C.). We apologize to the colleagues whose work is not specifically referenced owing to space limitations.
References
- 1.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 2.Taguchi A, White MF. Insulin-like signaling, nutrient homeostasis, and life span. Annu Rev Physiol. 2008;70:191–212. doi: 10.1146/annurev.physiol.70.113006.100533. [DOI] [PubMed] [Google Scholar]
- 3.Shaham O, et al. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol Syst Biol. 2008;4:214. doi: 10.1038/msb.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Numan S, Russell DS. Discrete expression of insulin receptor substrate-4 mRNA in adult rat brain. Brain Res Mol Brain Res. 1999;72:97–102. doi: 10.1016/s0169-328x(99)00160-6. [DOI] [PubMed] [Google Scholar]
- 5.Bjornholm M, et al. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia. 2002;45:1697–1702. doi: 10.1007/s00125-002-0945-z. [DOI] [PubMed] [Google Scholar]
- 6.Dearth RK, et al. Oncogenic transformation by the signaling adaptor proteins insulin receptor substrate (IRS)-1 and IRS-2. Cell Cycle. 2007;6:705–713. doi: 10.4161/cc.6.6.4035. [DOI] [PubMed] [Google Scholar]
- 7.White MF. Insulin signaling in health and disease. Science. 2003;302:1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- 8.Dhe-Paganon S, et al. Crystal structure of the pleckstrin homology-phosphotyrosine binding (PH-PTB) targeting region of insulin receptor substrate 1. Proc Natl Acad Sci USA. 1999;96:8378–8383. doi: 10.1073/pnas.96.15.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yenush L, et al. The pleckstrin homology and phosphotyrosine binding domains of insulin receptor substrate 1 mediate inhibition of apoptosis by insulin. Mol Cell Biol. 1998;18:6784–6794. doi: 10.1128/mcb.18.11.6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burks DJ, et al. IRS pleckstrin homology domains bind to acidic motifs in proteins. J Biol Chem. 1998;273:31061–31067. doi: 10.1074/jbc.273.47.31061. [DOI] [PubMed] [Google Scholar]
- 11.Songyang Z, et al. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 12.Songyang Z, Cantley LC. Recognition and specificity in protein tyrosine kinase-mediated signaling. Trends Biochem Sci. 1995;20:470–475. doi: 10.1016/s0968-0004(00)89103-3. [DOI] [PubMed] [Google Scholar]
- 13.Shoelson SE, et al. YMXM motifs of IRS-1 define the substrate specificity of the insulin receptor kinase. Proc Natl Acad Sci USA. 1992;89:2027–2031. doi: 10.1073/pnas.89.6.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawka-Verhelle D, et al. Insulin receptor substrate-2 binds to the insulin receptor through its phopshotyrosine-binding domain and through a newly identified domain comprising amino acids 591–786. J Biol Chem. 1996;271:5980–5983. doi: 10.1074/jbc.271.11.5980. [DOI] [PubMed] [Google Scholar]
- 16.Sawka-Verhelle D, et al. Tyr624 and Tyr628 in insulin receptor substrate-2 mediate its association with the insulin receptor. J Biol Chem. 1997;272:16414–16420. doi: 10.1074/jbc.272.26.16414. [DOI] [PubMed] [Google Scholar]
- 17.Wu J, et al. Structural and biochemical characterization of the KRLB region in insulin receptor substrate-2. Nat Struct Mol Biol. 2008;15:251–258. doi: 10.1038/nsmb.1388. [DOI] [PubMed] [Google Scholar]
- 18.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ueki K, et al. Positive and negative roles of p85 alpha and p85 beta regulatory subunits of phosphoinositide 3-kinase in insulin signaling. J Biol Chem. 2003;278:48453–48466. doi: 10.1074/jbc.M305602200. [DOI] [PubMed] [Google Scholar]
- 20.Ueki K, et al. Positive and negative regulation of phosphoinositide 3-kinase-dependent signaling pathways by three different gene products of the p85alpha regulatory subunit. Mol Cell Biol. 2000;20:8035–8046. doi: 10.1128/mcb.20.21.8035-8046.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Backer JM, et al. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 1992;11:3469–3479. doi: 10.1002/j.1460-2075.1992.tb05426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chagpar RB, et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 2010;107:5471–5476. doi: 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geering B, et al. Regulation of class IA PI3Ks: is there a role for monomeric PI3K subunits? Biochem Soc Trans. 2007;35:199–203. doi: 10.1042/BST0350199. [DOI] [PubMed] [Google Scholar]
- 24.Taniguchi CM, et al. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab. 2006;3:343–353. doi: 10.1016/j.cmet.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Bae SS, et al. Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. J Biol Chem. 2003;278:49530–49536. doi: 10.1074/jbc.M306782200. [DOI] [PubMed] [Google Scholar]
- 26.Cho H, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 27.George S, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semple RK, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest. 2009;119:315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19:R1046–R1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang J, Manning BD. The TSC1–TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaeschke A, et al. Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol. 2002;159:217–224. doi: 10.1083/jcb.jcb.200206108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tee AR, et al. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Proud CG. Cell signaling.mTOR, unleashed. Science. 2007;318:926–927. doi: 10.1126/science.1150653. [DOI] [PubMed] [Google Scholar]
- 35.Astrinidis A, Henske EP. Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease. Oncogene. 2005;24:7475–7481. doi: 10.1038/sj.onc.1209090. [DOI] [PubMed] [Google Scholar]
- 36.Hu C, et al. Molecular cloning and tissue distribution of PHAS-I, an intracellular target for insulin and growth factors. Proc Natl Acad Sci USA. 1994;91:3730–3734. doi: 10.1073/pnas.91.9.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pende M, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408:994–997. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- 38.Li S, et al. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci USA. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo S, et al. The reciprocal stability of FOXO1 and IRS2 creates a regulatory circuit that controls insulin signaling. Mol Endocrinol. 2006;20:3389–3399. doi: 10.1210/me.2006-0092. [DOI] [PubMed] [Google Scholar]
- 40.Ide T, et al. SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat Cell Biol. 2004;6:351–357. doi: 10.1038/ncb1111. [DOI] [PubMed] [Google Scholar]
- 41.Cheng Z, et al. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15:1307–1311. doi: 10.1038/nm.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong XC, et al. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–450. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 44.Gross DN, et al. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- 45.Montminy M, Koo SH. Diabetes: outfoxing insulin resistance? Nature. 2004;432:958–959. doi: 10.1038/432958a. [DOI] [PubMed] [Google Scholar]
- 46.Guo S, et al. IRS1-branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis. Mol Cell Biol. 2009;29:5070–5083. doi: 10.1128/MCB.00138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Purushotham A, et al. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White MF. Regulating insulin signaling and beta-cell function through IRS proteins. Can J Physiol Pharmacol. 2006;84:725–737. doi: 10.1139/y06-008. [DOI] [PubMed] [Google Scholar]
- 49.Cheng Z, White MF. Foxo1 in hepatic lipid metabolism. Cell Cycle. 2010;9:219–220. doi: 10.4161/cc.9.2.10567. [DOI] [PubMed] [Google Scholar]
- 50.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001;414:807–812. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 51.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 52.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 53.Petersen KF, et al. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turner N, Heilbronn LK. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol Metab. 2008;19:324–330. doi: 10.1016/j.tem.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 55.Kelley DE, et al. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 56.Petersen KF, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stump CS, et al. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci USA. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McKee EE, Grier BL. Insulin stimulates mitochondrial protein synthesis and respiration in isolated perfused rat heart. Am J Physiol. 1990;259:E413–E421. doi: 10.1152/ajpendo.1990.259.3.E413. [DOI] [PubMed] [Google Scholar]
- 59.Vianna CR, et al. Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metab. 2006;4:453–464. doi: 10.1016/j.cmet.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu S, et al. Insulin signaling regulates mitochondrial function in pancreatic beta-cells. PLoS One. 2009;4:e7983. doi: 10.1371/journal.pone.0007983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Riehle C, et al. Insulin receptor substrates (IRS) signaling are essential regulators of mitochondrial function and cardiomyocyte survival. Circulation. 2008;118:S444. [Google Scholar]
- 62.O’Neill BT, et al. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 2007;6:294–306. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schrauwen P. Of the fit and the fat: mitochondrial abnormalities and type 2 diabetes mellitus. J Clin Endocrinol Metab. 2007;92:1229–1231. doi: 10.1210/jc.2007-0295. [DOI] [PubMed] [Google Scholar]
- 64.Holloszy JO. Skeletal muscle ‘mitochondrial deficiency’ does not mediate insulin resistance. Am J Clin Nutr. 2009;89:463S–466S. doi: 10.3945/ajcn.2008.26717C. [DOI] [PubMed] [Google Scholar]
- 65.Goodpaster BH, et al. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. 2001;86:5755–5761. doi: 10.1210/jcem.86.12.8075. [DOI] [PubMed] [Google Scholar]
- 66.Short KR, et al. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes. 2003;52:1888–1896. doi: 10.2337/diabetes.52.8.1888. [DOI] [PubMed] [Google Scholar]
- 67.Turner N, et al. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes. 2007;56:2085–2092. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- 68.Hancock CR, et al. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA. 2008;105:7815–7820. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng Z, White MF. Targeting FOXO1 from the concept to metabolic diseases–lessons from mouse models. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Michael MD, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 71.Bonnard C, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yu T, et al. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laustsen PG, et al. Essential role of insulin and insulin-like growth factor 1 receptor signaling in cardiac development and function. Mol Cell Biol. 2007;27:1649–1664. doi: 10.1128/MCB.01110-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu Z, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 75.Daitoku H, et al. Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes. 2003;52:642–649. doi: 10.2337/diabetes.52.3.642. [DOI] [PubMed] [Google Scholar]
- 76.Liu HY, et al. Prolonged exposure to insulin suppresses mitochondrial production in primary hepatocytes. J Biol Chem. 2009;284:14087–14095. doi: 10.1074/jbc.M807992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Singh A, et al. Leptin-mediated changes in hepatic mitochondrial metabolism, structure, and protein levels. Proc Natl Acad Sci U S A. 2009;106:13100–13105. doi: 10.1073/pnas.0903723106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 79.Patti ME, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baur JA, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lagouge M, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 82.Feige JN, et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 83.Milne JC, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Converso DP, et al. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006;20:1236–1238. doi: 10.1096/fj.05-4204fje. [DOI] [PubMed] [Google Scholar]
- 85.Lumeng L, et al. Suppression of the mitochondrial oxidation of (−)-palmitylcarnitine by the malate–aspartate and alpha-glycerophosphate shuttles. J Biol Chem. 1976;251:277–284. [PubMed] [Google Scholar]
- 86.Motta MC, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 87.Yang Y, et al. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li Y, et al. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008;8:38–48. doi: 10.1016/j.cmet.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Balaban RS, et al. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 90.Anderson EJ, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Houstis N, et al. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 92.Goldstein BJ, et al. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005;7:1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Evans JL, et al. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005;7:1040–1052. doi: 10.1089/ars.2005.7.1040. [DOI] [PubMed] [Google Scholar]
- 94.Storozhevykh TP, et al. Mitochondrial respiratory chain is involved in insulin-stimulated hydrogen peroxide production and plays an integral role in insulin receptor autophosphorylation in neurons. BMC Neurosci. 2007;8:84. doi: 10.1186/1471-2202-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schmid E, et al. Redox priming of the insulin receptor beta-chain associated with altered tyrosine kinase activity and insulin responsiveness in the absence of tyrosine autophosphorylation. FASEB J. 1998;12:863–870. doi: 10.1096/fasebj.12.10.863. [DOI] [PubMed] [Google Scholar]
- 96.Schmid E, et al. Phosphorylation of the insulin receptor kinase by phosphocreatine in combination with hydrogen peroxide: the structural basis of redox priming. FASEB J. 1999;13:1491–1500. doi: 10.1096/fasebj.13.12.1491. [DOI] [PubMed] [Google Scholar]
- 97.Fritsche L, et al. How insulin receptor substrate proteins regulate the metabolic capacity of the liver – implications for health and disease. Curr Med Chem. 2008;15:1316–1329. doi: 10.2174/092986708784534956. [DOI] [PubMed] [Google Scholar]
- 98.Heneberg P. Use of protein tyrosine phosphatase inhibitors as promising targeted therapeutic drugs. Curr Med Chem. 2009;16:706–733. doi: 10.2174/092986709787458407. [DOI] [PubMed] [Google Scholar]
- 99.Qiu W, et al. Hepatic PTP-1B expression regulates the assembly and secretion of apolipoprotein B-containing lipoproteins: evidence from protein tyrosine phosphatase-1B overexpression, knockout, and RNAi studies. Diabetes. 2004;53:3057–3066. doi: 10.2337/diabetes.53.12.3057. [DOI] [PubMed] [Google Scholar]
- 100.Elchebly M, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 101.Haj FG, et al. Liver-specific protein-tyrosine phosphatase 1B (PTP1B) re-expression alters glucose homeostasis of PTP1B−/− mice. J Biol Chem. 2005;280:15038–15046. doi: 10.1074/jbc.M413240200. [DOI] [PubMed] [Google Scholar]
- 102.Andersen JN, et al. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barrett WC, et al. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 104.Loh K, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ristow M, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goldstein BJ, et al. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein tyrosine phosphatase 1B: possible facilitation by the formation of a ternary complex with the Grb-2 adaptor protein. J Biol Chem. 2000;275:4283–4289. doi: 10.1074/jbc.275.6.4283. [DOI] [PubMed] [Google Scholar]
- 107.Ueno M, et al. Regulation of insulin signalling by hyperinsulinaemia: role of IRS-1/2 serine phosphorylation and the mTOR/p70 S6K pathway. Diabetologia. 2005;48:506–518. doi: 10.1007/s00125-004-1662-6. [DOI] [PubMed] [Google Scholar]
- 108.Xue B, et al. Protein-tyrosine phosphatase 1B deficiency reduces insulin resistance and the diabetic phenotype in mice with polygenic insulin resistance. J Biol Chem. 2007;282:23829–23840. doi: 10.1074/jbc.M609680200. [DOI] [PubMed] [Google Scholar]
- 109.Delibegovic M, et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes. 2009;58:590–599. doi: 10.2337/db08-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gum RJ, et al. Reduction of protein tyrosine phosphatase 1B increases insulin-dependent signaling in ob/ob mice. Diabetes. 2003;52:21–28. doi: 10.2337/diabetes.52.1.21. [DOI] [PubMed] [Google Scholar]
- 111.Connor KM, et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J Biol Chem. 2005;280:16916–16924. doi: 10.1074/jbc.M410690200. [DOI] [PubMed] [Google Scholar]
- 112.Lee SR, et al. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 113.Leslie NR, et al. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu X, et al. Hyperglycemia potentiates H2O2 production in adipocytes and enhances insulin signal transduction: potential role for oxidative inhibition of thiol-sensitive protein-tyrosine phosphatases. Antioxid Redox Signal. 2005;7:526–537. doi: 10.1089/ars.2005.7.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kohli R, et al. Mitochondrial reactive oxygen species signal hepatocyte steatosis by regulating the phosphatidylinositol 3-kinase cell survival pathway. J Biol Chem. 2007;282:21327–21336. doi: 10.1074/jbc.M701759200. [DOI] [PubMed] [Google Scholar]
- 116.Radak Z, et al. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic Biol Med. 2008;44:153–159. doi: 10.1016/j.freeradbiomed.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 117.Rhoads RE. Signal transduction pathways that regulate eukaryotic protein synthesis. J Biol Chem. 1999;274:30337–30340. doi: 10.1074/jbc.274.43.30337. [DOI] [PubMed] [Google Scholar]
- 118.McClung JP, et al. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc Natl Acad Sci USA. 2004;101:8852–8857. doi: 10.1073/pnas.0308096101. [DOI] [PMC free article] [PubMed] [Google Scholar]