

Abstract
Background & objectives:
To study effects of drugs against rheumatoid arthritis (RA) synoviocytes or fibroblast like synoviocytes (FLS) are used. To overcome the drawbacks of using FLS, this study was conducted to show the validity of SW982 synovial cell line in RA study.
Methods:
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, Annexin V propidium iodide (PI) staining, mitochondrial membrane potential assay, Triton X-114 Phase partitioning, and immunolot for apoptosis signaling in SW982 human synovial cell line were performed.
Results:
Fluvastatin induced apoptosis in a dose- and time-dependent manner in TNFα -stimulated SW982 human synovial cells. A geranylgeranylpyrophosphate (GGPP) inhibitor, but not a farnesylpyrophosphate (FPP) inhibitor, induced apoptosis, and fluvastatin-induced apoptosis was associated with the translocation of isoprenylated RhoA and Rac1 proteins from the cell membrane to the cytosol. Fluvastatin-induced downstream apoptotic signals were associated with inhibition of the phosphoinositide 3-kinase (PI3K)/Akt pathway. Accordingly, 89 kDa apoptotic cleavage fragment of poly (ADP-ribose) polymerase (PARP) was detected.
Interpretation & conclusions:
Collectively, our data indicate that fluvastatin induces apoptotic cell death in TNFα-stimulated SW982 human synovial cells through the inactivation of the geranylgerenylated membrane fraction of RhoA and Rac1 proteins and the subsequent inhibition of the PI3K/Akt signaling pathway. This finding shows the validity of SW982 cell line for RA study.
Keywords: Apoptosis, fluvastatin, PI3K/Akt, Rac1, RhoA, SW982 human synovial cells
Rheumatoid arthritis (RA) is a synovial hyperplastic disorder associated with Th1-polarized immune disease, eventually leading to the development of pannus, progressive joint destruction, deformity and disability1,2. The synovium is the primary site of inflammation in RA, and fibroblast-like synoviocytes (FLS) play pivotal roles in both the initiation and perpetuation of RA3,4. The prolonged life span of activated synoviocytes accelerates synovial hyperplasia, pannus, and the destruction of cartilage and bone5. In response to inflammatory cytokines such as interleukin (IL)-1β, IL-6 and tumour necrosis factor-α (TNFα), activated synovial fibroblasts produce chemokines which promote inflammation, neovascularization, and cartilage degradation6,7,8. Of these cytokines, TNFα plays a major role in the activation and survival of synoviocytes in RA pathogenesis9,10. In that context, TNF inhibitors have been widely used in clinic as directly suppressing TNFα release.
However, simple TNF-α blockade fails to directly kill the inflamed FLS in the microenvironment of RA patient. Directing the apoptosis of inflamed FLS or synoviocytes might be an effective therapeutic strategy. Together, unraveling the apoptotic signaling in the inflamed FLS will confer insights into developing novel RA therapeutics.
Of the synovial apoptosis-inducers, statin has been one of the promising candidates11,12. The apoptosis might be ascribed to blocking the synthesis of isoprenoid intermediates such as geranylgeranylpyrophosphate (GGPP) and farnesylpyrophosphate (FPP). These intermediates are responsible for isoprenylation of the small GTP-binding proteins, including members of Rho, Rac, and Ras families13,14,15. These molecules play a pivotal role in intracellular signal transduction pathways such as synovial cell survival, apoptosis, and differentiation16,17. However, the apoptotic signaling of statins is not fully established and generalized owing to diverse, unique actions of every statin.
To screen and study the unique effects of diverse statins, primary FLSs from RA patients have been used. Despite the benefits of primary FLS, there are some drawbacks such as the difficulties to collect and establish, the lack of reproducibilities owing to the heterogenous clones, and inconsistent results because of non-standardized patient samples. To overcome this, we hypothesized that synovial cell line would be more suitable than primary synoviocytes. Of candidate cell lines, SW982 cell line has been used for examining the effects of anti-inflammatory drug. However, direct evidence and scientific rationale for alternative cell line to primary FLS is unclear. To clarify this, fluvastatin-induced apoptosis signaling was tested in SW982 cells. Fluvastatin has been shown as a novel therapeutic agent for RA12,18,19, and has also been reported to induce apoptosis in RA synoviocytes through blocking of protein geranylgeranylation12. However, how fluvastatin induces apoptosis signaling of TNFα inflamed synoviocytes is not fully understood. Specifically, phosphoinositide-3-kinase (PI3K)-Akt pathway triggered by fluvastatin in SW982 cells is unknown. To address this issue, adopting convenient, reproducible SW982 cell line, we investigated whether fluvastatin would trigger RhoA-Rac1 and PI3K-Akt signal pathway in TNFα-stimulated SW982 cells.
Material & Methods
The study was conducted in Department of Microbiology, Yonsei University, Wonju College of Medicine, Wonju, Republic of Korea.
Cell culture: The human synovial cell line SW982 was obtained from the American Type Culture Collection (Rockville, USA). The SW982 cells were routinely cultured in T-150 flasks (Corning) and grown in Dulbecco's modified Eagle medium (DMEM) with 2 mM L-glutamine, 10 per cent foetal bovine serum (FBS), and 1 per cent Pen-Strep (Invitrogen, USA) at 37°C, 5 per cent CO2.
Antibodies and reagents: Fluvastatin was purchased from Novartis (Basel, Switzerland) and was prepared as a 4 mg/ml (10 mM) stock solution. GGPP and FPP were obtained from Sigma-Aldrich (St. Louis, USA). Anti-β-actin, anti-PARP, anti-phospho-ERK1/2, anti-phospho-p38 MAPK, anti-phospho-Akt, and anti-phospho-Bad were obtained from Cell Signaling (San Diego, USA). The geranylgeranyl transferase inhibitor GGTI-298, the farnesyl transferase inhibitor FTI-277, and the Rho kinase inhibitor Y-27632 were purchased from Calbiochem (La Jolla, USA). Human recombinant TNFα was obtained from R&D Systems (Minneapolis, USA). Anti-RhoA and anti-Rac1 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, USA) and BD Biosciences (Ontario, Canada), respectively.
Western blot analysis: The SW982 cells were seeded in 35-mm plastic dishes (3 × 105 cells per dish). Before treatment with fluvastatin, cells were cultured for 24 h in the presence or absence of 10 ng/ml TNFα. Then, fluvastatin was added to the wells for different time periods. Cells were lysed in lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 % Triton X-100, 0.5 % sodium deoxycholate, 1 mM sodium orthovanadate, 1 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 μg/ml pepstatin A]. After centrifugation at 13,000 × g at 4°C for 30 min, the supernatant was collected; 20 μg of lysate from each sample was run on a 15 per cent SDS-polyacrylamide gel and then electrophoretically transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Millipore Corp, USA). The PVDF membranes were blocked in blocking buffer [tris buffered saline with tween 20 (TBST) containing 5% BSA] overnight at 4°C, incubated with primary antibodies overnight at 4°C, washed, and incubated with goat anti-rabbit horse radish peroxidase (HRP) or anti-mouse HRP (Sigma-Aldrich, St. Louis, USA) for 1 h at room temperature. The membrane was developed with enhanced chemiluminescence (ECL) substrate (Amersham Life Sciences, Arlington Heights, USA), and exposed to Biomax MS autoradiography X-ray film (Kodak, Rochester, USA).
Cell viability: The SW982 cells were plated in 100 μl at a concentration of 1 × 105 cells/ml in a 96-well plate. Before treatment with or without fluvastatin, these were cultured for 24 h in the presence or absence of 10 ng/ml TNFα. Fluvastatin was added to the wells, and the cells were cultured for various periods of time. Where indicated, SW982 cell culture media was supplemented with 10 μM GGPP or 10 μM FPP at the time of fluvastatin addition. Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays. In brief, MTT solution (50 μl) was added to each well. After 3 h, the medium was removed completely from each well, and then 200 μl of 40 mM HCl-isopropanol lysis buffer was added to each well. One hundred μl of cell lysate was transferred into each well of a 96-well plate. Absorbance at 570 nm was measured with a microplate reader (SpecraMax M2e; Bucher Biotech, Basel, Switzerland). Data are shown as values relative to the control with a control value of 100 per cent.
Annexin V/propidium iodide (PI) staining: As mentioned above, SW982 cells were cultured with or without fluvastatin (10 μM) and 10 ng/ml TNFα. To quantify cell death and apoptotic cell populations, SW982 cells were trypsinized, washed with phosphate buffer saline (PBS), and resuspended in a binding buffer (10 mM Hepes, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) prior to staining. Analysis of apoptosis was carried out by double-labelling for annexin V/propidium iodide (PI) (Pharmingen, San Diego, USA) and PI staining. A minimum of 10,000 cells/sample were analyzed on a FACScan flow cytometer (Becton Dickinson, USA) as described previously14.
Mitochondrial membrane potential assay: The mitochondrial membrane potentials (MMP) of the cells were determined by flow cytometry with the membrane-potential-sensitive dye, JC-1 (Molecular Probes Inc., USA). JC-1 is a dual-emission fluorescent dye that exhibits potential-dependent accumulation in mitochondria, leading to a fluorescence emission shift from green on the FL-1 detector (530 nm) to red on the FL-2 (590 nm) detector. Normal cells with high mitochondrial membrane potential show an increase in red fluorescence. Mitochondrial damage is indicated by a decrease in the red/green fluorescence intensity ratio. SW982 cells were cultured with or without fluvastatin (10 μM) and 10 ng/ml TNFα. Then, 1 × 106 cells were washed and loaded with JC-1 (10 μM) for 20 min at 37°C. The cells were subsequently washed and analyzed using a FACScan flow cytometer (Becton Dickinson, USA). The same incubation time was used for the control and the fluvastatin and/or TNFα-treated samples.
Triton X-114 Phase partitioning: SW982 cells were incubated for 24 h with 10 μM fluvastatin in the presence or absence of 10 μM GGPP or 10 μM FPP. Integral membrane proteins were extracted by Triton X-114 as described previously13. Briefly, 200 μl of lysis solution (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 1.0% Triton X-114) was added to each well of a six-well culture plate, and the plate was incubated on ice for 30 min. Samples were scraped from the plate and after 3 min incubation at 37°C, the samples were centrifuged at 300 g for 3 min to separate the phases. The upper aqueous phase was rinsed twice; Triton X-114 and buffer were then added to the aqueous and detergent phases, respectively, to yield equal volumes and approximately the same salt and detergent concentrations in both fractions. Samples were analyzed by immunoblotting, as described above.
Statistical analysis: Results are presented as mean ± standard deviation. The mean values among groups were analyzed and compared by one-way analysis of variance (ANOVA) followed by subsequent multiple comparison test (Tukey) with GraphPad Prism version 4.0 software packages (GraphPad Software, La Jolla, CA, USA). P values less than 0.05 were considered significant.
Results
Fluvastatin affects cell proliferation in a dose-dependent manner and induces apoptosis in by TNFα-stimulated SW982 human synovial cells: TNFα-stimulated SW982 cells were subjected to the escalated concentrations of fluvastatin for 24 h, and then cell viability was assessed using the MTT assay. Fluvastatin inhibited the proliferation of TNFα-stimulated SW982 cells. The stimulated SW982 cells were sensitive to fluvastatin, with viabilities of 85 ± 11 per cent at 1 μM, 57.6 ± 6.67 per cent at 10 μM, and 29 ± 6.56 per cent at 50 μM fluvastatin (Fig. 1). Further it was investigated whether the fluvastatin-induced cell death was due to apoptosis. Annexin V staining showed that treatment with fluvastatin significantly increased apoptosis of the stimulated SW982 human synovial cells in a dose-and time dependent manner (Fig. 2). The stimulated SW982 cells exhibited apoptotic frequencies of 10 ± 2 per cent at 1 μM, 50 ± 8 per cent at 10 μM, and 80 ± 11 per cent at 50 μM fluvastatin. These results were similar to the MTT assay results, indicating that fluvastatin induced apoptotic cell death in a dose-dependent manner.
Fig. 1.
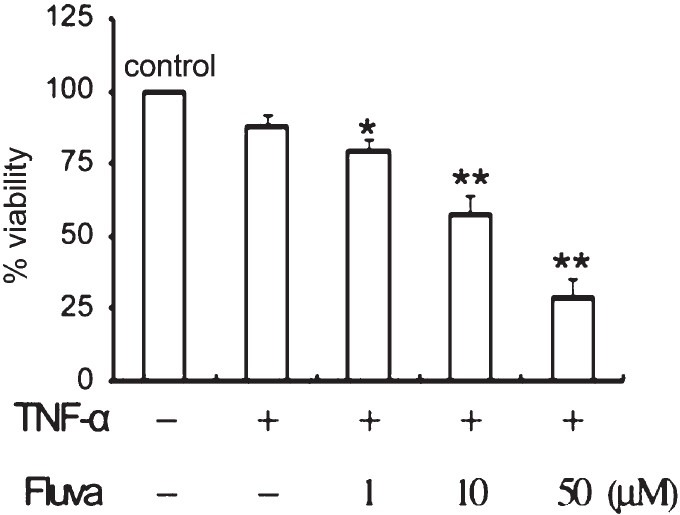
Reduction of cell viability by fluvastatin. TNFα-stimulated SW982 synovial cells were incubated with or without 0-50 μM fluvastatin for 24 h. Cell viability was determined by MTT assay. Data were obtained from duplicate experiments using three different samples. *P<0.05, **P<0.01 compared to control (medium only).
Fig. 2.
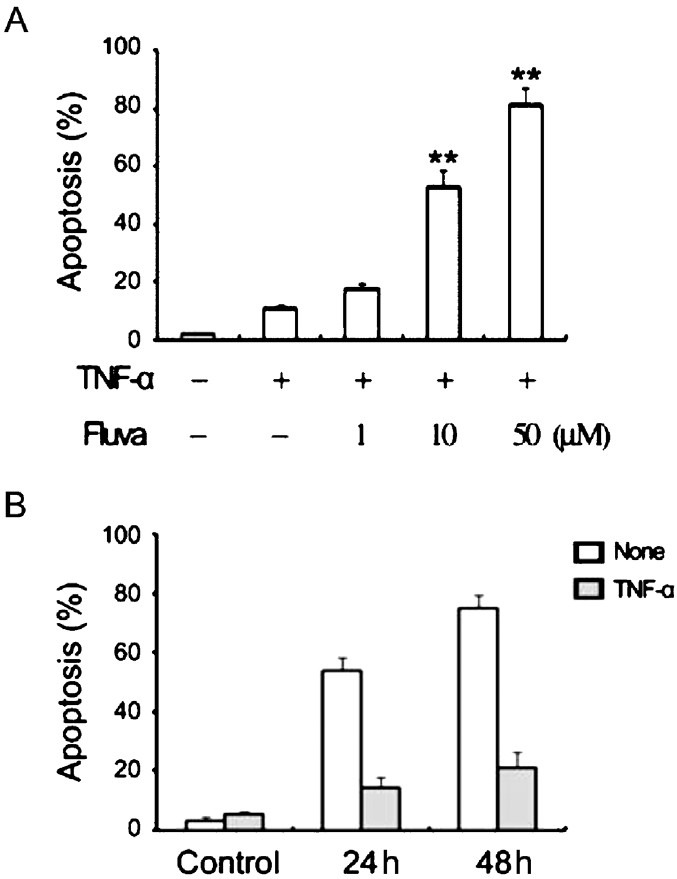
Influence of fluvastatin on the apoptosis of TNFα-stimulated SW982 synovial cells. Apoptosis was measured by flow cytometry after staining with annexin V. (A) The escalated fluvastatin concentrations (0-50 μM) resulted in a linear increase in the apoptotic response. (B) Cells were cultured for various durations (0-48 h) in the presence of 10 μM fluvastatin. **P<0.01compared to control (medium only).
Fluvastatin-induced apoptosis is associated with increased translocation of isoprenylated RhoA and Rac1 proteins from the cell membrane to the cytosol in TNFα-stimulated SW982 human synovial cells: Both FPP and GGPP are essential for the activation of a variety of intracellular proteins. Rho family proteins are located either in the cytoplasm or in the membrane, and these translocate between these two sites. Decreased expression of membrane-associated Rho family RhoA and Rac1 small G proteins was observed in the presence of fluvastatin in contrast to those of the control samples. The concentrations of RhoA and Rac1 increased in the cytoplasm, as determined by Triton X-114 partitioning. Supplementation of the culture medium with GGPP restored RhoA and Rac1 to the membrane.
To further ascertain the role of the RhoA protein in apoptosis, the effect of the RhoA kinase inhibitor Y-27362 was investigated. TNFα-stimulated SW982 human synovial cells were incubated in the presence or absence of Y-27632 at a concentration of 20 μM for 24 h. As shown in Fig. 3, inhibition of RhoA kinase resulted in a reduction in cell viability and an increase in apoptotic cell death. These findings suggested that fluvastatin-induced apoptosis was closely associated with RhoA signaling.
Fig. 3.
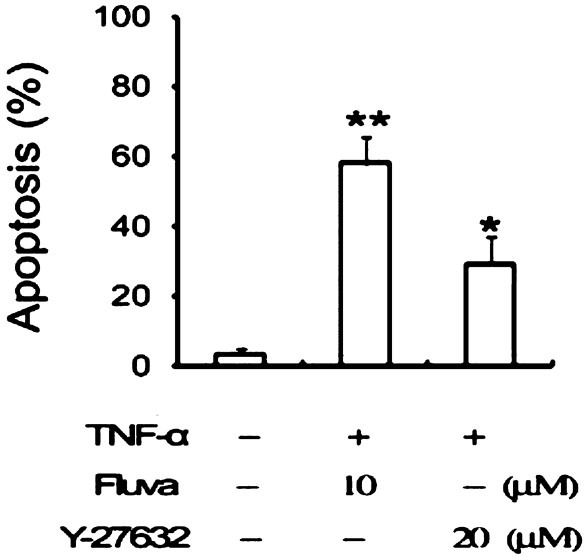
Effects of RhoA kinase inhibitors on apoptosis of TNF-α-stimulated SW982 cells. Cells were incubated for 48 h with medium alone, 10 μM fluvastatin, or 20 μM Y-27632. Apoptosis was measured by flow cytometer after staining with annexin V. *P<0.05, **P<0.01 compared to control (medium only).
A GGPP inhibitor, but not an FPP inhibitor, induces apoptosis in SW982 human synovial cells stimulated by TNFα: After a 24 h incubation with inhibitors (GGTI-298 or FTI-277), cell viability was determined, and apoptosis was measured. The GGPP inhibitor, GGTI-298, significantly decreased cell viability (Fig. 4A) and markedly increased apoptosis at a concentration of 20 μM (Fig. 4B). The FPP inhibitor, FTI-277, little affected cell viability or apoptotic cell death.
Fig. 4.
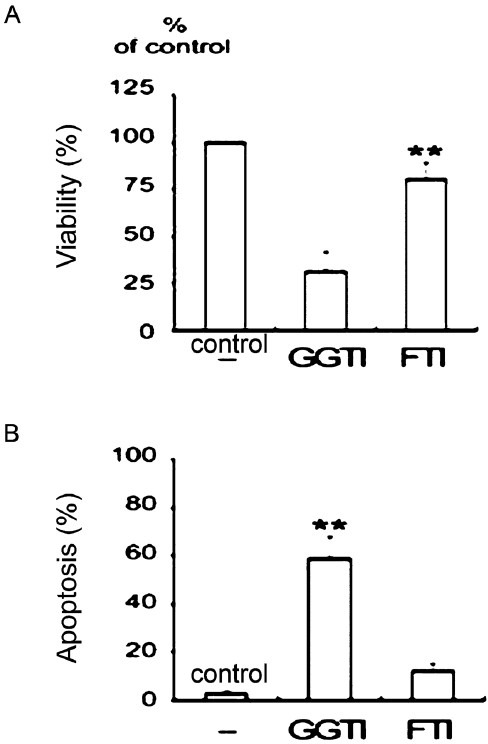
Effects of the geranylgeranyl transferase inhibitor GGTI-298 and the farnesyl transferase inhibitor FTI-277 on cell viability and apoptosis. TNFα-stimulated SW982 cells were incubated with 10 μM GGTI-298 or 10 μM FTI-277. The geranylgeranyl transferase inhibitor, but not the farnesyltransferase inhibitor, induced apoptosis. Data were obtained from duplicate experiments using three different samples. (A) Cell viability was determined by MTT assays. **P<0.01 compared between CGTI and FTI. (B) Apoptosis was measured by flow cytometry after staining with annexin V. **P<0.01 compared to control (medium only).
Fluvastatin-induced apoptosis is associated with the inhibition the PI3K/Akt pathway in TNFα-stimulated SW982 human synovial cells: To ascertain additional downstream effectors of Rho proteins, the PI3K/Akt and p38 MAPK pathways were examined. SW982 human synovial cells stimulated by TNFα were treated with fluvastatin, and the kinetics of PI3K/Akt and p38 MAPK phosphorylations were examined. After treatment with fluvastatin, the activation of Akt was suppressed, whereas the phosphorylations of ERK1/2 and p38 MAPK were sustained (Fig. 5A). Fluvastatin in TNFα-stimulated SW982 human synovial cells significantly reduced the expression of phosphorylated Bad. The PI3K/Akt inhibitor, LY294002 significantly enhanced apoptosis, whereas the ERK inhibitor, PD98059 and the p38 MAPK inhibitor, SB203580 failed to significantly induce apoptosis (Fig. 5B).
Fig. 5.
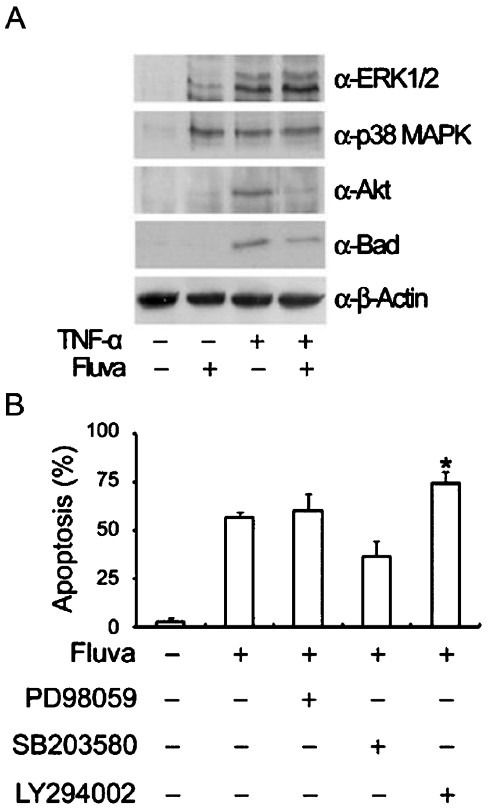
Effects of fluvastatin on the phosphorylations of ERK1/2, p38 MAPK, Akt, and Bad. After treatment with fluvastatin, phospho-Akt expression was suppressed whereas phosphorylations of ERK1/2 and p38 MAPK were sustained. Furthermore, phospho-Bad expression was significantly reduced. (A) TNFα-stimulated SW982 cells were treated with fluvastatin for 24 h. Cell lysates were subjected to SDS-PAGE and immunoblotted with anti-phospho-ERK1/2, anti-phospho-p38 MAPK, anti-phospho-Akt, or anti-phospho-Bad antibodies. Equal amounts of protein were loaded in each lane, and equal loading was confirmed by immunoblotting with an anti-β-actin antibody. (B) TNFα-stimulated SW982 cells were incubated for 24 h in the presence of 10 μM fluvastatin, 50 μM PD98059, 10 μM SB203580, or 15 μM LY294002. Apoptosis was measured by flow cytometry after staining with annexin V. The results are representative of two independents experiments. *P<0.05 compared to control (medium only).
Increased poly (ADP-ribose) polymerase (PARP) cleavage indicates that fluvastatin induces the apoptosis of SW982 human synovial cells stimulated by TNFα via caspase-dependent pathways: After a 24 h incubation, fluvastatin-treated cells displayed the disrupted mitochondrial transmembrane potentials (Fig. 6A) and markedly increased PARP cleavage activity (Fig. 6B).
Fig. 6.
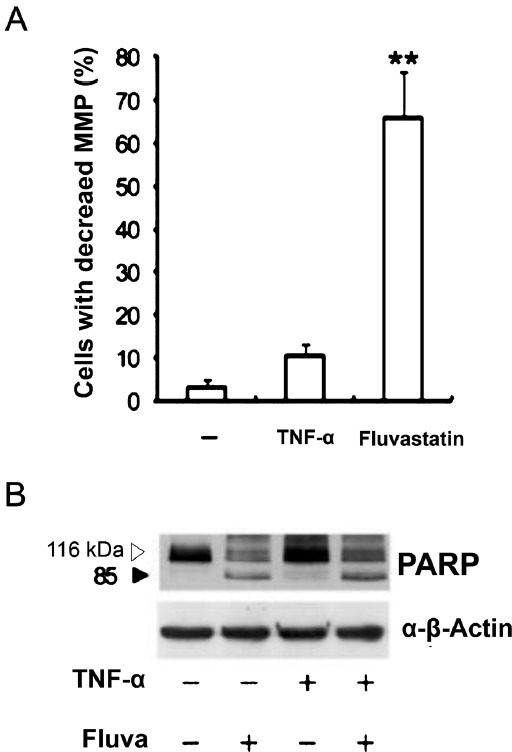
Effects of fluvastatin on mitochondrial transmembrane potentials (MMP) and PARP cleavage activity. TNFα-stimulated SW982 cells were cultured for 24 h in the presence of 10 μM fluvastatin. (A) Cells were stained with JC-1. Data are representative of three independent experiments. MMP, mitochondrial membrane potential. (B) The expression of cleaved PARP was detected by immunoblotting. The results are representative of two independent experiments. **P<0.01 compared to control (medium only).
Discussion
Our results showed that fluvastatin induced apoptosis via the inactivation of RhoA and Rac1 proteins and the inhibition of the PI3K/Akt pathway in TNFα-stimulated SW982 cells. Further, fluvastatin inhibited SW982 cells in a dose- and time-dependent fashion and induced apoptosis via the mitochondria and caspase pathway. These results were compatible with the apoptotic data of fluvastatin-treated synoviocytes from RA patient20. Our data plus numerous studies support that the human SW982 synovial cell line shares similar physiological and immunological properties such as cytokine expression and signaling profiles with primary human type B synovial cells21,22,23. Furthermore, TNFα stimulation of SW982 might lead to mimicking the inflammatory status of synovial cells, typically seen in RA patients. In that context, SW982 would be useful in comparing the efficacies of a wide array of statins including fluvastin for the treatment of RA. Fluvastatin triggered the PI3K/Akt/Bad signal path, as was evident by the decreased Akt and Bad phosphorylation as well as the increased apoptosis after fluvastatin and specific PI3K inhibitor treatment.
The Akt-dependent signal transduction pathway plays an essential role in preventing apoptosis in various cell death paradigms24,25,26. A growing body of evidence showed that FLS obtained from RA patients expressed higher levels of pAkt38, further incurring synovial hyperplasia and inflammation observed in RA patients27. Interestingly, pre-treatment with an Akt inhibitor, LY294002 enhanced apoptosis in TNFα-stimulated SW982 cells. These findings imply that fluvastatin might effectively reduce the pAkt levels and promote apoptosis of the inflamed synoviocytes in RA patients. Our data demonstrate that reduction of Akt phosphorylation is one of the critical events during fluvastatin-induced apoptosis. Fluvastatin inhibited the proliferation of TNF-α-stimulated SW982 cells through the inhibition of a geranylgeranyl pyrophosphate intermediate. This was in line with fluvastatin-induced apoptosis of the inflamed synoviocytes in RA patients, and further supported by the fact that pleiotropic effects of statins are closely linked to the inhibition of geranylgeranylation of small G proteins20. Such proteins are involved in the survival of non-malignant cells such as endothelial cells, osteoclasts, and cardiac myoblasts28,29,30. Taken together, our results suggest that fluvastatin-induced apoptotic cell death in TNFα-stimulated SW982 human synovial cells may be due to the inhibition of the geranylgeranylation of proteins.
To define the role of RhoA kinase, Y-27632 was used against TNFα-stimulated SW982 cell. Treatment with Y-27632 significantly induced apoptosis indicating that the RhoA/RhoA kinase might confer a survival signal for TNFα-stimulated SW982 cells. These findings were consistent with current studies that RhoA/Rac1-regulated Akt culminated in an anti-apoptotic signal25. However, we could not rule out the possibility that other geranylgeranylated proteins might also be involved. Through PARP cleavage and disrupted MMP, it was confirmed that fluvastatin-induced apoptosis might be mediated via caspase dependent pathway. Further, the decreased expression of proapoptotic molecule, Bad phosporylation strongly pointed out that fluvastatin might induce apoptosis of the inflamed human FLS via PI3K/Akt/Bad, mitochondria and caspase pathway.
In conclusion, our data indicate that fluvastatin induces apoptotic cell death in TNFα-stimulated SW982 human synovial cells through the inactivation of the geranylgerenylated membrane fraction of RhoA and Rac1 proteins and the subsequent inhibition of the PI3K/Akt signaling pathway. This finding confers evidence about the validity of SW982 cell line for RA study.
Acknowledgment
This study was supported by a research grant from Yonsei University Wonju College of Medicine (YUWCM-2007-06), Wonju, Republic of Korea.
References
- 1.Vieira-Sousa E, Gerlag DM, Tak PP. Synovial tissue response to treatment in rheumatoid arthritis. Open Rheumatol J. 2011;5:115–22. doi: 10.2174/1874312901105010115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komatsu N, Takayanagi H. Bone and cartilage destruction in RA and its intervention. Bone and cartilage destruction in rheumatoid arthritis. Clin Calcium. 2012;22:179–85. [PubMed] [Google Scholar]
- 3.Hitchon CA, El-Gabalawy HS. The synovium in rheumatoid arthritis. Open Rheumatol J. 2011;5:107–14. doi: 10.2174/1874312901105010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niedermeier M, Pap T, Korb A. Therapeutic opportunities in fibroblasts in inflammatory arthritis. Best Pract Res Clin Rheumatol. 2010;24:527–40. doi: 10.1016/j.berh.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Miller MC, Manning HB, Jain A, Troeberg L, Dudhia J, Essex D, et al. Membrane type 1 matrix metalloproteinase is a crucial promoter of synovial invasion in human rheumatoid arthritis. Arthritis Rheum. 2009;60:686–97. doi: 10.1002/art.24331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong XM, Wang JC, Shen Y, Xie JJ, Zhang JY, Jin J. Inhibition of inflammatory mediators and related signaling pathways by macrophage-stimulating protein in rheumatoid arthritis synovial fibroblasts. Inflamm Res. 2011;60:823–9. doi: 10.1007/s00011-011-0338-1. [DOI] [PubMed] [Google Scholar]
- 7.Scian R, Barrionuevo P, Giambartolomei GH, De Simone EA, Vanzulli SI, Fossati CA, et al. Potential role of fibroblast-like synoviocytes in joint damage induced by Brucella abortus infection through production and induction of matrix metalloproteinases. Infect Immun. 2011;79:3619–32. doi: 10.1128/IAI.05408-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imaizumi T, Matsumiya T, Yoshida H, Naraoka T, Uesato R, Ishibashi Y, et al. Tumor-necrosis factor-alpha induces retinoic acid-inducible gene-I in rheumatoid fibroblast-like synoviocytes. Immunol Lett. 2009;122:89–93. doi: 10.1016/j.imlet.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Caporali R, Pallavicini FB, Filippini M, Gorla R, Marchesoni A, Favalli EG, et al. Treatment of rheumatoid arthritis with anti-TNF-alpha agents: a reappraisal. Autoimmun Rev. 2009;8:274–80. doi: 10.1016/j.autrev.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2:364–71. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- 11.Yokota K, Miyoshi F, Miyazaki T, Sato K, Yoshida Y, Asanuma Y, et al. High concentration simvastatin induces apoptosis in fibroblast-like synoviocytes from patients with rheumatoid arthritis. J Rheumatol. 2008;35:193–200. [PubMed] [Google Scholar]
- 12.Nanke Y, Kawamoto M, Yago T, Chiba J, Yamanaka H, Kotake S. Geranylgeranylacetone, a non-toxic inducer of heat shock protein, induces cell death in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Mod Rheumatol. 2009;19:379–83. doi: 10.1007/s10165-009-0183-z. [DOI] [PubMed] [Google Scholar]
- 13.Ogata Y, Takahashi M, Takeuchi K, Ueno S, Mano H, Ookawara S, et al. Fluvastatin induces apoptosis in rat neonatal cardiac myocytes: a possible mechanism of statin-attenuated cardiac hypertrophy. J Cardiovasc Pharmacol. 2002;40:907–15. doi: 10.1097/00005344-200212000-00012. [DOI] [PubMed] [Google Scholar]
- 14.Itagaki M, Takaguri A, Kano S, Kaneta S, Ichihara K, Satoh K. Possible mechanisms underlying statin-induced skeletal muscle toxicity in L6 fibroblasts and in rats. J Pharmacol Sci. 2009;109:94–101. doi: 10.1254/jphs.08238fp. [DOI] [PubMed] [Google Scholar]
- 15.Matzno S, Yasuda S, Juman S, Yamamoto Y, Nagareya-Ishida N, Tazuya-Murayama K, et al. Statin-induced apoptosis linked with membrane farnesylated Ras small G protein depletion, rather than geranylated Rho protein. J Pharm Pharmacol. 2005;57:1475–84. doi: 10.1211/jpp.57.11.0014. [DOI] [PubMed] [Google Scholar]
- 16.Connor AM, Berger S, Narendran A, Keystone EC. Inhibition of protein geranylgeranylation induces apoptosis in synovial fibroblasts. Arthritis Res Ther. 2006;8:R94. doi: 10.1186/ar1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao C, Fernandes MJ, Turgeon M, Tancrede S, Di Battista J, Poubelle PE, et al. Specific and overlapping sphingosine-1-phosphate receptor functions in human synoviocytes: impact of TNF-alpha. J Lipid Res. 2008;49:2323–37. doi: 10.1194/jlr.M800143-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Fischetti F, Carretta R, Borotto G, Durigutto P, Bulla R, Meroni PL, et al. Fluvastatin treatment inhibits leucocyte adhesion and extravasation in models of complement-mediated acute inflammation. Clin Exp Immunol. 2004;135:186–93. doi: 10.1111/j.1365-2249.2003.02358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haruna Y, Morita Y, Yada T, Satoh M, Fox DA, Kashihara N. Fluvastatin reverses endothelial dysfunction and increased vascular oxidative stress in rat adjuvant-induced arthritis. Arthritis Rheum. 2007;56:1827–35. doi: 10.1002/art.22632. [DOI] [PubMed] [Google Scholar]
- 20.Nagashima T, Okazaki H, Yudoh K, Matsuno H, Minota S. Apoptosis of rheumatoid synovial cells by statins through the blocking of protein geranylgeranylation: a potential therapeutic approach to rheumatoid arthritis. Arthritis Rheum. 2006;54:579–86. doi: 10.1002/art.21564. [DOI] [PubMed] [Google Scholar]
- 21.Tsuji F, Oki K, Senda T, Horiuchi M, Mita S. Effects of mitogen-activated protein kinase inhibitors or phosphodiesterase inhibitors on interleukin-1-induced cytokines production in synovium-derived cells. Immunol Lett. 1999;68:275–9. doi: 10.1016/s0165-2478(99)00051-6. [DOI] [PubMed] [Google Scholar]
- 22.Kim KO, Park SY, Han CW, Chung HK, Yoo DH, Han JS. Effect of sildenafil citrate on interleukin-1beta-induced nitric oxide synthesis and iNOS expression in SW982 cells. Exp Mol Med. 2008;40:286–93. doi: 10.3858/emm.2008.40.3.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song JS, Kim CH, Heo JY, Cho YS. Rosiglitazone reduces a wide range of proinflammatory profiles in synovial fibroblast SW982 under spheroid culture. Immunol Lett. 2010;131:81–8. doi: 10.1016/j.imlet.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Liagre B, Leger DY, Vergne-Salle P, Beneytout JL. MAP kinase subtypes and Akt regulate diosgenin-induced apoptosis of rheumatoid synovial cells in association with COX-2 expression and prostanoid production. Int J Mol Med. 2007;19:113–22. [PubMed] [Google Scholar]
- 25.Del Re DP, Miyamoto S, Brown JH. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem. 2008;283:35622–9. doi: 10.1074/jbc.M804036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee YJ, Song JH, Oh MH, Lee YJ, Kim YB, Im JH, et al. ERK1/2 activation in quercetin-treated BEAS-2B cell plays a role in Nrf2-driven HO-1 expression. Mol Cell Toxicol. 2011;7:347–55. [Google Scholar]
- 27.Kang EH, Kim DJ, Lee EY, Lee YJ, Lee EB, Song YW. Downregulation of heat shock protein 70 protects rheumatoid arthritis fibroblast-like synoviocytes from nitric oxide-induced apoptosis. Arthritis Res Ther. 2009;11:R130. doi: 10.1186/ar2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo JT, Nakagawa H, Krecic AM, Nagai K, Hamilton AD, Sebti SM, et al. Inhibitory effects of mevastatin and a geranylgeranyl transferase I inhibitor (GGTI-2166) on mononuclear osteoclast formation induced by receptor activator of NF kappa B ligand (RANKL) or tumor necrosis factor-alpha (TNF-alpha) Biochem Pharmacol. 2005;69:87–95. doi: 10.1016/j.bcp.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 29.Maeda S, Sakamoto K, Matsuoka I, Iwamoto T, Kimura J. Lysophosphatidylcholine increases Na+/Ca2+ exchanger expression via RhoB-geranylgeranylation in H9c2 cells. J Pharmacol Sci. 2009;109:565–72. doi: 10.1254/jphs.08253fp. [DOI] [PubMed] [Google Scholar]
- 30.Tang D, Park HJ, Georgescu SP, Sebti SM, Hamilton AD, Galper JB. Simvastatin potentiates tumor necrosis factor alpha-mediated apoptosis of human vascular endothelial cells via the inhibition of the geranylgeranylation of RhoA. Life Sci. 2006;79:1484–92. doi: 10.1016/j.lfs.2006.04.019. [DOI] [PubMed] [Google Scholar]