

Abstract
Sleep disorders are nearly ubiquitous among patients with Parkinson’s disease (PD), and they manifest early in the disease process. While there are a number of possible mechanisms underlying these sleep disturbances, a primary dysfunction of the circadian system should be considered as a contributing factor. Our laboratory’s behavioral phenotyping of a well-validated transgenic mouse model of PD reveals that the electrical activity of neurons within the master pacemaker of the circadian system, the suprachiasmatic nuclei (SCN), is already disrupted at the onset of motor symptoms, although the core features of the intrinsic molecular oscillations in the SCN remain functional. Our observations suggest that the fundamental circadian deficit in these mice lies in the signaling output from the SCN, which may be caused by known mechanisms in PD etiology: oxidative stress and mitochondrial disruption. Disruption of the circadian system is expected to have pervasive effects throughout the body and may itself lead to neurological and cardiovascular disorders. In fact, there is much overlap in the non-motor symptoms experienced by PD patients and in the consequences of circadian disruption. This raises the possibility that the sleep and circadian dysfunction experienced by PD patients may not merely be a subsidiary of the motor symptoms, but an integral part of the disease. Furthermore, we speculate that circadian dysfunction can even accelerate the pathology underlying PD. If these hypotheses are correct, more aggressive treatment of the circadian misalignment and sleep disruptions in PD patients early in the pathogenesis of the disease may be powerful positive modulators of disease progression and patient quality of life.
Keywords: Circadian, dopamine, Parkinson’s disease, Non-motor symptoms of Parkinson’s disease, Sleep, Suprachiasmatic nucleus
Introduction
Epidemiological data indicates that sleep disorders are common in the developed world with an estimated 30 to 40% of the adult population reporting difficulty falling asleep at night and significant daytime sleepiness as a consequence (Hossain and Shapiro, 2002; Leger et al., 2000; Luckhaupt et al., 2010; Skaer and Sclar, 2010; Roenneberg, 2012). These data suggest that many of us are all too familiar with the symptoms of sleep deprivation, including feelings of fatigue, irritability, reduced concentration, reduced motor coordination (Acheson et al., 2007; Anderson et al., 2011; Durmer et al., 2005; Louter et al., 2012). There is also a growing awareness that sleep deprivation is associated with metabolic imbalances and compromised immune response (Bechtold et al., 2010; Litinski et al., 2009; Mullington et al., 2009). These changes can occur even with transient sleep deprivation in which case they return to baseline with sufficient restorative sleep. Unfortunately, in chronically ill patients, restorative sleep is often permanently impaired. There is increasing evidence that in the case of neurodegenerative disorders, sleep disorders are extremely common, if not ubiquitous, and occur early in the disease progression (Chokroverty, 2009; Gagnon et al., 2008). These sleep disturbances predate the onset of the cognitive and motor symptoms and have significant negative consequences for both patients and caregivers, and if recognized, may facilitate earlier diagnosis and treatment.
Parkinson’s disease (PD)
PD is the most common movement disorder among older adults, and is a leading cause of cognitive decline and dementia (Pontone et al., 2012; Williams-Gray et al., 2007). The classical triad of clinical features in PD consists of worsening resting tremor, rigidity, and bradykinesia. Pathologically, PD patients exhibit a progressive loss of dopaminergic neurons and the formation of Lewy bodies in the substantia nigra pars compacta (SNpc). Until relatively recently, it had been thought that it was this loss of pigmented, dopaminergic neurons in the SNpc that accounted for the symptoms of the disease. However, it is increasingly clear that PD is a multisystem disorder in which numerous brain structures are affected during the course of the illness (Braak et al., 2006; Jain, 2011; Jellinger, 2010). For example, the neuropsychiatric aspects of PD are particularly prominent, including cognitive impairment that progresses to dementia in approximately 30-40% of patients and depression in up to 40% of patients. Anxiety, apathy, personality changes, and sleep disorders are also common (Cummings and Mega, 2003; Blonder and Slevin, 2011). Other well established non-motor symptoms of PD include metabolic abnormalities, altered olfaction, cardiovascular dysfunction, gastrointestinal problems and sleep disturbances (Chaudhuri and Odin, 2010; Claassen et al., 2010; Menza et al., 2010; Park and Stacy, 2009; Ziemssen and Reichmann, 2010). One of the most interesting aspects of these non-motor symptoms of PD is that they may manifest decades prior to the onset of motor symptoms.
Sleep disorder are common in PD
Sleep disorders are extremely common in PD, with up to 90% of patients reporting primary insomnia, restless legs syndrome, hypersomnia, rapid eye movement (REM) sleep disorder (Ferreira et al., 2006; Matsui et al., 2006; Stevens et al., 2004; Thorpy and Adler, 2005). These latter two syndromes of REM sleep disorder and hypersomnia appear to occur well in advance of the motor symptoms of PD (Abbott et al., 2005; Boeve et al., 2007; Boeve et al., 2010; Claassen et al., 2010; Iranzo et al., 2009; Iranzo et al., 2010; Postuma et al., 2006; Schenck et al., 1996). The sleep disorders in PD patients are due to multiple causes, including motor disability, loss of function in the sleep control centers located in the brain stem, off-target effects of the drugs used to treat the motor symptoms, and possibly circadian dysfunction (Ahlskog, 2011; Diederich and McIntyre 2012; Mehta et al., 2008; Videnovic and Golombek 2012; Yong et al. 2011). Sleep is generally viewed as being regulated by the combination of two predominant, sometimes competing processes: one being a homeostatic load accumulated of sleep need based on time since last sleep episode, termed “Process S,” and another being a circadian-controlled rhythm in wakefulness or sleep, termed “Process C”. The potential impact of a disrupted circadian system in PD, has received relatively little attention in the literature. In this review, we will focus on the potential role of disrupted circadian rhythms in the pathogenesis of the non-motor symptoms in PD. While circadian disruption is unlikely to mediate these non-motor symptoms, we feel that it can be a contributing factor that may determine the severity of the symptoms. Since circadian disruptions are likely early in the disease progression, improved understanding of this aspect of PD will likely have important implications for improving early diagnosis and treatment.
Circadian system
In humans and other mammals, the circadian system is made up of a network of oscillators. The central clock is located in the suprachiasmatic nucleus (SCN). Neurons in this cell population receive light information from melanopsin-expressing retinal ganglion cells found in our retina. The axons of these ganglion cells make a direct synaptic connection onto cells in the SCN. These SCN neurons integrate this photic information with other timing cues to generate robust circadian oscillations that are synchronized to the environment. SCN outputs largely project to other hypothalamic regions, including the subparaventricular zone and other medial hypothalamic structures surrounding the SCN. These hypothalamic relay nuclei send projections throughout the nervous and endocrine systems, providing multiple pathways by which the SCN output conveys temporal information about the environment to the rest of the brain and body. Among the pathways that are particularly relevant for PD, SCN appears to regulate the levels of neural activity in the major subcortical arousal centers, including the locus coeruleus and raphe which increase their firing rate during the animal’s active period (e.g. Aston-Jones et al., 2001; Deurveilher and Semba, 2005). Through these multi-synaptic pathways, the SCN is well positioned to modulate the arousal levels of the nervous system. Additionally, output from physiological systems receiving circadian regulation can “feed-back” to alter the master clock. For example, both sleep states (Deboer et al., 2003) and locomotor activity (Yamazaki et al., 1998; Schaap and Meijer, 2001) feed-back to regulate SCN neural activity recorded in vivo. These physiological studies demonstrate the difficulty in attempting to disentangle the role of the circadian system and sleep on behavior and physiology, because it may not be possible to alter sleep without impacting the circadian system.
Signals from the SCN travel out via the hypothalamic-pituitary adrenal axis as well as through the autonomic nervous system to regulate independent circadian oscillators found throughout the body. The function of this circadian regulation is likely to vary with the tissue, with each of the major organ systems (e.g. heart, liver, islets) apparently having its own clockwork to regulate the transcription of genes that are important to the specific target organ (see Dibner et al., 2010; Mohawk et al., 2012). Using DNA microarray expression profiling it has been found that approximately 8 to 12% of genes display circadian oscillations (Hogenesch et al., 2003; Panda et al., 2002; Storch et al, 2002). Many of these genes are involved in key rate-limiting steps of biochemical pathways (Baggs and Hogenesch, 2010; Panda et al., 2002). For example, in the nervous system, a number of genes involved in peptide synthesis, secretion, and oxidative phosphorylation are transcribed with a circadian oscillation. The key point here is that the circadian timing controls the temporal patterning of molecular, cellular, and physiological processes throughout the body and the potential disruption of this timing system in PD would be expected to produce wide-spread symptoms.
Dysfunction in the circadian system may contribute to the etiology of the non-motor symptoms of PD
Several of the prominent non-motor symptoms of PD have a diurnal, temporal component that suggests an underlying circadian dysfunction. Most striking are the various types of sleep disruptions reported by PD patients: increased sleep latency, decreased sleep maintenance, fragmented sleep, and excessive daytime sleepiness. All of these symptoms may reflect alterations in the temporal patterning of sleep which often result from circadian dysfunction (Abbott et al., 2005; Dhawan et al., 2006; Mayer et al., 2011; Reid and Zee, 2009; Schulte and Winkelmann, 2011). Therefore, circadian disruption should be considered one of the factors which contributes to the insomnia and hypersomnia experienced by PD patients. Other sleep disorders seen so commonly in PD are less likely to involve the circadian system. In patients with REM sleep disorder, there is a loss of somatic muscle atonia during the REM phase of sleep which is thought to be due to damage to the medullar and pontine structures.Without this inhibition of muscular tone, patients with REM sleep disorder exhibit abnormal motor behavior during REM usually resulting in minor repetitive limb movements that can be ameliorated to some degree with melatonin or clonazepam (Arnulf et al., 2012: Kunz and Mahlberg, 2010; Mayer et al., 2011). While the circadian system controls the timing of REM sleep, we do not believe that there is any direct evidence that the SCN is directly involved in inhibiting motor tone during REM sleep. As far as we know, the possibility of this link has not been explored experimentally.
PD patients commonly exhibit disruption in other physiologic processes that are known to be influenced by the circadian system. For example, the autonomic nervous system is subject to circadian regulation; the balance between sympathetic and parasympathetic tone varies in synchrony with the daily circadian cycle (Jain and Goldstein, 2012). In healthy individuals, parasympathetic tone predominates during night-time/sleep and acts to reduce heart rate and blood pressure. Mechanistically, the central circadian clock in the SCN projects to the pre-autonomic neurons in the paraventricular nucleus (PVN) to effect these changes in autonomic tone (Buijs et al., 2003). Numerous studies have found that the circadian regulation of the autonomic nervous system is disrupted in PD patients (Kallio et al., 2000) and this disruption drives changes in blood pressure and heart rate. Approximately 30-40% of PD patients have orthostatic hypotension, which has been linked with cardiac sympathetic denervation as well as combined sympathetic/parasympathetic dysfunction of the arterial baroreflex (Jain, 2011). These cardiac symptoms are independent of the weakness associated with SNpc deterioration, are associated with Lewy body deposition in catecholaminergic neurons, and are thought to become apparent early in the course of the disease. Disruptions in the circadian system may well act in concert with the loss of the catecholaminergic neurons to cause the autonomic symptoms seen in PD.
To provide another example, urinary tract symptoms represent another manifestation of dysregulated autonomic nervous system control in PD patients. Urinary tract symptoms, such as nocturia, increased urgency and frequency of urination, and incontinence are nearly as common in PD patients as are sleep disturbances (Yeo et al., 2012). In one study, over 75% of PD patients complained of nocturia, while between 30 and 35% of patients complained of either urinary urgency or frequency (Ragab and Mohammed, 2011). Furthermore, this study showed no difference between urodynamic parameters in untreated PD patients and those who were treated with dopaminergic medications. This suggests that urinary tract symptoms are not adequately treated with these agents, which are the mainstay of pharmacologic treatment of PD. On one level, circadian disruption of sleep architecture contributes to more frequent nighttime waking which alone might increase the likelihood that a patient will decide to urinate. The SCN is also known to regulate the osmotic pressure sensing cells that are responsible for the nocturnal increase in arginine vasopression (also known as anti-diuretic hormone) secretion, which in turn reduces the volume of nighttime urine production (Colwell, 2010; Trudel and Bourque, 2010). In addition, disruption of the circadian control of the autonomic nervous system, which normally promotes nocturnal relaxation of the bladder wall and increased urethral sphincter tone, might lead to abnormal bladder contraction and relaxation of the urethral sphincter resulting in an increased risk of nocturia and urinary incontinence.
While it is often challenging to parse the relative contributions of insomnia, circadian dysfunction, and motor disturbances to a particular PD-associated symptom, little clinical attention has been directed toward normalizing circadian-controlled physiologic functions. A better appreciation of the broad effects that result from circadian rhythm dysregulation in PD patients might offer new insights into the etiology of non-motor symptoms and suggest novel therapeutic interventions. The circadian system represents a compelling target as a powerful potential modulator of disease progression and patient quality of life.
Dopaminergic treatments for the core motor symptoms of PD may contribute to the disruption of the sleep/wake cycle
Central DA is generally associated with arousal and a variety of evidence suggests that this transmitter is involved in the regulation of the sleep/wake cycle at multiple circuits. Overall, levels of DA appear to exhibit low amplitude, daily rhythm in humans (Poceta et al., 2009) and in mice (Hampp et al., 2008). Centrally, DA levels are modulated by monoamine oxidase A (MAO-A) and monoamine oxidase B (MAO-B), which are key enzymes that regulate the catabolism of several different monoamine neurotransmitters (Shih and Chen, 2004). Importantly, MAO-A appears to be a clock-controlled gene. A mutation in the circadian clock gene Period2 (Per2) in mice leads to reduced expression and activity of MAO-A in the mesolimbic dopaminergic system, which results in increased DA levels and changes in electrical activity in the striatum (Hampp et al., 2008). So any disruption of the circadian clockwork could be expected to alter levels of DA via MAO-A activity.
Since the motor symptoms are driven by the loss of dopaminergic neurons in the SNpc, the general treatment strategy has been to provide a blood-brain barrier-permeable metabolic precursor in the dopamine biosynthetic pathway with the goal of increasing synaptic concentrations of DA in the SNpc and striatum. DA and dopaminergic drugs, including L-DOPA, are well known to modulate clock gene expression (Hood et al., 2010; Imbesi et al., 2009). Furthermore, there is an extensive literature of rodent studies documenting the powerful circadian effects of dopaminergic drugs such as cocaine and methamphetamine, including profound disruption of sleep (Glass et al., 2012; Honma and Honma, 2009; Ironside et al., 2010). For example, continuous administration of methamphetamine in drinking water alters the rhythm of circadian activity in rats and mice (Honma et al., 1986; Tataroglu et al., 2006). Interestingly, methamphetamine treatment can even restore robust circadian activity in mice whose SCN had been electrolytically lesioned (Honma et al., 1987) demonstrating that dopaminergic drugs can modulate the circadian system even without a functional central circadian clock in the SCN.
Taken together, it seems likely that the very drugs that are prescribed to alleviate the motor symptoms of PD (or related disorders like restless leg syndrome which is similarly treated with dopamine agonists) are themselves contributing to the sleep disturbances in these patients (Rye, 2004; Santiago et al., 2010). Specifically, the data suggests that low doses of pharmacological agents that increase dopamine can improve sleep while higher doses delay the onset of sleep and decrease total sleep time (Diederich and McIntyre, 2012; Schafer and Greulich, 2000; Yong et al, 2011). Further work will be required to see if controlling the timing of the dosing of the L-DOPA and related compounds can minimize the undesirable sleep disruptions caused by dopaminergic medications (Wailke et al., 2011). For example, one simple prediction is that using pharmacological tools to reinforce the normal daily rhythm of DA with higher levels of DA activity during the day with lower levels at night may improve motor function without disrupting sleep and circadian function.
Several models of PD show sleep and possible circadian disruption
The loss of DA neurons may play a role in the circadian disruption observed in non-human primates. One of the most developed models of PD involves treating primates with the toxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) which produces a set of symptoms that resemble PD (Fox and Brotchie, 2010) and can be treated with dopaminergic drugs. The MPTP-treated primates exhibit an immediate disruption of the sleep/wake cycle (Vezoli et al., 2011) as well as alteration in REM sleep and increased daytime sleepiness (Almirall et al., 1999; Barraud et al., 2009; Verhave et al., 2011). Interestingly, mice treated with MPTP exhibit a significant loss of dopaminergic neurons (approximately 50%) without disruption of the circadian rhythms (Laloux et al., 2008; Tanaka et al., 2012). The injection with the toxin 6-hydroxydopamine produces a larger loss of neurons that does disrupt behavioral and clock gene expression rhythms in the rat (Gravotta et al., 2011).
Loss of function in dopaminergic cells has also been linked to non-motor symptoms, including sleep and circadian disruptions in other models. For example, mice deficient in vesicular monoamine transporter expression (VMAT2) exhibit severely diminished levels of the major monoamines, including DA, 5-HT and NE (Taylor et al, 2009). These mice exhibit many of the non-motor symptoms associated with PD, including altered latency to sleep and reduced amplitude of their diurnal rhythm. It would be interesting to examine the circadian behavior of these mice. In Drosophila, the targeted expression of mutant α-synuclein to monoamine (5HT, DA) expressing neurons resulted in altered sleep and circadian activity (Gajula Balija et al., 2011). It is worth noting that sleep disturbances have been reported in patients with mutations in the Parkin and DJ-1 genes (Kumru et al., 2004; Limousin et al., 2009; Lo Coco et al., 2009; Nishioka et al., 2009) which again highlights the importance of rigorously examining a range of PD models (Dawson et al., 2010; Magen et al., 2010) for possible circadian dysfunction.
α-synuclein over-expressing mice as model of synucleinopathies, including PD
One of the best studied models of PD and other synucleinopathies is a line of transgenic mice expressing human α-synuclein (aSyn) under the Thy-1 promoter: the α-synuclein over-expressing (Thy1-aSyn) mice (Rockenstein et al., 2002). Genetic mutations in, and duplication of, α-synuclein are strongly associated with familial forms of PD; and polymorphisms in this gene are associated with increased PD risk (Cookson et al, 2009; Pankratz et al., 2009; Ritz et al., 2012; Simón-Sánchez et al., 2009; Vekrellis et al., 2011; Winkler et al., 2007; Zarranz et al., 2005). Prior work has shown that at 2-3 months of age, Thy1-aSyn mice begin to exhibit progressive impairments in motor and non-motor symptoms that are analogous to those experienced by human PD patients; these include deficits in olfaction, cognition, and control of the autonomic nervous system (Fleming et al., 2004; Fleming et al, 2006; Fleming et al., 2008; Fleming and Chesselet, 2009). Thus, the Thy1-aSyn transgenic mouse provides an excellent model system to improve our understanding of the basic pathophysiologic mechanisms that cause PD, possibly including the early non-motor symptoms and circadian-related symptoms.
Thy1-aSyn mice show selective deficits in circadian-regulated behavior, including the temporal distribution of sleep and activity
As measured by wheel running activity under either a standard 12 hour light and 12 hour dark cycle (LD), or under a continuous dark cycle (DD), the observed circadian cycle of all Thy1-aSyn mice initially appeared to be grossly rhythmic (Kudo et al., 2011b). However, the Thy1-aSyn mice exhibited fragmented, weak (low power) rhythms under both LD and DD conditions. These deficits are clearly illustrated when the mice are placed in a skeleton photoperiod consisting of two 1-hr light exposures every 24-hrs. The mice synchronize nicely and these lighting conditions allow us to see the clock driven locomotor output without the direct activity-suppressing influence of light (Fig. 1). The peak amplitude of the behavioral rhythms and coherence (measured by fragmentation) under both LD and DD conditions progressively declined over the lifespan of both Thy1-aSyn and control wild-type (WT) mice.
Interestingly, the young (3 month old) Thy1-aSyn mice exhibited a circadian output similar to that of an old WT mouse. Our behavioral analysis demonstrates that by young adulthood, the Thy1-aSyn mouse exhibits disrupted daily circadian rhythms in wheel-running activity that worsened with increasing age and motor dysfunction, leading to greater deterioration in many critical parameters, such as amount and fragmentation of activity, as well as the power of rhythms. Distribution of sleep was altered at 6 months of age, when we observed a delay in the onset of behavioral sleep as measured by video recordings, whereas the total amount of sleep did not vary between the genotypes.
Other key circadian parameters are not altered in Thy1-aSyn mice
The core of the oscillatory clock that generates circadian rhythms in the SCN consists of an evolutionarily conserved transcriptional/translational feedback loop that drives rhythmic activity of key clock genes such as Per2 (Hastings et al., 2003), which in turn drive the oscillation of action potentials in SCN neurons that project to other regions of the brain. A normally functioning circadian clock in the SCN would cause SCN Per2 expression levels to be high during daylight and low in darkness. We were surprised to observe that Thy1-aSyn mice that have already started to show abnormal wheel-running behavior continued to show this normal WT pattern of oscillating PER2 expression (Fig. 2) (Kudo et al., 2011b). Furthermore, even at the relatively advanced age of 12 months, Thy1-aSyn mice maintained robust rhythmic oscillations in SCN PER2 expression.
Elimination of dopaminergic neurons by administration of the neurotoxin 6-hydroxydopamine has been shown to alter clock gene expression in the forebrain (Gravotta et al., 2011). Thus, possible alterations in clock gene expression later in disease progression or in brain areas outside the SCN should be evaluated in the Thy1-aSyn line at a time when changes in DA are observed. It is also worth noting that the Thy1-aSyn mice did not show any alteration in the free-running period of the behavioral rhythm in DD. Changes in the circadian period length are indicative of a disruption in the underlying circadian pacemaker system (Takahashi et al., 2008b). Therefore, our characterization of the Thy1-aSyn mice suggests that, at least during the early stages of disease, disruptions in periodicity are not the result of deficits in the molecular oscillations in the circadian system or its inputs, but in the downstream outputs of this system.
In Thy1-aSyn mice, the firing rate of SCN neurons is reduced early in the progression of PD
SCN neurons are spontaneously active and generate action potentials with peak activity during the day (Colwell, 2011). In the daytime, we found that the excitability of SCN neurons was significantly reduced in the Thy1-aSyn mice (Fig. 3) (Kudo et al., 2011b). At this age (3 months) we did not see evidence of cell loss within the SCN; however, we have not yet looked at older mice. In the Thy1-aSyn model, firing rate is also dramatically reduced in the striatal medium spiny neurons (Wu et al., 2010) so decreased electrical activity may be a common feature of this model. Neurons in the SCN drive the rhythmic output of the circadian system via regulation of the autonomic nervous system (Kalsbeek et al., 2006) and other neural and hormonal pathways (Dibner et al., 2010). The decrease in the daytime electrical activity that we observe in the SCN of Thy1-aSyn mice would be expected to weaken the temporal patterning of both the neural and hormonal outputs. Our observations are consistent with the hypothesis that decreased amplitude of efferent signals from the SCN could contribute to the non-motor symptoms in PD (Fig. 4).
One mechanism by which aSyn over-expression could reduce SCN neural output is through changes in synaptic transmission. aSyn is a pre-synaptic protein that regulates synaptic vesicle release, and its mis-expression alters synaptic transmission (Burre et al., 2010; Cabin et al., 2002; Wakabayashi et al., 1997). Recent work in a variety of mouse models of other neurodevelopmental and psychiatric disorders suggests that alterations in the balance between synaptic excitation and inhibition are a core pathophysiologic feature of these conditions (Dani et al., 2005; Gogolla et al., 2009; Milnerwood et al., 2010; Nelson and Turrigiano, 2008; Shepherd and Katz, 2011). Within the SCN circuit, the release of glutamate and the neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) from retinal ganglion cells mediates the effects of light on the retino-recipient SCN neurons (Colwell, 2011; Morin and Allen, 2006). For neurons contained within the SCN circuit, the neurotranmitter used is gamma-aminobutyric acid (GABA) (Moore and Speh, 1993; Okamura et al., 1989); most neurons receive a constant flux of GABA signaling (Itri et al., 2004; Jiang et al., 1997; Kim and Dudek, 1992; Strecker et al., 1997). The circadian symptoms of the Thy1-aSyn mice could be explained if aSyn over-expression tilted the balance toward increased inhibition within the SCN circuit.
An alternate explanation lies with the currents which underlie daily rhythms of spontaneous electrical activity of SCN. During the day, SCN neurons are relatively depolarized to keep them near the threshold for generating an action potential (-45 mV). This relatively depolarized resting potential is the result of excitatory drive provided by multiple cation currents (Jackson et al., 2004; Kononenko et al., 2004; Pennartz et al., 1997). Changes in these currents could underlie the decreased daytime firing observed in the Thy1-aSyn mice. In response to this depolarized membrane potential, SCN neurons exhibit sustained discharge for 4-6 hours in the subjective day without spike adaptation. Prior work suggests that 3 potassium (K+) currents, including fast delayed rectifier (FDR), subthreshold-operating A-type (IA) and large-conductance Ca2+ activated (BK) all play a critical role in the regulation of spontaneous action potential firing in SCN neurons during the day (See Colwell, 2011). Reduction in magnitude of the FDR and the BK currents would have the consequence of decreasing the daytime firing rate in the SCN (Kudo et al., 2011a; Montgomery and Meredith, 2012). Interestingly, a recent study (Farajnia et al., 2012) found that aging selectively impacts K+ (IA and FDR) currents in the SCN currents, which reduces the synchrony of the SCN cell population.
The decreased firing rate of SCN neurons in Thy1-aSyn mice may be due to mitochondrial dysfunction and oxidative stress
Although aSyn is primarily thought to be important for synaptic vesicle release and recycling, there is increasing evidence of its colocalization with the mitochondrial membrane (Li et al., 2007, Nakamura et al., 2008). Furthermore, mitochondrial function can be impaired upon mis-expression of aSyn (Martin et al., 2006; Xie and Chung, 2012) and conversely, the mitochondrial toxin MPTP leads to aSyn accumulation (Purisai et al., 2005). Other mouse genetic models of PD, also show altered mitochondrial function and increased oxidative stress (reviewed in Abou-Sleiman et al., 2006; Trancikova et al., 2012). SCN neurons require an energy-demanding sodium-potassium pump (Na+/K+-ATPase) to maintain the resting membrane potential during daytime peak firing (Wang and Huang, 2006). Without sufficient ATP, the neural membrane would depolarize, be unable to generate action potentials, thus dampening the output signals from the SCN. These data support the hypothesis that one cause of decreased electrical activity in the SCN is a decrease in mitochondrial function (Martin et al., 2006; Schapira, 2012).
One likely consequence of the circadian disruption seen in the Thy1-aSyn mice is an increase in oxidative stress and inflammation. Cellular metabolism results in the generation of by-products of oxygen (O2) known as reactive oxygen species (ROS). The reduction of O2 to H2O gives rise to superoxide anion, hydrogen peroxide, and hydroxyl radical which can all be damaging to the cell. Therefore, cells control the production of ROS and manage the negative consequences by the production of anti-oxidants. A long body of work from both plants and animals indicates that both the production of ROS as well as cellular anti-oxidants is temporally controlled by the circadian system (Edgar et al., 2012; Khapre et al., 2011; Kondratova and Kondratov, 2012; Hardeland et al., 2003; Lai et al., 2012; O’Neill and Reddy, 2011;Wang et al., 2012). Deletion of the critical clock gene BMAL1 leads to mitochondrial dysfunction, including increases in ROS in peripheral organs (Khapre et al., 2011; Kondratov et al., 2006; Lee et al., 2011). Mitochondrial ROS and oxidative stress have been consistently implicated in disease and age-related decline in tissues throughout the body (Balaban et al., 2005; Dai et al., 2010; Dutta et al., 2012; Labunskyy and Gladyshev, 2012; Liu et al., 2012; Wanagat et al., 2010). Although the molecular oscillator does not appear to be disrupted in the SCN of Thy1-aSyn mice, the effects of the overexpression of aSyn may disrupt ROS levels to affect neuronal firing. While oxidative stress damages a range of cellular processes, it is worth noting that K+ channels can be quite sensitive to oxidative damage (Cotella et al., 2012; Sesti et al., 2010). Therefore, in the SCN, an increase in oxidative damage to K+ channels may underlie the decrease in daytime firing observed in Thy1-aSyn mice.
Likewise, many immune parameters show daily and circadian variation, including levels of cytokines (e.g. Logan and Sarkar, 2012). The molecular clock is found in many of the key cells involved in the immune response and disruption of the circadian system alters the immune response (e.g. Castanon-Cervantes et al., 2010; Froy and Chapnik, 2007; Keller et al., 2009). Two recent studies have found that clock proteins can directly regulate the expression of proinflammatory cytokines through the NF-κB signaling pathway (Narasimamurthy et al., 2012; Spengler et al., 2012). Therefore, we speculate that a second consequence of circadian disruption in the Thy1-aSyn mice is changes in the NF-κB pathway which will result in pathological inflammation throughout the body. Within the SCN, increases in cytokines are known to disrupt rhythms in firing rate (Kwak et al., 2008; Lunkvist et al, 1998; 2010).
Increases in oxidative stress and inflammation due to circadian dysfunction in combination with aSyn aggregation could accelerate the pathology of PD. If this hypothesis is correct, circadian disruption may be a risk factor for PD. Many PD models consider a two-hit model in which genetic risk factors coincide with environmental perturbations to lead to the disease (Boger et al., 2010; Hawkes et al., 2007). We propose that circadian disruption and the resulting increase in chronic inflammation, mitochondrial dysfunction, oxidative stress, and DNA damage may be an environmental risk factor for developing PD, and may also serve to accelerate the pathology of the disease (Fig. 5).
Future directions
These types of disruptions of the circadian system that are caused by altered coupling within the SCN circuit are likely to have profound consequences on patient health (Bechtold et al., 2010; Karatsoreos et al., 2011; Hastings et al., 2003; Reddy and O’Neill, 2010; Takahashi et al., 2008a). There is mounting evidence that robust circadian rhythms are a necessary component to optimum health. In recent years, a wide range of studies have demonstrated that disruption of the circadian system leads to a cluster of symptoms, including cognitive deficits and psychiatric symptoms (Gerstner and Yin, 2010; Loh et al., 2010; Ruby et al., 2008; Wang et al., 2009), metabolic deficits (Gale et al., 2011; Marcheva et al., 2010; Turek et al., 2005), cardiovascular problems (Bray et al., 2008; Scheer et al., 2009), gastrointestinal problems (Gimble et al., 2011; Jain, 2011), and the increased risk for certain cancers (Monsees et al., 2012). Many of these same symptoms are described in patients with PD. Our results from the analysis of Thy1-aSyn mice support these observations. This combined with the observation that many of the symptoms predate the appearance of PD-related motor symptoms by years, raises the possibility that circadian dysfunction is not merely a symptom of PD, but rather is a core, perhaps even pathogenic, component of the disease.
Our hypothesis suggests that placing a greater emphasis on the development of pharmacological tools and behavioral interventions (Table 1) that can boost circadian output and synchrony of the SCN may be therapeutic in PD patients; perhaps especially in PD patients early during the course of the disease. Interventions should be designed to strengthen output and re-synchronize the central and peripheral circadian clocks, i.e. focus on the amplitude of the circadian output. New work suggests that the circadian system may be targeted with pharmacologic therapies and that high-throughput chemical screens can be applied to develop these potential treatments (Chen et al., 2012; Hirota et al., 2010). We would suggest that candidate molecules that boost amplitude may be particularly useful to counter the circadian dysfunction associated with PD. Together, the optimization of these strategies already in use and the development of new, targeted interventions aimed at restoring circadian functioning will provide promising avenues toward improving the prognosis of patients with PD.
Highlights.
Sleep disorders are common among PD patients and manifest early in the disease.
α-synuclein over-expressing (Thy1-aSyn) mice show circadian dysfunction.
In Thy1-aSyn mice, the electrical output of the central clock is compromised.
Circadian dysfunction may contribute to the pathology of PD.
Fig. 1.

Thy1-aSyn mice show an age-related decline in daily and circadian rhythms. Top panels show representative examples of wheel-running activity records from WT (left) and Thy1-aSyn (right) mice. Animals were initially entrained to 12:12 LD, then placed into a skeleton photoperiod (1: 11:1:11 LD). Each horizontal row represents the activity record for a 24-hr day that is then double plotted. Successive days are plotted from top to bottom. Gray shaded area represents darkness. Bottom panels show examples of average waveforms that illustrate the distribution of activity for WT (left) and Thy1-aSyn (right) mice. Besides the striking reduction in the amplitude of activity, the Thy1-aSyn mice exhibited a decrease in precision of the beginning of the nightly activity cycle and an increase in fragmentation of their activity. Data from Kudo et al., 2011b.
Fig. 2.
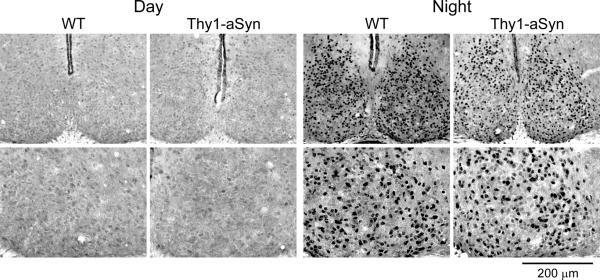
Thy1-aSyn mice did not show alterations in the PER2 rhythm within the SCN. Mice were held in DD and wheel running activity was measured to determine circadian phase. IHC was used to measure PER2 immunoreactivity in the SCN of Thy1-aSyn and WT controls. Tissue was collected in the subjective day or subjective night. Photomicrographs of SCN tissue of each genotype in low (10X) and higher (40X) magnification at 3 mo of age. Data from Kudo et al., 2011b.
Fig. 3.
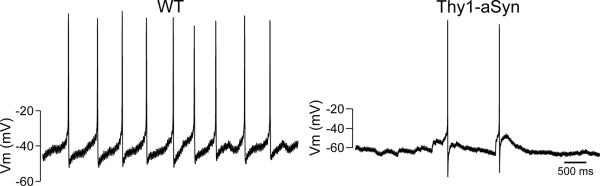
Thy1-aSyn mice show significantly reduced daytime spontaneous neural activity in the SCN. Using the current-clamp recording technique in the whole-cell patch clamp, the spontaneous firing rate in SCN neurons during the day was measured. The panels show representative examples of firing rate recorded from the WT and Thy1-aSyn mice at each time point. Data from Kudo et al., 2011b.
Fig. 4.

Circadian phenotype in Thy1-aSyn mice (3 mo) compared to middle aged WT mice (12 mo). In our laboratory, we measured the circadian phenotype of WT mice at 12 mo of age (Nakamura et al., 2011). We found significant reduction in activity output, including reduced amplitude, coherence and precision of wheel running activity rhythms. Physiological outputs of heart rate and body temperature are also reduced at this age. Finally, we found that the amplitude of SCN neural activity rhythms but not PER2 expression was reduced at middle age. The phenotype of the Thy1-aSyn mice at 3 mo of age is similar to that seen in a middle aged WT mouse. To date, we have not yet examined the physiological rhythms in the Thy1-aSyn mice.
Fig. 5.
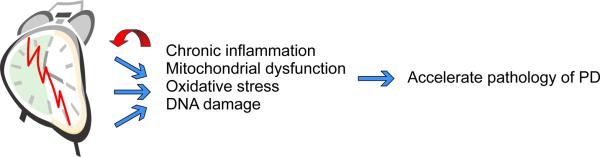
Potential mechanisms by which circadian dysfunction could accelerate the pathology of PD. The molecular clockwork regulates mitochondrial function, reactive oxygen species homeostasis, DNA repair and immune response. Dysfunction of this timing system is likely to contribute to chronic inflammation, mitochondrial dysfunction, and DNA damage. These processes are all thought to contribute to the pathology of PD and contribute to age-related changes in the brain. Therefore, we raise the possibility that circadian dysfunction due to genetic or environmental perturbations can accelerate the pathology of PD.
Table 1.
Possible Interventions to Strengthen the Circadian System
|
|
|
|
|
|
|
|
|
|
|
Acknowledgments
We acknowledge the support and encouragement from our colleagues at UCLA, including Drs. Chesselet and McCracken. We also thank Dr. Nurmi for insightful comments on a draft of the manuscript. Finally, we thank Ms. Donna Crandall for assistance with the graphics.
Abbreviations
- aSyn
α-synuclein
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- DD
Dark-Dark
- DA
Dopamine
- FDR
Fast delayed rectifier
- GABA
Gamma-aminobutyric acid
- BK
Large-conductance Ca2+ activated K+ channels
- LD
Light-Dark
- MAO
Monoamine oxidase
- PVN
Paraventricular nucleus
- PD
Parkinson’s disease
- Per2
Period2
- K+
Potassium
- REM
Rapid eye movement
- ROS
Reactive oxygen species
- RBD
REM sleep behavior disorder
- SNpc
Substantia nigra pars compacta
- SCN
Suprachiasmatic nuclei
- WT
Wild-type
References
- Abbott RD, Ross GW, White LR, Tanner CM, Masaki KH, Nelson JS, Curb JD, Petrovitch H. Excessive daytime sleepiness and subsequent development of Parkinson disease. Neurol. 2005;65:1442–1446. doi: 10.1212/01.wnl.0000183056.89590.0d. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat. Rev. Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- Acheson A, Richards JB, de Wit H. Effects of sleep deprivation on impulsive behaviors in men and women. Physiol. Behav. 2007;91:579–87. doi: 10.1016/j.physbeh.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Ahlskog JE. Pathological behaviors provoked by dopamine agonist therapy of Parkinson's disease. Physiol. Behav. 2011;104:168–72. doi: 10.1016/j.physbeh.2011.04.055. [DOI] [PubMed] [Google Scholar]
- Almirall H, Pigarev I, de la Calzada MD, Pigareva M, Herrero MT, Sagales T. Nocturnal sleep structure and temperature slope in MPTP treated monkeys. J. Neural Transm. 1999;106(11-12):1125–34. doi: 10.1007/s007020050228. [DOI] [PubMed] [Google Scholar]
- Anderson C, Platten CR. Sleep deprivation lowers inhibition and enhances impulsivity to negative stimuli. Behav Brain Res. 2011;217:463–466. doi: 10.1016/j.bbr.2010.09.020. [DOI] [PubMed] [Google Scholar]
- Antoch MP, Kondratov RV. Circadian proteins and genotoxic stress response. Circ Res, 2010;106:68–78. doi: 10.1161/CIRCRESAHA.109.207076. [DOI] [PubMed] [Google Scholar]
- Arnulf I. REM sleep behavior disorder: Motor manifestations and pathophysiology. Mov Disord. 2012;27:677–89. doi: 10.1002/mds.24957. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Chen S, Zhu Y, Oshinsky ML. A neural circuit for circadian regulation of arousal. Nat. Neurosci. 2001;4(7):732–8. doi: 10.1038/89522. [DOI] [PubMed] [Google Scholar]
- Baggs JE, Hogenesch JB. Genomics and systems approaches in the mammalian circadian clock. Curr. Opin. Genet. Dev. 2010;20(6):581–7. doi: 10.1016/j.gde.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Barone P, Amboni M, Vitale C, Bonavita V. Treatment of nocturnal disturbances and excessive daytime sleepiness in Parkinson's disease. Neurol. 2004;63(8 Suppl 3):35–8. doi: 10.1212/wnl.63.8_suppl_3.s35. [DOI] [PubMed] [Google Scholar]
- Barraud Q, Lambrecq V, Forni C, McGuire S, Hill M, Bioulac B, Balzamo E, Bezard E, Tison F, Ghorayeb I. Sleep disorders in Parkinson's disease: the contribution of the MPTP non-human primate model. Exp. Neurol. 2009;219(2):574–582. doi: 10.1016/j.expneurol.2009.07.019. [DOI] [PubMed] [Google Scholar]
- Bechtold DA, Gibbs JE, Loudon AS. Circadian dysfunction in disease. Trends Pharmacol. Sci. 2010;31:191–8. doi: 10.1016/j.tips.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Blonder LX, Slevin JT. Emotional dysfunction in Parkinson's disease. Behav. Neurol. 2011;24:201–17. doi: 10.3233/BEN-2011-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman-Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, Del Tredici K, Braak H. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007;130:2770–88. doi: 10.1093/brain/awm056. [DOI] [PubMed] [Google Scholar]
- Boeve BF. Predicting the future in idiopathic rapid-eye movement sleep behaviour disorder. Lancet Neurol. 2010;9:1040–1042. doi: 10.1016/S1474-4422(10)70221-0. [DOI] [PubMed] [Google Scholar]
- Boger HA, Granholm AC, McGinty JF, Middaugh LD. A dual-hit animal model for age-related parkinsonism. Prog. Neurobiol. 2010;90:217–29. doi: 10.1016/j.pneurobio.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Bohl JR, Müller CM, Rüb U, de Vos RA, Del Tredici K. The staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord. 2006;21:2042–51. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- Bray M, Shaw C, Moore M, Garcia R, Zanquetta M, Durgan D, Jeong W, Tsai J, Bugger H, Zhang D, Rohrwasser A, Rennison J, Dyck J, Litwin S, Hardin P, Chow C, Chandler M, Abel E, Young M. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ. Physiol. 2008;294:1036–1047. doi: 10.1152/ajpheart.01291.2007. [DOI] [PubMed] [Google Scholar]
- Boger HA, Granholm AC, McGinty JF, Middaugh LD. A dual-hit animal model for age-related parkinsonism. Prog Neurobiol. 2010;90(2):217–229. doi: 10.1016/j.pneurobio.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijs RM, La Fleur SE, Wortel J, Van Heyningen C, Zuiddam L, Mettenleiter TC, Kalsbeek A, Nagai K, Niijima A. The suprachiasmatic nucleus balances sympathetic and parasympathetic output to peripheral organs through separate preautonomic neurons. J Comp Neurol. 2003;2003;464:36–48. doi: 10.1002/cne.10765. [DOI] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–7. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002;22:8797–807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, Besing RC, Menaker M, Gewirtz AT, Davidson AJ. Dysregulation of inflammatory responses by chronic circadian disruption. J. Immunol. 2010;185(10):5796–805. doi: 10.4049/jimmunol.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri KR, Odin P. The challenge of non-motor symptoms in Parkinson's disease. Prog.Brain Res. 2010;184:325–41. doi: 10.1016/S0079-6123(10)84017-8. [DOI] [PubMed] [Google Scholar]
- Chen Z, Yoo SH, Park YS, Kim KH, Wei S, Buhr E, Ye ZY, Pan HL, Takahashi JS. Identification of diverse modulators of central and peripheral circadian clocks by high-throughput chemical screening. Proc. Natl. Acad. Sci. USA. 2012;109:101–6. doi: 10.1073/pnas.1118034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chokroverty S. Sleep and neurodegenerative diseases. Semin. Neurol. 2009;29:446–67. doi: 10.1055/s-0029-1237124. [DOI] [PubMed] [Google Scholar]
- Colwell CS. Preventing dehydration during sleep. Nature Neurosci. 2010;13:403–404. doi: 10.1038/nn0410-403. [DOI] [PubMed] [Google Scholar]
- Colwell CS. Linking neural activity and molecular oscillations in the SCN. Nat. Rev. Neurosci. 2011;12:553–69. doi: 10.1038/nrn3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassen DO, Josephs KA, Ahlskog JE, Silber MH, Tippmann-Peikert M, Boeve BF. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurol. 2010;75:494–9. doi: 10.1212/WNL.0b013e3181ec7fac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. Alpha-Synuclein and neuronal cell death. Mol. Neurodegener. 2009;4:9–9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotella D, Hernandez-Enriquez B, Wu X, Li R, Pan Z, Leveille J, Link CD, Oddo S, Sesti F. Toxic role of K+ channel oxidation in mammalian brain. J. Neurosci. 2012;32:4133–44. doi: 10.1523/JNEUROSCI.6153-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Mega MS. Oxford University Press; 2003. Neuropsychiatry and Behavioral Neuroscience. ISBN: 978-0195138580. [Google Scholar]
- Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110(8):1109–24. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA. 2005;102:12560–5. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–61. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deboer T, Vansteensel MJ, Détári L, Meijer JH. Sleep states alter activity of suprachiasmatic nucleus neurons. Nat. Neurosci. 2003;6(10):1086–90. doi: 10.1038/nn1122. [DOI] [PubMed] [Google Scholar]
- Deurveilher S, Semba K. Indirect projections from the suprachiasmatic nucleus to major arousal-promoting cell groups in rat: implications for the circadian control of behavioural state. Neurosci. 2005;130(1):165–83. doi: 10.1016/j.neuroscience.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Dhawan V, Healy DG, Pal S, Chaudhuri KR. Sleep-related problems of Parkinson's disease. Age Ageing. 2006;35:220–8. doi: 10.1093/ageing/afj087. [DOI] [PubMed] [Google Scholar]
- Diederich NJ, McIntyre DJ. Sleep disorders in Parkinson's disease: many causes, few therapeutic options. J. Neurol. Sci. 2012;314:12–19. doi: 10.1016/j.jns.2011.10.025. [DOI] [PubMed] [Google Scholar]
- Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 2010;72:517–49. doi: 10.1146/annurev-physiol-021909-135821. [DOI] [PubMed] [Google Scholar]
- Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E. Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ Res. 2012;110(8):1125–38. doi: 10.1161/CIRCRESAHA.111.246108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmer JS, Dinges DF. Neurocognitive consequences of sleep deprivation. Semin. Neurol. 2005;25:117–29. doi: 10.1055/s-2005-867080. [DOI] [PubMed] [Google Scholar]
- Edgar RS, Green EW, Zhao Y, van Ooijen G, Olmedo M, Qin X, Xu Y, Pan M, Valekunja UK, Feeney KA, Maywood ES, Hastings MH, Baliga NS, Merrow M, Millar AJ, Johnson CH, Kyriacou CP, O'Neill JS, Reddy AB. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485(7399):459–64. doi: 10.1038/nature11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farajnia S, Michel S, Deboer T, Vanderleest HT, Houben T, Rohling JH, Ramkisoensing A, Yasenkov R, Meijer JH. Evidence for neuronal desynchrony in the aged suprachiasmatic nucleus clock. J. Neurosci. 2012;32:5891–9. doi: 10.1523/JNEUROSCI.0469-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira JJ, Desboeuf K, Galitzky M, Thalamas C, Brefel-Courbon C, Fabre N, Senard JM, Montastruc JL, Sampaio C, Rascol O. Sleep disruption, daytime somnolence and 'sleep attacks' in Parkinson's disease, a clinical survey in PD patients and age-matched healthy volunteers. Eur. J. Neurol. 2006;13:209–14. doi: 10.1111/j.1468-1331.2006.01262.x. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Chesselet MF. Modeling non-motor symptoms of Parkinson's disease in genetic mouse models. In: Groenewegen HJ, Voorn P, Berendse HW, Mulder AB, Cools AR, editors. I.X. Basal Ganglia. Springer; New York: 2009. pp. 483–492. [Google Scholar]
- Fleming SM, Tetreault NA, Mulligan CK, Hutson CB, Masliah E, Chesselet MF. Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. Eur. J. Neurosci. 2008;28:247–56. doi: 10.1111/j.1460-9568.2008.06346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J. Neurosci. 2004;24:9434–40. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Hutson CB, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype alpha-synuclein. Neurosci. 2006;142:1245–53. doi: 10.1016/j.neuroscience.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froy O, Chapnik N. Circadian oscillation of innate immunity components in mouse small intestine. Mol Immunol. 2007;44(8):1954–60. doi: 10.1016/j.molimm.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Fox SH, Brotchie JM. The MPTP-lesioned non-human primate models of Parkinson's disease. Past, present, and future. Prog. Brain Res. 2010;184:133–57. doi: 10.1016/S0079-6123(10)84007-5. [DOI] [PubMed] [Google Scholar]
- Gagnon JF, Petit D, Latreille V, Montplaisir J. Neurobiology of sleep disturbances in neurodegenerative disorders. Curr. Pharm. Des. 2008;14:3430–45. doi: 10.2174/138161208786549353. [DOI] [PubMed] [Google Scholar]
- Gajula Balija MB, Griesinger C, Herzig A, Zweckstetter M, Jäckle H. Pre-fibrillar alpha-synuclein mutants cause Parkinson's disease-like non-motor symptoms in Drosophila. PLoS One. 2011;6(9):e24701. doi: 10.1371/journal.pone.0024701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale JE, Cox HI, Qian J, Block GD, Colwell CS, Matveyenko AV. Disruption of circadian rhythms accelerates development of diabetes through pancreatic beta-cell loss and dysfunction. J. Biol. Rhythms. 2011;26:423–33. doi: 10.1177/0748730411416341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner JR, Yin JC. Circadian rhythms and memory formation. Nat. Rev. Neurosci. 2010;11:577–88. doi: 10.1038/nrn2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimble JM, Floyd ZE. Metabolism: what causes the gut's circadian instincts? Curr. Biol. 2011;21:24–26. doi: 10.1016/j.cub.2011.07.009. [DOI] [PubMed] [Google Scholar]
- Glass JD, Brager AJ, Stowie AC, Prosser RA. Cocaine modulates pathways for photic and nonphotic entrainment of the mammalian SCN circadian clock. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012;302:740–50. doi: 10.1152/ajpregu.00602.2011. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Leblanc JJ, Quast KB, Sudhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 2009;1:172–81. doi: 10.1007/s11689-009-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravotta L, Gavrila AM, Hood S, Amir S. Global depletion of dopamine using intracerebroventricular 6-hydroxydopamine injection disrupts normal circadian wheel-running patterns and PERIOD2 expression in the rat forebrain. J. Mol Neurosci. 2011;45:162–71. doi: 10.1007/s12031-011-9520-8. [DOI] [PubMed] [Google Scholar]
- Hardeland R, Coto-Montes A, Poeggeler B. Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol. Int. 2003;20:921–62. doi: 10.1081/cbi-120025245. [DOI] [PubMed] [Google Scholar]
- Hampp G, Ripperger JA, Houben T, Schmutz I, Blex C, Perreau-Lenz S, Brunk I, Spanagel R, Ahnert-Hilger G, Meijer JH, Albrecht U. Regulation of monoamine oxidase A by circadian-clock components implies clock influence on mood. Curr. Biol. 2008;18:678–83. doi: 10.1016/j.cub.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Hastings M, Reddy A, Maywood E. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat. Rev. Neurosci. 2003;4:649–661. doi: 10.1038/nrn1177. [DOI] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H. Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Lee JW, Lewis WG, Zhang EE, Breton G, Liu X, Garcia M, Peters EC, Etchegaray JP, Traver D, Schultz PG, Kay SA. High-throughput chemical screen identifies a novel potent modulator of cellular circadian rhythms and reveals CKIα as a clock regulatory kinase. PLoS Biol. 2010;8:e1000559. doi: 10.1371/journal.pbio.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenesch JB, Panda S, Kay S, Takahashi JS. Circadian transcriptional output in the SCN and liver of the mouse. Novartis Found. Symp. 2003;253:171–80. [PubMed] [Google Scholar]
- Honma K, Honma S. The SCN-independent clocks, methamphetamine and food restriction. Eur J Neurosci. 2009;30:1707–17. doi: 10.1111/j.1460-9568.2009.06976.x. [DOI] [PubMed] [Google Scholar]
- Honma K, Honma S, Hiroshige T. Disorganization of the rat activity rhythm by chronic treatment with methamphetamine. Physiol. Behav. 1986;38:687–695. doi: 10.1016/0031-9384(86)90265-9. [DOI] [PubMed] [Google Scholar]
- Honma K, Honma S, Hiroshige T. Activity rhythms in the circadian domain appear in suprachiasmatic nuclei lesioned rats given methamphetamine. Physiol. Behav. 1987;40:767–774. doi: 10.1016/0031-9384(87)90281-2. [DOI] [PubMed] [Google Scholar]
- Hood S, Cassidy P, Cossette MP, Weigl Y, Verwey M, Robinson B, Stewart J, Amir S. Endogenous dopamine regulates the rhythm of expression of the clock protein PER2 in the rat dorsal striatum via daily activation of D2 dopamine receptors. J. Neurosci. 2010;30:14046–58. doi: 10.1523/JNEUROSCI.2128-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain JL, Shapiro CM. The prevalence, cost implications, and management of sleep disorders: an overview. Sleep Breath. 2002;6:85–102. doi: 10.1007/s11325-002-0085-1. [DOI] [PubMed] [Google Scholar]
- Imbesi M, Yildiz S, Dirim Arslan A, Sharma R, Manev H, Uz T. Dopamine receptor-mediated regulation of neuronal "clock" gene expression. Neurosci. 2009;158:537–44. doi: 10.1016/j.neuroscience.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iranzo A, Santamaria J, Tolosa E. The clinical and pathophysiological relevance of REM sleep behavior disorder in neurodegenerative diseases. Sleep Med. Rev. 2009;13:385–401. doi: 10.1016/j.smrv.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Iranzo A, Lomeña F, Stockner H, Valldeoriola F, Vilaseca I, Salamero M, Molinuevo JL, Serradell M, Duch J, Pavía J, Gallego J, Seppi K, Högl B, Tolosa E, Poewe W, Santamaria J. Decreased striatal dopamine transporter uptake and substantia nigra hyperechogenicity as risk markers of synucleinopathy in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol. 2010;9:1070–1077. doi: 10.1016/S1474-4422(10)70216-7. [DOI] [PubMed] [Google Scholar]
- Iranzo A. Sleep-wake changes in the premotor stage of Parkinson disease. J. Neurol. Sci. 2011;310:283–5. doi: 10.1016/j.jns.2011.07.049. [DOI] [PubMed] [Google Scholar]
- Ironside S, Davidson F, Corkum P. Circadian motor activity affected by stimulant medication in children with attention-deficit/hyperactivity disorder. J. Sleep Res. 2010;19:546–51. doi: 10.1111/j.1365-2869.2010.00845.x. [DOI] [PubMed] [Google Scholar]
- Itri J, Michel S, Waschek JA, Colwell CS. Circadian rhythm in inhibitory synaptic transmission in the mouse suprachiasmatic nucleus. J. Neurophysiol. 2004;92:311–319. doi: 10.1152/jn.01078.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Yao GL, Bean BP. Mechanism of spontaneous firing in dorsomedial suprachiasmatic nucleus neurons. J. Neurosci. 2004;24:7985–98. doi: 10.1523/JNEUROSCI.2146-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S. Multi-organ autonomic dysfunction in Parkinson disease. Parkinsonism and Related Disorders. 2011;17:77–83. doi: 10.1016/j.parkreldis.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Goldstein DS. Cardiovascular dysautonomia in Parkinson disease: From pathophysiology to pathogenesis. Neurobiol. Dis. 2012;46:572–80. doi: 10.1016/j.nbd.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Critical evaluation of the Braak staging scheme for Parkinson's disease. Ann. Neurol. 2010;67:550. doi: 10.1002/ana.21638. [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Yang Y, Liu ZP, Allen CN. Membrane properties and synaptic inputs of suprachiasmatic nucleus neurons in rat brain slices. J.Physiol. 1997;499:141–159. doi: 10.1113/jphysiol.1997.sp021917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio M, Haapaniemi T, Turkka J, Suominen K, Tolonen U, Sotaniemi K, Heikkila VP, Myllyla V. Heart rate variability in patients with untreated Parkinson’s disease. Eur. J. Neurol. 2000;7:667–672. doi: 10.1046/j.1468-1331.2000.00127.x. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, Palm IF, La Fleur SE, Scheer FAJL, Perreau-Lenz S, Ruiter M, Kreier F, Cailotto C, Buijs RM. SCN outputs and the hypothalamic balance of life. J. Biol. Rhythms. 2006;21:458–469. doi: 10.1177/0748730406293854. [DOI] [PubMed] [Google Scholar]
- Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, McEwen BS. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc. Natl. Acad. Sci. USA. 2011;108:1657–62. doi: 10.1073/pnas.1018375108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khapre RV, Kondratova AA, Susova O, Kondratov RV. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle. 2011;10(23):4162–9. doi: 10.4161/cc.10.23.18381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Mazuch J, Abraham U, Eom GD, Herzog ED, Volk HD, Kramer A, Maier B. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci USA. 2009;106(50):21407–12. doi: 10.1073/pnas.0906361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI, Dudek FE. Intracellular electrophysiological study of suprachiasmatic nucleus neurons in rodents: inhibitory synaptic mechanisms. J. Physiol. 1992;458:247–60. doi: 10.1113/jphysiol.1992.sp019416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratova AA, Kondratov RV. The circadian clock and pathology of the ageing brain. Nat. Rev. Neurosci. 2012;13(5):325–35. doi: 10.1038/nrn3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko NI, Medina I, Dudek FE. Persistent subthreshold voltage-dependent cation single channels in suprachiasmatic nucleus neurons. Neurosci. 2004;129:85–92. doi: 10.1016/j.neuroscience.2004.06.080. [DOI] [PubMed] [Google Scholar]
- Kudo T, Loh DH, Kuljis D, Constance C, Colwell CS. Fast delayed rectifier potassium current: critical for input and output of the circadian system. J Neurosci. 2011a;23(8):2746–55. doi: 10.1523/JNEUROSCI.5792-10.2011. 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Loh DH, Truong D, Wu Y, Colwell CS. Circadian dysfunction in a mouse model of Parkinson's disease. Exp. Neurol. 2011b;232:66–75. doi: 10.1016/j.expneurol.2011.08.003. [DOI] [PubMed] [Google Scholar]
- Kumru H, Santamaria J, Tolosa E, Valldeoriola F, Muñoz E, Marti MJ, Iranzo A. Rapid eye movement sleep behavior disorder in parkinsonism with parkin mutations. Ann. Neurol. 2004;56(4):599–603. doi: 10.1002/ana.20272. [DOI] [PubMed] [Google Scholar]
- Kunz D, Mahlberg R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J. Sleep Res. 2010;19:591–596. doi: 10.1111/j.1365-2869.2010.00848.x. [DOI] [PubMed] [Google Scholar]
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 2006;20:1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak Y, Lundkvist GB, Brask J, Davidson A, Menaker M, Kristensson K, Block GD. Interferon-gamma alters electrical activity and clock gene expression in suprachiasmatic nucleus neurons. J. Biol. Rhythms. 2008;23(2):150–9. doi: 10.1177/0748730407313355. [DOI] [PubMed] [Google Scholar]
- Labunskyy VM, Gladyshev VN. Role of Reactive Oxygen Species-Mediated Signaling in Aging. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2012.4891. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AG, Doherty CJ, Mueller-Roeber B, Kay SA, Schippers JH, Dijkwel PP. CIRCADIAN CLOCK-ASSOCIATED 1 regulates ROS homeostasis and oxidative stress responses. Proc. Natl. Acad. Sci. USA. 2012;109(42):17129–34. doi: 10.1073/pnas.1209148109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laloux C, Derambure P, Kreisler A, Houdayer E, Bruezière S, Bordet R, Destée A, Monaca C. MPTP-treated mice: long-lasting loss of nigral TH-ir neurons but not paradoxical sleep alterations. Exp. Brain Res. 2008;186(4):635–42. doi: 10.1007/s00221-008-1268-1. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim MS, Li R, Liu VY, Moore DD, Ma K, Yechoor VK. Loss of Bmal1 leads to uncoupling and impaired glucose-stimulated insulin secretion in B-cells. Islets. 2011;3(6):381–8. doi: 10.4161/isl.3.6.18157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger D, Bayon V. Societal costs of insomnia. Sleep Med. Rev. 2010;14(6):379–389. doi: 10.1016/j.smrv.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport. 2007;18:1543–1546. doi: 10.1097/WNR.0b013e3282f03db4. [DOI] [PubMed] [Google Scholar]
- Limousin N, Konofal E, Karroum E, Lohmann E, Theodorou I, Durr A, Arnulf I. Restless legs syndrome, rapid eye movement sleep behavior disorder, and hypersomnia in patients with two parkin mutations. Mov. Disord. 2009;24(13):1970–1976. doi: 10.1002/mds.22711. [DOI] [PubMed] [Google Scholar]
- Litinski M, Scheer FA, Shea SA. Influence of the Circadian System on Disease Severity. Sleep Med. Clin. 2009;4:143–163. doi: 10.1016/j.jsmc.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shi S, Gu Z, Du Y, Liu M, Yan S, Gao J, Li J, Shao Y, Zhong W, Chen X, Li C. Impaired autophagic function in rat islets with aging. Age (Dordr) 2012 doi: 10.1007/s11357-012-9456-0. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Coco D, Caruso G, Mattaliano A. REM sleep behavior disorder in patients with DJ-1 mutations and parkinsonism-dementia-ALS complex. Mov. Disord. 2009;24(10):1555–1556. doi: 10.1002/mds.22629. [DOI] [PubMed] [Google Scholar]
- Logan RW, Sarkar DK. Circadian nature of immune function. Mol. Cell Endocrinol. 2012;349(1):82–90. doi: 10.1016/j.mce.2011.06.039. [DOI] [PubMed] [Google Scholar]
- Loh D, Navarro J, Hagopian A, Wang L, Deboer T, Colwell C. Rapid changes in the light/dark cycle disrupt memory of conditioned fear in mice. PLoS One. 2010;5:e12546. doi: 10.1371/journal.pone.0012546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louter M, Aarden WC, Lion J, Bloem BR, Overeem S. Recognition and diagnosis of sleep disorders in Parkinson's disease. J. Neurol. 2012;259(10):2031–40. doi: 10.1007/s00415-012-6505-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckhaupt SE, Tak S, Calvert GM. The prevalence of short sleep duration by industry and occupation in the National Health Interview Survey. Sleep. 2010;33(2):149–59. doi: 10.1093/sleep/33.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundkvist GB, Christenson J, ElTayeb RA, Peng ZC, Grillner P, Mhlanga J, Bentivoglio M, Kristensson K. Altered neuronal activity rhythm and glutamate receptor expression in the suprachiasmatic nuclei of Trypanosoma brucei-infected rats. J. Neuropathol. Exp. Neurol. 1998;57(1):21–9. doi: 10.1097/00005072-199801000-00004. [DOI] [PubMed] [Google Scholar]
- Lundkvist GB, Sellix MT, Nygård M, Davis E, Straume M, Kristensson K, Block GD. Clock gene expression during chronic inflammation induced by infection with Trypanosoma brucei brucei in rats. J. Biol. Rhythms. 2010;25(2):92–102. doi: 10.1177/0748730409360963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen I, Chesselet MF. Genetic mouse models of Parkinson's disease. Prog. Brain Res. 2010;184:53–87. doi: 10.1016/S0079-6123(10)84004-X. [DOI] [PubMed] [Google Scholar]
- Marcheva B, Ramsey K, Buhr E, Kobayashi Y, Su H, Ko C, Ivanova G, Omura C, Mo S, Vitaterna M, Lopez J, Philipson L, Bradfield C, Crosby S, JeBailey L, Wang X, Takahashi J, Bass J. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Nishinaka K, Oda M, Hara N, Komatsu K, Kubori T, Udaka F. Excessive daytime sleepiness in Parkinson disease: a SPECT study. Sleep. 2006;29:917–20. doi: 10.1093/sleep/29.7.917. [DOI] [PubMed] [Google Scholar]
- Mayer G, Jennum P, Riemann D, Dauvilliers Y. Insomnia in central neurologic diseases--occurrence and management. 2011;15:369–378. doi: 10.1016/j.smrv.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Mehta SH, Morgan JC, Sethi KD. Sleep disorders associated with Parkinson's disease: role of dopamine, epidemiology, and clinical scales of assessment.". CNS Spectr. 2008;13(3 Suppl 4):6–11. doi: 10.1017/s1092852900017260. [DOI] [PubMed] [Google Scholar]
- Menza M, Dobkin RD, Marin H, Bienfait K. Sleep disturbances in Parkinson's disease. Mov. Disord. 25 Suppl. 2010;1:S117–122. doi: 10.1002/mds.22788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 2010;33:513–23. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012;35:445–62. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsees GM, Kraft P, Hankinson SE, Hunter DJ, Schernhammer ES. Circadian genes and breast cancer susceptibility in rotating shift workers. Int. J. Cancer. 2012;131(11):2547–52. doi: 10.1002/ijc.27564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JR, Meredith AL. Genetic activation of BK currents in vivo generates bidirectional effects on neuronal excitability. Proc Natl Acad Sci USA. 2012;109(46):18997–9002. doi: 10.1073/pnas.1205573109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin LP, Allen CN. The circadian visual system. Brain Res. Rev. 2006;51:1–60. doi: 10.1016/j.brainresrev.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Moore RY, Speh JC. GABA is the principal neurotransmitter of the circadian system. Neurosci. Lett. 1993;150:112–116. doi: 10.1016/0304-3940(93)90120-a. [DOI] [PubMed] [Google Scholar]
- Mullington JM, Haack M, Toth M, Serrador JM, Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog. Cardiovasc. Dis. 2009;51(4):294–302. doi: 10.1016/j.pcad.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, Edwards RH. Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. 2008;28:12305–12317. doi: 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB, Turrigiano GG. Strength through diversity. Neuron. 2008;60:477–82. doi: 10.1016/j.neuron.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimamurthy R, Hatori M, Nayak SK, Liu F, Panda S, Verma IM. Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc. Natl. Acad. Sci. USA. 2012;109(31):12662–7. doi: 10.1073/pnas.1209965109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Ross OA, Ishii K, Kachergus JM, Ishiwata K, Kitagawa M, Kono S, Obi T, Mizoguchi K, Inoue Y, Imai H, Takanashi M, Mizuno Y, Farrer MJ, Hattori N. Expanding the clinical phenotype of SNCA duplication carriers. Mov. Disord. 2009;24(12):1811–1819. doi: 10.1002/mds.22682. [DOI] [PubMed] [Google Scholar]
- Okamura H, Berod A, Julien JF, Geffard M, Kitahama K, Mallet J, Bobillier P. Demonstration of GABAergic cell bodies in the suprachiasmatic nucleus: in situ hybridization of glutamic acid decarboxylase (GAD) mRNA and immunocytochemistry of GAD and GABA. Neurosci. Lett. 1989;102:131–6. doi: 10.1016/0304-3940(89)90067-0. [DOI] [PubMed] [Google Scholar]
- O'Neill JS, Reddy AB. Circadian clocks in human red blood cells. Nature. 2011;469(7331):498–503. doi: 10.1038/nature09702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109(3):307–20. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH. Genome-wide association study for susceptibility genes contributing to familial Parkinson disease. Hum.Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A, Stacy M. Non-motor symptoms in Parkinson's disease. J. Neurol. 256 Suppl. 2009;3:293–8. doi: 10.1007/s00415-009-5240-1. [DOI] [PubMed] [Google Scholar]
- Pennartz CM, Bierlaagh MA, Geurtsen AM. Cellular mechanisms underlying spontaneous firing in rat suprachiasmatic nucleus: involvement of a slowly inactivating component of sodium current. J. Neurophysiol. 1997;78:1811–25. doi: 10.1152/jn.1997.78.4.1811. [DOI] [PubMed] [Google Scholar]
- Poceta JS, Parsons L, Engelland S, Kripke DF. Circadian rhythm of CSF monoamines and hypocretin-1 in restless legs syndrome and Parkinson's disease. Sleep Med. 2009;10(1):129–33. doi: 10.1016/j.sleep.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Pontone GM, Palanci J, Williams JR, Bassett SS. Screening for DSM-IV-TR cognitive disorder NOS in Parkinson's disease using the Mattis Dementia Rating Scale. Int J Geriatr Psychiatry. 2012 doi: 10.1002/gps.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma RB, Lang AE, Massicotte-Marquez J, Montplaisir J. Potential early markers of Parkinson disease in idiopathic REM sleep behavior disorder. Neurol. 2006;66:845–851. doi: 10.1212/01.wnl.0000203648.80727.5b. [DOI] [PubMed] [Google Scholar]
- Purisai MG, McCormack AL, Langston WJ, Johnston LC, Di Monte DA. Alpha-synuclein expression in the substantia nigra of MPTP-lesioned non-human primates. 2005;20:898–906. doi: 10.1016/j.nbd.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Ragab MM, Mohammed ES. Idiopathic Parkinson's disease patients at the urologic clinic. Neurourol Urodyn. 2011;30:1258–61. doi: 10.1002/nau.20983. [DOI] [PubMed] [Google Scholar]
- Reddy AB, O'Neill JS. Healthy clocks, healthy body, healthy mind. Trends Cell Biol. 2010;20:36–44. doi: 10.1016/j.tcb.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid K, Zee P. Circadian rhythm disorders. Semin. Neurol. 2009;29:393–405. doi: 10.1055/s-0029-1237120. [DOI] [PubMed] [Google Scholar]
- Ritz B, Rhodes SL, Bordelon Y, Bronstein J. a-Synuclein Genetic Variants Predict Faster Motor Symptom Progression in Idiopathic Parkinson Disease. PLoS ONE. 2012;7:e36199. doi: 10.1371/journal.pone.0036199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 2002;68:568–78. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Roenneberg T. Harvard University Press; 2012. Chronotypes, Social . Jet Lag, and Why You're So Tired. ISBN: 0674065859. [Google Scholar]
- Ruby NF, Hwang CE, Wessells C, Fernandez F, Zhang P, Sapolsky R, Heller HC. Hippocampal-dependent learning requires a functional circadian system. Proc. Natl. Acad. Sci. USA. 2008;105:15593–8. doi: 10.1073/pnas.0808259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye DB. The two faces of Eve: dopamine's modulation of wakefulness and sleep. Neurol. 2004;63(8 Suppl 3):S2–7. doi: 10.1212/wnl.63.8_suppl_3.s2. [DOI] [PubMed] [Google Scholar]
- Santiago PL, Rossi M, Cardinali DP, Merello M. Activity-rest rhythm abnormalities in Parkinson's disease patients are related to dopaminergic therapy. Int. J. Neurosci. 2010;120:11–6. doi: 10.3109/00207450903326179. [DOI] [PubMed] [Google Scholar]
- Schaap J, Meijer JH. Opposing effects of behavioural activity and light on neurons of the suprachiasmatic nucleus. Eur. J. Neurosci. 2001;13(10):1955–62. doi: 10.1046/j.0953-816x.2001.01561.x. [DOI] [PubMed] [Google Scholar]
- Schafer D, Greulich W. Effects of parkinsonian medication on sleep. J. Neurol. 2000;247(Suppl 4):24–27. doi: 10.1007/pl00007770. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial diseases. Lancet. 2012;379:1825–34. doi: 10.1016/S0140-6736(11)61305-6. [DOI] [PubMed] [Google Scholar]
- Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc. Natl. Acad. Sci. USA. 2009;106:4453–8. doi: 10.1073/pnas.0808180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck CH, Bundlie SR, Mahowald MW. Delayed emergence of a parkinsonian disorder in 38% of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurol. 1996;46:388–393. doi: 10.1212/wnl.46.2.388. [DOI] [PubMed] [Google Scholar]
- Schulte EC, Winkelmann J. When Parkinson's disease patients go to sleep: specific sleep disturbances related to Parkinson's disease. J. Neurol. 2011;258(Suppl 2):328–335. doi: 10.1007/s00415-011-5933-0. [DOI] [PubMed] [Google Scholar]
- Sesti F, Liu S, Cai SQ. Oxidation of potassium channels by ROS: a general mechanism of aging and neurodegeneration? Trends Cell Biol. 2010;20:45–51. doi: 10.1016/j.tcb.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Katz DM. Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: focus on Mecp2 and Met. Curr. Opin. Neurobiol. 2011;21:827–833. doi: 10.1016/j.conb.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JC, Chen K. Regulation of MAO-A and MAO-B gene expression. Curr. Med. Chem. 2004;11(15):1995–2005. doi: 10.2174/0929867043364757. [DOI] [PubMed] [Google Scholar]
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaer TL, Sclar DA. Economic implications of sleep disorders. Pharmacoeconomics. 2010;28:1015–23. doi: 10.2165/11537390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Spengler ML, Kuropatwinski KK, Comas M, Gasparian AV, Fedtsova N, Gleiberman AS, Gitlin II, Artemicheva NM, Deluca KA, Gudkov AV, Antoch MP. Core circadian protein CLOCK is a positive regulator of NF-κB-mediated transcription. Proc. Natl. Acad. Sci. USA. 2012;109(37):E2457–65. doi: 10.1073/pnas.1206274109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens S, Cormella CL, Stepanski EJ. Daytime sleepiness and alertness in patients with Parkinson disease. Sleep. 2004;27:967–72. doi: 10.1093/sleep/27.5.967. [DOI] [PubMed] [Google Scholar]
- Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002;417(6884):78–83. doi: 10.1038/nature744. [DOI] [PubMed] [Google Scholar]
- Strecker GJ, Wuarin JP, Dudek FE. GABAA-mediated local synaptic pathways connect neurons in the rat suprachiasmatic nucleus. J. Neurophysiol. 1997;78:2217–2220. doi: 10.1152/jn.1997.78.4.2217. [DOI] [PubMed] [Google Scholar]
- Takahashi J, Hong H, Ko C, McDearmon E. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat. Rev. Genet. 2008a;9:764–775. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi JS, Shimomura K, Kumar V. Searching for genes underlying behavior: lessons from crcadian rhythms. Science. 2008b;322:909–12. doi: 10.1126/science.1158822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Yamaguchi E, Takahashi M, Hashimura K, Shibata T, Nakamura W, Nakamura TJ. Effects of age-related dopaminergic neuron loss in the substantia nigra on the circadian rhythms of locomotor activity in mice. Neurosci Res. 2012 doi: 10.1016/j.neures.2012.09.005. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Tataroglu O, Davidson AJ, Benvenuto LJ, Menaker M. The methamphetamine-sensitive circadian oscillator (MASCO) in mice. J. Biol. Rhythms. 2006;21:185–194. doi: 10.1177/0748730406287529. [DOI] [PubMed] [Google Scholar]
- Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM, Weinshenker D, Greene JG, Miller GW. Nonmotor symptoms of Parkinson's disease revealed in an animal model with reduced monoamine storage capacity. J. Neurosci. 2009;29(25):8103–13. doi: 10.1523/JNEUROSCI.1495-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TN, Caudle WM, Miller GW. VMAT2-Deficient Mice Display Nigral and Extranigral Pathology and Motor and Nonmotor Symptoms of Parkinson's Disease. Parkinsons Dis. 2011;124165 doi: 10.4061/2011/124165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel E, Bourque CW. Central clock excites vasopressin neurons by waking osmosensory afferents during late sleep. Nat. Neurosci. 2010;13:467–74. doi: 10.1038/nn.2503. [DOI] [PubMed] [Google Scholar]
- Thorpy MJ, Adler CH. Parkinson's disease and sleep. Neurol. Clin. 2005;23:1187–208. doi: 10.1016/j.ncl.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Trancikova A, Tsika E, Moore DJ. Mitochondrial dysfunction in genetic animal models of Parkinson's disease. Antioxid Redox Signal. 2012;16:896–919. doi: 10.1089/ars.2011.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L. Pathological roles of α-synuclein in neurological disorders. Lancet Neurol. 2011;10:1015–25. doi: 10.1016/S1474-4422(11)70213-7. [DOI] [PubMed] [Google Scholar]
- Verhave PS, Jongsma MJ, Van den Berg RM, Vis JC, Vanwersch RA, Smit AB, Van Someren EJ, Philippens IH. REM sleep behavior disorder in the marmoset MPTP model of early Parkinson disease. Sleep. 2011;34(8):1119–1125. doi: 10.5665/SLEEP.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezoli J, Fifel K, Leviel V, Dehay C, Kennedy H, Cooper HM, Gronfier C, Procyk E. Early presymptomatic and long-term changes of rest activity cycles and cognitive behavior in a MPTP-monkey model of Parkinson's disease. PLoS One. 2011;6(8):e23952. doi: 10.1371/journal.pone.0023952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videnovic A, Golombek D. Circadian and sleep disorders in Parkinson's disease. Exp. Neurol. 2012 doi: 10.1016/j.expneurol.2012.08.018. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wailke S, Herzog J, Witt K, Deuschl G, Volkmann J. Effect of controlled-release levodopa on the microstructure of sleep in Parkinson's disease. Eur. J. Neurol. 2011;18:590–596. doi: 10.1111/j.1468-1331.2010.03213.x. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H. NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson's disease. Neurosci. Lett. 1997;239:45–8. doi: 10.1016/s0304-3940(97)00891-4. [DOI] [PubMed] [Google Scholar]
- Wanagat J, Dai DF, Rabinovitch P. Mitochondrial oxidative stress and mammalian healthspan. Mech. Ageing Dev. 2010;131(7-8):527–35. doi: 10.1016/j.mad.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LM, Dragich JM, Kudo T, Odom IH, Welsh DK, O'Dell TJ, Colwell CS. Expression of the circadian clock gene Period2 in the hippocampus: possible implications for synaptic plasticity and learned behaviour. ASN Neuro. 2009;1:e00012. doi: 10.1042/AN20090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Huang RC. Effects of sodium pump activity on spontaneous firing in neurons of the rat suprachiasmatic nucleus. J. Neurophysiol. 2006;96:109–18. doi: 10.1152/jn.01369.2005. [DOI] [PubMed] [Google Scholar]
- Wang TA, Yu YV, Govindaiah G, Ye X, Artinian L, Coleman TP, Sweedler JV, Cox CL, Gillette MU. Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science. 2012;337(6096):839–42. doi: 10.1126/science.1222826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Gray CH, Foltynie T, Brayne CE, Robbins TW, Barker RA. Evolution of cognitive dysfunction in an incident Parkinson’s disease cohort. Brain. 2007;130:1787–1798. doi: 10.1093/brain/awm111. [DOI] [PubMed] [Google Scholar]
- Winkler S, Hagenah J, Lincoln S, Heckman M, Haugarvoll K, Lohmann-Hedrich K, Kostic V, Farrer M, Klein C. alpha-Synuclein and Parkinson disease susceptibility. Neurol. 2007;69:1745–50. doi: 10.1212/01.wnl.0000275524.15125.f4. [DOI] [PubMed] [Google Scholar]
- Wu N, Joshi PR, Cepeda C, Masliah E, Levine MS. Alpha-synuclein overexpression in mice alters synaptic communication in the corticostriatal pathway. J. Neurosci Res. 2010;88:1764–1776. doi: 10.1002/jnr.22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Chung KK. Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson's disease. J Neurochem. 2012 doi: 10.1111/j.1471-4159.2012.07769.x. Published online. [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Kerbeshian MC, Hocker CG, Block GD, Menaker M. Rhythmic properties of the hamster suprachiasmatic nucleus in vivo. J. Neurosci. 1998;18(24):10709–23. doi: 10.1523/JNEUROSCI.18-24-10709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo L, Singh R, Gundeti M, Barua JM, Masood J. Urinary tract dysfunction in Parkinson's disease: a review. Int Urol Nephrol. 2012;44:415–24. doi: 10.1007/s11255-011-9969-y. [DOI] [PubMed] [Google Scholar]
- Yong MH, Fook-Chong S, Pavanni R, Lim LL, Tan EK. Case control polysomnographic studies of sleep disorders in Parkinson's disease. PLoS One. 2011;6:e22511. doi: 10.1371/journal.pone.0022511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Fernández-Bedoya A, Lambarri I, Gómez-Esteban JC, Lezcano E, Zamacona J, Madoz P. Abnormal sleep architecture is an early feature in the E46K familial synucleinopathy. Mov. Disord. 2005;20(10):1310–1315. doi: 10.1002/mds.20581. [DOI] [PubMed] [Google Scholar]
- Ziemssen T, Reichmann H. Cardiovascular autonomic dysfunction in Parkinson's disease. J Neurol Sci. 2010;289(1-2):74–80. doi: 10.1016/j.jns.2009.08.031. [DOI] [PubMed] [Google Scholar]