

Abstract
Many mechanisms of and treatments for type 1 diabetes studied in the NOD mouse model have not been replicated in human disease models. Thus, the field of diabetes research remains hindered by the lack of an in vivo system in which to study the development and onset of autoimmune diabetes. To this end, we characterized a system using human CD4+ T cells pulsed with autoantigen-derived peptides. Six weeks after injection of as few as 0.5 × 106 antigen-pulsed cells into the NOD-Scid Il2rg−/− mouse expressing the human HLA-DR4 transgene, infiltration of mouse islets by human T cells was seen. Although islet infiltration occurred with both healthy and diabetic donor antigen-pulsed CD4+ T cells, diabetic donor injections yielded significantly greater levels of insulitis. Additionally, significantly reduced insulin staining was observed in mice injected with CD4+ T-cell lines from diabetic donors. Increased levels of demethylated β-cell–derived DNA in the bloodstream accompanied this loss of insulin staining. Together, these data show that injection of small numbers of autoantigen-reactive CD4+ T cells can cause a targeted, destructive infiltration of pancreatic β-cells. This model may be valuable for understanding mechanisms of induction of human diabetes.
Introduction
The development of type 1 diabetes involves a combination of genetic and environmental factors governing susceptibility to and/or protection from disease (1). NOD mice, the most widely studied model of human type 1 diabetes, share a number of disease characteristics, including autoantigens, the chronicity of the autoimmunity, and major histocompatibility complex (MHC) homology, but significant differences between the two still remain (e.g., the time of progression from insulitis to clinical diabetes, the sex bias of disease incidence) (2). Because of these differences and others, many mechanisms and treatments that have been verified in NOD mice have failed to translate to successful treatments in humans (3,4). Therefore, developing model systems in which human cells involved in diabetes can be directly studied is imperative.
The antigens involved in type 1 diabetes have largely been identified through autoantibodies found in individuals at risk for and with the disease. They include preproinsulin (PPI), GAD65, and islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) as well as other antigens recognized by polyclonal antibodies (islet cell antibodies) (5). T cells directed against these antigens are believed to cause β-cell destruction, but little direct evidence shows that this is the case. The technical problems in studying the functions of autoreactive T cells include difficulties in growing and maintaining autoantigen-reactive lines and the lack of a suitable model system in which they can be studied.
Previous studies have analyzed histopathology (6–8) and T-cell tetramer staining (9) of pancreata from cadaveric diabetic donors. In these studies, CD8+ T cells that are reactive with IGRP were detected by immunohistochemical staining. However, staining of prediabetic insulitic lesions in humans is still conspicuously missing from the literature. Better visualization and understanding of these earliest events are of great significance because it is unknown how the cellular composition of these lesions may have changed up to the point of clinical type 1 diabetes diagnosis, let alone over a lifetime of disease in an individual. Knowledge of these very early events could allow for the design of therapeutics aimed at the prevention as well as the treatment of type 1 diabetes.
In the current study, we tested whether CD4+ T cells derived from HLA-matched diabetic and healthy donors and expanded on diabetes antigens could cause insulitis and β-cell destruction in NOD mice devoid of endogenous T cells, B cells, and natural killer cells (NOD-Scid Il2rγ−/− mice, referred to herein as NSG mice) and transgenic for human HLA-DR4 (10,11) (referred to herein as NSG.DR4 mice). Parallel injections of peripheral blood mononuclear cells (PBMCs) from diabetic or healthy control individuals were also performed, allowing for direct comparisons of both the extent of insulitis and the nonspecific organ involvement of the two systems.
We show that injections of antigen-pulsed expanded CD4+ T cells from patients with type 1 diabetes result in varying degrees of islet infiltration from peri-insulitis to severe insulitis. In these mice, there was a significant loss of insulin and increased levels of demethylated Ins1 DNA and caspase-3 staining compared with control mice, reflecting β-cell death. Of note, we isolated increased numbers of mouse CD45+ cells from the pancreata of mice injected with diabetic donor CD4+ T cells, suggesting that in this model, cells from diabetic patients are able to establish an inflammatory environment in which murine leukocytes collaborate. These studies are the first to our knowledge to show β-cell destruction mediated by human cells in a hybrid humanized mouse system. This model will be useful for studies of early insulitis and β-cell destruction mediated by human immune cells.
Research Design and Methods
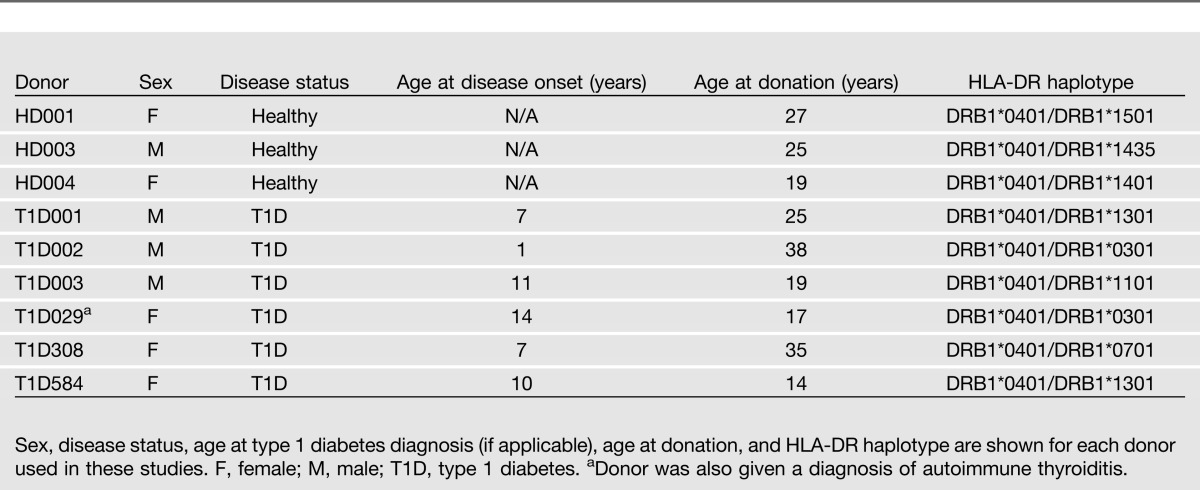
HLA Haplotype Determination
PBMCs were collected from patients with type 1 diabetes and nondiabetic donors through leukopheresis or whole-blood collection. Lymphocytes were isolated through Ficoll gradient. DNA was isolated from each prospective donor (Qiagen DNeasy Blood & Tissue Kit), and MHC haplotype was determined using the DRDQ 2 Test SSP UniTray Kit (Invitrogen). Only donors determined to be DRB1*0401 were used for further analysis. HLA-DR typing and disease status of donors are shown in Table 1. Written informed consent was obtained from all donors. The use of human cells was approved by the Yale Institutional Review Board.
Table 1.
Summary of HLA-DR4 donors
Magnetic-Activated Cell Sorting
To isolate CD4+ T cells, PBMCs were incubated with a depleting antibody cocktail and processed according to the manufacturer’s instructions (CD4+ T Cell Isolation Kit II, human; Miltenyi Biotech). The effluent population was considered CD4+ T-cell enriched and the population remaining on the column to be CD4+ T-cell depleted; this second population was used as donor-autologous antigen presenting cells (APCs). Purity of the isolated CD4+ T-cell population was routinely >93% by a fluorescence-activated cell sorter (FACS).
Peptides and T-Cell Culture
Peptides used for T-cell priming, as previously described (12,13), were produced by the Keck Biotechnology Research Laboratory, Yale University, as follows: IGRP23–35 (YTFLNFMSNVGDP), IGRP247–259 (DWIHIDTTPFAGL), and PPI76–90, 88S (SLQPLALEGSLQSRG). For generation of antigen-pulsed lines, previously described protocols were amended (12–14). APCs generated by magnetic-activated cell sorting were adhered to plastic for 2 h. Adherent cells were collected and irradiated (5,000 rad) after being loaded with 10 μg/mL peptide in AIM V medium (Invitrogen). Twenty units interleukin (IL)-2 were replenished every 4 days until immortalization.
Cultures were restimulated every 2 weeks with irradiated, peptide-pulsed, donor-matched APCs at ratios of 10:1 (CD4:APC). After four stimulations, CD4+ T cells were infected with a human telomerase (hTERT)–expressing murine leukemia virus (MuLV)–based retroviral vector (15,16) 3 days poststimulation to generate continuous cell lines. Cells were incubated in T-cell medium with virus plus 8 μg/mL polybrene (hexadimethrin bromide; Sigma) for 6 h.
Transfer of T Cells
NSG.DR4 mice were generated by breeding our NS.DR4 mice to NSG mice (The Jackson Laboratory). Six- to 8-week-old NSG.DR4 mice were injected with 1.0–3.0 × 106 HLA-DR4 donor PBMCs or 0.5–5.0 × 106 antigen-pulsed CD4+ T cells. For a given donor and line, there were no differences in tissue reconstitution or insulitis scores observed in conjunction with variations in the number of cells injected. The use of animals was approved by the Yale University Institutional Animal Care and Use Committee. Mice were killed 6 weeks postinjection. Single-cell preparations were made from the blood, spleen, and pancreas. The percent reconstitution (Fig. 1D) with human cells was calculated based on the results of staining with human anti-CD45 (APC) and mouse anti-CD45 (fluorescein isothiocyanate; BD Biosciences) as shown in equation 1:
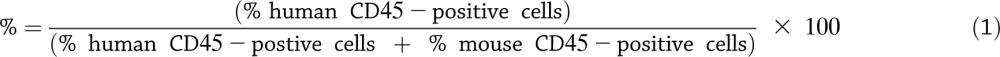 |
Figure 1.

Injection of antigen-specific CD4+ T-cell lines into NSG.DR4 mice. A: CD4+ T cells from two healthy and two diabetic donors were stained for tetramer (as described in Research Design and Methods) at two time points: 24 h after isolation (when they had not yet been APC/peptide stimulated) and 2 weeks after APC/peptide stimulation. Percentages of CD4+ T cells staining tetramer positive are shown. After 24 h, tetramer staining fell in the range of 0.06–0.20%. Tetramer staining after peptide pulsing for 2 weeks ranged between 0.53 and 3.80%, with antigen-specific fold inductions in the range of 4–43 times. B: Mismatched tetramer staining of antigen-expanded lines was also performed to determine that the increased tetramer staining at 2 weeks was due to a true expansion of antigen-specific cells and not to nonspecific staining. Mismatched tetramer staining was found to be at or below background levels; an example of such staining (from donor T1D029) is shown. C: Six- to 8-week-old NSG.DR4 mice were injected with 1.0–3.0 × 106 HLA-DR4 donor PBMCs (41 T1D and 15 healthy control mice) or 0.5–5.0 × 106 antigen-pulsed CD4+ T cells (87 T1D and 21 healthy control mice) in 150 μL PBS or PBS alone through a retro-orbital intravenous route. Injection numbers were determined based on cell availability, and no significant differences in reconstitution were observed over either range. Mice were killed 6 weeks postinjection. Single-cell preparations were made from the blood (top left), spleen (top right), and pancreas (bottom) of each mouse and were stained for expression of both human and mouse CD45 as described in Research Design and Methods. The percentage of live cells staining positive for human CD45 is graphed along the y-axis. ○, T1D CD4 line injections; □, HD CD4 line injections; ●, T1D PBMC injections; ■, HD PBMC injections. PBMC injections from all donors and CD4+ T-cell line injections from donors HD003, HD004, T1D001, T1D029, and T1D584 are shown. Each data point represents one animal, and significance was determined using nonparametric ANOVA (overall P < 0.0001). *P < 0.05; ***P < 0.001. HD, healthy donor; T1D, type 1 diabetes; wk, weeks.
Characterization of mouse cells in the pancreas was done using anti-mouse CD11b (PE; BD Biosciences) and anti-mouse CD11c (PeCy7; eBioscience).
Histology: Preparation and Analysis
Pancreatic tissues were stained for human CD45RO and insulin. For CD45RO, antigen retrieval was followed by incubation with a 1:50 dilution of mouse anti-human CD45RO (Invitrogen) for 60 min and a biotinylated goat anti-mouse IgG (Kirkegaard & Perry Laboratories, Inc.) for 30 min. For insulin staining, a 1:50 dilution of guinea pig anti-insulin (Invitrogen) was incubated for 60 min followed by peroxidase rabbit anti-guinea pig (Invitrogen; 1:200 in PBS).
Insulin Staining Quantification
Insulin staining of pancreas sections was performed as described previously for all mice that showed positive insulitis scores. Photographs of all islets from one section were then taken for each such mouse. ImageJ software (http://rsbweb.nih.gov/ij) was used to measure the total area of each islet. The total area of each islet staining positive for insulin was then determined with the Analyze Particles function, taking into account pixel size 500–∞ and circularity 0.00–1.00. The percent positive insulin staining was then determined for each islet as follows in equation 2:
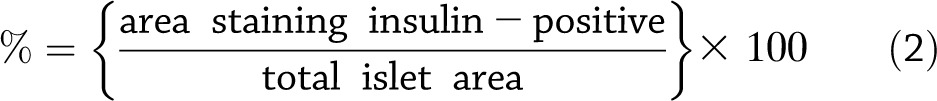 |
Analyzing and Scoring Tissue
For each mouse, every islet on one slide was scored according to the amount of CD45RO+ cell invasion, using guidelines shown in Supplementary Table 1. For scoring the small intestine, only very-well-oriented villi were considered. Slides were scored as described in Supplementary Table 1. Scoring of graft-versus-host disease (GVHD) in the liver was based on the number of portal tracts affected/involved, using guidelines in Supplementary Table 1. Scoring of the acinar tissue was performed to determine the nature of any pancreatic infiltration, using guidelines in Supplementary Table 1. Balding and skin conditions at the time of harvest were scored using guidelines in Supplementary Table 1.
Tetramer Staining and Flow Cytometry
IGRP247–259 and PPI76–90, 88S tetramers were produced by the Tetramer Core Laboratory, Benaroya Research Institute at Virginia Mason, using methods previously described (17). Fourteen days after generation, 5 × 106 antigen-pulsed cells were stained with tetramer (1-μL tetramer/105 cells) in AIM V medium plus 2% human serum for 2.5 h at a concentration of 1,000 cells/μL. Cells were then washed and stained with anti-human CD25-fluorescein isothiocyanate and anti-human CD4-APC (BD Biosciences) for an additional 45 min. Cells were analyzed/sorted on the BD FACS Vantage SE using the following guidelines: CD4+ cells were first gated, and cells were then examined using CD25 and tetramer staining. Cells staining tetramer positive and CD25mid were then collected as the final target population.
Analyzing β-Cell Death
Relative levels of serum-derived demethylated Ins1 DNA were measured by RT-PCR, using methods previously described (18). Caspase-3 staining was performed by Yale Pathology Tissue Services, using rabbit anti-caspase-3 (abCam) and biotin-SP–conjugated donkey anti-rabbit IgG (Jackson Immunoresearch). For each islet evaluated, the total number of cells as well as the number of cells staining caspase-3 positive were counted, and these values were used to calculate the percentage of caspase-3–positive cells in each islet.
Statistical Analysis
GraphPad Prism software was used for statistical analysis. Statistical significance was determined using the unpaired t test or ANOVA as noted, except in the case of the linear regression analysis shown in Fig. 5B.
Figure 5.

Insulin loss and β-cell death in mice injected with CD4+ T-cell lines from T1D donors. A: The cumulative analysis of insulin staining in mice with islet scores >0. The percentage of each islet staining positive for insulin (see research design and methods) is graphed along the y-axis; each data point represents one islet. The percent insulin-positive mean, number of mice analyzed, and number of islets analyzed for each cohort are as follows: T1D CD4+ lines, 47.6%, 32 mice, 206 islets; healthy CD4+ lines, 57.3%, 6 mice, 44 islets; T1D PBMC, 51.7%, 27 mice, 147 islets; healthy PBMC, 56.8%, 14 mice, 50 islets; PBS only (noninjected) control, 62.0%, 3 mice, 23 islets; and STZ treated, 3.0%, 3 mice, 4 islets. Control, STZ-treated mice were injected intraperitoneally for 5 consecutive days with 40 mg/kg body weight STZ. PBMC injections from all donors and CD4+ T-cell line injections are shown from donors HD004, T1D001, T1D029, and T1D584. Significance was determined using parametric ANOVA (overall P < 0.0001). *P < 0.05; **P < 0.01; ****P < 0.0001. B: In mice injected with CD4+ T-cell lines from T1D donors, average insulin staining is significantly reduced in those with higher average islet scores (P = 0.0150), with a slope of −4.044 ± 1.570. Each data point represents one mouse (n = 32). CD4+ T-cell line injections from donors T1D001, T1D029, and T1D584 are shown. C: There was an increase in β-cell death in recipients of T1D donor antigen-expanded cell lines. The relative level of demethylated Ins1 DNA (i.e., Δ = Ctmeth – Ctdemeth) was measured by RT-PCR from 150 μL serum isolated from mice 6 weeks after receiving cell lines from healthy control or T1D donors by methods previously described (18). Control STZ-treated mice were injected intraperitoneally with 200 mg/kg body weight STZ and harvested 24 h later. The percent increase in Δ was compared with the same for control NSG mice treated with PBS. The percent increase in Δ for mice treated with STZ ranged from 38 to 118%. ●, healthy control CD4+ T-cell line injections; ■, T1D CD4+ T-cell line injections. CD4+ T-cell line injections from donors HD004, T1D001, T1D029, and T1D584 are shown. Significance was determined using the unpaired t test. *P < 0.05. D: There was an increase in the percentage of islet cells staining caspase-3 positive in mice injected with CD4+ T-cell lines from T1D donors. Mice with islet scores >0 were included in the analysis. For each such mouse, the total number of cells as well as the number of caspase-3–staining cells were counted for each islet. The percentage of each islet staining caspase-3 positive is shown along the y-axis. Each data point represents one islet. For T1D CD4 lines, 32 mice, 111 islets; healthy CD4 lines, 6 mice, 53 islets; T1D PBMC, 27 mice, 137 islets; healthy PBMC, 14 mice, 29 islets; and PBS only (noninjected) control, 3 mice, 11 islets. PBMC injections from all donors and CD4+ T-cell line injections from donors HD004, T1D001, T1D029, and T1D584 are shown. Significance was determined using parametric ANOVA (overall P < 0.0001). *P < 0.05; ***P < 0.001; ****P < 0.0001. n/s, not significant; T1D, type 1 diabetes.
Results
Reconstitution of NSG.DR4 Mice With PBMCs or Antigen-Pulsed CD4+ T-Cell Lines From Healthy and Diabetic Donors
We stimulated antigen-reactive T cells from PBMCs from healthy control or type 1 diabetic HLA-DR4–positive donors using diabetes antigens PPI76–90, 88S, IGRP23, and IGRP247. Tetramer staining was used to identify the percentages of antigen-specific cells in pulsed lines after 2 weeks (Fig. 1A and 1B). Cells were immortalized (15,16), and 3H-based proliferation assays were used to verify continued antigen-specific T-cell expansion in the lines postimmortalization (Supplementary Fig. 1).
NSG.DR4 mice were injected with 1.0–3.0 × 106 HLA-DR4 donor PBMCs or 0.5–5.0 × 106 HLA-DR4 donor antigen-pulsed CD4+ cells and were killed 6 weeks postinjection. Single-cell preparations were made from the blood, spleen, and pancreas of each mouse, and cells were stained for expression of both human and mouse CD45 to determine the extent of reconstitution of a given tissue.
For both healthy and diabetic donors, the average level of human CD45 cell reconstitution of blood (P < 0.001) (Fig. 1C, top left) and spleen (P < 0.001) (Fig. 1C, top right) was greater in mice injected with PBMCs than in those injected with antigen-pulsed CD4+ T-cell lines. We observed differences in the infiltration of cells into the pancreas when the NSG.DR4 mice received CD4+ T-cell lines expanded with islet antigens. There was significantly greater pancreatic infiltration of human CD45+ cells in recipients of the CD4+ lines from diabetic donors than from healthy donors (P < 0.05) (Fig. 1C, bottom).
Comparison of Insulitis in Mice Reconstituted With PBMCs or CD4+ T Cells From Healthy Control Subjects or Patients With Type 1 Diabetes
We next sought to determine whether the human CD45+ cells detected in the pancreas were homing to the islets; thus, human CD45RO staining of pancreatic sections was performed. Examples of human lymphocyte infiltration of mouse islets are shown in Fig. 2A. The accumulated islet scores of all diabetic and healthy control donor CD4+ T-cell–injected mice are shown in Fig. 2B. Injection of diabetic donor T cells yielded significantly higher insulitis scores (P < 0.05). Furthermore, severe insulitis was only observed with diabetic donor CD4+ T-cell injections. The differences in the cellular infiltrates were not due to general differences in the viability of the CD4+ T cells before transfer because the lines responded to antigen stimulation and expanded with similar kinetics. In addition, the expansion of the CD4+ T cells did not reflect a difference in MHC gene dosing because all the diabetic and healthy control donors were heterozygous for DR4 (Table 1).
Figure 2.

Lymphocytic infiltration of the pancreatic islets at termination. A: Islet scoring was based on human CD45RO staining of paraffin slides as described in Research Design and Methods. Shown are examples of pancreata from mice injected with T1D CD4+ T-cell lines, highlighting various levels of insulitis. Human CD45RO+ lymphocytes are stained dark red. B: Cumulative scoring of islets is shown for injections of T1D CD4+ T-cell lines (3 donors, 87 mice, 277 islets), healthy control CD4+ T-cell lines (2 donors, 21 mice, 99 islets), T1D PBMCs (6 donors, 41 mice, 131 islets), and healthy control PBMCs (3 donors, 15 mice, 64 islets). Each data point represents one islet. PBMC injections from all donors and CD4+ T-cell line injections from donors HD003, HD004, T1D001, T1D029, and T1D584 are shown. Statistical significance was determined using nonparametric ANOVA (overall P < 0.0001). *P < 0.05; ***P < 0.001. HD, healthy donor; T1D, type 1 diabetes.
Lymphocytic Infiltration of the Extraislet Pancreas and Other Organs
To determine whether the infiltrates we had seen with the diabetes antigen-specific CD4+ cell lines were specific for islets, we determined whether there were infiltrates in nonislet tissues (small intestine, liver, pancreatic acinar tissue, and skin). Tissues were scored for each mouse with any islet scores >0 (Fig. 3). There was evidence of a minor GVHD-like response in the small intestine of mice injected with PBMCs; this was not the case in mice injected with CD4+ T-cell lines (Fig. 3A). Liver infiltration and pancreatic acinar tissue infiltration were considerable in mice injected with PBMCs but not in mice receiving CD4+ T-cell lines (Fig. 3B and 3C). The majority of mice (>70%) injected with PBMCs showed some level of balding at the time of sacrifice (Fig. 3D), but this was not seen in the recipients of the CD4+ T-cell lines.
Figure 3.

Lymphocytic infiltration outside the pancreatic islets. For evaluation of nonspecific tissue infiltration by lymphocytes, a 6-cm section of small intestine was removed just below the duodenum and a 0.5 × 1.0 cm section was removed from the tip of the median lobe of liver at the termination of experiments. Shown is lymphocytic infiltration of the small intestine (A), liver (B), and pancreatic acinar tissue (C) and skin health (D) at termination in mice injected with PBMCs (n = 41) or CD4+ (n = 39) T-cell lines. Only mice with islet scores >0 were included in the analysis. Healthy control and diabetic donors were grouped together for analysis because no differences were observed between the grouped cohorts. For liver, small intestine, and pancreatic acinar tissue, scoring was based on hematoxylin-eosin staining of paraffin slides as described in Research Design and Methods. ○, CD4 line injections; ●, PBMC injections. PBMC injections from all donors and CD4+ T-cell line injections from donors HD003, HD004, T1D001, T1D029, and T1D584 are shown. Significance was determined using nonparametric ANOVA.
Variations in Peptide Response in Type 1 Diabetic Donors
We next compared the islet infiltration with CD4+ T-cell lines grown on various diabetes-associated peptides and found that the islet infiltration varied by cell donor and by islet antigen (Fig. 4). With injections from patient T1D001, for example, PPI88S-pulsed cells show the greatest infiltration of mouse islets. CD4+ T cells from donor T1D584 also showed a dominant response to PPI88S, although all three peptide-pulsed lines caused insulitis in mice. Donor T1D029 showed a dominant response to IGRP23, with only minor insulitis associated with the PPI88S line injection and no infiltration with the IGRP247 line injection. These findings also indicate that variation in donor responses are islet or insulin peptide driven and not merely a by-product of nonspecific in vitro stimulation because donor lines were prepared and stimulated in parallel and injected in equal numbers and yet showed different infiltration capabilities in vivo.
Figure 4.

Lymphocytic infiltration of the islets at termination by donor. Examples of lymphocytic infiltration of the pancreatic islets at termination in mice injected with PBMC (n = 56) or CD4+ T-cell lines (n = 108). Each data point represents one individual islet. A breakdown of the islet scores resulting from CD4+ line injections for each donor is shown. ●, IGRP23-pulsed CD4 T-cell lines; ■, IGRP247-pulsed CD4 T-cell lines; ▲, PPI88S-pulsed CD4 T-cell lines; ▼, IGRP23/IGRP247 combination–pulsed CD4 T-cell lines. Statistical significance was determined using nonparametric ANOVA (T1D001 overall P = 0.0142, T1D029 overall P = 0.002, T1D584 overall P = 0.0019, HD004 overall P = 0.0048). *P < 0.05; **P < 0.01.
Insulin Staining Is Reduced in Islets Exhibiting Insulitis
Next, we determined whether the significant pancreatic infiltration affected insulin production in the islets. ImageJ (19) was used to analyze all mice with islet scores >0, and the percentage of the total islet area that was positive for insulin staining was determined for each islet.
On average, the insulin-positive area from islets in untreated control mice was 62%, consistent with previously published observations (20). There was no statistically significant decrease in the insulin-positive area in mice receiving PBMCs from healthy control donors, but there was a small reduction in the insulin-positive area in recipients of PBMCs from type 1 diabetes donors (P < 0.01) (Fig. 5A). Recipients of antigen-pulsed diabetic CD4+ T-cell lines showed less insulin staining than those in any other category examined (mean insulin positive 47.6%) and significantly less (P < 0.0001) than those from healthy donors, which did not cause a significantly reduced expression of insulin.
We performed linear regression analysis to determine whether there was a relationship between the average percentage of insulin positivity and the average islet score of mice injected with CD4+ T-cell lines from diabetic donors (Fig. 5B). This analysis showed that average insulin staining was negatively correlated with insulitis scores (r2 = 0.176, P = 0.015). However, none of the recipient mice developed hyperglycemia.
Analysis of the Mechanism of Islet Destruction in the Recipients of Antigen-Reactive CD4+ T Cells
Because the mice did not develop overt diabetes, it was not clear whether the infiltrating cells were actually killing β-cells or, alternatively, whether the reduced insulin staining was due to functional impairment of the β-cells, resulting in decreased insulin staining (21). To determine whether the transfer of the CD4+ T cells from patients with type 1 diabetes caused β-cell death, we used a quantitative PCR-based assay to detect circulating β-cell–derived demethylated Ins1 DNA released from dying β-cells. We previously showed that this assay can detect β-cell death in streptozotocin (STZ)-induced diabetes in mice and in NOD mice before hyperglycemia (18). Serum samples from 12 mice injected with CD4+ T-cell lines from type 1 diabetic donors and resulting in marked insulin loss were compared with serum samples from mice injected with healthy donor CD4+ T-cell lines with insulitis scores >0 (none of the healthy donor line–injected mice showed marked insulin loss). Control samples were taken from age-matched mice injected with PBS only as well as from STZ-treated mice. For CD4+ T-cell– and STZ-injected mice, the percent increase in Δ = (Ctmeth − Ctdemeth) was compared with the same for control NSG mice treated with PBS. There were significantly greater levels of demethylated Ins1 DNA in the serum of recipients of antigen-expanded CD4+ T-cell lines from type 1 diabetic donors compared with recipients of CD4+ T-cell lines injected from healthy donors (P < 0.05) (Fig. 5C). Caspase-3 staining of pancreatic sections corroborated these results. Mice injected with CD4+ T-cell lines from diabetic donors had the highest percentage of caspase-3–positive cells per islet (Fig. 5D).
To better elucidate the mechanism of β-cell killing observed in mice injected with type 1 diabetic donor CD4+ T-cell lines, we next isolated PPI88S antigen-specific clones from healthy and type 1 diabetic donor CD4+ T-cell lines, using a modified version of previously described methods (14). Fourteen clones from healthy donors and four from patients with type 1 diabetes were isolated and verified as antigen specific by 3H-based proliferation assays (data not shown). We compared the cytokine production by the clones in response to phorbol myristic acid/ionomycin stimulation and found that the average levels of granulocyte-macrophage colony-stimulating factor, interferon γ, and tumor necrosis factor α production were comparable in cells from type 1 diabetic and healthy donors, but the cells from healthy donors produced higher levels of IL-4, IL-5, and IL-13 (Supplementary Fig. 2).
Antigen-specific CD4+ T-cells with a Th2-like cytokine phenotype have been shown to be less capable of recruiting cells to the pancreas than cells with a Th1-like phenotype and to be unable overall to promote islet destruction in NOD mouse (22–25). Consequently, we hypothesized that a difference in cytokine production on a cellular level leads to a less inflammatory environment in mice injected with healthy donor cells. A hallmark of any inflammatory lesion is the recruitment of antigen-nonspecific cells. Therefore, we looked for another indicator of inflammation—the presence of mouse CD45+ cells—in mice injected with type 1 diabetic versus healthy donor CD4+ T-cell lines. We evaluated overall pancreatic cell counts and mouse CD45 staining (by FACS) of pancreas at harvest. We found that mice injected with type 1 diabetic CD4+ T cells show a marked increase in mouse CD45 cell homing to the pancreas compared with mice injected with healthy donor CD4+ T cells (P = 0.016) (Fig. 6A).
Figure 6.

Mouse CD45 cells were recruited to the pancreas by CD4+ T cells from T1D donors. A: Recruitment of mouse leukocytes to the pancreas was determined by cell count in one-half a pancreas at the time of harvest in conjunction with mouse CD45 staining by FACS. The number of mouse CD45 cells (in millions) is shown along the y-axis. Recruitment of mouse leukocytes was significantly higher in mice injected with T1D CD4 lines (87 mice) than in mice injected with HD CD4 lines (21 mice, P = 0.0160). Each point represents one mouse. CD4+ T-cell line injections from donors HD003, HD004, T1D001, T1D029, and T1D584 are shown. B: Single-cell suspensions were isolated and pooled from mice injected with T1D001 PPI88S lines. Cells were stained for expression of mouse CD45 (left panel), the CD45+ (middle panel), and CD45− (right panel) populations further analyzed for CD11b and CD11c expression to better characterize the mouse cells recruited to the pancreas in this model. Of the cells staining mouse CD45+, 34% also stained CD11b+CD11c+, identifying them as dendritic cells (middle panel). The majority of the remaining CD45+ cells stained CD11b+ (middle panel). No CD11b or CD11c staining was detected in the population staining CD45– (right panel). C: F4/80 staining (left panel) shows large quantities of pancreas-infiltrating F4/80-positive macrophages and/or dendritic cells in a mouse injected with a CD4+ T-cell line from donor T1D001. An NSG.DR4 mouse injected with cells from HD004 (middle panel) shows little or no F4/80 staining, similar to an NSG.DR4 mouse injected with PBS only (right panel). All panels, 40× magnification. HD, healthy donor; T1D, type 1 diabetes.
FACS analysis was performed on cells recovered from the pooled pancreata of mice injected with T1D001 PPI88S lines. Approximately 90% of mouse CD45+ cells analyzed were CD11b+, and approximately one-third of these cells were also CD11c+. These CD11b+CD11c+ cells correspond to a mouse islet antigen-presenting dendritic cell population previously described by Calderon and Unanue (26) (Fig. 6B). The remaining CD11b+CD11c– population likely contains pancreas-infiltrating macrophages, which have previously been identified in diabetic islet infiltrates (27).
Histologic staining of mice injected with diabetic donor CD4+ T cells revealed large numbers of F4/80-positive–staining cells in the islets (Fig. 6C, left). Similar staining was also performed in healthy donor–injected and PBS-injected NSG.DR4 mice; in these cases, F4/80-positive cells were present only in very small numbers or not at all (Fig. 6C, centerand right).
Discussion
We characterized a system using autoantigen-pulsed human CD4+ T cells whose homing was readily observed in a mouse expressing the human HLA-DR4 transgene. We observed infiltration of mouse islets by human cells; severe insulitis was only observed in mice injected with CD4+ T-cell lines from diabetic donors (Fig. 2). The infiltration was specific for the islets (Fig. 3) and was associated with reduced insulin staining (Fig. 5A). Insulin staining in mice injected with healthy donor CD4+ T cells was not reduced in this system. Because only mice with positive insulitis scores were included in this analysis, it can be concluded that although healthy donor T cells can infiltrate the pancreas, they do not disrupt insulin production in this model. Although other groups have observed islet infiltration using injection of either human PBMCs alone or human PBMCs followed by a human CD8+ T-cell clone in the NSG HLA-A2–positive mouse (28,29), the current study, to our knowledge, is the first demonstration of CD4+ T-cell–associated β-cell death in a humanized mouse model.
Some insulitis was observed with transfer of diabetic donor PBMCs, although insulin staining was significantly more decreased (and caspase-3 staining significantly more increased) with diabetic donor CD4+ T-cell line transfer than with PBMC transfer. However, because of high levels of infiltration of the exocrine pancreas associated with PBMC transfer (as well as advanced balding and increased levels of disease in the liver and intestine), it seems that the actions of PBMCs are nonspecific in nature in this model. A more appropriate label for the infiltration observed in the pancreata of PBMC-injected mice would thus be GVHD as opposed to insulitis.
It was possible that reduced insulin staining in diabetic donor CD4+ T-cell line–injected mice was the consequence of inflammatory infiltrates and not the result of β-cell killing because dysfunctional β-cells that can recover with immune therapy have been identified previously by our group (21). However, along with reduced insulin staining and increased caspase-3 staining, we found increased levels of circulating demethylated β-cell–derived DNA in recipients of CD4+ T-cell lines from patients with type 1 diabetes, indicating that the cellular infiltrates contributed to β-cell killing (Fig. 5C). Islet destruction and insulin loss may have occurred before the 6-week termination point of our experiments. Because the window for demethylated DNA detection in the process of β-cell death may be limited, the levels of β-cell death may be even greater than estimated by the analyses discussed herein.
Despite the reduced insulin-positive area and evidence of β-cell killing, the mice did not develop overt hyperglycemia, suggesting that additional cells (e.g., CD8+ T cells) may be required for complete β-cell destruction. Van Belle et al. (30) showed in the human insulin promoter-lymphocytic choriomeningitis mouse model of type 1 diabetes that autoreactive CD8+ T cells depend on CD4+ T-cell–mediated recruitment to the pancreatic islets. Indeed, the importance of CD4+ T cells and MHC class II expression in type 1 diabetes pathogenesis is well documented. Several groups determined that allelic variations in MHC class II genes result in odds ratios for diabetes susceptibility much greater than any other known gene or chromosomal region (31–33). Specifically, HLA-DR4 expression (especially in conjunction with coexpression of HLA-DR3/HLA-DQ8) results in greatly increased diabetes susceptibility (34). Furthermore, Arif et al. (35) demonstrated that CD4+ T cells appear in a greater percentage of islets and in significantly greater numbers in individuals recently diagnosed with type 1 diabetes as opposed to patients with long-term diabetes, highlighting their importance early in disease. Although CD4+ T cells do not appear capable of initiating fulminant diabetes in the current model, the findings reinforce the potential of diabetes antigen-reactive CD4+ T cells to establish a critical pathogenic environment in the pancreatic islets.
To this end, we observed by FACS significantly increased levels of mouse CD45+ cells in the pancreata of mice injected with type 1 diabetic donor CD4+ T cells relative to mice injected with healthy donor CD4+ T cells (Fig. 6A). In a healthy, intact pancreas, a baseline level of mouse sentinel leukocyte homing is present in the islets (36). In the current model, the combination of large numbers of antigen-specific infiltrating human CD45RO cells (Fig. 2B) and a proinflammatory cytokine environment (Supplementary Fig. 2) appears to lead to increased mouse leukocyte homing to and accumulation in the islet, which in turn could lead to antigen release by dying β-cells and to further recruitment of murine cells capable of antigen presentation and β-cell killing. This explanation would also account for the lack of mouse CD45+ cell recruitment in mice injected with healthy donor cells because healthy donor cells did not accumulate significantly in the islets and clones derived from these cells displayed a more Th2-like cytokine profile relative to type 1 diabetic clones.
We also observed a dominant antigenic response leading to insulitis that differed among subjects (Fig. 4), possibly reflecting an increased proportion of antigen-specific cells or growth advantage of cells with particular antigen specificity. It has been argued that insulin autoimmunity is a base requirement for initiation of type 1 diabetes (37–39). We observed that a mutant PPI (PPI88S) peptide was often the dominant epitope tested in terms of the levels of insulitis observed. In one diabetic donor (T1D029), however, the IGRP23 epitope clearly dictated the dominant response. In light of these observations, the roles of different autoimmune targets are theoretically different among individuals or even wax and wane over the course of disease in a given individual so that autoimmune responses (or a lack thereof) targeting different antigens may direct the course of disease at various time points. Furthermore, this observation is a proof of concept for our model. If the insulitis observed was simply due to nonspecific activation in vitro before injection, one would expect to find comparable levels of infiltration with all three lines from the same donor; this was clearly not the case.
The results of experiments outlined in this article show that injection of autoantigen-pulsed CD4+ T cells leads to targeted infiltration of the pancreas, insulin loss, and β-cell killing. This model system may be useful for testing the ability of immune modulatory agents to prevent β-cell destruction. Such interventions could include coinjection of donor-matched regulatory T cells (40), cytokine therapy (30,41,42), or chemokine/chemokine receptor blockade (43,44). Furthermore, by defining the dominant antigens recognized by pathologic T cells, a personalized approach to immune therapy may be developed.
Article Information
Acknowledgments. The authors thank Thomas Gibson (University of Connecticut Health Center) for assistance with histology and immunostaining, and William Kwok and Eddie James (Benaroya Institute at Virginia Mason) for MHC class II tetramers.
Funding. This work was supported by a JDRF Center grant (JDRF 2007-1059), a National Institutes of Health Interdisciplinary Immunology Training Program grant (T32-AI-07019), and a National Institutes of Health grant (P30-DK-045835).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. A.A.V.M. designed and performed experiments and wrote the manuscript. S.E.M. and J.L. performed experiments. J.A.G. consulted on scoring pathology and reviewed and edited the manuscript. L.W., N.H.R., and K.C.H. contributed to the discussion and reviewed and edited the manuscript. A.L.M.B. designed experiments and wrote the manuscript. A.L.M.B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1141/-/DC1.
References
- 1.Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 1986;314:1360–1368 [DOI] [PubMed] [Google Scholar]
- 2.Kikutani H, Makino S. The murine autoimmune diabetes model: NOD and Related strains. In Advances in Immunology Dixon FJ, Ed. Washington, DC, Academic Press, 1992, p. 285–322 [DOI] [PubMed] [Google Scholar]
- 3.Roep BO. Are insights gained from NOD mice sufficient to guide clinical translation? Another inconvenient truth. Ann N Y Acad Sci 2007;1103:1–10 [DOI] [PubMed] [Google Scholar]
- 4.von Herrath M, Nepom GT. Remodeling rodent models to mimic human type 1 diabetes. Eur J Immunol 2009;39:2049–2054 [DOI] [PubMed] [Google Scholar]
- 5.Eisenbarth GS, Moriyama H, Robles DT, et al. Insulin autoimmunity: prediction/precipitation/prevention type 1A diabetes [published correction appears in Autoimmun Rev 2006;5:364]. Autoimmun Rev 2002;1:139–145 [DOI] [PubMed] [Google Scholar]
- 6.Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia 1986;29:267–274 [DOI] [PubMed] [Google Scholar]
- 7.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009;155:173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965;14:619–633 [DOI] [PubMed] [Google Scholar]
- 9.Coppieters KT, Dotta F, Amirian N, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 2012;209:51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wicker LS, Chen SL, Nepom GT, et al. Naturally processed T cell epitopes from human glutamic acid decarboxylase identified using mice transgenic for the type 1 diabetes-associated human MHC class II allele, DRB1*0401. J Clin Invest 1996;98:2597–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woods A, Chen HY, Trumbauer ME, Sirotina A, Cummings R, Zaller DM. Human major histocompatibility complex class II-restricted T cell responses in transgenic mice. J Exp Med 1994;180:173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Danke NA, Berger D, et al. Islet-specific glucose-6-phosphatase catalytic subunit-related protein-reactive CD4+ T cells in human subjects. J Immunol 2006;176:2781–2789 [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Danke N, Roti M, et al. CD4+ T cells from type 1 diabetic and healthy subjects exhibit different thresholds of activation to a naturally processed proinsulin epitope. J Autoimmun 2008;31:30–41 [DOI] [PubMed] [Google Scholar]
- 14.Raddassi K, Kent SC, Yang J, et al. Increased frequencies of myelin oligodendrocyte glycoprotein/MHC class II-binding CD4 cells in patients with multiple sclerosis. J Immunol 2011;187:1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barsov EV, Andersen H, Coalter VJ, Carrington M, Lifson JD, Ott DE. Capture of antigen-specific T lymphocytes from human blood by selective immortalization to establish long-term T-cell lines maintaining primary cell characteristics. Immunol Lett 2006;105:26–37 [DOI] [PubMed] [Google Scholar]
- 16.Barsov EV. Selective immortalization of tumor-specific T cells to establish long-term T-cell lines maintaining primary cell characteristics. Methods Mol Biol 2009;511:143–158 [DOI] [PubMed] [Google Scholar]
- 17.Novak EJ, Liu AW, Nepom GT, Kwok WW. MHC class II tetramers identify peptide-specific human CD4(+) T cells proliferating in response to influenza A antigen. J Clin Invest 1999;104:R63–R67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akirav EM, Lebastchi J, Galvan EM, et al. Detection of β cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci U S A 2011;108:19018–19023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brissova M, Fowler MJ, Nicholson WE, et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 2005;53:1087–1097 [DOI] [PubMed] [Google Scholar]
- 21.Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, Herold KC. Effects of autoimmunity and immune therapy on β-cell turnover in type 1 diabetes. Diabetes 2006;55:3238–3245 [DOI] [PubMed] [Google Scholar]
- 22.Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science 1995;268:1185–1188 [DOI] [PubMed] [Google Scholar]
- 23.Healey D, Ozegbe P, Arden S, Chandler P, Hutton J, Cooke A. In vivo activity and in vitro specificity of CD4+ Th1 and Th2 cells derived from the spleens of diabetic NOD mice. J Clin Invest 1995;95:2979–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradley LM, Asensio VC, Schioetz LK, et al. Islet-specific Th1, but not Th2, cells secrete multiple chemokines and promote rapid induction of autoimmune diabetes. J Immunol 1999;162:2511–2520 [PubMed] [Google Scholar]
- 25.Hill NJ, Van Gunst K, Sarvetnick N. Th1 and Th2 pancreatic inflammation differentially affects homing of islet-reactive CD4 cells in nonobese diabetic mice. J Immunol 2003;170:1649–1658 [DOI] [PubMed] [Google Scholar]
- 26.Calderon B, Unanue ER. Antigen presentation events in autoimmune diabetes. Curr Opin Immunol 2012;24:119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoh N, Hanafusa T, Miyazaki A, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest 1993;92:2313–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitfield-Larry F, Young EF, Talmage G, et al. HLA-A2-matched peripheral blood mononuclear cells from type 1 diabetic patients, but not nondiabetic donors, transfer insulitis to NOD-scid/γc(null)/HLA-A2 transgenic mice concurrent with the expansion of islet-specific CD8+ T cells. Diabetes 2011;60:1726–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Unger WWJ, Pearson T, Abreu JRF, et al. Islet-specific CTL cloned from a type 1 diabetes patient cause beta-cell destruction after engraftment into HLA-A2 transgenic NOD/scid/IL2RG null mice. PLoS ONE 2012;7:e49213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Belle TL, Nierkens S, Arens R, von Herrath MG. Interleukin-21 receptor-mediated signals control autoreactive T cell infiltration in pancreatic islets. Immunity 2012;36:1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies JL, Kawaguchi Y, Bennett ST, et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature 1994;371:130–136 [DOI] [PubMed] [Google Scholar]
- 32.Noble JA, Valdes AM, Cook M, Klitz W, Thomson G, Erlich HA. The role of HLA class II genes in insulin-dependent diabetes mellitus: molecular analysis of 180 Caucasian, multiplex families. Am J Hum Genet 1996;59:1134–1148 [PMC free article] [PubMed] [Google Scholar]
- 33.Todd JA, Walker NM, Cooper JD, et al. Genetics of Type 1 Diabetes in Finland. Wellcome Trust Case Control Consortium Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 2007;39:857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aly TA, Ide A, Jahromi MM, et al. Extreme genetic risk for type 1A diabetes. Proc Natl Acad Sci U S A 2006;103:14074–14079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arif S, Moore F, Marks K, et al. Peripheral and islet interleukin-17 pathway activation characterizes human autoimmune diabetes and promotes cytokine-mediated β-cell death. Diabetes 2011;60:2112–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calderon B, Suri A, Miller MJ, Unanue ER. Dendritic cells in islets of Langerhans constitutively present β cell-derived peptides bound to their class II MHC molecules. Proc Natl Acad Sci U S A 2008;105:6121–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moriyama H, Abiru N, Paronen J, et al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A 2003;100:10376–10381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakayama M, Abiru N, Moriyama H, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005;435:220–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krishnamurthy B, Dudek NL, McKenzie MD, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest 2006;116:3258–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bluestone JA, Tang Q. Therapeutic vaccination using CD4+CD25+ antigen-specific regulatory T cells. Proc Natl Acad Sci U S A 2004;101(Suppl. 2):14622–14626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grinberg-Bleyer Y, Baeyens A, You S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 2010;207:1871–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hulme MA, Wasserfall CH, Atkinson MA, Brusko TM. Central role for interleukin-2 in type 1 diabetes. Diabetes 2012;61:14–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christen U, von Herrath MG. Initiation of autoimmunity. Curr Opin Immunol 2004;16:759–767 [DOI] [PubMed] [Google Scholar]
- 44.Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MBA. Among CXCR3 chemokines, IFN-γ-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-γ (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J Immunol 2003;171:6838–6845 [DOI] [PubMed] [Google Scholar]