

Abstract
We assessed whether epigenetic histone posttranslational modifications are associated with the prolonged beneficial effects (metabolic memory) of intensive versus conventional therapy during the Diabetes Control and Complications Trial (DCCT) on the progression of microvascular outcomes in the long-term Epidemiology of Diabetes Interventions and Complications (EDIC) study. We performed chromatin immunoprecipitation linked to promoter tiling arrays to profile H3 lysine-9 acetylation (H3K9Ac), H3 lysine-4 trimethylation (H3K4Me3), and H3K9Me2 in blood monocytes and lymphocytes obtained from 30 DCCT conventional treatment group subjects (case subjects: mean DCCT HbA1c level >9.1% [76 mmol/mol] and progression of retinopathy or nephropathy by EDIC year 10 of follow-up) versus 30 DCCT intensive treatment subjects (control subjects: mean DCCT HbA1c level <7.3% [56 mmol/mol] and without progression of retinopathy or nephropathy). Monocytes from case subjects had statistically greater numbers of promoter regions with enrichment in H3K9Ac (active chromatin mark) compared with control subjects (P = 0.0096). Among the patients in the two groups combined, monocyte H3K9Ac was significantly associated with the mean HbA1c level during the DCCT and EDIC (each P < 2.2E-16). Of note, the top 38 case hyperacetylated promoters (P < 0.05) included >15 genes related to the nuclear factor-κB inflammatory pathway and were enriched in genes related to diabetes complications. These results suggest an association between HbA1c level and H3K9Ac, and a possible epigenetic explanation for metabolic memory in humans.
Introduction
Long-term microvascular and macrovascular complications are a major cause of morbidity and mortality in type 1 diabetes patients. Several genetic studies (1–3) have identified loci associated with diabetic nephropathy, and recent evidence suggests that diabetes and its complications may also involve epigenetic factors (4–7). Epigenetic mechanisms triggered by hyperglycemia and related diabetic conditions may be particularly important since long-term complications appear to develop in some patients with diabetes even after improved glucose control.
The Diabetes Control and Complications Trial (DCCT) (1983–1993) demonstrated that intensive therapy aimed at near-normal levels of glycemia (HbA1c level) profoundly reduced the development and progression of microvascular complications in patients with type 1 diabetes compared with conventional therapy aimed at maintaining clinical well-being (8). After DCCT end, the cohort was followed up annually in the observational Epidemiology of Diabetes Interventions and Complications (EDIC) study (1994 to present) when all subjects were encouraged to practice intensive therapy. Since year 4 of the EDIC, both groups have maintained mean HbA1c levels of ∼8% (64 mmol/mol). Surprisingly, despite nearly equivalent glycemic control during the EDIC, complications continued to develop in subjects in the original DCCT conventional therapy group at significantly greater rates than in participants in the original intensive therapy group (8–14). This phenomenon has been termed “metabolic memory.” Similar “legacy effects” of prior strict glycemic control were also observed in the UK Prospective Diabetes Study (15,16).
Thus, an early period of better glycemic control appears to have sustained influence on long-term complications, and the adverse effects of early hyperglycemia may be “imprinted” on target tissues over time. Recently, epigenetic mechanisms have been implicated in the damaging effects of hyperglycemia and in experimental models of metabolic memory (17–25).
Epigenetics refers to mechanisms that can modify gene expression and phenotype without changes in the underlying DNA sequence (26,27). Although different cell types in a person are assumed to have identical DNA sequences, they possess distinct differences in their epigenetic information, such as DNA methylation and post-translational modifications (PTMs) of histone proteins contained in the chromatin. Epigenetic modifications can occur when individuals are exposed to environmental factors, such as infections and nutritional changes, and can predispose them to diseases such as diabetes (28). Nucleosomes, the basic subunits of chromatin, consist of octamers of histones H2A, H2B, H3, and H4, wrapped by DNA. PTMs of histones (e.g., acetylation, methylation, phosphorylation, and ubiquitylation) form an epigenetic layer together with DNA methylation (29–31), which affects gene transcription. Acetylation of histones at lysine residues is generally associated with transcriptionally active genes, whereas lysine methylation leads to gene activation or repression, depending on the specific site and level of methylation (29,30,32). Alterations in histone PTMs and their interactions with other nuclear proteins at gene promoters or other regulatory regions can lead to relatively stable epigenetic changes that alter chromatin structure. In turn, this can lead to long-term dysregulated gene expression and disease progression.
Recent studies using vascular and inflammatory cells treated in vitro with high glucose (HG), or target cells and tissues derived from models of diabetes complications, provide strong evidence that alterations in epigenetic histone PTMs play key roles in diabetes-induced inflammation and vascular complications, and potentially in the metabolic memory phenomenon (17–25,33–41). However, studies have not yet been performed directly in humans with diabetes and metabolic memory. To examine whether epigenetic mechanisms are related to glycemic history, the progression of complications and metabolic memory in human diabetes, we explored variations in the profiles of key histone PTMs at promoter regions in peripheral blood lymphocytes and monocytes obtained from selected EDIC cohort subjects.
Research Design and Methods
The human research study protocol was approved by the Institutional Review Board at City of Hope National Medical Center and the EDIC clinics.
Subjects and Experimental Design
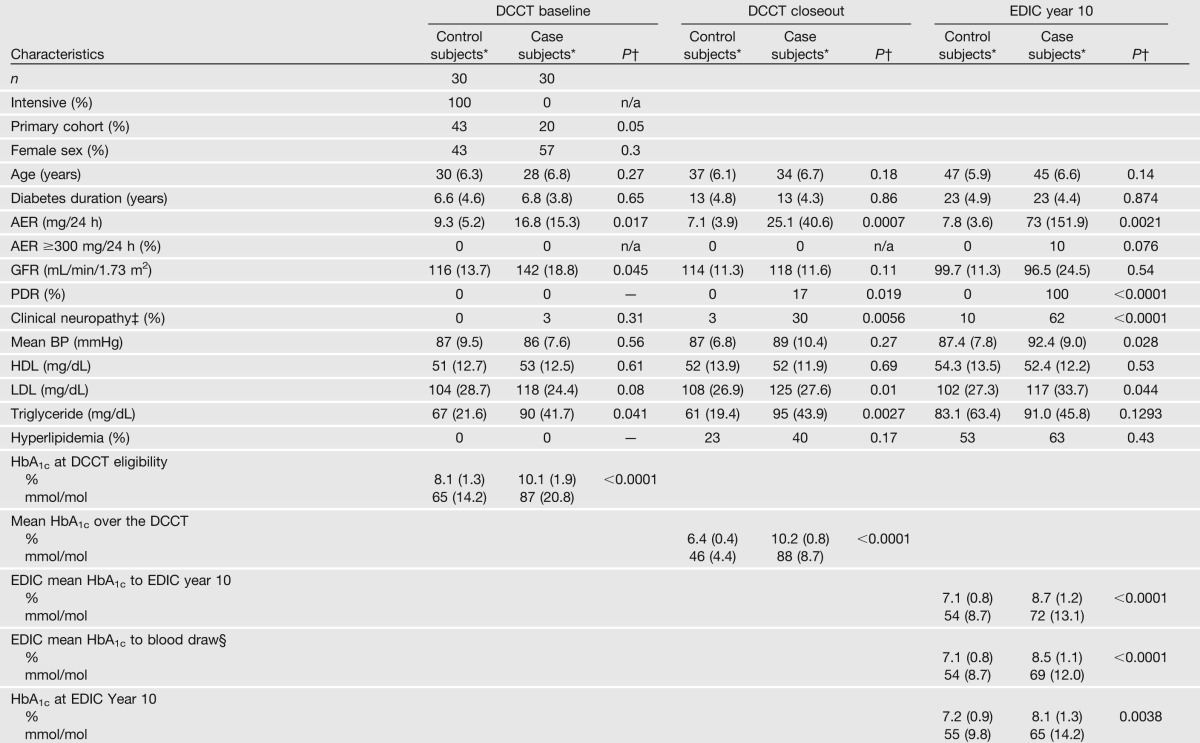
The DCCT enrolled 1,441 subjects who were followed up for an average of 6.5 years until study closeout in 1993. Consenting subjects, of whom 60 were selected for this study, were then followed up annually during the EDIC. A case group of 30 subjects was selected from the former DCCT conventional therapy group who had a DCCT mean HbA1c level of >9.1% (76 mmol/mol), and progression of five or more steps from DCCT closeout to year 10 in the EDIC on the Early Treatment Diabetic Retinopathy Study scale of retinopathy severity or scatter laser in the EDIC, and/or an albumin excretion rate (AER) of >300 mg/24 h or end-stage renal disease at EDIC year 9/10 among those subjects free of scatter laser and albuminuria during the DCCT (some subjects had both retinal and renal complications). A control group of 30 subjects was selected from the former DCCT intensive therapy group who had a DCCT mean HbA1c level of <7.3% (56 mmol/mol), and no progression in Early Treatment Diabetic Retinopathy Study scale from DCCT closeout through EDIC year 9/10 and an AER of ≤30 mg/24 h in all DCCT/EDIC years (Table 1). All subjects were white nonsmokers. The epigenetics laboratory was masked to the case/control group identity of each patient until all profiling and initial data analyses were completed.
Table 1.
Clinical characteristics of case and control subjects at DCCT baseline, DCCT closeout (end of randomized therapy), and at EDIC year 10, and HbA1c at different times
Blood Cell Isolation
Blood samples (50 mL) were freshly collected from each patient at participating EDIC clinics and shipped overnight to the City of Hope National Medical Center using ThermaSure Ambient Gel Packs (Sebra) to maintain the temperature between 15 and 25°C. Peripheral blood mononuclear cells were isolated using endotoxin-free Ficoll-Paque PLUS (catalog no. 17-1440-03; GE Healthcare Life Sciences). Monocytes were purified by CD14-positive selection (catalog no. 18088; STEMCELL Technologies), and flow-through fractions were collected, washed, and used as the lymphocyte fractions.
Chromatin Preparation, Chromatin Immunoprecipitation Assays, and Chromatin Immunoprecipitation-Chip Experiments
Lymphocytes and monocytes from each participant were cross-linked in 1% formaldehyde, sonicated to shear DNA, and stored at −80°C for chromatin immunoprecipitation (ChIP) assays. ChIP assays were performed as previously described (17,21) using the following histone PTM antibodies: anti-histone H3 lysine-9 acetylation (H3K9Ac) (ab10812; Abcam); H3 lysine-4 trimethylation (H3K4Me3) (active motif 39169); and H3K9Me2 (active motif 39375). ChIP-enriched DNA samples and no antibody controls were purified from monocytes and lymphocytes of each subject. Five sets of ChIP-enriched DNA samples (H3K9Ac, H3K4Me3, and H3K9Me2 in lymphocytes; and H3K9Ac and H3K4Me3 in monocytes) from the 60 patients were obtained (299 samples total; one monocyte H3K4Me3 ChIP from the control group failed). Each sample was then labeled and hybridized to Roche Nimblegen human 720 K RefSeq promoter tiling arrays (ChIP-chip) to profile these histone PTMs at promoter regions (−3,200 to 800 base pairs [bp] relative to the transcription start sites [TSSs] of 22,542 RefSeq genes) genome wide. Hybridizations were performed at the Clinical Microarray Core, University of California, Los Angeles, or the Functional Genomics Core, Beckman Research Institute. Therefore, five ChIP-chip data sets were generated from five sample sets. The experimental design and data analysis pipeline are shown in Supplementary Figs. 1 and 2.
Real-Time Quantitative PCR
ChIP-quantitative PCRs (qPCRs) were performed using the histone H3K9Ac ChIP-enriched DNA from each individual (n = 60) and normalized to Input DNA as previously described (17,21). Primers were designed to amplify the following promoter regions for ChIP-qPCR validations: STAT1 (5′-GGTCATTCAGAGACCCCAGA-3′ and 5′-TTAAAGCCCAGCCCAAATAC-3′); TNFα (5′-ACCACAGCAATGGGTAGGAG-3′ and 5′-GAGGTCCTGGAGGCTCTTTC-3′); IL1A (5′-TCACTTGAGGCCCAGAGTTT-3′ and 5′-AAGCTGCTTTCCTCCCAGAT-3′); and SPI1 (5′-GCTTCCCTACCACCAGAAGA-3′ and 5′-TGGGGAAACTGAGAAACCTG-3′).Total RNA and polyA RNA were prepared as described previously (18).
Bioinformatic Analysis of ChIP-Chip Data
Extensive quality assessments were first applied to each array/data set using methods listed in Supplementary Fig. 3. Probe-level Cy5/Cy3 log2 ratios on each array were shifted to render the Tukey biweight mean of log2 ratios to 0. The complete data from all the samples from case and control subjects in each data set were then subjected to quantile normalization. For H3K4Me3 and H3K9Me2, Loess normalization was initially applied to each sample to correct dye bias found on some of the samples by MA plots. Regions enriched with histone modifications (i.e., containing at least four consecutive probes with log2 ratio greater than the 85th percentile of all the probe-level log2 ratios on each array) were identified using TAMAL (42) in each of the 60 samples. Commonly H3K9 acetylated regions among all these samples were then designated as regions acetylated in at least 25% of samples and at least 350 bp in length. The associations between acetylation and HbA1c levels at different time periods (Table 1) in these commonly acetylated regions were analyzed using Pearson correlation coefficients (see Fig. 2 legend). Null data were generated in the same 6,248 regions after random sample permutation (n = 100). Furthermore, case hypermodified regions in commonly modified regions in at least 50% of the case subjects were identified by multiple linear regression analysis with and without adjusting for age and gender (see Fig. 3 legend). Hypermodified regions in control subjects were similarly identified at commonly modified regions in the control subjects. The comparisons were not adjusted for batches because of good technical reproducibility (Supplementary Fig. 4), the sample processing (Supplementary Fig. 1), as well as the normalization applied for systemic bias correction. The false discovery rate (FDR) was calculated as the number of falsely discovered regions (estimated by the median number of regions of 100 sample permutations using the same analysis procedure described above) divided by the number of observed regions.
Figure 2.

Association of monocyte H3K9 acetylation levels with HbA1c at different time periods. A: The five time periods from which documented HbA1c values for the case and control groups were used. Each EDIC participant’s history of type 1 diabetes (T1D) is divided into three phases: Pre-DCCT, DCCT, and EDIC. The pre-DCCT phase is from the date of type 1 diabetes diagnosis (Dx) to the date of entry into the DCCT in 1983. DCCT is from entry into DCCT to the end of DCCT in 1993 within which HbA1c was measured quarterly. EDIC is from 1993 (start of EDIC) to current, during which HbA1c level was documented annually. All our study participants had blood drawn in the EDIC year 16/17 (2009–2010), and the HbA1c level measured at this time is termed the “blood draw.” HbA1c levels in the other four periods (DCCT, EDIC, DCCT/EDIC, and Pre-DCCT/DCCT/EDIC) are represented by mean values across specified periods. B: Association of acetylation levels with HbA1c level at H3K9 commonly acetylated regions. Commonly acetylated regions across all 60 samples were defined as regions enriched with H3K9Ac in at least 15 samples (25%) that were at least 350 bp in length. The average acetylation level at each of the resulting 6,248 commonly acetylated regions was calculated in each sample, and its relationship with HbA1c level at different time periods (color coded) is represented by Pearson correlation coefficient. Sample permutation (n = 100) was performed to generate the correlation coefficients under the null condition. The cumulative densities of the correlation coefficients are plotted (color solid lines, original data; black dashed line, permuted data), and a shift to the right indicates a higher positive association. The statistical significances of distances between two empirical distributions were tested using Kolmogorov-Smirnov tests. The time periods are color-coded as indicated.
Figure 3.

Identification of group-specific promoter regions depicting the largest differences in the levels of histone H3K9Ac in monocytes. The H3K9 commonly acetylated regions in case and control subjects were first identified. The average log2 ratio of at least four probes within these regions in case and control subjects were compared, and the hyperacetylated regions in case or control subjects were identified using multiple linear regression with one-sided test before and after adjusting for age and gender. The analyses were performed using custom R codes built on top of the functions implemented in “preprocessing Core” and “GenomicRanges” bioconductor packages, as well a “stats” package. A: The volcano plot of 4,644 commonly acetylated regions found in case subjects. B: The volcano plot of 4,286 commonly acetylated regions found in control subjects. The y-axis indicates the −log10 (P value), and the x-axis indicates the log2 ratio differences between the two groups. Each dot represents one group-specific commonly acetylated region. Regions considered to be hypermodified (fold change ≥1.1 and nominal P < 0.05) in case or control subjects are highlighted as small empty circles. C: Venn diagrams showing the comparison of the hyperacetylated promoters in the case and control groups, with and without adjustment for age and gender.
Data analyses (Supplementary Fig. 2) were performed using custom R codes on top of Bioconductor and “stats” packages, unless otherwise specified. Hierarchical clustering was generated by Cluster version 3.0 and visualized by Java TreeView version 2.1.
Data Deposition
The ChIP-chip data sets have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (Gene Expression Omnibus accession no. GSE47385).
Results
Case and Control Subjects
The characteristics of the selected 30 case and 30 control subjects are summarized in Table 1. As a result of the design, the case subjects had higher AERs and estimated glomerular filtration rates (evidence of hyperfiltration), even at DCCT baseline; and higher prevalences of retinopathy, nephropathy, and neuropathy during the DCCT and EDIC. While case and control subjects were not selected on the basis of lipid level and blood pressure, the levels among case subjects were also higher. Since the case and control subjects were selected in part on the basis of different levels of HbA1c during the DCCT, the HbA1c level was higher among case subjects than control subjects during the DCCT (10.2% vs. 6.4% [88 vs. 46 mmol/mol]) (Table 1). Owing to a modest within-person (intraclass) correlation of HbA1c levels of 0.62 over the DCCT and 0.64 over the 10 years of EDIC, the case subjects also had higher HbA1c levels at DCCT eligibility testing (10.1% vs. 8.1% [87 vs. 65 mmol/mol]), during the 10 years of the EDIC (8.5% vs. 7.1% [69 vs. 54 mmol/mol]), and at the time of blood draw for this study (EDIC year 16/17) (8.0% vs. 7.4% [64 vs. 57 mmol/mol]).
Global Comparison of Key Histone Modifications Between Case and Control Subjects
ChIP-enriched DNA samples were obtained from monocytes and lymphocytes of each of the 60 subjects with antibodies to histone H3K9Ac (a modification usually associated with active gene expression), H3K4Me3 (normally associated with active promoters), and H3K9Me2 (associated with repressed genes). Five ChIP-chip data sets were thereby generated as described in Research Design and Methods. From these data, regions with differences in histone modifications were identified as described in Research Design and Methods. The distribution of the number of regions enriched with histone modifications in the case and control subjects is shown in Fig. 1 for each of the five chromatin marks evaluated. Case subjects had a significantly higher average number of regions enriched with H3K9Ac in monocytes than did control subjects (6,470 vs. 6,105; Fig. 1A, Wilcoxon rank sum exact two-sided test, P = 0.0096; P = 0.048 adjusting for five tests). For the other chromatin marks studied, the average number of enriched regions showed no significant difference between case and control subjects: monocyte H3K4Me3 (6,284 vs. 6,310); lymphocyte H3K9Ac (6,979 vs. 7,153); lymphocyte H3K4Me3 (7,686 vs. 7,699); and lymphocyte H3K9Me2 (4,979 vs. 5,150) (Fig. 1B–E).
Figure 1.

Distribution of the number of regions enriched with the histone PTMs in the case and control groups. For each histone modification mark studied, the promoter regions of genes modified by the specific chromatin marks were identified in each sample using TAMAL. Modified regions were defined as regions that had at least four consecutive probes with modification levels in the form of log2 ratios greater than the 85th percentile of all the probes on the Nimblegen promoter array. The distribution of the number (#) of histone-modified regions across the case and control groups are shown by box plots for H3K9Ac (A) and H3K4Me3 (B) in monocytes and H3K9Ac (C), H3K4Me3 (D), and H3K9Me2 (E) in lymphocytes. The box plot function in R was used to generate the plot where the middle lines within the boxes represent the median value, and the top and bottom whiskers are the maximum and minimum values excluding outliers that are over 1.5 times the interquantile range. Outliers are represented by empty circles. Two-sided Wilcoxon rank sum exact tests were used to compare the number of modified regions between the control subjects and case subjects in each data set. The increased number of H3K9 acetylated regions in monocytes from case vs. control subjects was statistically significant (*P = 0.0096).
Association of Monocyte H3K9Ac Levels With HbA1c Levels at Different Time Periods
For each of the 60 EDIC subjects, mean HbA1c levels were measured over four time periods and also at the time of blood draw (EDIC years 16/17) for this study (Table 1) (schematically depicted in Fig. 2A). HbA1c was measured upon study entry, then quarterly during the DCCT, and annually during the EDIC. The four periods are as follows: DCCT mean, representing the mean level of quarterly HbA1c measurements during the DCCT; EDIC mean, representing the mean of annual HbA1c measurements during the EDIC up to the time of blood draw for the current epigenetics study; the DCCT/EDIC combined mean, representing the mean level during the DCCT and EDIC periods combined; and the pre-DCCT/DCCT/EDIC combined mean, representing an estimate of the actual mean HbA1c level since diagnosis up to the time of the blood draw. The DCCT/EDIC combined mean is the time-weighted average of the quarterly DCCT HbA1c values (weighted by 3 months) and the annual EDIC values (weighted by 12 months). The pre-DCCT/DCCT/EDIC HbA1c level likewise is a time-weighted average including the baseline HbA1c weighted by the months duration of diabetes on entry.
We assessed the correlation between HbA1c levels over these different time periods with the H3K9Ac levels in the 6,248 commonly acetylated regions across all 60 samples (as defined in Research Design and Methods). The empirical distributions of correlation coefficients with HbA1c level at different time periods (colored solid lines), as well as under the null condition (black dashed line) in all these regions, are shown in Fig. 2B (blue curve for EDIC phase is not visible because of overlap by the curves for the DCCT/EDIC and pre-DCCT/DCCT/EDIC periods). Notably, when compared with null conditions, H3K9Ac levels showed highly significant, positive associations (right shifts) with HbA1c levels at all time periods (P < 2.2E-16, Kolmogorov-Smirnov two-sided test).
Identification of Group-Specific Promoter Regions Displaying the Largest Differences in the H3K9Ac Levels
We then searched for specific genomic regions that had the largest differences in monocyte H3K9Ac levels in case versus control subjects. We first identified 4,644 H3K9 commonly acetylated regions in case subjects, and 4,286 regions in the control subjects (see Research Design and Methods and Fig. 3 legend for details). Among these, 3,780 case commonly acetylated regions overlap with 3,784 control regions.
For each of the 4,644 commonly acetylated regions among the case subjects, the H3K9Ac levels were compared in case versus control subjects using a one-sided t test. Of these, 35 regions had nominally significantly higher H3K9Ac levels in the case versus the control subjects (nominal P < 0.05 without correction for the 4,644 tests, and fold change ≥1.1) (Fig. 3A). The median FDR of these identified regions was estimated to be 0.31 by sample permutation (n = 100), which means that ∼70% of the genes in this list are true positives. Because of the potential effect of demographic factors (principally age and gender) on H3K9Ac levels, we also analyzed the 4,644 regions that were acetylated among the case subjects using linear regression models adjusting for age and gender. Using the same criteria, 39 regions (Table 2) were identified that had higher H3K9Ac levels in the case versus the control subjects (nominal P < 0.05) with estimated FDR at 33%. These regions were located in the promoters of 38 genes (−3,200 to 800 bp relative to TSS) compared with the 35 genes identified without adjustment, 26 being in common (Fig. 3C).
Table 2.
Case or control H3K9 hyperacetylated regions in monocytes
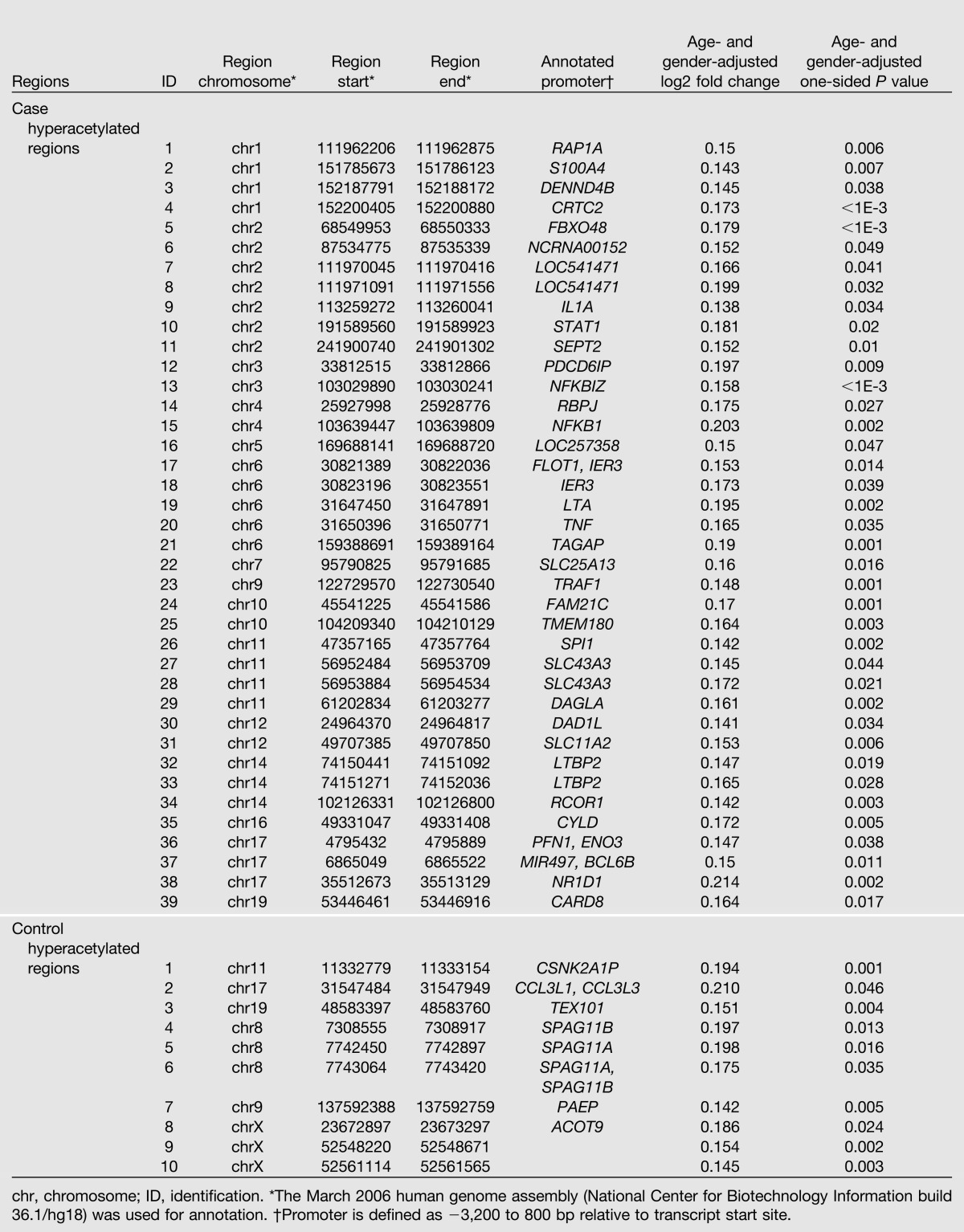
Similarly, among the 4,286 control commonly acetylated regions, 6 regions (located in the promoters of 7 RefSeq genes) and 10 regions (including 8 covering 8 RefSeq promoters; Table 2) were hyperacetylated in control subjects (relative to case subjects, nominal P < 0.05) without and with adjusting for age and gender (Fig. 3B). Between the two lists, five genes are in common (Fig. 3C). However, the median FDR estimated by permutation was 100%.
Similar analyses were also performed on the other four data sets, including the monocyte H3K4Me3, and the lymphocytes H3K9Ac, H3K4Me3, and H3K9Me2. Using the same criteria applied to the monocyte H3K9Ac, the case and control hypermodified regions were identified and summarized for these four data sets (Supplementary Table 1). Because the estimated FDR was 100% for each of these four identified lists, we focused our subsequent analyses only on the monocyte H3K9Ac data set. There was also no overlap between the regions found in the five data sets, suggesting that in general the differences in PTMs occur independent of each other.
Analysis of Monocyte Histone H3K9 Hyperacetylated Regions/Promoters in Case and Control Subjects
To visualize the differences between the case and control groups, and examine whether these regions share similar acetylation patterns among these samples, we performed hierarchical clustering analysis on the case or control hyperacetylated regions identified after adjusting for age and gender (which include 39 annotated case hyperacetylated regions and 8 annotated control hyperacetylated regions) (Fig. 4A). As seen, the acetylation levels were higher in general in the case subjects than in the control subjects at the case hyperacetylated regions. Among these regions, many were hyperacetylated in the same set of case subjects. Some heterogeneity can also be noted.
Figure 4.

Regions with variations in histone H3K9Ac levels in monocytes of the case and control groups, and the biological functions related to the annotated promoters in these regions. A: Hierarchical clustering of hyperacetylated regions in case and control groups. The H3K9 hyperacetylated regions (age- and gender-adjusted nominal P < 0.05, fold change ≥1.1) in the case and control groups were pooled, and the acetylation levels were calculated for each region by averaging the log2 ratios of probes falling into these regions. The acetylation levels were mean centered across the 60 samples, and hierarchical clustering of these regions was generated using the average linkage method with Pearson correlation as the similarity metric and visualized by Java TreeView version 2.1. Blue represents acetylation levels above the mean, and yellow represents acetylation levels below the mean. The annotated genes containing a hyperacetylated region in their promoters are shown on the right. For regions located in the promoters of more than one gene, all the corresponding gene symbols are listed (separated by commas). For multiple regions located within the same gene promoter, the same gene symbol is listed for each region. Thus, 39 case hyperacetylated regions (spanning the promoters of 38 genes) and 8 control hyperacetylated regions (spanning the promoters of 8 genes) are depicted. B: IPA of H3K9 hyperacetylated promoters in case subjects (in silico analyses). The 38 genes identified in case subject samples that have H3K9 hyperacetylated regions in their promoters (−3,200 to 800 bp of TSS) were imported into IPA for identification of overrepresented canonical pathways. The 162 pathways in IPA knowledge-based database were sorted by P values (Fisher’s exact test P values adjusted by the Bonferroni-Holm [B-H] method), and the top 10 pathways are shown. Blue bars represent −log10 (P value) of pathways. The B-H–adjusted P values of the enrichments of all the 10 pathways are <0.0025. IL, interleukin; IRF, interferon regulator factor; PKR, RNA-dependent protein kinase; Hyper, hyperacetylation.
We used in silico analysis to further evaluate the potential functional relevance of the 38 genes that have 39 case hyperacetylated regions in their promoters (Table 2). Ingenuity pathway analysis (IPA) revealed that these genes were enriched for several diabetes and diabetes complication–related pathways, including tumor necrosis factor receptor (TNFR)-2 signaling, macrophage and dendritic cell functions, interferon regulator factor signaling and pattern recognition, apoptosis, reactive oxygen species, and antiviral responses (Fig. 4B). Of note, genes related to the nuclear factor-κB (NF-κB) pathway, which is well-known to be associated with inflammation, immunity, and diabetes complications, were clearly enriched in this gene list. These included NF-κB pathway or target genes (TNF, IL1A, DENND4B, CYLD, IER3, and TRAF1), transcription factors (NFKB1, NFKBIZ, and STAT1), and regulatory genes (CYLD, PU.1, IER3, and TRAF1). Furthermore, motif analysis by transcription factor affinity prediction (43) showed that the top five enriched motifs (BH-adjusted P < 0.00005) with high affinities to the promoters of the 38 case-hyperacetylated genes (−500 to 100 bp relative to TSS) are all related to NF-κB.
Evaluation of Association Between Group-Specific Hyperacetylated Regions and HbA1c Levels at Different Time Periods
We then assessed the relationship between HbA1c levels over different time periods and the 39 case and 8 control hyperacetylated regions located in RefSeq promoters (Table 2). Among the 60 case and control group subjects combined, we calculated the Pearson correlation coefficients of HbA1c measures with the H3K9Ac levels of these 47 regions, and visualized the resulting correlation coefficients by heat maps (Fig. 5). We found that the acetylation levels in most of these regions were positively (in case hyperacetylated regions) or negatively (in control hyperacetylated regions) correlated with average HbA1c levels. The associations with mean HbA1c levels during EDIC, DCCT/EDIC combined, or the pre-DCCT/DCCT/EDIC combined periods were much greater than at the time of blood draw or during the DCCT only. HbA1c level from the pre-DCCT/DCCT/EDIC combined period had the highest association, while association at the time of blood draw was lowest. Notably, these results suggest that H3K9Ac levels of case/control hyperacetylated regions are associated with long-term HbA1c levels over the lifetime of diabetes, including the DCCT and EDIC study phases.
Figure 5.

Correlation of HbA1c with acetylation levels at case/control H3K9 hyperacetylated regions. Pearson correlation coefficients of acetylation levels with HbA1c levels at regions depicting hyperacetylation in case and control subjects (same regions shown in Fig. 4A) at the different time periods across 60 samples are shown as a heat map. The rows represent the case/control hyperacetylated regions, and the columns represent different time periods (labels on top of the heat map). Hyper, hyperacetylation.
Additional analyses also assessed correlations of HbA1c levels with the H3K9Ac levels at these same regions separately among the case and the control groups, with predominantly positive correlations among case subjects at case hyperacetylated regions, and inverse correlations among case subjects at control hyperacetylated regions. Correlations among control subjects were somewhat weaker. Details are shown by heat maps in Supplementary Fig. 5 and its legend.
Validation of Hyperacetylated Region at the STAT1 Gene Promoter
To validate the case hyperacetylated regions (Table 2), we chose STAT1, TNF, IL1A, and SPI1. The H3K9Ac differences between case and control subjects in the STAT1 promoter region from the ChIP-chip experiments are illustrated as a representative example in Fig. 6A by plotting the acetylation levels of the 30 case subjects (red dots) and 30 control subjects (blue dots) within the STAT1 promoter. The hyperacetylated region located at 2,800–2,400 bp upstream of the STAT1 TSS is highlighted. The average acetylation enrichment is clearly higher in case subjects (red line) than in control subjects (blue line). To validate the ChIP-chip microarray data, we performed follow-up ChIP-qPCRs with ChIP-enriched DNA samples from each of the 60 participants. There was a significant difference between case and control subjects in the H3K9Ac level at the STAT1 promoter region (one-sided P = 0.018, Wilcoxon rank sum exact test; Fig. 6B). Similar significant differences were also obtained for TNF, IL1A, and SPI1 promoter acetylations (Fig. 6C–E) (P < 0.001, P = 0.007, and P = 0.002, respectively). A positive correlation was also seen between data from ChIP-chips and ChIP-qPCRs for STAT1 (Pearson r = 0.46, P = 1.9 × 10−4) (Supplementary Fig. 6).
Figure 6.

Validation of ChIP-chip data of histone H3K9 hyperacetylated regions in case versus control subjects. A: Probe level H3K9Ac levels at the promoter region (−3,200 to 800 bp of TSS) of the STAT1 gene (from ChIP-chip data). This region contained 38 probes. For each probe, each dot represents one sample, with red dots representing the case subjects and blue dots representing the control subjects. The red line represents the average probe log2 ratio of samples in all of the case subjects, and the blue line represents the average probe log2 ratio of all of the control subjects. The dotted rectangle highlights the hyperacetylated region in the case subjects compared with control subjects. B–E: Results of ChIP-qPCR validation of the case hyperacetylated region located in the promoter/enhancer regions of the indicated genes STAT1, TNFα, IL1A, and SPI1. The histone H3K9Ac ChIPs were prepared from monocytes of each individual participant (30 case and 30 control subjects). Standardized ChIP-qPCRs were then used to quantify the amount of specific modified ChIP-enriched DNA in monocyte samples from each patient. Results shown were obtained using PCR primers designed to amplify the promoter/enhancer regions of the indicated four genes selected from Table 2. Data shown are the average of samples run in duplicate. One-sided P values (case vs. control subjects) were calculated by Wilcoxon rank sum exact test: STAT1, P = 0.018; TNFα, P < 0.001; IL1A, P = 0.007; and SPI1, P = 0.002.
Discussion
The objective of this study was to explore whether epigenetic differences could be associated in part with glycemic history and complications, and metabolic memory in individuals with type 1 diabetes by studying patients from the DCCT/EDIC cohort. As a first step toward understanding the link between epigenetics and metabolic memory in humans, we examined whether there were specific differences in genome-wide PTMs of promoter histones in monocytes and lymphocytes between subjects whose complications progressed and those whose complications did not (case vs. control subjects). Among case subjects drawn from the conventional group with a mean HbA1c level of 10.2% (88 mmol/mol) during the DCCT, complications progressed markedly in the EDIC, during which the mean HbA1c level was 8.7% (72 mmol/mol). Conversely, among control subjects drawn from the intensive therapy group with a mean HbA1c level of 6.4% (46 mmol/mol) during the DCCT, complications showed no progression in the EDIC, during which the mean HbA1c level rose to 7.1% (54 mmol/mol). The conventional group of the entire DCCT/EDIC cohort showed progression of complications during the EDIC even though the HbA1c level improved from ∼9% (75mmol/mol) during the DCCT to ∼8% (64 mmol/mol) during the EDIC, and the intensive therapy group showed substantially less progression even though the HbA1c level rose from ∼7% (53 mmol/mol) during the DCCT to ∼8% (64 mmol/mol) during the EDIC. Thus, our case and control subjects represent the extremes of the respective original conventional and intensive therapy groups in which the metabolic memory effect was initially documented (9,12,13), and thus provided a good platform for this study.
The rationale for studying monocytes and lymphocytes was as follows. These cells have inflammatory and immune response properties and can be obtained in a relatively noninvasive fashion during a regular visit. Next, the epigenome, unlike the genome, is cell type–specific. Moreover, evidence shows that inflammation and abnormal immune responses accompanied by monocyte and lymphocyte infiltration play major pathogenic roles in diabetic retinopathy and nephropathy (5–7), two complications evident in our case group, but not in the control group. Finally, reports (18,34,44,45) show that hyperglycemia itself can induce histone lysine acetylation and methylation in monocytes at inflammatory- and diabetes-related gene loci.
As the first study to profile multiple histone modifications at promoters genome-wide in lymphocytes and monocytes of participants from a diabetes clinical trial, we analyzed the initial overall correlations across the five histone PTM data sets. This revealed high correlations between the same active marks (H3K9Ac and H3K4Me3) in different cell types (monocytes and lymphocytes), modest correlations between the two active marks in the same cell types, and poor correlations between the repressive mark (H3K9Me2) and either of the active marks (Supplementary Figs. 7 and 8). Notably, through identification of promoter histone–modified regions genome-wide, we found significantly greater H3K9Ac-enriched regions in the monocytes of case subjects than in those of control subjects, whereas the other PTMs showed no significant differences. Furthermore, we identified 38 H3K9 hyperacetylated promoters in case subjects, none of which was differentially modified in the other PTM data sets. Exploratory evaluation of the functional relevance of these 38 hyperacetylated hotspots revealed they were enriched for several diabetes and diabetes complication–related pathways, including TNFR2 signaling. Of note, recent studies show that circulating TNFR1 and TNFR2 levels predict renal function decline in type 1 diabetes (46,47). Of the affected gene promoters detected in case subjects, many were regulated by the NF-κB pathway, including key inflammatory genes, transcriptional factors, and others involved in inflammatory and immunological pathways known to operate in diabetes complications (5–7). This suggests that systemic deregulation induced by histone acetylation at multiple levels in the NF-κB pathway could be related to metabolic memory.
The associations of HbA1c with H3K9Ac levels of the case-control groups combined were further analyzed. The mean HbA1c level over extended periods (e.g., DCCT and EDIC combined) was significantly associated with H3K9Ac levels in monocytes among >6,000 commonly acetylated regions across 60 samples. Although this may be expected since we observed hyperacetylation in case subjects, and the case-control groups were defined on the basis of differential HbA1c levels during DCCT, interestingly, additional analyses separately among case and control subjects in case hyperacetylated regions showed somewhat stronger correlations with HbA1c levels among case subjects (Supplementary Fig. 5). These results are consistent with those of prior analyses from the DCCT (48,49) showing a nonlinear relationship between HbA1c level and the progression of complications, such that the risk increases exponentially with increasing HbA1c levels.
Our current studies link diabetes complications to the histone acetylation of a set of genes related to inflammation in vivo. Moreover, the acetylation level was correlated with the history of Hb1Ac levels. These results, together, support a possible epigenetic mechanism for metabolic memory, whereby prolonged hyperglycemia may have a cumulative effect over time on H3K9 hyperacetylation in key genome regions that once acetylated, may remain relatively stable and are transmitted by unknown mechanisms. One obvious question here is whether the hyperacetylation of the promoters correlates with increased expression of the corresponding genes. We did not observe significant correlations between them when we profiled gene expression (Affymetrix) in 12 monocyte samples (7 case and 5 control subjects) (data not shown). This could be due to the limited number of samples tested. However, because we also did not find any significant differences in levels of circulating cytokines (serum) between 30 case and 30 control subjects at blood draw (data not shown), we speculate that, unlike the demonstrated epigenetic H3K9Ac levels, gene expression levels at the time of blood draw may not clearly reflect differences between case and control subjects. But, the acetylation could sensitize or render genes more susceptible to self-perpetuating changes in expression in response to external stimuli. Because H3K9Ac level is associated with relaxed chromatin and active genes, higher H3K9Ac levels occurring around a set of complication-related gene promoter regions may lead to a more open or poised chromatin state at these regions. Support also comes from our recent transcriptomic profiling of monocytes, which showed that most genes induced by HG have high levels of promoter H3K9Ac even in the normoglycemic state and are thus preselected or poised for activation by hyperglycemia (45). Interestingly, among our 38 case hyperacetylated promoters, the expression of TNF, LTA, STAT1, and CARD8 were all induced by HG (45).
Thus, the most likely scenario is that these genes are more poised for activation in the case group compared with the control group, contributing to various diabetes complications in the long term. This could be a consequence of the early exposure to hyperglycemia (measured by HbA1c level), which is known to be associated with increased rates of long-term diabetes complications. Recent evidence indicates that the enzymes mediating histone modifications (writers) are more likely to be inherited epigenetically than the modification itself (50), suggesting that, in our study, key histone H3K9 acetyltransferases may be playing a role. It is also likely that observed acetylation differences are due to either DNA methylation or other heritable epigenetic changes at distant transcription factors, or microRNA-encoding regions. An epigenetic mechanism is also supported by previous experimental models of metabolic memory (17–25,34–40). Further investigation is needed to better understand the biological meaning of these phenotypes (hyperacetylation), and their association with diabetes complications and metabolic memory.
Our results suggest an association between epigenetic changes and microvascular complications, and that hyperglycemia-induced epigenetic changes can potentially explain part of the metabolic memory phenomenon. However, alone they do not prove it, because from a cross-sectional study of this nature one can only infer correlations, not causation. A limitation is that we did not select samples of case and control subjects with different levels of HbA1c during the DCCT, but equivalent levels of HbA1c at DCCT baseline and during EDIC. Thus, hyperglycemia prior to the DCCT or during the EDIC could be partly responsible for epigenetic changes that affected the course of complications during the DCCT (or even earlier) and the EDIC. Second, given that case and control subjects were selected on the basis of HbA1c levels (high vs. low) during the DCCT and progression of complications (yes vs. no) during the EDIC, any differences in epigenetic changes in case versus control subjects would be expected to also be accompanied by correlations of those changes with HbA1c level. Third, the case and control subjects also differ with respect to other characteristics, such as blood pressure and lipid levels (Table 1), which in turn may have caused some epigenetic differences, and the resulting associations with HbA1c level may be secondary to the association of HbA1c with these factors. Unfortunately, the sample size is relatively small to conduct multiple linear regression analyses adjusting for any impact of these different factors to provide a more definitive assessment of these relationships. Ideally, an epigenetic profiling of a much larger fraction of the cohort would be needed to address these issues.
In conclusion, we conducted comprehensive epigenomic profiling using cells from two selected subsets of DCCT/EDIC participants who experienced different rates of complications following a period with different levels of hyperglycemia to explore an epigenetic mechanism for metabolic memory in individuals with type 1 diabetes. Our results suggest that this metabolic memory phenomenon can in part be explained by increased epigenetic differences at key complication-related genes among individuals with higher HbA1c levels that may contribute to further progression of complications during EDIC.
Article Information
Acknowledgments. The authors thank Dr. David Nathan (Harvard Medical School, Boston, MA) and the Epidemiology of Diabetes Interventions and Complications (EDIC) publications committee for many helpful discussions and critical comments; the Juvenile Diabetes Research Foundation for their invaluable support; and all of the study participants for their generosity and interest in the study. The authors also thank Dr. Margaret Morgan (City of Hope National Medical Center) for her help with the manuscript. The authors thank Dr. Rose Gubitosi-Klug (Case Western University), all the EDIC coordinators, and Stephan Villavicencio (George Washington University) for their valuable assistance throughout the project.
Funding. This work was supported by grants from the Juvenile Diabetes Research Foundation (JDRF17-2008-900 and 17-2012-480) and the National Institutes of Health (R01-DK-065073 and R01-DK-058191, to R.N.). The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Study Group has been supported by National Institutes of Health U01 Cooperative Agreement grants (1982–1993 and 2011–2016), and by contracts (1982–2011) with the Division of Diabetes, Endocrinology, and Metabolic Diseases of the National Institute of Diabetes and Digestive and Kidney Diseases grants (U01-DK-094176 and U01-DK-094157).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. F.M. designed and performed the experiments, researched the data, and wrote and edited the manuscript. Z.C., S.G., A.P., and J.M.L. designed the experiments, researched the data, and wrote and edited the manuscript. L.Z. performed experiments and researched the data. X.W., S.M.L., P.C., O.K., and W.S. researched the data. A.R. and D.M.H. contributed to the discussion and reviewed the manuscript. G.L. researched data and reviewed and edited the manuscript. R.N. designed the experiments, researched the data, wrote and edited the manuscript, and supervised the project. The DCCT/EDIC Research Group reviewed the manuscript. R.N. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1251/-/DC1.
F.M., Z.C., and S.G. contributed equally to this study.
See accompanying article, p. 1460.
References
- 1.Pezzolesi MG, Katavetin P, Kure M, et al. Confirmation of genetic associations at ELMO1 in the GoKinD collection supports its role as a susceptibility gene in diabetic nephropathy. Diabetes 2009;58:2698–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pezzolesi MG, Poznik GD, Mychaleckyj JC, et al. DCCT/EDIC Research Group Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes 2009;58:1403–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandholm N, Salem RM, McKnight AJ, et al. DCCT/EDIC Research Group New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet 2012;8:e1002921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leung A, Schones DE, Natarajan R. Using epigenetic mechanisms to understand the impact of common disease causing alleles. Curr Opin Immunol 2012;24:558–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villeneuve LM, Reddy MA, Natarajan R. Epigenetics: deciphering its role in diabetes and its chronic complications. Clin Exp Pharmacol Physiol 2011;38:451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reddy MA, Natarajan R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res 2011;90:421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper ME, El-Osta A. Epigenetics: mechanisms and implications for diabetic complications. Circ Res 2010;107:1403–1413 [DOI] [PubMed] [Google Scholar]
- 8.Diabetes Control and Complications Trial Research Group The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993;329:977–986 [DOI] [PubMed] [Google Scholar]
- 9.Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med 2000;342:381–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genuth S. Insights from the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study on the use of intensive glycemic treatment to reduce the risk of complications of type 1 diabetes. Endocr Pract 2006;12(Suppl. 1):34–41 [DOI] [PubMed] [Google Scholar]
- 11.Cleary PA, Orchard TJ, Genuth S, et al. DCCT/EDIC Research Group The effect of intensive glycemic treatment on coronary artery calcification in type 1 diabetic participants of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study. Diabetes 2006;55:3556–3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA 2002;287:2563–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA 2003;290:2159–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nathan DM, Cleary PA, Backlund JY, et al. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005;353:2643–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008;359:1577–1589 [DOI] [PubMed] [Google Scholar]
- 16.Chalmers J, Cooper ME. UKPDS and the legacy effect. N Engl J Med 2008;359:1618–1620 [DOI] [PubMed] [Google Scholar]
- 17.Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes 2008;57:3189–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miao F, Wu X, Zhang L, Yuan YC, Riggs AD, Natarajan R. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J Biol Chem 2007;282:13854–13863 [DOI] [PubMed] [Google Scholar]
- 19.Pirola L, Balcerczyk A, Tothill RW, et al. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 2011;21:1601–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 2008;205:2409–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miao F, Chen Z, Zhang L, et al. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J Biol Chem 2012;287:16335–16345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun G, Reddy MA, Yuan H, Lanting L, Kato M, Natarajan R. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol 2010;21:2069–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong Q, Kowluru RA. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J Cell Biochem 2010;110:1306–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci USA 2008;105:9047–9052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan PS, Kanwar M, Kowluru RA. Resistance of retinal inflammatory mediators to suppress after reinstitution of good glycemic control: novel mechanism for metabolic memory. J Diabetes Complications 2010;24:55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bird A. Perceptions of epigenetics. Nature 2007;447:396–398 [DOI] [PubMed] [Google Scholar]
- 27.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science 2010;330:612–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007;8:253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenuwein T, Allis CD. Translating the histone code. Science 2001;293:1074–1080 [DOI] [PubMed] [Google Scholar]
- 30.Kouzarides T. Chromatin modifications and their function. Cell 2007;128:693–705 [DOI] [PubMed] [Google Scholar]
- 31.Razin A, Riggs AD. DNA methylation and gene function. Science 1980;210:604–610 [DOI] [PubMed] [Google Scholar]
- 32.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 2005;6:838–849 [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Reddy MA, Miao F, et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem 2008;283:26771–26781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miao F, Gonzalo IG, Lanting L, Natarajan R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem 2004;279:18091–18097 [DOI] [PubMed] [Google Scholar]
- 35.Brasacchio D, Okabe J, Tikellis C, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009;58:1229–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan H, Reddy MA, Sun G, et al. Involvement of p300/CBP and epigenetic histone acetylation in TGF-β1-mediated gene transcription in mesangial cells. Am J Physiol Renal Physiol 2013;304:F601–F613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kadiyala CS, Zheng L, Du Y, et al. Acetylation of retinal histones in diabetes increases inflammatory proteins: effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J Biol Chem 2012;287:25869–25880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol 2010;6:665–675 [DOI] [PubMed] [Google Scholar]
- 39.Zhong Q, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes 2011;60:1304–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caramori ML, Kim Y, Moore JH, et al. Gene expression differences in skin fibroblasts in identical twins discordant for type 1 diabetes. Diabetes 2012;61:739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ko YA, Mohtat D, Suzuki M, et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol 2013;14:R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res 2006;16:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas-Chollier M, Hufton A, Heinig M, et al. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat Protoc 2011;6:1860–1869 [DOI] [PubMed] [Google Scholar]
- 44.Yun JM, Jialal I, Devaraj S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J Nutr Biochem 2011;22:450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miao F, Chen Z, Zhang L, et al. RNA-sequencing analysis of high glucose-treated monocytes reveals novel transcriptome signatures and associated epigenetic profiles. Physiol Genomics 2013;45:287–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gohda T, Niewczas MA, Ficociello LH, et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol 2012;23:516–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lopes-Virella MF, Baker NL, Hunt KJ, Cleary PA, Klein R, Virella G, DCCT/EDIC Research Group Baseline markers of inflammation are associated with progression to macroalbuminuria in type 1 diabetic subjects. Diabetes Care 2013;36:2317–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DCCT Research Group The absence of a glycemic threshold for the development of long-term complications: the perspective of the Diabetes Control and Complications Trial. Diabetes 1996;45:1289–1298 [PubMed] [Google Scholar]
- 49.DCCT Research Group The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the Diabetes Control and Complications Trial. Diabetes 1995;44:968–983 [PubMed] [Google Scholar]
- 50.Petruk S, Sedkov Y, Johnston DM, et al. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 2012;150:922–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DCCT Research Group The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Intern Med 1995;122:561–568 [DOI] [PubMed] [Google Scholar]