

Summary
The central role of phosphoinositide 3-kinase (PI3K) activation in tumor cell biology has prompted a sizeable effort to target PI3K and/or downstream kinases such as AKT and mTOR in cancer. However, emerging clinical data show limited single agent activity of PI3K/AKT/mTOR inhibitors at tolerated doses. One exception is the response to PI3Kδ inhibitors in chronic lymphocytic leukemia, where a combination of cell-intrinsic and -extrinsic activities drive efficacy. Here we review key challenges and opportunities for clinical development of PI3K/AKT/mTOR inhibitors. Through a greater focus on patient selection, increased understanding of immune modulation, and strategic application of rational combinations, it should be possible to realize the potential of this promising class of targeted anti-cancer agents.
Introduction
The signaling network defined by PI3K, AKT and the mechanistic target of rapamycin (mTOR) controls most hallmarks of cancer: cell cycle, survival, metabolism, motility, and genomic instability1. The pathway also contributes to cancer-promoting aspects of the tumor environment such as angiogenesis and inflammatory cell recruitment (Fig. 1)2–4. The lipid second messenger produced by PI3K enzymes, phosphatidylinositol-3,4,5-trisphosphate (PIP3), is elevated constitutively in most cancer cells and recruits cytoplasmic proteins to membrane-localized “onco” signalosomes5,6. The oncogenic signaling proteins recruited in this way include members of the AGC kinase family (e.g. AKT, Fig. 1), TEC family tyrosine kinases, and various modulators of small GTPase activity7. Cancer genetic studies suggest that the pathway is the most frequently altered in human tumors: the PIK3CA gene encoding the PI3K catalytic isoform p110α is the second most frequently mutated oncogene, and PTEN encoding the major PIP3 phosphatase is among the most mutated tumor suppressor genes8,9. In accord, a recent genomic study of head and neck cancer found the PI3K pathway to be the most frequently mutated10. Indeed, even in cancer cells expressing normal PI3K and PTEN genes, other lesions are present that activate the PI3K signaling network (i.e. activated tyrosine kinases, RAS, AKT; loss of LKB1 (STK11), INPP4B, TSC)11. This strong genetic evidence and the druggability of various components in the network provided the original rationale and excitement for targeting PI3K/AKT/mTOR signaling in oncology. The signaling network was seen as an opportunity to combat tumor complexity and genomic heterogeneity through a central, common oncogenic driver fundamental to all cancer cells. However, counterbalancing this opportunity is the challenge of targeting enzymes that are also active and critical in normal cells and tissues.
Figure 1. Targets in the signaling network and their role in tumor biology.

This diagram shows a highly simplified scheme of the signaling pathway leading from PI3K to AKT to mTOR. The four isoforms of class I PI3K are shown in dark gray boxes. Blue italics illustrate cancer cell-intrinsic functions of isoforms: p110α is a frequent genetic driver (PIK3CA mutations); basal activity of p110β is implicated in tumors with PTEN loss; p110δ has a fundamental role in survival of normal B cells and malignancies of this lineage. PI3K and mTOR drive tumor metastasis by promoting cell motility and epithelial-mesenchymal transition (EMT). Purple arrows represent cell extrinsic functions of various components in the network. p110α drives angiogenesis. p110γ, p110δ and p110β have important functions in inflammatory cells. p110δ and mTOR control key aspects of adaptive immunity including lymphocyte activation, differentiation and tolerance. Drugs in clinical development that target nodes in this network are listed in Supplementary Table 1.
Groundbreaking structural studies of PI3K enzymes12–18, together with extensive medicinal chemistry efforts19–21 have led to the discovery of compounds targeting one or more nodes in the network. Several of them harbor favorable drug properties and suppress tumor growth in preclinical models of cancer20–24. The challenge is to translate these findings into a meaningful activity at acceptable tolerability in cancer patients. The early results from trials in advanced solid tumors are rather sobering, showing limited single agent activity of PI3K and mTOR inhibitors25, 222, especially when compared to agents targeting driver oncogenes such as BCRABL, ALK or BRAF. Pharmacology plays an important part in clinical efficacy, in that doses high enough and over a long enough exposure period to achieve cancer eradication might not be tolerated due to mechanism based on-target toxicities. Yet the pathway itself might not be as essential to cancer cells as originally proposed, at least at an advanced stage of tumorigenesis. Indeed, blockade of the pathway generally fails to induce cancer cell death and selects for compensatory pathways that maintain survival and restore tumor growth26–28. Furthermore, refinement of genetically engineered mouse models suggests that PIK3CA mutants expressed at endogenous levels do not strongly drive tumor development like some other oncogenes29,30. In essence, “oncogene addiction” to PI3K/AKT/mTOR signaling is not absolute. Therefore, unleashing the full potential of PI3K/AKT/mTOR inhibitors in oncology will require earlier treatment, dose/schedule optimization, and rational combinations with other therapeutic approaches.
It will also be important to identify biomarkers that can guide patient selection, and to determine which tumor types/genetic profiles benefit from blocking single nodes/isoforms versus multiple targets. Encouragingly, the p110δ-selective inhibitor GS-1101 (formerly CAL-101 and currently in phase 3 development) produces dramatic responses in some B cell malignancies31,32. This proves the principle that a potent and selective PI3K inhibitor can improve survival of selected cancer patient populations. Yet, GS-1101 has an unusual mechanism of action: the drug is not directly cytotoxic to malignant B lymphoma cells and its efficacy arises in part from modulating the tumor immune environment31,32,223. This illustrates the importance of understanding PI3K pathway biology in immune cells and in physiological models of tumor immunity (or immunology). The success of antibody therapies targeting immune checkpoints (CTLA-4, PD-1)33,34 emphasizes the potential of targeting immune-inhibitory pathways in cancer and the importance of evaluating immune effects of small molecule kinase inhibitors.
The goal of this review is to reset both expectations and directions. Our understanding of the complexity of the PI3K/AKT/mTOR signaling network and its role in cancer has significantly increased, establishing the pathway as a challenging yet viable target in oncology. Much can be learned from clinical failures and the limited successes to date, to chart a course for next generation strategies. In our opinion, enthusiasm and commitment towards targeting such an important pathway in cancer should not be reduced.
Overview of the PI3K/AKT/mTOR signaling network
Key features of the PI3K/AKT/mTOR signaling network that illustrate both the promise and the challenges for targeting the pathway in cancer have been previously discussed11,21,35,36. Below, we will provide a brief overview of the functions and signaling mechanisms of members of the family of PI3K enzymes, highlighting their roles in cancer and issues faced in therapeutically targeting them.
There are eight mammalian PI3K enzymes, grouped into three classes36. The most important in cancer are the four class I enzymes, termed PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ. These are heterodimers of a catalytic subunit of 110 kDa (p110α, p110β, p110γ or p110δ) and a regulatory subunit. The catalytic isoforms share considerable sequence homology and produce the same lipid product (PIP3), and each can receive activation inputs from both tyrosine kinases and from GTPase signaling36,37. However, the details of these inputs differ (see Box 1). The distinct activation mechanisms of the class I PI3K isoforms suggest that each has unique biological functions, a model supported by abundant evidence from targeted gene inactivation in mice36,38–41. It follows that targeting single isoforms might have therapeutic effects. On the other hand, functional redundancy in maintaining cell survival has been documented in various cell types including cancer cells42. Furthermore, mouse genetic models have caveats and do not always accurately predict the response to acute target inhibition by pharmacological agents.
Box 1: Inputs from GTPases and tyrosine kinases to PI3K.
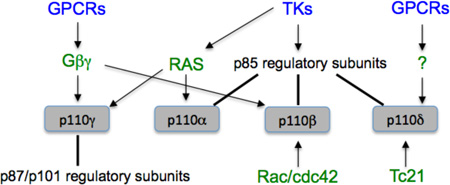
Each of the class I PI3K catalytic isoforms has a segment known as the RAS-binding domain (RBD). Knock-in mouse studies established p110α as a bona fide downstream effector of oncogenic RAS73, and demonstrated that the RBD of p110γ is required for PI3K signaling in neutrophils206. Recent work indicates that the RBD of p110β does not bind to RAS or its close relatives; instead, the RBD of p110β interacts with GTP-bound Rac/cdc42, establishing p110β as a novel effector of these GTPases37. The RBD of p110δ interacts with the Tc21 small G protein207,208. Tyrosine kinases activate p110α, p110β and p110δ via interaction of their regulatory subunits with tyrosine phosphorylated (pTyr) peptide motifs, and can activate p110γ in some cell types via RAS86. GPCRs directly stimulate p110β and p110γ via βγ subunits of heterotrimeric G proteins206,209, and can activate p110δ in B cells by an unknown mechanism210,211.
Of the four class I catalytic isoforms, only PIK3CA (encoding p110α) is frequently mutated in human cancer8,11. Although many PIK3CA mutations exist, there are two hotspots that cause elevated PI3K enzyme activity through distinct mechanisms15. Transforming mutations in the ubiquitously expressed gene PIK3CB (encoding p110β) are rare43, perhaps due to this isoform’s distinct mode of interaction with regulatory subunits18. Mutations in class I regulatory subunit genes (PIK3R1 or PIK3R2) are also found in cancer cells and cause increased PI3K activity44,45. In cell transformation assays, p110α plays a dominant role in the oncogenic potential of PIK3R1 mutants46. This observation provides further support for a unique role of the p110α isoform in tumorigenesis. In addition, p110α has a cell-extrinsic role in tumor angiogenesis (Fig. 1) and possibly stromal fibroblasts3,4, another potential advantage for targeting this isoform. In cancer cells with wildtype PI3K genes, there are usually oncogenic lesions in upstream tyrosine kinases (TKs) and/or RAS that cause constitutive signaling through PI3K11. Loss of lipid phosphatases PTEN and/or INPP4B is an alternative path to elevated PI3K lipid products, but inactivation of these tumor suppressors is not mutually exclusive with mutations in PI3K or RAS44,47. Indeed, a mouse model demonstrated that loss of PTEN cooperates with PIK3CA mutations to cause ovarian tumors29. PIK3CA mutations or PTEN loss can also co-exist with oncogenic TKs48,49.
The mTOR serine/threonine kinase functions at two distinct nodes in the PI3K signaling network (Fig. 2A)50,51. mTOR complex-2 (TORC2) phosphorylates key residues to activate AKT and other kinases. TORC2 appears to have basal activity that is stimulated by growth factors and through association with ribosomes52. mTOR complex-1 (TORC1) is a central regulator of cellular metabolism and biosynthesis, and is subject to complex regulation by growth factors, nutrients and cellular stresses51. When conditions are favorable for cell growth, TORC1 phosphorylates several substrates to promote anabolic processes (ribosome biogenesis, translation, and synthesis of lipids and nucleotides) and suppress catabolic processes (e.g. autophagy)51. One of the key control nodes for TORC1 activity is the TSC complex containing TSC1, TSC2 and TBC1D7 proteins53,54. By phosphorylating TSC2, AKT suppresses the inhibitory effect of the TSC complex on TORC1. Although the MTOR gene is not frequently mutated in human tumors, there is evidence for “non-oncogene addiction” to mTOR function in cancer cells. For example, tissue specific deletion of mTOR in mouse prostate inhibits tumor formation driven by PTEN loss without disrupting normal prostate tissue55,56. Also, mTOR catalytic inhibitors can achieve anti-leukemic effects at doses that preserve the function of normal bone marrow and peripheral lymphocytes57,58.
Figure 2. Complexity, crosstalk and feedback in the PI3K/AKT/mTOR signaling network.

(A) The TORC1-TORC2 network and key feedback mechanisms. The mTOR serine-threonine kinase forms two multi-protein complexes whose defining subunits are raptor (TORC1) and rictor (TORC2). TORC2 activity is stimulated by association with ribosomes and by growth factors through a poorly defined mechanism, which may involve PI3K. TORC2 promotes stability and activity of AKT and other kinases including serum- and glucocorticoid-induced kinases (SGKs) and protein kinase C (PKC). TORC1 is a signal integrator whose activity is tuned by diverse inputs. Growth factors, energy sensors and cellular stress converge at the level of the TSC complex (TSC1/TSC2/TBC1D7), a negative regulator of TORC1 with GAP activity towards the Rheb GTPase. Amino acids regulate TORC1 through the Ragulator and GATOR complexes. TORC1 promotes anabolic programs through many substrates, of which three classes are shown: S6 kinases, eukaryotic initiation factor-4E (eIF4E)-binding proteins (4E-BPs), and autophagy regulators (ULK1, etc.). TORC1 activity exerts feedback control on growth factor signaling. One canonical feedback pathway is initiated by S6 kinase-1 (S6K1), a TORC1 substrate, which phosphorylates adaptor proteins of the insulin receptor substrate (IRS) family to attenuate growth factor receptor signaling to PI3K and RAS. In parallel, TORC1 suppresses growth factor receptor signaling by phosphorylating the GRB10 adaptor protein. AKT activity triggers a feedback mechanism that suppresses growth factor receptor expression and signaling. Through phosphorylation and inactivation of Forkhead Box Subgroup O (FOXO) transcription factors, active AKT reduces the transcription of FOXO target genes including several growth factor receptors. (B) Redundancy and feedback between the RAS-RAF-MEK-ERK and PI3K/AKT/mTOR signaling networks. ERK and downstream kinase RSK can compensate for AKT in the activation of TORC1 via inhibitory TSC phosphorylation; GSK3 and AMPK phosphorylation of TSC2 increase its ability to suppress TORC1 activity. MNK kinases phosphorylate eIF4E to provide a distinct signal to increase cap-dependent translation. ERK and mTOR independently promote accumulation of MYC oncoproteins. Mutual feedback inhibition is a feature of the two pathways: MEK activity suppresses PI3K signaling by promoting PTEN membrane localization, while TORC1 activity suppresses RAS activation through mechanisms shown in Figure 3.
Feedback control is a common feature of cellular signaling systems, and the PI3K/mTOR network provides many examples (Fig. 2A). An important consequence of feedback is that inhibitors of AKT or mTOR tend to cause elevated expression and activity of growth factor receptors, leading to increased PI3K activity and RAS signaling, and alternative survival pathways in cancer cells59,60. There are several potential strategies to overcome the “rebound” signaling in response to PI3K/AKT/mTOR inhibitors, including vertical inhibition of several signaling nodes and combination approaches.
There is also crosstalk between elements of the PI3K signaling network and components of other oncogenic pathways (Fig. 2B). A key consequence is that PI3K and AKT are not the dominant regulators of TSC1/2 and TORC1 in some cells. ERK and RSK are two effector kinases downstream of RAS that can promote TORC1 activity by phosphorylating TSC2 on residues distinct from AKT phospho-acceptor sites61–63. GSK3 and AMPK can also phosphorylate TSC264. Another example of crosstalk is that ERK and TORC1 provide distinct and complementary inputs to eIF4E, a central regulator of cap-dependent mRNA translation65,66. The PI3K/AKT/mTOR and RAS/RAF/MEK/ERK networks also converge to stabilize protein expression of the MYC oncoprotein67. Therefore, oncogenic compensation by RAS can severely limit the anti-cancer efficacy of PI3K/AKT/mTOR inhibitors. Conversely, active PI3K signaling is likely a central mechanism of resistance to various targeted therapies.
Clinical trial results and associated challenges
There are six general classes of agents in clinical trials that target the PI3K/AKT/mTOR network: pan-class I PI3K inhibitors, isoform-selective PI3K inhibitors, rapamycin analogs (rapalogs), active-site mTOR inhibitors, pan-PI3K/mTOR inhibitors, and AKT inhibitors. Supplementary Table 1 lists many of the compounds currently in oncology clinical trials according to clinicaltrials.gov. Rapalogs are not broadly effective as single agents, though they have obtained FDA approval for treatment of a few tumor types where modest therapeutic effects can be achieved. Clinical trial data for the other five classes remain largely unpublished; however, results presented at conferences allow some preliminary conclusions. The most impressive results have been achieved with the p110δ-selective inhibitor GS-1101 (Idelalisib), which causes dramatic responses in chronic lymphocytic leukemia (CLL) and certain other B cell malignancies (Box 2). Overall, other agents targeting PI3K/AKT/mTOR have not yielded broad responses when given to advanced cancer patients at tolerated doses. By comparison, early trials of currently approved drugs targeting oncogenes like BCR-ABL, mutant B-RAF or ALK revealed marked single agent activity even in phase I trials, albeit in prospectively selected patient populations. A recent review by Tabernero and colleagues provided a detailed discussion of emerging clinical trial data for PI3K pathway inhibitors, including safety profiles and pharmacodynamic markers25. Below we highlight four central challenges/issues in the field of PI3K/AKT/mTOR drug development arising from clinical studies so far.
Box 2: Selective inhibitors of PI3Kδ or the PI3K effector BTK.
The PI3Kδ isoform is mainly expressed in immune cells and is absent from most solid tumors. Gene targeting in mice has established essential functions for PI3Kδ in mature B cells, and in other immune cell types41,212. A key downstream effector of PI3Kδ in B cells is BTK, a member of the TEC family of nonreceptor tyrosine kinases. PI3Kδ and BTK are activated by signals from the B cell receptor (BCR), chemokines and cytokines to drive survival, proliferation and adhesion to supportive stromal cells. However, activating mutations in PI3Kδ and BTK are not present in B cell tumors, and inhibitors of these enzymes were initially developed for application in immune diseases. Unexpectedly, phase I clinical trials of a PI3Kδ inhibitor (CAL-101, renamed GS-1101) and a BTK inhibitor (PCI-32765) showed dramatic and durable responses in a subset of human patients with indolent B cell malignancies31,32,69,224. Even greater efficacy was achieved in combination studies with Rituximab and/or Bendamustine. Both the PI3Kδ inhibitor and BTK inhibitors have shown acceptable safety profiles. These compounds, now called Idelalisib and Ibrutinib, have progressed to phase II/III trials and are likely to be the first FDA-approved agents targeting the PI3K pathway. The FDA has granted Breakthrough Therapy Designation for Ibrutinib in three diseases: mantle cell lymphoma, Waldenström’s macroglobulinemia, and chronic lymphocytic leukemia with deletion at chromosome 17p (note added in proof: Ibrutinib was approved Nov. 13, 2013, for relapsed mantle cell lymphoma).
Pan-PI3K versus isoform-selective inhibition
Several pan-class I PI3K inhibitors in clinical trials target all four class I PI3K isoforms with similar potencies (Supp Table 1). The main argument for pan-PI3K inhibitors is that most cancer cells express multiple PI3K isoforms with redundant functions in oncogenic signaling42. Another factor driving early development of pan-PI3K compounds was that these efforts proceeded before PI3K isoform structures were available to aid the design of isoform-selective compounds. However, pan-PI3K inhibitors are blunt tools that are not specifically aligned with the disease biology and context. The main concern with pan-PI3K inhibitors is that doses needed to fully block all class I PI3Ks for extended periods might not be tolerated. For this reason, it is possible that trials to date have missed an “all or nothing” threshold for tumor responses due to dose-limiting toxicities. A related concern is that the first-in-class compounds that have entered oncology trials are not sufficiently selective for PI3K. Compared to isoform-selective inhibitors, compounds targeting all class I PI3Ks seem more commonly to have off-target effects on members of the PI3K-related kinase (PIKK) family (mTOR, DNA-PK, ATM, ATR) and other cell components. For example, BKM120 at concentrations needed to fully inhibit PI3K (5X the IC50) has off-target effects on tubulin and causes general cellular toxicity68. Filling the competitive landscape with inadequate compounds might have discouraged later entry of “bestin- class” agents with the needed selectivity to deliver on the potential of the target biology.
Isoform-selective PI3K inhibitors (Supp Table 1) have the potential to block the relevant target more completely, while limiting toxicities associated with broader inhibition profiles. Indeed, GS-1101 is well tolerated in most patients at doses that maintain drug exposure levels sufficient to suppress p110δ activity to a level that translates into anti-tumor activity32. Yet the therapeutic activity of p110δ inhibitors was unexpected. These compounds deviate from the traditional paradigm for targeting a kinase that is required for the cancer cell but not its normal counterpart. In this case, the target (PIK3CD) is not mutated in cancer but is required for survival of normal B cells (Fig. 1). GS-1101 efficacy derives from an unusual confluence of factors: a very selective drug with a target whose expression is restricted, and a dual role for the target in the cancer cell and the tumor immune environment. This emerging paradigm for leukemia/lymphoma treatment also applies to Ibrutinib, an inhibitor of the tyrosine kinase BTK that acts downstream of p110δ in B cells69,224.
Other than p110δ inhibitors, the most advanced isoform-specific compounds are selective for p110α (Supp Table 1). The prevalence of PIK3CA mutations in human cancer provides a potentially rapid and cost-effective development path analogous to BRAF or ALK inhibitors. Yet questions remain about the best patient selection strategy for p110α-selective inhibitors. One approach is to design “basket” trials grouping patients with PIK3CA mutant tumors across several histologies, and let the data guide expanded trials. This idea builds on experience attained from the use of BRAF inhibitors, which provided efficacy in BRAF mutant melanoma but not colorectal cancer70,71. Similarly, GS-1101 efficacy in CLL emerged from empirical testing in a broad range of B cell malignancies31,32. Another approach is to include tumor types that are PIK3CA wildtype but in which p110α plays a critical signaling role (e.g. HER2, KRAS, PIK3R1)46,72,73. With either approach, drugs targeting p110α should be tested in patients at an earlier stage of disease with less tumor complexity and reduced toxicity load of prior treatments.
There is also an opportunity, so far untapped, to develop irreversible p110α inhibitors, as this is the only isoform with a reactive cysteine residue near the ATP binding site74. The clinical success of the covalent BTK inhibitor Ibrutinib provides encouragement for this pharmacological approach69,224. Another way to improve the therapeutic index might be to develop inhibitors that are selective for the common PIK3CA “hotspot” mutant enzymes such as H1047R, E542K and E545K. However, such compounds would lose cell-extrinsic activity (e.g. angiogenesis), they would not act on wild-type p110α downstream of receptors and RAS, and they might select for other mutants. Agents targeting hotspot PIK3CA mutants might find an alternative use in the treatment of inherited overgrowth syndromes caused by somatic PIK3CA mutations75–77. Another clinical use of isoform-selective agents outside of oncology might be p110δ inhibitors in patients with newly identified immunodeficiency syndromes caused by activating mutations in PIK3CD78,79.
Some studies suggest that p110β activity is essential in cancer cells lacking PTEN (Fig. 1), particularly in prostate and breast cancer80–82, suggesting that p110β inhibitors would be more effective than p110α inhibitors in patients with PTEN-deficient tumors. However, another study reported that p110α and p110β have overlapping functions in various PTEN-deficient tumor models83. p110α also has the aforementioned role in tumor angiogenesis3,4. Ultimately, the success of targeting p110β alone in PTEN mutant advanced tumors might depend on whether the tumor also harbors mutations in upstream receptors or RAS that activate p110α.
Arguments can be made for compounds targeting two of the four class I isoforms, and now it seems technically feasible due to advances at the level of structural biology and medicinal chemistry. A dual p110α/p110β inhibitor might work in tumors lacking PTEN, or in PIK3CA mutant tumors that have grown resistant to single p110α inhibition. Yet, targeting both p110α and p110β is likely to recapitulate most of the toxicity profile seen with pan-PI3K inhibitors. A compound targeting p110α and p110δ might overcome resistance to GS-1101 in B cell malignancies, since in some cases resistance correlates with elevated p110α expression and activity84. A dual p110α/δ inhibitor, BAY 80-6946, was recently described225. Combined targeting of p110γ and p110δ has potential in T cell leukemias, in which p110γ and p110δ have redundant functions85. The compound IPI-145 is selective for p110γ and p110δ (though 10-fold more potent towards p110δ), has activity in autoimmunity models226,227, and is in clinical trials for both B and T cell malignancies.
Most solid tumor cells express p110α and/or p110β. but usually not p110γ or p110δ. Nevertheless, inhibiting p110γ or p110δ may suppress tumor viability by modulating leukocyte subsets in the tumor environment (Fig. 1). In mouse models, blocking p110γ activity reduces recruitment of inflammatory cells to tumor sites and suppresses tumor growth86. Whether p110γ inhibition can shrink established tumors is uncertain, but this approach could be employed to prevent regrowth or metastasis. Inhibiting p110δ suppresses the function of regulatory T cells, enabling increased cytotoxic T cell responses to tumors (Khaled Ali and Bart Vanhaesebroeck, personal communication). Thus, targeting p110α with p110δ in solid tumors seems a particularly promising approach that would have cell-intrinsic anti-cancer effects while promoting a favorable immune environment and avoiding some toxicities of pan-PI3K inhibition. A downside of pan-PI3K inhibitors is that they suppress function of mouse and human lymphocytes to a much greater degree than p110α inhibition alone, or p110α/δ combinations87. It is likely that compounds with seemingly subtle differences in potency against different isoforms will provide significantly different efficacy and tolerability based on cancer cell-extrinsic effects.
Single node versus pan PI3K/mTOR inhibition
mTOR is structurally related to PI3Ks, and many ATP-competitive compounds inhibit mTOR and PI3K with similar potencies. In fact, the broadly used experimental PI3K inhibitors wortmannin and LY294002 also inhibit mTOR directly88–90. Several pan-PI3K/mTOR inhibitors with improved pharmacological properties are now in clinical trials (Supp Table 1). The rationale for this compound class is to overcome crosstalk and feedback through “vertical” inhibition of the pathway at three key nodes: PI3K, TORC1 and TORC2 (Fig. 2A). This approach circumvents a limitation of selective PI3K inhibitors, that other inputs maintain considerable TORC1 activity even when PI3K and AKT are shut off91. Pan-PI3K/mTOR inhibitors should also prevent the rebound activation of PI3K that occurs in cells treated with rapalogs or active-site mTOR inhibitors.
It seems unlikely that pan-PI3K/mTOR inhibitors will provide a better efficacy window compared to targeting single nodes as there is obviously potential for greater toxicity at effective doses. Another consideration is that pan-PI3K/mTOR inhibitors do not always provide better efficacy than selective active-site mTOR inhibitors in preclinical tumor models58. Combined targeting of mTOR and one PI3K isoform (for example, p110α in PIK3CA mutant tumors) might improve tolerability relative to pan-PI3K/mTOR inhibitors and increase efficacy compared to single PI3K inhibition. The greater efficacy of a combination of the RAF and MEK inhibitors (Dabrafenib/Trametinib) in metastatic melanoma compared to monotherapy provides proof of concept in a different oncogenic pathway92. In addition to suppressing intra-pathway feedback, such approaches may achieve synergistic suppression of key downstream effectors using synchronized doses that partially inhibit the two upstream targets. A phase 1b trial (NCT01899053) has been initiated (by Millennium Pharmaceuticals) to test a combination of inhibitors selective for p110α and mTOR (MLN1117 and MLN0128). Providing further support for such combinations, resistance of PIK3CA mutant breast cancers to BYL719 correlated with persistent TORC1 signaling93. In the Dabrafenib/Trametinib trial, pERK was a reliable downstream PD marker and we would anticipate that pS6 and p4EBP1 offer similar potential as PD markers for TORC1 activity.
Rapalogs versus TOR-KIs
Rapalogs (everolimus, temsirolimus, deforolimus) are structural analogs of rapamycin with improved pharmacological properties94. Their acceptable safety profile95 has allowed the completion of a large number of trials testing rapalogs as single agents or in combination. Although most single agent trials have not demonstrated therapeutic benefit, rapalog monotherapy does significantly extend survival for some cancer patients. Currently one or more rapalogs are FDA-approved for use in renal cell carcinoma, mantle cell lymphoma and neuroendocrine tumors.
Incomplete mTOR inhibition contributes to the limited efficacy of single agent rapalogs in cancer (see Box 3). A second strategy to target mTOR is through ATP-competitive inhibitors that completely block mTOR kinase activity in both complexes, TORC1 and TORC220,96,97 (Box 3). Termed active-site mTOR inhibitors, these compounds cause greater suppression of biosynthetic pathways compared to rapamycin, and generally cause a more marked cytostatic effect in cell lines20,96–98. Active-site mTOR inhibitors have shown cytotoxic effects in some but not all preclinical cancer models58,99,100. A key question for clinical development is whether active-site mTOR inhibitors should be tested first in malignancies in which rapalogs have some clinical benefit, since mTOR is a validated target in those diseases. An alternative is to test more broadly to see whether complete TORC1/TORC2 inhibition is effective in diseases where partial TORC1 inhibition is not. It is important to note that the most encouraging preclinical results with active-site mTOR inhibitors have been achieved in combination with TKIs in human tumor xenograft models in mice58,101. These findings argue for early evaluation of active-site mTOR inhibitors in combination with other targeted agents. A note of caution is that advanced tumors often have an increased ratio of eIF4E to 4EBPs, a known mechanism of resistance to active-site mTOR inhibitors102,103. Therefore, high eIF4E expression might be a useful negative prognostic biomarker for patient selection.
Box 3: Two distinct classes of mTOR inhibitors.
Rapalogs act through an allosteric mechanism and cause only partial inhibition of TORC1, with more marked effects on certain TORC1 substrates (i.e. S6K1) than others (i.e. 4E-BPs). This profile causes weak inhibition of capdependent translation and releases negative feedback, leading to “rebound” activation of upstream signaling. Rapalogs do not directly inhibit TORC2, allowing continual survival signaling by AKT and other TORC2 substrates. In accord, extensive cell line surveys consistently show that rapamycin and analogs are cytostatic and not cytotoxic. Active-site mTOR inhibitors fully block phosphorylation of all known TORC1 and TORC2 substrates.

Recent findings have renewed interest in clinical application of rapalogs. A large combination trial (BOLERO-2) of everolimus with anti-estrogen therapy (aromatase inhibitors) showed a significant survival benefit for patients with hormone receptor-positive breast cancer104. Rapalogs are useful in the treatment of subependymal giant cell tumors and angiomyolipomas in patients with tuberous sclerosis caused by inherited mutations in TSC1 or TSC2105,106. Another important study used deep genomic sequencing of a rare responder tumor to identify TSC1 loss as a biomarker of everolimus sensitivity in bladder cancer107. Novel tumor suppressors (NPRL2, DEPDC5) have been identified in the GATOR1 complex that regulates amino acid sensing by TORC1, and human cancer cell lines lacking these components are very sensitive to rapamycin108,109. Additional clinical studies are needed to determine whether loss of TSC or GATOR1 components predict sensitivity to rapalogs in a broad range of tumors. Finally, two recent studies identified TORC1 signaling in resistance to PI3K or BRAF inhibitors228,229.
The strong immunosuppressive properties of rapamycin have led to extensive investigation of mTOR function in the immune system. Genetic and pharmacological studies in mice have shown remarkable complexity of mTOR function in different immune cell types and at different stages of cell activation110–112. Although mTOR blockade reduces proliferation and effector differentiation of CD4 T cells, mTOR inhibition enhances generation of CD8 T cell memory113–115. In addition, mTOR inhibition can augment inflammatory cytokine production by innate immune cells116. Remarkably, mTOR inhibition can either promote or suppress the function of regulatory T cells depending on the timing and experimental conditions115,117,118. Therefore, modulating the schedule of mTOR inhibitor therapy has the potential to promote anti-tumor immune responses while providing some tumor intrinsic activity.
Tolerability and alternative targets
A recurring theme above is the challenge of achieving a therapeutic window for compounds targeting PI3K and/or mTOR. PI3K signaling is linked to many physiological processes, and mTOR is a non-redundant sensor of nutrients and growth factors in dividing cells51,119,120. For these reasons, many investigators have evaluated other targets in the PI3K/mTOR signaling network. The best studied of these is AKT. This kinase is commonly overexpressed or mutated in tumors and was first discovered as the oncogene of a transforming virus121. Since AKT is one of many PI3K effectors linked to cell-specific physiological functions, it is conceivable that direct AKT inhibition would attack cancer cells with greater selectivity than PI3K inhibition. However, the data so far suggest that this might not be the case. AKT inhibitors cause severe rash similar to some TKIs, and cause hyperglycemia in both mice and humans122,123. Genetic studies in mice have shown that the AKT2 isoform is required for insulin signaling124, and most clinical AKT inhibitors block both the AKT1 and AKT2 isoforms125. On the other hand, some AKT inhibitor candidates have off-target effects and a recent study suggested that a pharmacologically optimized AKT inhibitor causes only transient and reversible hyperglycemia126. Hyperglycemia can be managed with approved drugs like metformin, and can also be a useful biomarker of target modulation. Interestingly, metformin and related compounds have been suggested to provide direct anti-tumor effects by activating AMP kinase leading to reduced TORC1 signaling127.
Several other cellular components associated with the PI3K/mTOR network might be useful targets for anti-cancer therapeutics (Table 1). Based on the central role of cap-dependent translation in cancer cells, drugs targeting eIF4E have been developed and show promise in preclinical studies128. For example, the compound 4EGI-I interferes with the eIF4E-eIF4G interaction and has anticancer activity in cell lines129. Selective inhibitors of S6 kinases have been identified130–132, and could attack cancer by restricting protein synthesis and other anabolic growth processes mediated by S6Ks. Supporting this concept, genetic targeting of S6K1 delayed leukemogenesis in a PTEN loss model133. MNK and PIM kinases also promote protein synthesis and are under evaluation as targets in oncology134,135. MNK kinases phosphorylate eIF4E to increase cap-dependent translation and promote survival136, and PIM kinases increase translation through phosphorylating several substrates including eIF4B135. Inhibiting RAS function in RAS-driven cancers would be expected to diminish signaling through PI3K as well as ERK and other RAS effectors. Though targeting RAS has long been an unrealized dream in molecular medicine, novel strategies have recently been137,138 described.
Table 1.
Emerging Targets within the PI3K signaling network
| Target | Upstream activators | Effectors/Substrates | Tool Compounds (Refs.) |
|---|---|---|---|
| eIF4E | mTORC1, MNK | Cap-dependent translation | 4EGI-I129 4ei-1213 |
| S6K | mTORC1, PDK1 | S6, PDCD4, eIF4B, eEF2K, SKAR | PF-4608671132 DG2131 LYS6K2130 |
| MNK | ERK | eIF4E |
CGP57380214 AST 487215 Cercosporamide216 |
| PIM | Growth factor- mediated increase in transcription |
eIF4B, 4EBP1, BAD, p27 | SMI-4a217 ETP-45299218 SGI-1776219 Pimi-14J220 K00135221 |
Emerging rational combination strategies
The typical drug development path for a targeted anti-cancer drug involves establishing single agent efficacy before testing the drug in combination. However the initial results from PI3K/AKT/mTOR inhibitor trials suggest that deep and sustained responses to single agents are infrequent. Given limited resources, some promising drugs might be halted in development because they do not significantly prolong survival of a selected patient population. An alternative approach would be to initiate combination trials as soon as pharmacodynamic (PD) activity can be established at a tolerated dose. Development of robust and informative PD markers remains a challenge, as discussed in reference25. Moreover, it is essential to choose rational combinations that are most likely to provide synergy (a “1 + 1 = 3” effect), to overcome the expected increases in toxicity and justify the costs and complexity of combination trials. The BOLERO-2 clinical trial combining everolimus with endocrine therapy provided proof of principle that the PI3K/AKT/mTOR pathway can be targeted in rational combinations to achieve real therapeutic benefit to a large patient population104. What other combinations can be envisioned (Table 2)?
Table 2.
Selected PI3K Pathway Combination Strategies
| Target for combo | Tumor stratification | References |
|---|---|---|
| TKs | Active or overexpressed TK | 58,139–144 |
| MEK | Active RTK, RAS mutant | 26,149,150,155 |
| BRAFV600E | Melanoma, colon | 156,157 |
| MYC | MYC amplification, NOTCH mutant | 27,28,163–165,167 |
| Autophagy | Glioma, leukemia, others | 168–170 |
| PARP | TNBC | 173,174 |
| Aromatase inhibitors | ER-positive | 104,181 |
| BCL2 antagonists | Leukemia, lymphoma, others | 188–190 |
There is ample mechanistic rationale to test combinations of PI3K/AKT/mTOR inhibitors with TKIs. It is useful to consider this issue from two perspectives (Fig. 3). First, cancers harboring active or overexpressed receptor tyrosine kinases (RTKs) such as EGFR or HER2 can display resistance to TKIs through PI3K signaling139,140. This knowledge provides justification for adding PI3K/AKT/mTOR inhibitors to initial TKI treatments to prevent the emergence of resistance, even in tumors with a high initial response rate to TKIs. In line with this view, a review from Rexer and Arteaga discussed strategies to incorporate PI3K inhibitors into treatment regimens for HER2+ breast cancer141. Such approaches should be considered for other TK-driven cancers, as combining GDC-0941 with imatinib produced more durable remissions than imatinib alone in a xenograft model of gastrointestinal stromal tumor (GIST) driven by BCRABL142. A second consideration is that single agent PI3K/AKT/mTOR inhibitors increase RTK expression through FOXO-mediated feedback27,60. From this point of view, TKIs act as the second agent to augment the efficacy of a PI3K/AKT inhibitor. Supporting this concept, targeting EGFR-family receptors with lapatinib increased the efficacy of a p110α-selective inhibitor in PIK3CA mutant breast cancer cells143. A challenge is that the feedback tends to increase expression of multiple RTKs, such that selective TKIs would have minimal efficacy. In hematopoietic malignancies driven by non-receptor TKs such as BCR-ABL or JAK2, PI3K/AKT/mTOR inhibitors strongly synergize with TKIs58,144. A possible explanation is that blood cancer survival is maintained in part by cytokines and stromal cell contacts that signal through the PI3K pathway. There is also evidence for interdependence of PI3K signaling and the JAK/STAT pathway, with agents targeting STAT3 or upstream kinases holding promise to enhance efficacy of PI3K inhibitors145.
Figure 3. Two arguments for combining TKIs with PI3K/AKT/mTOR inhibitors.

Left: In cancers driven by activated tyrosine kinases, TKI resistance can develop through alternative pathways that maintain PI3K signaling such as compensatory growth factor (GF) receptors, PTEN loss, PIK3CA mutation or RAS activation. Combined targeting of PI3K can prevent or overcome drug resistance. Right: In cancers driven by lesions in PI3K or PTEN, inhibiting PI3K or AKT or TORC1/TORC2 can cause elevated GF receptor signaling through FOXO-dependent gene expression. Adding a TKI can ameliorate this compensatory signaling mechanism.
Targeting the RAS/RAF/MEK/ERK cascade is an attractive strategy for combination therapies with PI3K/AKT/mTOR inhibitors (Fig. 2B). Both networks can promote cell proliferation and survival, and there is extensive crosstalk between the pathways. Thus, TORC1 inhibition tends to increase ERK phosphorylation146,147 whereas MEK inhibition reduces PTEN membrane localization and increases AKT activity148. Synergy of a MEK inhibitor with the dual PI3K/mTOR inhibitor NVP-BEZ235 was shown first in a KRAS-driven lung cancer model26. Similar findings have been observed in many subsequent reports including a study of NRASmutant melanoma cells149. One mechanism for synergistic cell killing appears to be through complementary effects on pro-apoptotic proteins: MEK/ERK inhibition stabilizes BIM, whereas PI3K/AKT inhibition upregulates PUMA via FOXO transcription factors150. Both pathways also converge on the pro-apoptotic protein BAD151. Such combinations might also achieve synergy at the level of metastasis suppression. MEK inhibitors can suppress the epithelial-mesenchymal transition (EMT) that is a critical step in the evolution of metastatic tumor cells152,153. Active-site mTOR inhibitors decrease translation of mRNAs encoding proteins involved in prostate cancer invasion and EMT154. A major concern is whether a therapeutic window can be achieved with combinations of PI3K/AKT/mTOR and MEK inhibitors155. To overcome likely toxicities, it might be necessary to experiment with dose and schedule, such as high dose intermittent treatments or alternating sequences. Validation of downstream or parallel effectors (such as eIF4E, S6K, MNK, PIM and RSK kinases) might lead to more tolerable combinations with anticancer efficacy. One setting in which toxicity should be minimized is in colorectal cancers harboring B-RAFV600E. Selective inhibitors of mutant B-RAF (vemurafinib) are well tolerated but are ineffective due to compensatory signaling. Combining B-RAF inhibitors with a PI3K/mTOR inhibitor caused apoptosis and tumor regression in a model of colorectal cancer driven by mutant B-RAF156. A very recent study showed persistent TORC1 activity in vemurafinib-resistant melanomas157, providing rationale for also combining vemurafinib with mTOR inhibitors in this setting.
The MYC oncogene is frequently amplified in cancer, and can confer resistance to PI3K/AKT/mTOR inhibitors independent of the RAS pathway27,28. Recent breakthroughs indicate that it might be possible to suppress the MYC transcriptional program indirectly by targeting BET proteins (bromo and extra terminal (BET) proteins) such as BRD2 and BRD4, which are transcriptional regulators required for efficient expression of MYC158–161. Inhibition of the BET-histone interaction by small molecules blocking the bromo-domain binding site (so called BET inhibitors) can downregulate expression of MYC and its target genes in tumor cells158–161. Combining BET inhibitors with PI3K/AKT/mTOR inhibitors is a sensible strategy, particularly in hematopoietic malignancies where MYC cooperation with PI3K has been established162. MYC is the defining oncogene of Burkitt lymphoma, and there is evidence for a cooperative role for PI3K activation in human specimens and in a mouse model163,164. Inhibiting mTOR or eIF4E strongly impaired MYC lymphomagenesis in mice165. In T-cell acute lymphoblastic leukemia (T-ALL), two common lesions are loss of PTEN and activating NOTCH mutations that elevate MYC activity166,167. Hence, either NOTCH inhibitors or BET inhibitors could be combined with PI3K/AKT/mTOR inhibitors in T-ALL trials.
Blocking autophagy might provide another avenue to augment cancer cell killing by PI3K/AKT/mTOR inhibitors168. Autophagy is a process by which cells recycle organelles and macromolecules to survive under conditions of starvation or other stresses. Inhibition of mTOR causes an autophagy response comparable to nutrient starvation. In glioma, leukemia and other cancer cell types, chemical inhibitors of autophagy potentiate apoptosis by active-site mTOR inhibitors or dual PI3K/mTOR inhibitors169,170. A combination trial of temsirolimus with an autophagy inhibitor in renal cell carcinoma is underway168. However, current autophagy inhibitors are nonspecific agents that generally act by inhibiting lysosomal degradation. Discovery of compounds inhibiting specific components of the autophagy machinery will be helpful for testing the potential of combination approaches with PI3K/AKT/mTOR inhibitors. A related issue is how PI3K/AKT/mTOR inhibitors will affect the response to emerging therapies targeting cancer cell metabolism. Considering that the PI3K/AKT/mTOR pathway drives many of the metabolic hallmarks of cancer cells, it is possible that pathway inhibition will reduce sensitivity to metabolic interventions.
Emerging evidence connects the PI3K/AKT/mTOR network to maintenance of genome integrity. PI3K is involved in sensing double strand breaks171,172 and in maintaining expression of BRCA1 and BRCA2 that participate in homologous recombination173. Exploiting these findings, two groups showed that PI3K inhibitors increase DNA damage and sensitize triple-negative breast cancer (TNBC) cells to inhibitors of polyADP-ribose polymerase (PARP)173,174. A phase I trial of the PARP inhibitor olaparib with the panPI3K inhibitor BKM120 has been initiated, enrolling patients with TNBC or high-grade serous ovarian cancer. Paradoxically, PTEN also has a role in protecting cells from genotoxic stress mediated by a nuclear pool of the phosphatase175. Elevated PI3K survival signaling in PTEN-deficient cells protects them from accumulated DNA damage. This property renders PTEN-deficient tumors sensitive to the combination of PI3K inhibitors and DNA damaging agents in preclinical studies175. Loss of PTEN might also sensitize to PARP inhibitors, similar to BRCA1-deficient tumors. It is worth noting that several DNA repair enzymes are members of the PIKK family: ATM, ATR and DNA-PK176. Some inhibitors developed against class I PI3Ks have off-target effects on PIKK family members, and this property might enhance synergy with DNA-damaging agents177. Inhibiting mTOR can also promote DNA damage through suppression of the FANCD2 and other mechanisms178–180. Thus, an important area for continuing study is to investigate how inhibitors acting at different levels of the PI3K/AKT/mTOR network affect the cellular response to radiation and chemotherapeutic drugs that are currently the standard of care in many cancers. Also worth considering is that off-target effects on DNA-PK and ATM are probably a liability rather than an advantage, if not combined with DNA damaging agents or radiation, by increasing genomic instability that tends to accelerate drug resistance.
The BOLERO-2 trial illustrated the potential of PI3K/AKT/mTOR inhibitors to prevent or overcome targeted therapy in hormone-dependent cancers. There is evidence for positive feedback between hormone receptors and the PI3K network181. The ligand-bound estrogen receptor (ER) interacts directly with PI3K, augmenting PI3K/AKT activity. In turn, AKT and S6K1 can phosphorylate the hormone receptor to increase its activity. Hormone-dependent cancers frequently exhibit high basal PI3K activity through loss of PTEN, PIK3CA mutation or other mechanisms182,183. Thus, inhibitors acting at multiple levels of the PI3K/AKT/mTOR network might supplement the anti-cancer effects of hormonal therapy. In advanced prostate cancer with loss of PTEN, it would be interesting to test a p110β inhibitor in combination with an androgen receptor antagonist. There is also rationale for a dual p110β/δ inhibitor in this setting, based on a report that B cell infiltrates sustain prostate cancer survival after hormone withdrawal184. This strategy would act via tumor intrinsic effects (p110β) together with extrinsic effects on the immune infiltration (p110δ).
A conceptually simple approach to sensitize cancer cells to PI3K pathway-targeted agents is to combine with agents that increase mitochondrial priming for death. BCL2 family members (BCL2, BCL-XL, MCL-1, A1) maintain mitochondrial integrity by blocking the pro-apoptotic function of BAX and BAK185. A large family of pro-apoptotic proteins homologous to BCL2 can sequester the pro-survival proteins or directly activate BAX and BAK186. Priming refers to suppressing the activity of pro-survival factors at the mitochondria, such as BCL2 and MCL-1, relative to pro-apoptotic proteins like BIM and PUMA187. ABT-263, a small molecule inhibitor of BCL2 and BCL-XL, has entered clinical trials for cancer and shown some promise in CLL185. By increasing mitochondrial priming, BCL2 antagonists should lower the threshold for apoptosis in response to PI3K/AKT/mTOR inhibition (Figure 4). A growing body of work supports the synergistic anti-tumor effects of PI3K/AKT/mTOR inhibitors combined with BCL2 antagonists150,188–190.
Figure 4. Rationale for BCL2 antagonist combination.

The balance of pro-survival and proapoptotic BCL2 family members at the mitochondria is a primary factor controlling cell survival versus apoptosis. PI3K/AKT/mTOR signaling suppresses expression and activity of multiple pro-apoptotic proteins (i.e. BAD, BIM, PUMA, and death receptors) and can increase expression of pro-survival factors (MCL-1). However, PI3K/AKT/mTOR inhibition does not necessarily tip the balance towards apoptosis. Combining with small molecule antagonists of pro-survival proteins (BCL2, BCL-XL) increases mitochondrial “priming” for death, lowering the threshold for apoptosis induction by PI3K/AKT/mTOR inhibitors.
Future directions
Building on the discussions above, we envision four key strategies that will maximize the potential of PI3K/AKT/mTOR inhibitors in oncology.
Biomarker identification through next generation sequencing
A limited response rate with a single agent strategy at an early stage of development should not necessarily mean that a clinical trial has failed, especially when targeting a genetically validated target or disease biology. The advent of next generation sequencing allows significant knowledge to be gained from the rare responders in a trial. The “n = 1 response” matters. An exciting report from Solit and colleagues used exome sequencing to identify TSC1 inactivation in a rare bladder cancer that responded to everolimus107. Targeted sequencing of additional tumors from everolimus trials showed a significant delay in recurrence for samples with TSC1 mutations96. As sequencing costs decline and technologies improve, it should be feasible to apply this approach in trials of PI3K/AKT/mTOR inhibitors as single agents or in combinations. This idea should not replace patient selection based on the drug target, cancer genetics and disease biology. It should, however, be applied in parallel to select additional genetic markers for subsequent trials.
Initial emphasis on hematologic malignancies
Leukemia, lymphoma and myeloma are a diverse set of cancers that are individually less common than solid tumors like lung, breast and colon cancer. Few blood cancers carry activating mutations in RAS or PI3K. Nevertheless, there are several reasons to devote resources to PI3K/mTOR inhibitor trials in blood cancer. For one thing, leukemia and lymphoma models often show non-oncogene addiction to mTOR58,165. In addition, blood cancers generally express p110δ and/or p110γ that should in principle confer responsiveness to agents targeting these isoforms, as illustrated by GS-1101. Unlike many solid tumors, hematopoietic cancer cells are in constant contact with the immune system and might be especially sensitive to immune enhancing effects of PI3K/mTOR inhibitors. It is also easier to access tumor cells for PD monitoring in patients with blood cancers, and plasma analysis can provide useful information about immune modulation191. Lastly, treating rare blood cancers effectively can be rewarding, as proven by BCR-ABL inhibitors that have saved the lives of an ever-expanding population of CML patients who must continue on therapy.
Harness immune effects
Though cancer is a genetic disease of aberrant cells, it is also a chronic immune disease (Fig. 1)33,192. The immune system restrains tumorigenesis but eventually the tumor enforces a state of immune tolerance and exhaustion. Recently there has been exciting progress treating human tumors with immunotherapies to overcome tolerance and exhaustion33,34,193. There is also an increasing appreciation for how small molecules targeting the cancer cell affect the immune context of the tumor194,195. The efficacy of GS-1101 emphasizes how a drug targeting both the tumor and the immune system can act as an all-in-one combination therapy. How is it possible to harness anti-tumor immunity through a pathway defined by a target of the immunosuppressive drug rapamycin?
Extensive studies of the PI3K/AKT/mTOR network in immune cells have shown that PI3K activation is not a simple on/off switch110,111,196,197. Inhibiting the pathway can either suppress or enhance immune responses, through effects on diverse subsets of innate and adaptive immune cells. In theory it should be possible to implement treatment regimens that increase immune rejection of tumors in concert with direct anti-tumor effects. A significant factor limiting progress in this area is that preclinical drug development programs mainly use xenograft tumor models. These models are convenient and are useful to assess drug pharmacology, providing valuable information about PD in the context of PK and general tolerability. Yet these models overlook any modulation of adaptive immunity, since growing human tumor cells in mice requires host strains lacking functional lymphocytes. Interactions of xenograft cells with innate immune components might also fail to recapitulate events in the development of human tumors. For these reasons it is essential to test candidate inhibitors in genetically engineered mouse (GEM) models, and to extensively monitor infiltration and activity of diverse immune subsets including macrophages, T cells and natural killer cells. Such systems would be useful to test ideas such as “dialing in” activity against p110δ and/or p110γ to create a more favorable immune environment. One can even imagine that inhibiting p110δ and/or p110γ alone in solid tumors would provide significant therapeutic benefit and tolerability without any direct effect on the PI3K isoforms expressed within the cancer cell (based on the work of Ali and Vanhaesebroeck, personal communication).
It will also be crucial to determine which agents targeting PI3K/AKT/mTOR enhance or suppress the efficacy of emerging immunotherapies and cancer vaccines. In mouse models, both PI3K and mTOR inhibitors can enhance the efficacy of immune-directed therapies198–200. It is relevant to consider that isoform-selective agents minimize immune suppressive effects on lymphocytes compared to pan-class I inhibitors201. Hence, pan-PI3K inhibitors are more likely to enforce or accelerate the immune exhaustion state. Eventually, the best combination therapies might turn out to be isoform-selective PI3K or mTOR inhibitors with immunotherapies or cancer vaccines. Matching patients to the right combinations will require knowledge of the genomic driver and the immune fingerprint of the tumor.
Combination trials
Above, we proposed several rational combinations to increase cancer cell killing by PI3K/AKT/mTOR inhibitors. However, the process of drug development always faces tension between what should be done and what can be done. Developing combination therapies costs more resources and time and might ultimately result in challenges for reimbursement. On the other hand, experience with BRAF inhibitors shows that combinations will be justified even for therapies that provide an impressive initial response in selected patients. In the short term, the plan should be to prioritize approaches based on feasibility, pragmatism and the likelihood of a meaningful therapeutic advance. It makes sense to start by combining PI3K/AKT/mTOR inhibitors with approved targeted agents that are standard of care for specific malignancies. Using a companion drug whose safety profile and optimal dosing is well understood will reduce the complexity of the combination trial. It will also allow the incorporation of PI3K/AKT/mTOR inhibitors into treatment regimens at earlier stages of disease, rather than only in patients with relapsed or refractory tumors. The limited success of single agents to date might be explained in part by the polygenic and polyclonal nature of advanced tumors, some of which is caused by prior therapies. Preclinical data support testing of several combinations with approved TKIs: BCR-ABL inhibitors in Ph+ leukemias and GIST142, EGFR inhibitors in lung and colon cancer139,140, and agents targeting HER2/ErbB3 in breast cancer202. Adding a PI3K inhibitor to a BRAF inhibitor might enhance efficacy in melanoma and produce responses in colorectal cancers carrying mutant B-RAF156,203,204.
Sometimes key biological insights from preclinical data can justify combination trials of two experimental agents. Agents that show synthetic lethality with PI3K pathway inhibitors in cancer cell lines and patient-derived xenografts, but not in normal cells, should be given priority for clinical testing. An example discussed above is the combination of PI3K and PARP inhibitors for TNBC (Table 2), in which trials were quickly initiated after remarkable preclinical results173,174.
In cases where GEM models exist for driving oncogenes and tumor types, it should be possible to design synchronous “co-clinical” trials that help in identifying genetic and pharmacodynamic markers of responsiveness205. These are especially powerful when paired with studies using patient-derived primary tumor tissue analysis. Regulatory approval of immunotherapies in certain cancers also sets the stage for testing the immune enhancing potential of PI3K or mTOR inhibitors.
Conclusions
The rationale for targeting the PI3K/AKT/mTOR network in cancer remains anchored on a solid foundation of cancer genetics and cell biological studies. Despite many challenges, measurable advances have been achieved in the clinic. Rapalogs are useful in some advanced cancers and as adjuvants to hormone therapy in breast cancer. Inhibitors of PI3Kδ and BTK are on track for FDA approval in certain B cell malignancies. Other agents are advancing through development. Nevertheless, early hopes have been tempered by the realization that targeting PI3K/AKT/mTOR alone will not be a cure-all for diverse cancers.
How can we reset strategies to maximize the potential of PI3K/AKT/mTOR inhibitors? Previous experience with successful oncology drug development shows the importance of: (1) targeting genetic drivers in selected patient populations; (2) understanding the biology of crosstalk and feedback to employ effective combinations; (3) stimulating an immune environment that favors tumor eradication. Thoughtful application of these principles will light the path towards effective cancer control by PI3K/AKT/mTOR inhibitors.
Supplementary Material
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Beagle B, Fruman DA. A lipid kinase cousin cooperates to promote cancer. Cancer Cell. 2011;19:693–695. doi: 10.1016/j.ccr.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graupera M, Potente M. Regulation of angiogenesis by PI3K signaling networks. Exp Cell Res. 2013;319:1348–1355. doi: 10.1016/j.yexcr.2013.02.021. [DOI] [PubMed] [Google Scholar]
- 4.Hirsch E, Ciraolo E, Franco I, Ghigo A, Martini M. PI3K in cancer-stroma interactions: bad in seed and ugly in soil. Oncogene. 2013 doi: 10.1038/onc.2013.265. Epub July 29. [DOI] [PubMed] [Google Scholar]
- 5.Vanhaesebroeck B, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 6.Zhao JJ, Roberts TM. PI3 kinases in cancer: from oncogene artifact to leading cancer target. Science's STKE : signal transduction knowledge environment. 2006;2006:pe52. doi: 10.1126/stke.3652006pe52. [DOI] [PubMed] [Google Scholar]
- 7.Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 8.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 9.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature reviews. Molecular cell biology. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 10.Lui VW, et al. Frequent Mutation of the PI3K Pathway in Head and Neck Cancer Defines Predictive Biomarkers. Cancer discovery. 2013;3:761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature reviews. Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 12.Berndt A, et al. The p110delta structure: mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat Chem Biol. 2010;6:244. doi: 10.1038/nchembio0310-244b. [DOI] [PubMed] [Google Scholar]
- 13. Huang CH, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–1748. doi: 10.1126/science.1150799. One of two hallmark structural studies of p110alpha.
- 14. Miled N, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–242. doi: 10.1126/science.1135394. One of two hallmark structural studies of p110alpha.
- 15.Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4 doi: 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- 16. Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. The first x-ray structure of a class I PI3K.
- 17.Wu H, et al. Regulation of Class IA PI 3-kinases: C2 domain-iSH2 domain contacts inhibit p85/p110alpha and are disrupted in oncogenic p85 mutants. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20258–20263. doi: 10.1073/pnas.0902369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X, et al. Structure of lipid kinase p110beta/p85beta elucidates an unusual SH2-domain-mediated inhibitory mechanism. Molecular cell. 2011;41:567–578. doi: 10.1016/j.molcel.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 20.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Workman P, Clarke PA, Raynaud FI, van Montfort RL. Drugging the PI3 Kinome: From Chemical Tools to Drugs in the Clinic. Cancer Res. 2010;70:2146–2157. doi: 10.1158/0008-5472.CAN-09-4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agarwal R, Carey M, Hennessy B, Mills GB. PI3K pathway-directed therapeutic strategies in cancer. Current opinion in investigational drugs. 2010;11:615–628. [PubMed] [Google Scholar]
- 23.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Yap TA, et al. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nature Reviews Clinical Oncology. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 26. Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature medicine. 2008;14:1351–1356. doi: 10.1038/nm.1890. First proof of concept in vivo for co-targeting PI3K and MEK.
- 27.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E699–E708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu P, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nature Medicine. 2011;17:1116–1120. doi: 10.1038/nm.2402. Reversible PIK3CA model showed mechanisms of relapse.
- 29.Kinross KM, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122:553–557. doi: 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tikoo A, et al. Physiological levels of Pik3ca(H1047R) mutation in the mouse mammary gland results in ductal hyperplasia and formation of ERalpha-positive tumors. PLoS ONE. 2012;7:e36924. doi: 10.1371/journal.pone.0036924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fruman DA, Rommel C. PI3Kdelta Inhibitors in Cancer: Rationale and Serendipity Merge in the Clinic. Cancer discovery. 2011;1:562–572. doi: 10.1158/2159-8290.CD-11-0249. [DOI] [PubMed] [Google Scholar]
- 32.Macias-Perez IM, Flinn IW. GS-1101: a delta-specific PI3K inhibitor in chronic lymphocytic leukemia. Current hematologic malignancy reports. 2013;8:22–27. doi: 10.1007/s11899-012-0142-1. [DOI] [PubMed] [Google Scholar]
- 33.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 34.Riley JL. Combination Checkpoint Blockade - Taking Melanoma Immunotherapy to the Next Level. N Engl J Med. 2013;369:187–189. doi: 10.1056/NEJMe1305484. [DOI] [PubMed] [Google Scholar]
- 35.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 37. Fritsch R, et al. RAS and RHO Families of GTPases Directly Regulate Distinct Phosphoinositide 3-Kinase Isoforms. Cell. 2013;153:1050–1063. doi: 10.1016/j.cell.2013.04.031. Discovered that RAC and cdc42, and not RAS contribute to activation of p110beta.
- 38.Fruman DA. Towards an understanding of isoform specificity in phosphoinositide 3-kinase signalling in lymphocytes. Biochem Soc Trans. 2004;32:315–319. doi: 10.1042/bst0320315. [DOI] [PubMed] [Google Scholar]
- 39.Hawkins PT, Stephens LR, Suire S, Wilson M. PI3K signaling in neutrophils. Curr Top Microbiol Immunol. 2010;346:183–202. doi: 10.1007/82_2010_40. [DOI] [PubMed] [Google Scholar]
- 40.Okkenhaug K, Ali K, Vanhaesebroeck B. Antigen receptor signalling: a distinctive role for the p110delta isoform of PI3K. Trends Immunol. 2007;28:80–87. doi: 10.1016/j.it.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okkenhaug K, Fruman DA. PI3Ks in Lymphocyte Signaling and Development. Curr Top Microbiol Immunol. 2011;346:57–85. doi: 10.1007/82_2010_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Foukas LC, Berenjeno IM, Gray A, Khwaja A, Vanhaesebroeck B. Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11381–11386. doi: 10.1073/pnas.0906461107. Provided evidence for redundant functions of PI3K isoforms in cell proliferation and survival.
- 43.Dbouk HA, et al. Characterization of a tumor-associated activating mutation of the p110beta PI 3-kinase. PLoS ONE. 2013;8:e63833. doi: 10.1371/journal.pone.0063833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheung LW, et al. High Frequency of PIK3R1 and PIK3R2 Mutations in Endometrial Cancer Elucidates a Novel Mechanism for Regulation of PTEN Protein Stability. Cancer discovery. 2011;1:170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaiswal BS, et al. Somatic Mutations in p85alpha Promote Tumorigenesis through Class IA PI3K Activation. Cancer Cell. 2009;16:463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15547–15552. doi: 10.1073/pnas.1009652107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wee S, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–4293. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 48.Ludovini V, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2011;6:707–715. doi: 10.1097/JTO.0b013e31820a3a6b. [DOI] [PubMed] [Google Scholar]
- 49.Suda K, Mizuuchi H, Maehara Y, Mitsudomi T. Acquired resistance mechanisms to tyrosine kinase inhibitors in lung cancer with activating epidermal growth factor receptor mutation--diversity, ductility, and destiny. Cancer metastasis reviews. 2012;31:807–814. doi: 10.1007/s10555-012-9391-7. [DOI] [PubMed] [Google Scholar]
- 50.Cybulski N, Hall MN. TOR complex 2: a signaling pathway of its own. Trends Biochem Sci. 2009;34:620–627. doi: 10.1016/j.tibs.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 51.Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 53.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dibble CC, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012;47:535–546. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guertin DA, et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. One of two papers genetically validating mTOR as a selective cancer target in prostate cancer.
- 56. Nardella C, et al. Differential Requirement of mTOR in Postmitotic Tissues and Tumorigenesis. Sci Signal. 2009;2:ra2. doi: 10.1126/scisignal.2000189. One of two papers genetically validating mTOR as a selective cancer target in prostate cancer.
- 57.Evangelisti C, et al. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia : official journal of the Leukemia Society of America, Leukemia Research Fund, U.K. 2011;25:781–791. doi: 10.1038/leu.2011.20. [DOI] [PubMed] [Google Scholar]
- 58.Janes MR, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandarlapaty S, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rodrik-Outmezguine VS, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer discovery. 2011;1:248–259. doi: 10.1158/2159-8290.CD-11-0085. Detailed analysis of feedback effects of mTOR kinase inhibitors and role of FOXO transcription factors.
- 61.Ballif BA, et al. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:667–672. doi: 10.1073/pnas.0409143102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 63.Ma L, et al. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 2007;67:7106–7112. doi: 10.1158/0008-5472.CAN-06-4798. [DOI] [PubMed] [Google Scholar]
- 64.Tabernero J, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:1603–1610. doi: 10.1200/JCO.2007.14.5482. [DOI] [PubMed] [Google Scholar]
- 65. She QB, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. Provided evidence for convergence of PI3K/AKT and RAS/ERK signals at the level of 4E-BPs.
- 66.Wang X, et al. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Molecular and cellular biology. 2007;27:7405–7413. doi: 10.1128/MCB.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee T, Yao G, Nevins J, You L. Sensing and integration of Erk and PI3K signals by Myc. PLoS computational biology. 2008;4:e1000013. doi: 10.1371/journal.pcbi.1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brachmann SM, et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol Cancer Ther. 2012;11:1747–1757. doi: 10.1158/1535-7163.MCT-11-1021. [DOI] [PubMed] [Google Scholar]
- 69.Advani RH, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Corcoran RB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer discovery. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prahallad A, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 72.Garrett JT, et al. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110alpha inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-13-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gupta S, et al. Binding of Ras to Phosphoinositide 3-Kinase p110alpha Is Required for Ras-Driven Tumorigenesis in Mice. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. Knockin mouse defined role for p110alpha in RAS transformation.
- 74.Nacht M, et al. Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase PI3Kalpha. Journal of medicinal chemistry. 2013;56:712–721. doi: 10.1021/jm3008745. [DOI] [PubMed] [Google Scholar]
- 75.Lee JH, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nature genetics. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lindhurst MJ, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nature genetics. 2012;44:928–933. doi: 10.1038/ng.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Riviere JB, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nature genetics. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Angulo I, et al. Phosphoinositide 3-Kinase delta Gene Mutation Predisposes to Respiratory Infection and Airway Damage. Science. 2013;342:866–871. doi: 10.1126/science.1243292. One of two recent papers identifying human immunodeficiency patients with gain-of-function mutations affecting p110delta.
- 79. Lucas CL, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. 2013 doi: 10.1038/ni.2771. Epub Oct 28. One of two recent papers identifying human immunodeficiency patients with gain-of-function mutations affecting p110delta.
- 80. Jia S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. Provide first genetic evidence for p110beta function in tumorigenesis.
- 81.Torbett NE, et al. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J. 2008;415:97–110. doi: 10.1042/BJ20080639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wee S, et al. PTEN-deficient cancers depend on PIK3CB. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Berenjeno IM, et al. Both p110alpha and p110beta isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J. 2012;442:151–159. doi: 10.1042/BJ20111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iyengar S, et al. P110alpha-mediated constitutive PI3K signaling limits the efficacy of p110delta-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood. 2013;121:2274–2284. doi: 10.1182/blood-2012-10-460832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Subramaniam PS, et al. Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell. 2012;21:459–472. doi: 10.1016/j.ccr.2012.02.029. Provided proof of concept for dual targeting of p110gamma and p110delta in T cell leukemia.
- 86. Schmid MC, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19:715–727. doi: 10.1016/j.ccr.2011.04.016. Provided evidence that p110gamma activity in myeloid cells acts downstream of diverse receptors, and promotes solid tumors even though the isoform is not expressed in the cancer cells.
- 87.So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442:465–481. doi: 10.1042/BJ20112092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brunn GJ, et al. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. Embo J. 1996;15:5256–5267. [PMC free article] [PubMed] [Google Scholar]
- 89.Gharbi SI, et al. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J. 2007;404:15–21. doi: 10.1042/BJ20061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Knight ZA, et al. A Pharmacological Map of the PI3-K Family Defines a Role for p110alpha in Insulin Signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kharas MG, et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest. 2008;118:3038–3050. doi: 10.1172/JCI33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Elkabets M, et al. mTORC1 Inhibition Is Required for Sensitivity to PI3K p110alpha Inhibitors in PIK3CA-Mutant Breast Cancer. Science translational medicine. 2013;5:196ra99. doi: 10.1126/scitranslmed.3005747. Evidence that mTORC1 preserves survival in PIK3CA mutant cells treated with p110alpha inhibitors.
- 94.Yuan R, Kay A, Berg WJ, Lebwohl D. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. Journal of hematology & oncology. 2009;2:45. doi: 10.1186/1756-8722-2-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sankhala K, et al. The emerging safety profile of mTOR inhibitors, a novel class of anticancer agents. Target Oncol. 2009;4:135–142. doi: 10.1007/s11523-009-0107-z. [DOI] [PubMed] [Google Scholar]
- 96.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nature reviews. Drug discovery. 2011;10:868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 97.Janes MR, Fruman DA. Targeting TOR dependence in cancer. Oncotarget. 2010;1:69–76. doi: 10.18632/oncotarget.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gentzler RD, Altman JK, Platanias LC. An overview of the mTOR pathway as a target in cancer therapy. Expert Opin Ther Targets. 2012 doi: 10.1517/14728222.2012.677439. Epub April 12. [DOI] [PubMed] [Google Scholar]
- 99.Chresta CM, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 100.Yu K, et al. Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res. 2010;70:621–631. doi: 10.1158/0008-5472.CAN-09-2340. [DOI] [PubMed] [Google Scholar]
- 101.Garcia-Garcia C, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:2603–2612. doi: 10.1158/1078-0432.CCR-11-2750. [DOI] [PubMed] [Google Scholar]
- 102.Alain T, Sonenberg N, Topisirovic I. mTOR inhibitor efficacy is determined by the eIF4E/4E-BP ratio. Oncotarget. 2012;3:1491–1492. doi: 10.18632/oncotarget.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Martineau Y, et al. Pancreatic tumours escape from translational control through 4E-BP1 loss. Oncogene. 2013 doi: 10.1038/onc.2013.100. Epub April 8. [DOI] [PubMed] [Google Scholar]
- 104. Baselga J, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. Clinical study establishing rapalog combination with anti-estrogen therapy in breast cancer.
- 105.Bissler JJ, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Krueger DA, et al. Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology. 2013;80:574–580. doi: 10.1212/WNL.0b013e3182815428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Iyer G, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012;338:221. doi: 10.1126/science.1226344. Demonstrated that genome sequencing of rare responders can identify predictive biomarkers for rapalog sensitivity.
- 108.Bar-Peled L, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Panchaud N, Peli-Gulli MP, De Virgilio C. Amino Acid Deprivation Inhibits TORC1 Through a GTPase-Activating Protein Complex for the Rag Family GTPase Gtr1. Sci Signal. 2013;6:ra42. doi: 10.1126/scisignal.2004112. [DOI] [PubMed] [Google Scholar]
- 110.Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annual review of immunology. 2012;30:39–68. doi: 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zeng H, Chi H. mTOR and lymphocyte metabolism. Curr Opin Immunol. 2013;25:347–355. doi: 10.1016/j.coi.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Araki K, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Delgoffe GM, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Delgoffe GM, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nature immunology. 2011;12:295–303. doi: 10.1038/ni.2005. Informative dissection of mTOR complex functions in T cell differentiation, determined using genetic and pharmacological approaches.
- 116.Katholnig K, Linke M, Pham H, Hengstschlager M, Weichhart T. Immune responses of macrophages and dendritic cells regulated by mTOR signalling. Biochem Soc Trans. 2013;41:927–933. doi: 10.1042/BST20130032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Procaccini C, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–941. doi: 10.1016/j.immuni.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zeng H, et al. mTORC1 couples immune signals and metabolic programming to establish T-cell function. Nature. 2013;499:485–490. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 120.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 121.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 122.Rhodes N, et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008;68:2366–2374. doi: 10.1158/0008-5472.CAN-07-5783. [DOI] [PubMed] [Google Scholar]
- 123.Yap TA, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:4688–4695. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 124.Cho H, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 125.Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert opinion on investigational drugs. 2010;19:1355–1366. doi: 10.1517/13543784.2010.520701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lin J, et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:1760–1772. doi: 10.1158/1078-0432.CCR-12-3072. [DOI] [PubMed] [Google Scholar]
- 127.Vakana E, Altman JK, Platanias LC. Targeting AMPK in the treatment of malignancies. Journal of cellular biochemistry. 2012;113:404–409. doi: 10.1002/jcb.23369. [DOI] [PubMed] [Google Scholar]
- 128.Lindqvist L, Pelletier J. Inhibitors of translation initiation as cancer therapeutics. Future medicinal chemistry. 2009;1:1709–1722. doi: 10.4155/fmc.09.122. [DOI] [PubMed] [Google Scholar]
- 129.Moerke NJ, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 130.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Okuzumi T, et al. Inhibitor hijacking of Akt activation. Nat Chem Biol. 2009;5:484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pearce LR, et al. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) Biochem J. 2010;431:245–255. doi: 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- 133.Tandon P, et al. Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proc Natl Acad Sci U S A. 2011;108:2361–2365. doi: 10.1073/pnas.1013629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Merkel AL, Meggers E, Ocker M. PIM1 kinase as a target for cancer therapy. Expert opinion on investigational drugs. 2012;21:425–436. doi: 10.1517/13543784.2012.668527. [DOI] [PubMed] [Google Scholar]
- 135.Yang J, et al. eIF4B Phosphorylation by Pim Kinases Plays a Critical Role in Cellular Transformation by Abl Oncogenes. Cancer Res. 2013;73:4898–4908. doi: 10.1158/0008-5472.CAN-12-4277. [DOI] [PubMed] [Google Scholar]
- 136.Wendel HG, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21:3232–3237. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. One of two papers identifying promising new approaches to target oncogenic RAS.
- 138. Zimmermann G, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. One of two papers identifying promising new approaches to target oncogenic RAS.
- 139.Chakrabarty A, et al. Trastuzumab-resistant cells rely on a HER2-PI3K-FoxO-survivin axis and are sensitive to PI3K inhibitors. Cancer Res. 2013;73:1190–1200. doi: 10.1158/0008-5472.CAN-12-2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Donev IS, et al. Transient PI3K inhibition induces apoptosis and overcomes HGFmediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:2260–2269. doi: 10.1158/1078-0432.CCR-10-1993. [DOI] [PubMed] [Google Scholar]
- 141.Rexer BN, Arteaga CL. Optimal Targeting of HER2-PI3K Signaling in Breast Cancer: Mechanistic Insights and Clinical Implications. Cancer Res. 2013;73:3817–3820. doi: 10.1158/0008-5472.CAN-13-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Floris G, et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:620–630. doi: 10.1158/1078-0432.CCR-12-2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Young CD, et al. Conditional loss of ErbB3 delays mammary gland hyperplasia induced by mutant PIK3CA without affecting mammary tumor latency, gene expression or signaling. Cancer Res. 2013;73:4075–4085. doi: 10.1158/0008-5472.CAN-12-4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fiskus W, et al. Dual PI3K/AKT/mTOR Inhibitor BEZ235 Synergistically Enhances the Activity of JAK2 Inhibitor against Cultured and Primary Human Myeloproliferative Neoplasm Cells. Mol Cancer Ther. 2013;12:577–588. doi: 10.1158/1535-7163.MCT-12-0862. [DOI] [PubMed] [Google Scholar]
- 145.Vogt PK, Hart JR. PI3K and STAT3: a new alliance. Cancer discovery. 2011;1:481–486. doi: 10.1158/2159-8290.CD-11-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Carracedo A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kinkade CW, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zmajkovicova K, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43–55. doi: 10.1016/j.molcel.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Posch C, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4015–4020. doi: 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bean GR, et al. PUMA and BIM are required for oncogene inactivation-induced apoptosis. Sci Signal. 2013;6:ra20. doi: 10.1126/scisignal.2003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu Y, et al. Rapamycin induces Bad phosphorylation in association with its resistance to human lung cancer cells. Mol Cancer Ther. 2012;11:45–56. doi: 10.1158/1535-7163.MCT-11-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ellenrieder V, et al. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001;61:4222–4228. [PubMed] [Google Scholar]
- 153.Mulholland DJ, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72:1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hsieh AC, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shimizu T, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:2316–2325. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 156.Coffee EM, et al. Concomitant BRAF and PI3K/mTOR Blockade Is Required for Effective Treatment of BRAFV600E Colorectal Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:2688–2698. doi: 10.1158/1078-0432.CCR-12-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Corcoran RB, et al. TORC1 Suppression Predicts Responsiveness to RAF and MEK Inhibition in BRAF-Mutant Melanoma. Science translational medicine. 2013;5:196ra98. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dawson MA, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mertz JA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zuber J, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dominguez-Sola D, Dalla-Favera R. Burkitt lymphoma: much more than MYC. Cancer Cell. 2012;22:141–142. doi: 10.1016/j.ccr.2012.07.018. [DOI] [PubMed] [Google Scholar]
- 163. Sander S, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22:167–179. doi: 10.1016/j.ccr.2012.06.012. Establishes animal model for Burkitt's lymphoma, requiring both MYC and active PI3K.
- 164.Schmitz R, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–120. doi: 10.1038/nature11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Pourdehnad M, et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11988–11993. doi: 10.1073/pnas.1310230110. Evidence that MYC-driven lymphoma is addicted to mTOR activity.
- 166.Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nature reviews. Cancer. 2006;6:347–359. doi: 10.1038/nrc1880. [DOI] [PubMed] [Google Scholar]
- 167.Guo D, Teng Q, Ji C. NOTCH and phosphatidylinositide 3-kinase/phosphatase and tensin homolog deleted on chromosome ten/AKT/mammalian target of rapamycin (mTOR) signaling in T-cell development and T-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2011;52:1200–1210. doi: 10.3109/10428194.2011.564696. [DOI] [PubMed] [Google Scholar]
- 168.Shanware NP, Bray K, Abraham RT. The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Annual review of pharmacology and toxicology. 2013;53:89–106. doi: 10.1146/annurev-pharmtox-010611-134717. [DOI] [PubMed] [Google Scholar]
- 169.Carayol N, et al. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A. 2010;107:12469–12474. doi: 10.1073/pnas.1005114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fan QW, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Science signaling. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem. 2007;282:21206–21212. doi: 10.1074/jbc.M703042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7491–7496. doi: 10.1073/pnas.0914242107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ibrahim YH, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer discovery. 2012;2:1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Juvekar A, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer discovery. 2012;2:1048–1063. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Bassi C, et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341:395–399. doi: 10.1126/science.1236188. Identified novel SUMOylation and nuclear function of PTEN.
- 176.Lempiainen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. Embo J. 2009;28:3067–3073. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Munck JM, et al. Chemosensitization of cancer cells by KU-0060648, a dual inhibitor of DNA-PK and PI-3K. Mol Cancer Ther. 2012;11:1789–1798. doi: 10.1158/1535-7163.MCT-11-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Khalaileh A, et al. Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res. 2013;73:1811–1820. doi: 10.1158/0008-5472.CAN-12-2014. [DOI] [PubMed] [Google Scholar]
- 179.Shen C, et al. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double strand breaks. Cancer Res. 2013;73:3393–3401. doi: 10.1158/0008-5472.CAN-12-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guo F, et al. mTOR regulates DNA damage response through NF-kappaB-mediated FANCD2 pathway in hematopoietic cells. Leukemia. 2013;27:2040–2046. doi: 10.1038/leu.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:4452–4461. doi: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Li J, et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 183.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 184.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010;464:302–305. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:3127–3135. doi: 10.1200/JCO.2011.37.0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Letai A, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 187.Certo M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 188.Coloff JL, et al. Akt-dependent glucose metabolism promotes mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011;71:5204–5213. doi: 10.1158/0008-5472.CAN-10-4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Davids MS, et al. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012;120:3501–3509. doi: 10.1182/blood-2012-02-414060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rahmani M, et al. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013;73:1340–1351. doi: 10.1158/0008-5472.CAN-12-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Hoellenriegel J, et al. The phosphoinositide 3'-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118:3603–3612. doi: 10.1182/blood-2011-05-352492. Provide mechanism for CAL-101 (GS-1101) efficacy and included pharmacodynamic data from clinical studies.
- 192.Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nature reviews. Cancer. 2012;12:237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88. doi: 10.1016/j.immuni.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 196.Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev. 2009;228:253–272. doi: 10.1111/j.1600-065X.2008.00750.x. [DOI] [PubMed] [Google Scholar]
- 197.Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annual review of immunology. 2013;31:675–704. doi: 10.1146/annurev-immunol-032712-095946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jiang Q, et al. mTOR Kinase Inhibitor AZD8055 Enhances the Immunotherapeutic Activity of an Agonist CD40 Antibody in Cancer Treatment. Cancer research. 2011;71:4074–4084. doi: 10.1158/0008-5472.CAN-10-3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Li Q, et al. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity. 2011;34:541–553. doi: 10.1016/j.immuni.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Marshall NA, et al. Immunotherapy with PI3K inhibitor and Toll-like receptor agonist induces IFN-gamma+IL-17+ polyfunctional T cells that mediate rejection of murine tumors. Cancer Res. 2012;72:581–591. doi: 10.1158/0008-5472.CAN-11-0307. Shows that PI3K inhibitors can enhance adjuvant activity of TLR agonists to improve dendritic cell-based tumor vaccines in mice.
- 201.So L, et al. Selective inhibition of phosphoinositide 3-kinase p110alpha preserves lymphocyte function. J Biol Chem. 2013;288:5718–5731. doi: 10.1074/jbc.M112.379446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yao E, et al. Suppression of HER2/HER3-mediated growth of breast cancer cells with combinations of GDC-0941 PI3K inhibitor, trastuzumab, and pertuzumab. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:4147–4156. doi: 10.1158/1078-0432.CCR-08-2814. [DOI] [PubMed] [Google Scholar]
- 203.Mao M, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Paraiso KH, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nardella C, Lunardi A, Patnaik A, Cantley LC, Pandolfi PP. The APL paradigm and the "co-clinical trial" project. Cancer discovery. 2011;1:108–116. doi: 10.1158/2159-8290.CD-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Suire S, et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol. 2006;8:1303–1309. doi: 10.1038/ncb1494. [DOI] [PubMed] [Google Scholar]
- 207.Delgado P, et al. Essential function for the GTPase TC21 in homeostatic antigen receptor signaling. Nat Immunol. 2009;10:880–888. doi: 10.1038/ni.1749. [DOI] [PubMed] [Google Scholar]
- 208.Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Molecular and cellular biology. 2004;24:4943–4954. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dbouk HA, et al. G Protein-Coupled Receptor-Mediated Activation of p110beta by Gbetagamma Is Required for Cellular Transformation and Invasiveness. Sci Signal. 2012;5:ra89. doi: 10.1126/scisignal.2003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Durand CA, et al. Phosphoinositide 3-kinase p110delta regulates natural antibody production, marginal zone and B-1 B cell function, and autoantibody responses. J Immunol. 2009;183:5673–5684. doi: 10.4049/jimmunol.0900432. [DOI] [PubMed] [Google Scholar]
- 211.Reif K, et al. Cutting edge: differential roles for phosphoinositide 3-kinases, p110gamma and p110delta, in lymphocyte chemotaxis and homing. J Immunol. 2004;173:2236–2240. doi: 10.4049/jimmunol.173.4.2236. [DOI] [PubMed] [Google Scholar]
- 212.Puri KD, Gold MR. Selective inhibitors of phosphoinositide 3-kinase delta: modulators of B-cell function with potential for treating autoimmune inflammatory diseases and B-cell malignancies. Frontiers in immunology. 2012;3:256. doi: 10.3389/fimmu.2012.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ghosh B, et al. Nontoxic chemical interdiction of the epithelial-to-mesenchymal transition by targeting cap-dependent translation. ACS chemical biology. 2009;4:367–377. doi: 10.1021/cb9000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Knauf U, Tschopp C, Gram H. Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Molecular and cellular biology. 2001;21:5500–5511. doi: 10.1128/MCB.21.16.5500-5511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Lim S, et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2298–E2307. doi: 10.1073/pnas.1301838110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Konicek BW, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011;71:1849–1857. doi: 10.1158/0008-5472.CAN-10-3298. [DOI] [PubMed] [Google Scholar]
- 217.Lin YW, et al. A small molecule inhibitor of Pim protein kinases blocks the growth of precursor T-cell lymphoblastic leukemia/lymphoma. Blood. 2010;115:824–833. doi: 10.1182/blood-2009-07-233445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Blanco-Aparicio C, et al. Pim 1 kinase inhibitor ETP-45299 suppresses cellular proliferation and synergizes with PI3K inhibition. Cancer letters. 2011;300:145–153. doi: 10.1016/j.canlet.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 219.Chen LS, Redkar S, Bearss D, Wierda WG, Gandhi V. Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood. 2009;114:4150–4157. doi: 10.1182/blood-2009-03-212852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Song JH, Kraft AS. Pim kinase inhibitors sensitize prostate cancer cells to apoptosis triggered by Bcl-2 family inhibitor ABT-737. Cancer Res. 2012;72:294–303. doi: 10.1158/0008-5472.CAN-11-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pogacic V, et al. Structural analysis identifies imidazo[1,2-b]pyridazines as PIM kinase inhibitors with in vitro antileukemic activity. Cancer Res. 2007;67:6916–6924. doi: 10.1158/0008-5472.CAN-07-0320. [DOI] [PubMed] [Google Scholar]
- 222.Klempner SJ, Myers AP, Cantley LC. What a Tangled Web We Weave: Emerging Resistance Mechanisms to Inhibition of the Phosphoinositide 3-Kinase Pathway. Cancer Discov. 2013 doi: 10.1158/2159-8290.CD-13-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Burger JA. Targeting the microenvironment in chronic lymphocytic leukemia is changing the therapeutic landscape. Current opinion in oncology. 2012;24:643–649. doi: 10.1097/CCO.0b013e3283589950. [DOI] [PubMed] [Google Scholar]
- 224.Byrd JC, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Liu N, et al. BAY 80-6946 Is a Highly Selective Intravenous PI3K Inhibitor with Potent p110alpha and p110delta Activities in Tumor Cell Lines and Xenograft Models. Molecular cancer therapeutics. 2013;12:2319–2330. doi: 10.1158/1535-7163.MCT-12-0993-T. [DOI] [PubMed] [Google Scholar]
- 226.Winkler DG, et al. PI3K-delta and PI3K-gamma Inhibition by IPI-145 Abrogates Immune Responses and Suppresses Activity in Autoimmune and Inflammatory Disease Models. Chemistry & biology. 2013;20:1364–1374. doi: 10.1016/j.chembiol.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 227.Boyle DL, Kim HR, Topolewski K, Bartok B, Firestein GS. Novel dual phosphoinositide 3-kinase-delta,gamma inhibitor: Potent anti-inflammatory effects and joint protection in models of rheumatoid arthritis. J Pharmacol Exp Ther. 2013 doi: 10.1124/jpet.113.205955. [DOI] [PubMed] [Google Scholar]
- 228.Elkabets M, et al. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Science translational medicine. 2013;5:196ra199. doi: 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Corcoran RB, et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Science translational medicine. 2013;5:196ra198. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.