

Abstract
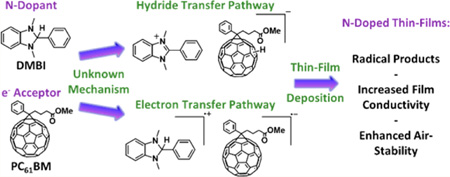
The discovery of air-stable n-dopants for organic semiconductor materials has been hindered by the necessity of high-energy HOMOs and the air sensitivity of compounds that satisfy this requirement. One strategy for circumventing this problem is to utilize stable precursor molecules that form the active doping complex in situ during the doping process or in a postdeposition thermal- or photo-activation step. Some of us have reported on the use of 1H-benzimidazole (DMBI) and benzimidazolium (DMBI-I) salts as solution- and vacuum-processable n-type dopant precursors, respectively. It was initially suggested that DMBI dopants function as single-electron radical donors wherein the active doping species, the imidazoline radical, is generated in a postdeposition thermal annealing step. Herein we report the results of extensive mechanistic studies on DMBI-doped fullerenes, the results of which suggest a more complicated doping mechanism is operative. Specifically, a reaction between the dopant and host that begins with either hydride or hydrogen atom transfer and which ultimately leads to the formation of host radical anions is responsible for the doping effect. The results of this research will be useful for identifying applications of current organic n-doping technology and will drive the design of next-generation n-type dopants that are air stable and capable of doping low-electron-affinity host materials in organic devices.
INTRODUCTION
The controllable doping of inorganic semiconductors has been critical to the development of modern electronics. Similarly, it is envisioned that the development of efficient doping methods for organic materials will aid in the improvement of existing organic electronic devices and in the design of new organic-based electronic devices.1 A particular challenge in developing n-dopants is that typically to dope electron-transport materials of interest by direct one-electron transfer, dopants must have low ionization energies, which leads to instability in air. The earlier reported n-type dopants for organic semiconductors were reducing metals such as Li, Na, and Cs.2,3 However, the applicability of these dopants is limited by their high reactivity and diffusivity and by the requirement that they be vacuum-deposited. Molecular dopants such as cobaltocenes4,5 and Ru(terpy)26 are anticipated to be more stable with respect to diffusion, but do not avoid the problem of air sensitivity. One approach to circumvent this problem is to couple the electron transfer from the dopant to the host material with a chemical reaction. The Leo group reported several precursor molecular n-dopants that were air-stable in a series of papers;1,7,8 these include the use of organic salts, which are assumed to decompose on heating to liberate the corresponding highly reducing organic radicals, and the use of hydride-reduced forms of organic cations, such as leuco crystal violet (LCV). Another recent approach to air-stable dopants involves coupling of electron transfer to the cleavage of dimeric organometallic sandwich compounds; these react with organic electron acceptors, including relatively weak acceptors such as 6,13-bis(triisopropylsilylethynyl)pentacene, to form monomeric cations and acceptor radical anions.9,10 Bao et al. has developed n-dopants based on reduced 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazoles (DMBI; Figure 1) and oxidized benzimidazolium (DMBI-I) salts as solution- and vacuum-processable n-type dopants, respectively.11,12 C60 films n-doped using DMBI-I exhibited conductivities as high as 5 S/cm. More recently, the tuning of the workfunction of graphene was accomplished by the careful addition of DMBI dopants.13 In order to design new and improved dopants in a rational manner, a detailed understanding of the doping kinetics and more generally the mechanism of the doping reaction would be valuable; however, with the exception of the organometallic dimers mentioned above,14 there has been very little study of the mechanism of solution-phase doping reactions. Furthermore, the hypothesized n-doping mechanism of DMBI dopants has been discussed in research reports following the initial communication,15 and comparative studies between DMBI dopants and electron transfer n-dopants have been reported.16 However, there is still a lack of understanding of the doping mechanism. Although reduced heterocycles have been studied extensively as hydride donors, the goal of the present study is to shed light on the mechanism by which free carriers are generated by organic hydride donors when doped into semiconducting hosts such as PC61BM (Figure 1).
Figure 1.

Structure of DMBI dopants and PC61BM. The naming system for the dopants is such that the phenyl substituent position and identity precede DMBI for all compounds where R3 = H (e.g., p-MeO–DMBI where R1 = H and R2 = MeO). The names of compounds for which R3 = D are prefixed by “d-“ (e.g., for R3 = D, R1 = H, R2 = NMe2 the name is d-NMe2-DMBI). In the particular cases of p-NMe2-DMBI and d-p-NMe2-DMBI, their names have been further abbreviated to N-DMBI and d-N-DMBI respectively.
In the initial report on n-doping PC61BM with 4-(1,3-dimethyl-2,3-dihydro-1H-benzoimidazol-2-yl)phenyl)-dimethylamine (p-NMe2-DMBI, shortened to N-DMBI) it was speculated that N-DMBI undergoes a C–H bond homolysis in an annealing step and that the resulting imidazoline radical functions as a single-electron donor (Figure 1). However, the mechanism was not studied. Furthermore, it was unclear whether the doping reaction began in solution or after the film was deposited and what effect a solution-phase reaction might have on the doping efficacy. To better understand the doping process of DMBI derivatives, the doping products with PC61BM were characterized by electron spin resonance, ESR, and UV–vis–near-IR spectroscopy. Here, we report that the rate law, kinetic isotope effect, and linear free-energy relationships together most strongly support the conclusion that the dopant functions as a hydride atom donor to the host material; this hydride transfer is then followed by an electron-transfer step between host molecules. Additionally, the energy-level alignment was probed by ultraviolet and inverse photoelectron spectroscopy (UPS and IPES, respectively) to better understand the energetics of the doping process and to confirm the n-doping by the shift of the Fermi level, EF, toward the LUMO. These mechanistic results are then discussed in the context of the doping of organic semiconductors. Importantly, the mechanism of DMBI dopants is distinct from that of electron transfer dopants, and the consequences of this distinction should be considered when employing DMBI or related hydride donor dopants, especially when the goal is to compare DMBI dopants to other n-dopants where electron transfer is not coupled to C–H bond cleavage. Furthermore, because the doping reaction for DMBI dopants is coupled to a hydride or hydrogen atom transfer, the energetics of the doping process cannot be solely determined by the alignment of the DMBI imidazoline SOMO and the host LUMO. Rather, the feasibility of doping depends on the free energy of electron transfer from DMBI• to the host and on the difference in the strengths of the DMBI–H bond and the C–H bond formed on hydride or hydrogen-radical reduction of the acceptor. The results of this study will clarify the ambiguity regarding the doping mechanism of DMBI dopants and help define the scope and limitations of current organic n-doping technology based upon organic materials in which electron transfer is coupled to cleavage of a C–H bond.
RESULTS
Mechanisms under Consideration
Several mechanisms have been reported in the literature for reductions by organic hydrides; these are shown in Figure 2 for the case of DMBI and PC61BM.17,18 We have performed experiments designed to help elucidate which mechanism(s) is(are) operative in generating anionic species when DMBI dopants react with organic semiconductors. To emphasize the –H group of DMBI dopants, we use DMBI-H here to represent DMBI in the reduced state, DMBI+ to represent the benzimidazolium form, and DMBI• to represent the neutral imidazoline radical. PC61BM and other fullerenes are known to undergo exergonic hydrogenation reactions with a wide variety of hydrogen-atom donors, and C60 is known to accept multiple hydrogen atoms.19,20 Thus, PC61BM hydrides, PC61BMH and higher order open- or closed-shell hydrides PC61BMHx, are likely products of the doping reaction; indeed, analogous derivatives of C60 have been detected using IR spectroscopy after photoinduced doping with LCV.7 For the doping reaction of PC61BM, DMBI+ and PC61BM•− are also reasonable products, as radical anions have been found in similarly n-doped thin films.8 All four reaction mechanisms proposed may yield the same final products, and evidence for their formation is presented in the following section. Another possibility is that a hydride is transferred to the carbonyl group of PC61BM rather than to the fullerene cage; however, as noted below, we have no evidence for the formation of the aldehyde expected to be formed on subsequent loss of methoxide from the ester–hydride adduct. Moreover, DMBI derivatives do not react with simple esters, such as benzyl acetate and ethyl acetate, and are capable of doping C60, where no ester group is present. Table 1 outlines the distinguishing characteristics of the four reaction mechanisms for the solution reaction of DMBI and PC61BM, based on the assumption that the first step in each case is rate-determining. Some of the expected results if the second steps of these mechanisms were rate determining are noted in the Table 1 footnotes. The assumption of a rate-determining first step is supported by the mechanistic data presented below and is consistent with general observations reported in the literature. A thorough discussion of the mechanisms and data justifying the assumption of a rate-determining first step will be presented below.
Figure 2.

Four reaction mechanisms for the n-doping reaction of PC61BM with DMBI derivatives. In each case the hydrogen reduced side product PCBHx will be a radical, as indicated, if x is odd, but a closed-shell species, when x is even.
Table 1.
Comparison of Proposed Mechanisms Shown in Figure 2 for the n-Doping Reaction of PC61BM with N-DMBI, Assuming in Each Case That the First Step Is Rate Determining
| mech | rate- determining step (RDS) |
obsd rate law | isotope effect (MH vs MD) |
influence of solvent dielectric (ε)a |
LFERb |
|---|---|---|---|---|---|
| I | electron transfer | d[DMBI-H]/dt = −k[DMBI-H] [PC61BM] | weak | potentially strong | σ, σ+ |
| II | H− transfer | d[DMBI-H]/dt = −k[DMBI-H] [PC61BM] | primary | potentially strong | σ, σ+ |
| III | H• transfer | d[DMBI-H]/dt = −k[DMBI-H] [PC61BM] | primary | unknown | σ• |
| IV | single molecule dissociation | d[DMBIH]/dt = −k[DMBI-H] | primary | weak | σ• |
ε is the solvent dielectric constant.
Linear free-energy relationship parameters: σ, widely used Hammett parameter; σ+, resonance parameter gauging stabilization of positive charge; σ•, a parameter gauging effects on radical stability. In all cases, a rate-determining second step will lead to a more complex rate law and different isotope effects. Use of the steady-state approximation leads to the following rate laws and isotope effects.
Mechanism I: rate = k1k2[DMBI-H][PCBM]2/(k−1[PCBM•−]); primary isotope effect (where k1 and k2 are rate constants for the first and second steps and k−1 is the rate constant for the reverse of the first step). Mechanism II: rate = k1k2[DMBI-H][PCBM]2/(k−1[DMBI+]); isotope effect reduced to a minor equilibrium effect on k1/k−1. Mechanism III: rate = k1k2[DMBI-H][PCBM]2/(k−1[PCBMH•]); isotope effect as for II. In mechanism IV, the steady-state approximation cannot be readily applied, since both DMBI• and the H• radical should be treated as highly reactive intermediates.
Reaction Products
The doping products were characterized by ESR and UV–vis–near-IR spectroscopy and by mass spectrometry. Thin films of PC61BM and C60 were deposited on the walls of 3 mm quartz ESR tubes by evaporation of deoxygenated toluene solutions under vacuum at 70 °C; solutions were prepared under a nitrogen atmosphere (O2 <0.3 ppm). Undoped C60 and PC61BM films and solutions exhibited no appreciable radical signal by ESR spectroscopy and no UV–vis–near-IR absorptions attributable to radical anions. However, codeposition of C60 with o-MeO-DMBI revealed a strong signal consistent with previous reports for the S = 1/2 C60 radical anion. The observed signal for doped C60 consisted of two superimposed signals one at 298 K at a g value of 1.999 with a peak-to-peak line width of 32.3 G and the other at a g value of 2.001 with a peak-to-peak line width of 4.1 G; various explanations for the double signals have been discussed in the literature.21,22 In the present case, the ratio of the two signals could be controlled by varying the mole percent of the dopant, which strongly suggests that the signals originate from distinct chemical environments that arise at low and high doping concentrations, respectively (Supporting Information, Figure S1). Similarly, the doping of PC61BM revealed an isotropic radical with a g value of 1.998 (Figure 3). Double integration of constant C60 mass samples with varying dopant concentrations were used to estimate the spin concentration by comparison with solutions of the stable nitroxyl radical (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) in toluene. At moderate concentrations a linear relationship between the spin density and doping concentration was revealed (Supporting Information, Figures S2 and S3). It is important to be aware that the measured spin concentration is not necessarily equivalent to the radical product yield because of possible spin-pairing, broadening, or quenching effects.23 However, the observation of a linear relationship between the dopant and spin concentrations indicates the formation of the latter may be attributed to the former, and the substantial amount of spin present implies that the fullerene radicals are not minor byproducts of the doping reaction. This conclusion was further substantiated by the UV–vis–near-IR spectra of doped solutions of PC61BM.
Figure 3.

X-band ESR spectra of undoped and o-MeO-DMBI doped PC61BM.
The black and red lines in Figure 4 are the UV–vis–near-IR spectra of N-DMBI and PC61BM in chlorobenzene, respectively. Both reactants have no significant absorption between 700 and 1200 nm. After N-DMBI and PC61BM were mixed in chlorobenzene, a new band with a maximum absorption at 1030 nm began to form. The spectrum of the mixed solution after reaction overnight at room temperature is shown as the dotted blue spectrum of Figure 4. The band shape of the blue spectrum is consistent with the previously reported absorption spectra of C60•− and PC61BM•− in solution.24 The identity of the 1030 nm band was further confirmed by its similarity to that seen when PC61BM was treated with [Ru(Cp*)(TEB)]2 (Cp* = pentamethylcyclopentadienyl; TEB = 1,3,5-triethylbenzene) in chlorobenzene; [Ru(Cp*)(TEB)]2 is a strong electron donor that cleanly reduces acceptors such as fullerenes without the formation of side products (Supporting Information, Figure S5).9,14
Figure 4.

UV–vis–near-IR spectra of N-DMBI, PC61BM, and N-DMBI/PC61BM at 298 K. Conditions: [N-DMBI] = 4.8 × 10−4 M, [PCBM] = 2.2 × 10−4 M, the mixture [N-DMBI] = 2.4 × 10−4 M and [PCBM] = 5.5 × 10−4 M.
At higher concentrations of DMBI dopants and PC61BM or C60 in toluene black insoluble precipitates form; these were shown to contain fullerene radical anions by ESR spectroscopy (Supporting Information, Figure S4). The composition of the precipitates that formed between N-DMBI and PC61BM or C60 was investigated by DART-TOF-MS and C,H,N elemental analysis. DART-TOF-MS identified a peak consistent with PC61BM in the negative-ion mode, and DMBI cations were identified in positive-ion ESI spectra. However, it should be stated that an m/z ratio corresponding to a particular species is not unambiguous evidence for the presence of that species in the doped film, since the species may arise by the ionization or decomposition of another species. For instance, the observed DMBI+ ion may in principle arise from DMBI-H, although this would suggest that DMBI coprecipitated with PC61BM without undergoing a reaction, which seems unlikely, given the UV–vis–near-IR and ESR results. Elemental analysis of the precipitates gave a C,N ratio consistent with a near 1:1 mixture of dopant cation to PC61BM, further supporting the assignment of these ions. (Anal. Calcd: C, 90.72; H, 2.99; N, 3.57; C:N ratio, 25.4. Found: C, 86.97; H, 2.73; N, 2.84; C:N, 30.62.) The observed C:N ratio differs from a perfect 1:1 salt possibly due to the presence of excess PC61BM or chlorobenzene trapped in the precipitate.
Negative ion MALDI-TOF spectra of the concentrated filtrate left from the precipitation reaction between N-DMBI and PC61BM show a strong peak at m/z 910.1053, again attributed to PC61BM. However, minor deviations in the isotopic envelope for this peak from that calculated for PC61BM and, more significantly, differences in the isotopic envelopes between samples formed using N-DMBI and its deuterated analogue d-N-DMBI may be due to the presence of PCBMHx/PCBMDx species (Supporting Information, Figures S6–S11). Overlap with the PC61BM isotopic envelope and the possibility of hydrogen-transfer reactions in the ionization process, however, preclude specific identification of the value(s) of x in this case. In addition, the mass spectra do not show alcohol or aldehyde products resulting from hydride reduction of the ester.
To summarize the product analysis data, radical anions of fullerenes were identified in the doping products in solution by UV–vis–near-IR and ESR spectroscopy, and in thin films by ESR spectroscopy. Quantitative ESR showed that the radicals are a major product and that the density of radicals is directly proportional to the dopant density at moderate to low doping concentrations. Mass spectra of the insoluble precipitate of the doping reaction contained both PC61BM and N-DMBI ions, and the C:N ratio as determined by C,H,N elemental analysis provides evidence for a near 1:1 stoichiometry for these two components. Evidence for PC61BM hydrides may be found by comparing the observed isotope ratio for the concentrated filtrate remaining after the doping reaction to the expected isotope distribution for PC61BM. Similarly, the Leo group has positively identified fullerene C–H bond stretches in the IR spectra of LCV-doped thin films of C60.7 The above product characterizations served as the starting point for identifying the reaction mechanism(s) by which DMBI dopes fullerenes to produce charge carriers.
Reaction Kinetics
Having already established the products of the doping reaction that lead ultimately to the generation of charge carriers in DMBI doped fullerene thin films, a thorough mechanistic understanding of this process was sought. The growth of the PC61BM•− absorbance band around 1030 nm in the solution reaction of DMBI derivatives and PC61BM was used to monitor the rate of the reaction, d[PC61BM•−]/dt. The initial reaction rates, (d[PC61BM•−]/dt)t=0, of mixtures of N-DMBI and PC61BM at various reactant concentrations were determined, and the results are presented in Table 2. It is clear that when doubling the concentration of either reactant, the initial reaction rate doubles. This is consistent with the following rate expression:
−d[PC61BM]/dt = k[DMBI-H][PC61BM]
Table 2.
Initial Reaction Rates for the Mixture Solution of N-DMBI and PC61BM in Chlorobenzene
| [N-DMBI] (M) | [PC61BM] (M) | initial rate (au)a | normalized rateb |
|---|---|---|---|
| 2.40 × 10 −3 | 5.50 × 10−4 | 8.01 × 10−4 | 8.31 |
| 1.20 × 10−3 | 5.50 × 10−4 | 3.72 × 10−4 | 3.86 |
| 2.40 × 10−3 | 2.75 × 10−4 | 3.86 × 10−4 | 4.00 |
| 1.20 × 10−3 | 2.75 × 10−4 | 1.79 × 10−4 | 1.86 |
| 1.20 × 10−3 | 1.38 × 10−4 | 9.64 × 10−5 | 1.00 |
Initial rate obtained by plotting the evolution of the intensity of the fullerene radical anion absorption with time in the first few percent of the reaction.
The normalized rate is the rate relative to that of the slowest reaction.
This expression is further supported by fitting of the temporal evolution of the PC61BM•− absorption, which is good when PC61BM is in excess. This rate expression suggests a reaction mechanism in which the first step of the reaction between N-DMBI and PC61BM is bimolecular, an observation that is consistent with mechanisms I–III, where the first step in each case is rate determining. This result is inconsistent with mechanism IV or with the more complicated rate laws that are predicted when the second step is rate determining (Table 1). In mechanisms I–III, the first step involves transfer of an electron, hydride anion, and hydrogen radical, respectively. As discussed earlier, a primary kinetic isotope effect (KIE) would be expected for the first step of mechanisms II and III but not for that of mechanism I. The synthesis of deuterated N-DMBI (d-N-DMBI) is discussed in detail in the Supporting Information. The reaction between PC61BM and d-N-DMBI indeed exhibited a strong kinetic isotope effect (kH/kD) of 8.6 at room temperature, implying that C–H bond cleavage and/or formation takes place during the rate-determining step. This result is inconsistent with mechanism I and suggests that DMBI dopants react first via donation of either a hydride or hydrogen atom. Furthermore, the observed primary KIE justifies the assumption of a rate-determining first step for mechanisms II–IV.
A series of para-substituted DMBI compounds were synthesized and reacted with PC61BM in chlorobenzene at 85 °C (Table 3). The rate constants of the reaction between the various substituted DMBI derivatives and PC61BM correlate strongly with both Hammett σ and σ+ parameters25 but do not strongly correlate with any radical σ scales (Figure 5).26 The ρ value for the σ scale was −1.41, which indicates that in the transition state of the reaction significant positive charge develops on the C-2 of the imidazoline ring. The magnitude of the observed ρ value is comparable to those reported for other hydride-transfer processes.27 The lack of a correlation with any radical σ scale does not completely rule out mechanism II, as these scales only correlate with nonpolar homolysis reactions. Some radical hydrogen abstractions correlate well with polar LFER scales as a consequence of the polar nature of the transition state when proton and electron transfers are asynchronous.26 In the present case an asynchronous electron/proton net H• transfer in which the electron is largely transferred in the transition state, but the proton is not, would not lead to development of a large positive charge on C-2 in the transition state and only a weak dependence of the rate on the identity of the para substituent (i.e., a small negative value of ρ) would be expected. In the reverse case, where proton transfer is followed by electron transfer, a negative charge would be developed on C-2 and a positive value of ρ would be expected. Only a H• transfer approximated as asynchronous hydride transfer/reverse electron transfer could potentially give the observed LFER, and this seems unlikely, given the relatively poor electron-donating and -accepting abilities expected for PCBMH− and DMBI+, respectively.
Table 3.
LFER Values Obtained from the Literature25,28 and Plotted against the Reaction Constant Ratios for Various p-X-N-DMBI Compounds for the Doping Reaction with PC61BM in Chlorobenzene (ClBz) at 85 °C
| log(kX/kH) | |||
|---|---|---|---|
| X group | σ | excess PC61BM in ClBz | excess DMBI in ClBz |
| H | 0.00 | 0.00 | 0.00 |
| NO2 | 0.78 | −1.53 | −2.10 |
| MeO | −0.27 | 0.30 | 0.43 |
| F | 0.06 | −0.30 | −0.25 |
| Cl | 0.23 | −0.40 | −0.32 |
| NMe2 | −0.83 | 0.81 | 0.87 |
| CN | 0.66 | −1.00 | −0.99 |
| CH3 | −0.17 | 0.21 | 0.37 |
| CF3 | 0.54 | −0.89 | −0.78 |
Figure 5.

Hammett plots for the reaction between PC61BM and para-substituted DMBI derivatives in chlorobenzene.
If the rate-determining step involves formation of ionic intermediates, a strong dependence of the reaction rate on the solvent polarity would be expected, although presumably polar radical-transfer reactions of the type mentioned above could also exhibit polarity dependence. Unfortunately, few solvents are capable of solvating both starting materials and the DMBI+/PC61BM•− product, thereby limiting our ability to study solvent effects for this reaction. The rate constants in chlorobenzene and toluene were found to be similar; however, both of these solvents have rather low polarities (ε = 5.62 and 2.38, respectively). Consequently, our results do not distinguish between hydride and hydrogen-atom transfer. However, the implications of our results are the same regardless; namely, that acceptor hydrogenation is important for the action of DMBI and related dopants, and thus the thermodynamics and kinetics of the doping process cannot be understood by considering only the dopant ionization energy and host electron affinity, as is the case for dopants that operate through simple one-electron transfer.
Arrhenius and Eyring plots based on variable-temperature reaction constants are displayed in Figure 6. The parameters obtained from the Arrhenius and Eyring plots are given in Table S1 (Supporting Information). Significantly, the negative value of ΔS‡ (−92 J mol−1 K−1 for N-DMBI/PC61BM in chlorobenzene) is consistent with an associative bimolecular reaction. The free energy for direct electron transfer from N-DMBI to PC61BM, as in mechanism I, was estimated to be 1.18 eV at 300 K from electrochemical data (Supporting Information, Table S2 and Figures S12–S16), meaning that ΔG‡ for such a reaction should be somewhat larger. However, the value of ΔG‡ extracted from the variable-temperature rate data is 0.63 eV, significantly lower than the free energy for electron transfer, which is inconsistent with the reaction proceeding by mechanism I.
Figure 6.

Arrhenius and Eyring plots of reaction constants at variable temperatures for the reaction of N-DMBI and PC61BM in chlorobenzene.
Doping Energetics
To confirm that charge carriers are generated in thin films by the reaction between PC61BM and DMBI derivatives, UPS and IPES spectra (Supporting Information, Figure S17) were recorded for PC61BM spin-coated on ITO, undoped or doped with 1 wt % o-MeO-DMBI. The ionization energy (IE), electron affinity (EA), and single-particle gap (IE–EA) for PC61BM were found to be 6.08, 3.7, and 2.4 eV, respectively, in good agreement with previously reported values.29 Upon doping PC61BM with 1 wt % of o-MeO-DMBI, the line shapes of the spectra did not substantially change except for a shift toward higher binding energy. This is equivalent to an upward movement of EF in the gap, indicative of n-doping, and the generation of free electrons. This conclusion is further substantiated by the observed conductivity increase that arises when PC61BM is doped by DMBI derivatives. The results are summarized in Figure 7. Most relevant to the current study, the energy difference between the bottom edge of the LUMO and EF, measured with the combination of UPS and IPES, decreases from 0.72 to 0.21 eV, which corresponds to a roughly 500 meV shift of EF. It should be noted that the initial EF position in the undoped film is not particularly significant, as it depends on the nature and work function of the substrate; the significance lies in the movement of EF upon doping and its ultimate position in the gap of the semiconductor. The ionization energy of o-MeO-DMBI was also measured on Au and was found to be 4.26 eV, consistent with its air stability (Supporting Information, Figure S18).
Figure 7.

Energy level alignment of undoped and 1 wt % o-MeO-DMBI doped PC61BM.
DISCUSSION
For a chemical transformation with competing reaction mechanisms, different mechanisms might dominate under specific reaction conditions. For example, different mechanisms are observed for the reaction between TIPS-pentacene and 1,2,3,4,5-pentamethylrhodocene dimer when the reactant concentration ratio is changed from <0.1 to >10.14 The initial reaction rates for various p-X-DMBI and PC61BM were measured at two extreme concentration ratios, with either reactant in large excess (Table 2). No evidence for such a change in the mechanism was found for the reaction between p-X-DMBI and PC61BM. This result also shows that the hydrogenated fullerene products are not interfering with the interpretation of the initial reaction rates.
Fullerenes are capable of accepting as many as six electrons, which may complicate the interpretation of kinetic data. To control the possibility of multiple reductions, PC61BM was employed in large excess such that the anion product can only be PC61BM•− (any PC61BM2− would comproportionate with remaining PC61BM to yield PC61BM•−). Additionally, the same absorbance features are present when PC61BM is in large excess and when the dopant is in large excess, further confirming that the same radical anion product is formed, although different hydrogen-reduced fullerenes may be formed. Furthermore, the absorption spectra in this study compared well with known spectra for fullerene radical anions but not with that of fullerene dianions.24
It is possible that the DMBI dopants operate via a different mechanism depending on the host material used. For instance, Cheng and co-workers oxidized DMBI-H with Fe3+ and confirmed the formation of DMBI-H•+ by an ESR and deuterium labeling study.17 Thus, electron transfer may precede C–H bond homolysis when strongly electron-accepting hosts (oxidants) that have low affinities for hydride or hydrogen atoms are employed. Electron transfer was found to precede C–H bond homolysis for the reaction between the related LCV dopant and the strong electron acceptor 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ).30 These results demonstrate that the doping mechanism may depend on the dopant/host combination, and it is unlikely that a single mechanism will account for all dopant/host combinations.
The dependence of the reaction rate on both the host and dopant concentrations has important implications for the utility of DMBI dopants, because this implies that the imidazoline radical does not independently form. The relatively high ionization energy of o-MeO-DMBI makes direct electron transfer unfavorable to many common organic semiconductors, and a drastic variation in the ionization energy of the DMBI dopants reported herein is unlikely. For some acceptor materials, such as fullerenes, a reaction is coupled to the electron transfer event, thereby enabling the n-doping to proceed. In such cases the feasibility of doping depends on the free energy of electron transfer from DMBI• to the host and on the difference in the strengths of the DMBI-H and the hydrogenated acceptor C–H bonds. However, with other materials, such as TIPS-pentacene, it is evident from solution and thin film absorption spectra that the dopant is not actually transferring an electron to the host material, possibly because the barrier to break the C–H bond is too high to initiate the n-doping reaction and/or TIPS-pentacene is not a sufficiently strong hydride acceptor; instead, it is proposed that only trap sites with higher electron affinities are filled. Notably, trap filling may be advantageous for OTFT applications where a conductive film is not desired and where trap filling by a dopant has been shown to enhance mobilities and permit control over the threshold voltage.31–34 This attribute of DMBI dopants was exploited in another work, where it was found that TIPS-pentacene can be doped with o-MeO-DMBI to obtain high-mobility n-channel transistors without increasing the bulk conductivity, as is commonly observed when doping organic transistors.35
CONCLUSION
DMBI dopants have been shown to effectively n-dope fullerenes, carbon nanotubes, and graphene to give highly conductive electron-transport materials. They have also been used to improve the air stability of n-channel OTFTs, to switch the transport characteristics of p-channel materials in OTFT configurations, and have been employed in electron-transport layers for inverted solar cells.36 The utility that has already been demonstrated for DMBI dopants provides the motivation to study the fundamental reaction mechanism by which they generate charge carriers; the goal of this research was to illuminate this mechanism and to spur further developments in the field of air-stable molecular n-dopants. It was found that PC61BM and C60 spontaneously react with DMBI dopants to form fullerene radical anions, stable organic cations, and hydrogen-reduced fullerenes. The doping of the PC61BM films was confirmed by UPS and IPES spectroscopy. Initial rates of the reaction of substituted and deuterium-labeled DMBI dopants were compared at various concentrations of the dopant and host to determine the reaction order and nature of the rate-determining step. The most probable reaction mechanism involves a bimolecular hydride transfer from the dopant to PC61BM in the rate-determining step, although a hydrogen-atom transfer with polar character cannot be definitively ruled out. Electron transfer to form the detected PC61BM radical anions must succeed the hydrogen/hydride atom transfer event, and most simply would occur via electron transfer between the hydride-reduced PC61BM and unreduced PC61BM. It is important to recognize that the mechanism of DMBI dopants is distinct from that of electron-transfer dopants, and this distinction should be considered when employing DMBI or related hydride-donor dopants. Most importantly, the efficacy of doping is inadequately described by the offset between the ionization energy of the DMBI-H donor or DMBI• radical and the electron affinity of the host material, because DMBI-H dopants act first as hydride/hydrogen-atom donors. However, our results do not exclude the possibility of different mechanisms in other host materials.
EXPERIMENTAL SECTION
General Methods
Spectrophotometric grade (99.9%) chlorobenzene was purchased from Alfa Aesar, dried over calcium hydride, distilled, deoxygenated by three cycles of freeze–pump–thaw, and stored in a nitrogen-filled glovebox. Toluene was passed through an alumina column, refluxed over phosphorus pentoxide, distilled, and deoxygenated by the freeze–pump–thaw method prior to use. 1H and 13C NMR spectra were recorded in deuterated chloroform or dimethyl sulfoxide on Varian 500, 400, or 300 MHz NMR spectrometers. The chemical shifts (δ) are reported in parts per million (ppm) relative to a tetramethylsilane (TMS, 0.00 ppm) internal standard. Electrospray ionization mass spectrometry (ESI-MS) and direct analysis in real time mass spectrometry (DART-TOF-MS) were performed at the University of Florida mass spectrometry facilities. Elemental analysis was performed by Robertson Microlit.
Electron Spin Resonance (ESR) Detection Experiments
The ESR measurements were performed on an X-band Bruker EMX operating at 9.44 GHz with a Bruker SHQ cavity with variable temperature control. The spectra were recorded under nonsaturating microwave power conditions. The magnitude of modulation was chosen to optimize the resolution and the signal-to-noise ratio of the observed spectra. The g value was calibrated against an external 2,2-diphenyl-1-picrylhydrazyl (DPPH) standard. Spin quantification experiments were performed on thin films of doped C60 prepared by evaporation of toluene solutions on the walls of the ESR tubes. Double integrals of the spectra were compared to those of external (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) standards in toluene.
Ultraviolet–Visible–/Near-Infrared (UV–Vis–Near-IR) Spectroscopy Measurements
The samples for UV–vis–near-IR measurements were prepared in a glovebox and then transferred into polytetrafluoroethylene (PTFE) stopcock sealed quartz cuvettes (175–2700 nm) with path lengths of 1 mm. The cuvettes were then taken to a Varian Cary 5E ultraviolet–visible–near-infrared spectrometer to monitor the reaction progress. A Quantum Northwest TC 125 temperature controller was used to control the temperature of the sample cuvette holder.
Ultraviolet and Inverse Photoelectron Spectroscopy (UPS and IPES)
For the photoelectron spectroscopy measurements the spin-cast samples were transferred into an ultrahigh-vacuum system. Samples containing the n-dopant were transferred via a transport system without air exposure, while the more stable films were exposed to air for a few seconds during the loading process. For the UPS measurements, either the He I line (21.22 eV) or the He II line (40.8 eV) from a discharge lamp were used with an experimental resolution of 150 meV. The IPES measurements were performed in the same setup in the isochromat mode at an energy resolution of 400 meV using a setup described elsewhere.37
Preparation of α-Deuteriodimethylaminobenzaldehyde
A small dry argon-flushed vial was charged with deuterated dimethylformamide (260 µL, 3.3 mmol) and then cooled to 0 °C. To this was slowly added phosphoryl chloride (150 µL, 1.65 mmol) dropwise, after which the solution was warmed to room temperature. Dimethylaniline (210 µL, 1.65 mmol) was then added, and the solution was heated to 90 °C for 2 h with stirring under argon. The reaction mixture was then diluted with ice and neutralized to pH of 6–8 with 10 wt % aqueous sodium acetate solution. The solids that formed were collected by vacuum filtration and washed with water until the color cleared. The crude product was stored under argon at −20 °C and used without further purification.
General Procedure for Preparation of Benzimidazoles
A 20 mL vial was charged with N,N′-dimethyl-o-phenylenediamine (136 mg, 1.00 mmol) in methanol (2 mL); to this was added the appropriate aromatic aldehyde (1.00 mmol). One drop of glacial acetic acid was then added, and the solution was sonicated at room temperature until precipitation was observed. The crude product was then collected by vacuum filtration and recrystallized from methanol/H2O to give the corresponding pure 1,3-dimethyl-2-arylbenzimidazoles.17
Supplementary Material
ACKNOWLEDGMENTS
B.D.N. gratefully acknowledges the National Defense Science and Engineering Graduate Research Fellowship and the National Science Foundation for a Graduate Research Fellowship. This research was also funded by the National Science Foundation Materials Network Program (NSF-DMR-1209468) and Air Force Office of Scientific Research (FA9550-12-1-0190). The work at Georgia Tech was supported by the National Science Foundation through the Science and Technology Center Program (DMR-0120967) and by the Office of Naval Research (N00014-11-1-0313). The work at Princeton University was supported by the National Science Foundation (DMR-1005892). The work at the University of California–Santa Cruz was supported by the National Institutes of Health (NIH grant GM065790). We thank Dr. Swagat Mohapatra for synthesis of the [Ru(Cp*)(TEB)]2 used.
Footnotes
ASSOCIATED CONTENT
S Supporting Information
Text, figures, and tables giving a complete characterization summary for all synthesized materials (13C and 1H NMR, C,H,N elemental analysis, high-resolution mass spectra), quantitative electron spin resonance ESR spectra, mass spectra for doping products, UV–vis–near-IR comparison to [Ru-(Cp*)(TEB)]2, electrochemical data, Arrenhius and Eyring parameters, UPS and IPES data for PC61BM, and o-MeO-DMBI, and 1H and 13C NMR spectra for the synthesized compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Lüssem B, Riede M, Leo K. Phys. Status Solidi A. 2013;210:9–43. [Google Scholar]
- 2.Schneider WG, Helfrich W. Phys. Rev. Lett. 1965;14:229–232. [Google Scholar]
- 3.Kido J, Matsumoto T. Appl. Phys. Lett. 1998;73:18–21. [Google Scholar]
- 4.Chan CK, Kahn A, Zhang Q, Barlow S, Marder SR. J. Appl. Phys. 2007;102:014906. [Google Scholar]
- 5.Chan CK, Zhao W, Barlow S, Marder S, Kahn A. Org. Electron. 2008;9:575–581. [Google Scholar]
- 6.Walzer K, Maennig B, Pfeiffer M, Leo K. Chem. Rev. 2007;107:1233–1271. doi: 10.1021/cr050156n. [DOI] [PubMed] [Google Scholar]
- 7.Li F, Werner A, Pfeiffer M, Leo K, Liu X. J. Phys. Chem. B. 2004;108:17076–17082. [Google Scholar]
- 8.Li F, Pfeiffer M, Werner A, Harada K, Leo K, Hayashi N, Seki K, Liu X, Dang X-D. J. Appl. Phys. 2006;100:023716. [Google Scholar]
- 9.Qi Y, Mohapatra SK, Bok Kim S, Barlow S, Marder SR, Kahn A. Appl. Phys. Lett. 2012;100:083305. [Google Scholar]
- 10.Guo S, Kim SB, Mohapatra SK, Qi Y, Sajoto T, Kahn A, Marder SR, Barlow S. Adv. Mater. 2012;24:699–703. doi: 10.1002/adma.201103238. [DOI] [PubMed] [Google Scholar]
- 11.Wei P, Oh JH, Dong G, Bao Z. J. Am. Chem. Soc. 2010;132:8852–8853. doi: 10.1021/ja103173m. [DOI] [PubMed] [Google Scholar]
- 12.Wei P, Menke T, Naab BD, Leo K, Riede M, Bao Z. J. Am. Chem. Soc. 2012;134:3999–4002. doi: 10.1021/ja211382x. [DOI] [PubMed] [Google Scholar]
- 13.Wei Peng, Liu N, Lee HR, Adijanto E, Ci L, Naab BD, Zhong JQ, Park J, Chen W, Cui Y, Bao Z. Nano Lett. 2013;13:1890–1897. doi: 10.1021/nl303410g. [DOI] [PubMed] [Google Scholar]
- 14.Guo S, Mohapatra SK, Romanov A, Timofeeva TV, Hardcastle KI, Yesudas K, Risko C, Brédas J-L, Marder SR, Barlow S. Chem. Eur. J. 2012;18:14760–14772. doi: 10.1002/chem.201202591. [DOI] [PubMed] [Google Scholar]
- 15.Lu M, Nicolai HT, Wetzelaer G-JaH, Blom PWM. Appl. Phys. Lett. 2011;99:173302. [Google Scholar]
- 16.Menke T, Wei P, Ray D, Kleemann H, Naab BD, Bao Z, Leo K, Riede M. Org. Electron. 2012;13:3319–3325. [Google Scholar]
- 17.Zhu X-Q, Zhang M-T, Yu A, Wang C-H, Cheng J-P. J. Am. Chem. Soc. 2008;130:2501–2516. doi: 10.1021/ja075523m. [DOI] [PubMed] [Google Scholar]
- 18.Fukuzumi S, Nishizawa N, Tanaka T. J. Org. Chem. 1984;49:3571–3578. [Google Scholar]
- 19.Henderson CC, Cahill PA. Science. 1993;2790:1–3. [Google Scholar]
- 20.Jin C, Hettich R, Compton R, Joyce D, Blencoe J, Burch T. J. Phys. Chem. 1994;98:4215–4217. [Google Scholar]
- 21.Watanabeb T. Synth. Met. 1999;103:2374–2375. [Google Scholar]
- 22.Hwang YL, Yang CC, Hwang KC. J. Phys. Chem. A. 1997;101:7971–7976. [Google Scholar]
- 23.Chen S-G, Branz HM, Eaton SS, Taylor PC, Cormier RA, Gregg BA. J. Phys. Chem. B. 2004;108:17329–17336. [Google Scholar]
- 24.Konarev DV, Drichko NV, Graja A. J. Chem. Phys. 1998;95:2143–2156. [Google Scholar]
- 25.C H, Leo A, RW T. Chem. Rev. 1991;91:165–195. [Google Scholar]
- 26.Heberger K. J. Phys. Org. Chem. 1994;7:244–250. [Google Scholar]
- 27.Zhu X-Q, Li X-T, Han S-H, Mei L-R. J. Org. Chem. 2012;77:4774–4783. doi: 10.1021/jo3005952. [DOI] [PubMed] [Google Scholar]
- 28.Dust JM, Arnold DR. J. Am. Chem. Soc. 1983;105:1221–1227. [Google Scholar]
- 29.Akaike K, Kanai K, Yoshida H, Tsutsumi J, Nishi T, Sato N, Ouchi Y, Seki K. J. Appl. Phys. 2008;104:023710. [Google Scholar]
- 30.Yamamoto S, Sakurai T, Yingjin L, Sueishi Y. Phys. Chem. Chem. Phys. 1999;1:833–837. [Google Scholar]
- 31.Kim JH, Yun SW, An B-K, Han YD, Yoon S-J, Joo J, Park SY. Adv. Mater. 2013;25:719–724. doi: 10.1002/adma.201202789. [DOI] [PubMed] [Google Scholar]
- 32.Abe Y, Hasegawa T, Takahashi Y, Yamada T, Tokura Y. Appl. Phys. Lett. 2005;87:153506. [Google Scholar]
- 33.Soeda J, Hirose Y, Yamagishi M, Nakao A, Uemura T, Nakayama K, Uno M, Nakazawa Y, Takimiya K, Takeya J. Adv. Mater. 2011;23:3309–3314. doi: 10.1002/adma.201101027. [DOI] [PubMed] [Google Scholar]
- 34.Olthof S, Singh S, Mohapatra SK, Barlow S, Marder SR, Kippelen B, Kahn A. Appl. Phys. Lett. 2012;101:253303. [Google Scholar]
- 35.Naab BD, Himmelberger S, Diao T, Vandewal K, Wei P, Lussem B, Salleo A, Bao Z. Adv. Mater. 2013;25:4663–4667. doi: 10.1002/adma.201205098. [DOI] [PubMed] [Google Scholar]
- 36.Cho N, Yip H-L, Davies JA, Kazarinoff PD, Zeigler DF, Durban MM, Segawa Y, O’Malley KM, Luscombe CK, Jen AK-Y. Adv. Energy Mater. 2011;1:1148–1153. [Google Scholar]
- 37.Wu CI, Hirose Y, Sirringhaus H, Kahn A. Chem. Phys. Lett. 1997;272:43–47. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.