

Abstract
Aldehyde dehydrogenase 1A1 (ALDH1A1) is a member of a superfamily of detoxification enzymes found in various tissues that participate in the oxidation of both aliphatic and aromatic aldehydes. In the brain, ALDH1A1 participates in the metabolism of catecholamines including dopamine (DA) and norepinephrine, but is uniquely expressed in a subset of dopaminergic (DAergic) neurons in the ventral mesencephalon where it converts 3,4-dihydroxyphenylacetaldehyde, a potentially toxic aldehyde, to 3,4-dihydroxyphenylacetic acid, a non toxic metabolite. Therefore, loss of ALDH1A1 expression could be predicted to alter DA metabolism and potentially increase neurotoxicity in ventral mesencephalic DA neurons. Recent reports of reduced levels of expression of both Aldh1a1 mRNA and protein in the substantia nigra (SN) of Parkinson's disease patients suggest possible involvement of ALDH1A1 in this progressive neurodegenerative disease. The present study used an Aldh1a1 null mouse to assess the influence of ALDH1A1 on the function and maintenance of the DAergic system. Results indicate that the absence of Aldh1a1 did not negatively affect growth and development of SN DA neurons nor alter protein expression levels of tyrosine hydroxylase, the DA transporter or vesicular monoamine transporter 2. However, absence of Aldh1a1 significantly increased basal extracellular DA levels, decreased KCl and amphetamine stimulated DA release and decreased DA re-uptake and resulted in more tyrosine hydroxylase expressing neurons in the SN than in wildtype animals. These data suggest that in young adult animals with deletion of the Aldh1a1 gene there is altered DA metabolism and dysfunction of the DA transporter and DA release mechanisms.
Keywords: Aldehyde dehydrogenase, Microdialysis, Dopamine, Mouse
1. Introduction
Aldehyde dehydrogenase 1 (ALDH1A1) and aldehyde dehydrogenase 2 (ALDH2) are members of a diverse family of enzymes that participates in the oxidation of a variety of aldehydes. In the brain, ALDH1A1 is significantly involved in the metabolism of biogenic aldehydes, norepinephrine, dopa-mine (DA) and gamma-aminobutyric acid (Maring et al., 1985). 3,4-Dihydroxyphenylacetaldehyde (DOPAL), a potentially toxic metabolite of DA is formed by the oxidative deamination of DA catalyzed by monoamine oxidases (Burke et al., 2003). DOPAL, an intermediate in the degradation of DA, is then oxidized to 3,4-dihydroxyphenylacetic acid (DOPAC) by ALDH1A1 or reduced to 3,4-dihydroxyphenylethanol (DOPET) by aldose/ aldehyde reductase. Potentially damaging oxygen radicals are formed by this process (Adams et al., 2001). Although impaired DA transmission may influence ALDH activity and/or changes in ALDH-mediated metabolism may affect DA levels in nerve cell bodies and terminal fields in the basal ganglia and limbic system (Galter et al., 2003), data directly supporting this are lacking.
While ALDH1A1 is strongly and specifically expressed in ventral mesencephalic DA neurons in rodents (Westerlund et al., 2005) and man (Galter et al., 2003), its functional significance to the DA system and its possible relevance to Parkinson's disease (PD) is incompletely understood. mRNA encoding cytosolic ALDH1A1 is highly expressed in human substantia nigra (SN) and ventral tegmental area (VTA) dopaminergic (DAergic) neurons, with the virtual absence of this gene in neighboring non-DAergic cells (Galter et al., 2003). In brains from PD patients, there was a markedly lower expression of Aldh1a1 mRNA in tyrosine hydroxylase (TH)-positive and DA transporter (DAT)-positive neurons in the SN, compared to non-PD controls. In contrast, Aldh1a1 expression was not significantly decreased in TH- or DAT-positive neurons in the VTA in most PD patients (Galter et al., 2003). Based on these data, it was suggested that a down-regulation of Aldh1a1 in SN DA neurons in PD might reflect a type of compensatory response of these cells to decrease the rate of DA degradation. However, reduced ALDH1A1 expression in the SN could also contribute to the development of PD rather than be a consequence of PD. With reduced ALDH1A1 expression, it is possible that the degradation of DA would be slowed at the level of DOPAL. An accumulation of DOPAL could be potentially toxic to DA neurons and could render these neurons more susceptible to aldehyde toxicity and degeneration.
Although current evidence suggests that ALDH1A1 is important for DA neuron function, there is little detail available on this, primarily due to absence of an appropriate animal model for study. The present investigation characterizes the influence of ALDH1A1 on DAergic function using an Aldh1a1 (Raldh1) null mouse model (Fan et al., 2003) utilizing in vivo microdialysis and post-mortem evaluations of pre-and post-synaptic DAergic markers.
2. Results
All mice used were genotyped by PCR to confirm the presence of the Neo cassette which deleted the expression of the ALDH1A1 protein in the Aldh1a1−/− animals (Fig. 1A). Western blotting was used to visualize the effective deletion of ALDH1A1 protein in the striatum (Fig. 1B) and immunohistochemistry verified the lack of ALDH1A1 expression in neurons in the SN in Aldh1a1−/− animals (Fig. 1C).
Fig. 1.

Expression of Aldh1a1 mRNA and protein in Aldh1a1−/− and wildtype (WT) mice. (A) Representative genotyping gel. Aldh1a1−/− animals did not contain a functional copy of the Aldh1a1 gene, but were positive for the Neo insert used to disrupt gene expression (NEO cassette, 600 bp; Aldh1a1, 250 bp). (B) Western blot analysis of ALDH1A1 protein in the striatum showed a significant loss of ALDH1A1 expression in the Aldh1a1−/− mice compared to normal expression levels in WT mice. Loading controls (using GAPDH) for all lanes are shown. (C) Immunohistochemical analysis of ALDH1A1 expression in the substantia nigra pars compacta showed significant loss ALDH1A1-immunoreactivity when compared to WT animals.
2.1. Microdialysis
Basal extracellular fluid (ECF) DA levels in the striatum in awake Aldh1a1−/− mice (23.39±4.43 nM) were significantly higher than those observed in wildtype animals (7.59±0.86 nM; t(12)=3.023, p=0.0106) (Fig. 2A). Perfusion of the probe with 120 mM KCl resulted in significant release of DA in both Aldh1a1−/− animals (402.0 ± 71.49 nM) and wildtype control animals (383.4 ± 100.5 nM). However, Aldh1a1−/− animals had significantly less KCl-stimulated DA release (67%) than did wildtype animals (t(12)=2.814, p=0.0156), relative to basal ECF DA levels (Fig. 2B). Perfusion of the probe with 50 μM D-amphetamine also resulted in significant release of DA in both Aldh1a1−/− animals (232.7±75.85 nM) and wildtype control animals (218.4±48.05 nM). There was significantly less (68%) amphetamine-stimulated DA release in Aldh1a1−/− animals compared to wildtype animals (t(12)=2.857, p=0.0144), relative to basal ECF DA levels (Fig. 2C).
Fig. 2.

Analysis of dopamine (DA) levels in the striatum of freely moving awake mice by in vivo microdialysis. (A) Basal extracellular levels of DA were significantly higher in the Aldh1a1−/− mice than wildtype (WT) mice. (B) KCl-stimulated DA release was significantly reduced in Aldh1a1−/− mice compared to WT mice. (C) Amphetamine stimulated DA release was significantly reduced in Aldh1a1−/− mice compared to WT mice. *p<0.05 vs. wildtype. (n=8 and 6 respectively for Aldh1a1−/− and WT).
2.2. Tissue DA levels
There were no statistically significant differences in the levels of striatal DA measured in post-mortem tissue from Aldh1a1−/− or wildtype animals (Fig. 3A). Aldh1a1−/− animals had a significantly lower level of striatal DOPAC when compared to wildtype animals (t(13)=2.257, p=0.0419; Fig. 3B). However, an index of DA turnover (DOPAC/DA ratio) showed no statistically significant differences between Aldh1a1−/− animals and wild-type controls (Fig. 3C).
Fig. 3.
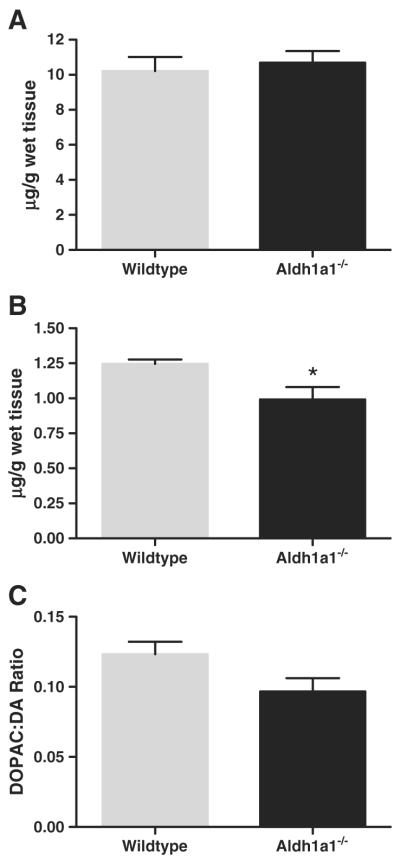
Comparison of tissue dopamine (DA) and metabolite levels in the striatum of Aldh1a1−/− and wildtype mice. (A) Striatal DA levels in Aldh1a1−/− and wildtype (WT) mice were not significantly different. (B) The level of 3,4-dihydroxyphenylacetic acid (DOPAC) in the Aldh1a1−/− mouse was significantly lower than in WT mice (*p<0.05). (C) Dopamine turnover was reduced in Aldh1a1−/− mice when compared to WT, but the difference was not statistically significant. (n=10 and 6 respectively for Aldh1a1−/− and WT).
2.3. Tyrosine hydroxylase positive neuron number
Stereological estimates of the number of tyrosine hydroxylase-immunoreactive (TH-IR) neurons in the substantia nigra pars compacta indicated more TH-IR neurons in Aldh1a1−/− animals (5377±52.5) compared to wildtype controls (4590±145.9; t(6)= 5.075, p=0.0023; Fig. 4). Stereological estimates of Nissl stained neurons in the substantia nigra pars compacta were not significantly different between Aldh1a1−/− animals (5929± 501.3) compared to wildtype controls (5886±286.0; Fig. 4— all values are mean±SEM).
Fig. 4.

Photomicrographs of ALDH1A1 and tyrosine hydroxylase immunoreactivity in the substantia nigra of Aldh1a1−/− and wildtype (WT) mice. All images were taken at 4× magnification, with higher 40× magnification inset for both tyrosine hydroxylase photomicrographs. (n=6 and 6 respectively for Aldh1a1−/− and WT).
2.4. Striatal protein analysis
Striatal tissue was analyzed by Western blot to assess the levels of TH, DAT, and vesicular monoamine transporter 2 (VMAT2) protein. Expression levels of TH in Aldh1a1−/− animals (0.80±0.04 o.d.) were not significantly different from wildtype animals (0.87±0.04 o.d.). Expression levels of DAT also showed no statistically significant change in Aldh1a1−/− animals compared to wildtype controls (1.96±0.08 o.d. vs. 1.91±0.06 o.d. respectively). Similarly, VMAT2 expression levels were not statistically significantly different in Aldh1a1−/− animals compared to wildtype controls (0.94±0.06 o.d. vs. 0.75±0.02 o.d. respectively) although this trended towards significance (p=0.052) (Fig. 5, all values are stated as mean±SEM.).
Fig. 5.

Striatal protein expression in Aldh1a1−/− and wildtype (WT) mice. There were no significant differences in any of the analyzed proteins between the Aldh1a1−/− and WT mice. TH=tyrosine hydroxylase; DAT=dopamine transporter; VMAT2=vesicular monoamine transporter 2. GAPDH or β-actin was used as loading controls. (n=6 and 6 respectively for Aldh1a1−/− and WT).
2.5. Dopamine uptake
Dopamine uptake in striatal homogenates from Aldh1a1−/− animals, measured during the linear phase, was significantly slowed compared to uptake measured in striatal homogenates from wildtype controls. Striatal DA uptake in Aldh1a1−/− striatal homogenates was 30.29%±5.72 less than that observed in wildtype controls (t(12)=4.672, p=0.0005) (Fig. 6).
Fig. 6.
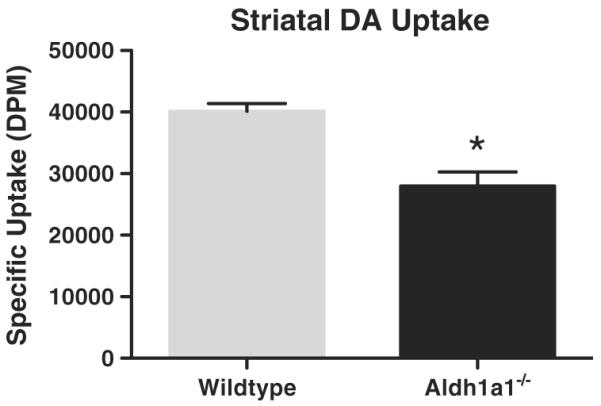
Analysis of striatal dopamine uptake in Aldh1a1−/− and wildtype (WT) mice. There was a significant decrease in DA uptake in striatal homogenates from Aldh1a1−/− animals compared to wildtype control animals. *p<0.001. (n=7 and 7 respectively for Aldh1a1−/− and WT).
3. Discussion
The results of the present study show that absence of ALDH1A1 results in higher than normal extracellular fluid levels of DA in the striatum and alterations in DA release in response to KCl or amphetamine stimulation. Tissue levels of DA, as well as TH, DAT and VMAT2 protein expression in the striatum were not affected by Aldh1a1 gene deletion. Dopamine uptake rate was significantly reduced in Aldh1a1−/− animals compared to wildtype controls. These findings suggest that loss of Aldh1a1 in young animals affects at least some functional dynamics of DA neurons. However, the absence of Aldh1a1 does not appear to negatively impact the initial growth and development of SN DAergic neurons nor materially impact their survival in young animals. However, absence of Aldh1a1 in SN DA neurons does influence the ability to release DA in response to a physiological signal (KCl) and to clear released DA. Surprisingly, the number of TH-IR neurons in the SN was higher in Aldh1a1−/− mice than in wildtype animals, while overall neuronal numbers remained unchanged from wildtype controls. Even though there was a significant increase TH-IR cell numbers in the SN, there were no increases in levels of the striatal protein markers TH, DAT or VMAT. Although the reasons for these findings are presently unclear, it is possible that despite a larger number of cells expressing detectable levels of TH in the SN, the number of DAergic terminals in the striatum has not changed and/or a defective intracellular transport of proteins may exist between the SN and the striatum in the Aldh1a1−/− mice. Taken together these data suggest that the expression of ALDH1A1 is not critical to the development or survival of DA neurons in the SN of young animals but does affect the functioning of the DA system.
It is not entirely clear at this time how the findings from these animals relate to findings in Parkinson's disease brain. These animals had no ALDH1A1 for their entire lives and it is not clear when Parkinson's patients may develop reduced levels of ALDH1A1. However, ALDH1A1 function is highly dependent upon the availability of its cofactor NAD+ (Meyer et al., 2004), which is formed in the mitochondria, and perturbations of mitochondrial function, specifically complex I inhibition have been suggested to play a role in the processes underlying the loss of the SN DAergic neurons (Olanow and Tatton, 1999; Schapira et al., 1998). Profound decreases in NAD+ levels, leading to decreased processing of aldehydes may further compromise the function of these neurons and increase their susceptibility to degeneration. Thus, some of what we have observed in these young adult Aldh1a1−/− animals may represent at least in part a response of the nigrostriatal system to the lifetime absence of ALDH1A1. What happens to the nigrostriatal system in these animals as they age is not known but is the subject of on-going follow-up studies.
Aldehyde dehydrogenase 1A1 is a member of a superfamily of detoxification enzymes found in brain and other tissues that participate in the oxidation of both aliphatic and aromatic aldehydes of endogenous and exogenous cellular origin. In the brain, aldehyde dehydrogenases were first implicated in the oxidation of catecholamine-derived aldehydes in the early 1960s (Erwin and Deitrich, 1966), and are now known to be significantly involved in the metabolism of numerous biogenic aldehydes including DA, norepinephrine and gamma-aminobutyric acid. Specifically, ALDH1A1 is uniquely and highly expressed within the DAergic neurons of the SN and VTA of both rodents and man (Galter et al., 2003; Grimm et al., 2004). As such, ALDH1A1 is the primary catalyst of the irreversible oxidation of the biogenic aldehyde DOPAL to DOPAC, which is itself formed by the oxidative deamination of DA by monoamine oxidases (MAO) (Lamensdorf et al., 2000; Tank et al., 1981), a process that also produces potentially damaging oxygen radicals (Adams et al., 2001). Modification of the processing of DOPAL by ALDH1A1 could have significant impact on the long-term survival of DAergic neurons. Although absence of the Aldh1a1 gene had no negative impact on the number of DAergic neurons in the SN in young (8–12 week old) animals, it is possible that as these animals age, the long-term effects of an increased level of DOPAL could be potentially toxic to DA neurons and could render these neurons more vulnerable to aldehyde toxicity and degeneration. Since the primary role of ALDH1A1 in nigrostriatal DA neurons is the oxidation of DOPAL to DOPAC, thereby removing a neurotoxic intermediary aldehyde from the cytoplasm before it can undergo conversion to even greater toxic compounds such as tetrahydropapaveroline (THP), an ALDH1A1 deficiency could have marked long-term effects on DA neurons. Over prolonged periods of time, ALDH1A1 deficiency could lead to behavioral/phenotypic and neurodegenerative changes consistent with PD that would occur with increasing age. Also, it is possible that other ALDHs may have a redundant function in oxidation of DOPAL to DOPAC that prevents more severe effects in Aldh1a1 mutants.
A loss of function mutation in the Aldh1a1 gene could be a risk factor for PD either alone or in conjunction with environmental risk factors. Aldh1a1 mRNA levels are specifically down-regulated in DA neurons in PD brain (Galter et al, 2003; Grunblatt et al., 2004; Mandel et al., 2005). It is not possible to know if this decrease in Aldh1a1 gene expression preceded the onset of PD or was a consequence of the degenerative process in PD. However, in the periphery, ALDH1A1 is expressed in the digestive tract and implicated in a variety of detoxification reactions. Impaired function of the Aldh1a1 gene in the lining of the gastrointestinal tract could result in toxic compounds, including aldehydes, entering the circulation, entering the brain and potentially, affecting SN DAergic neurons. This, combined with the already discussed influences of loss of the Aldh1a1 gene on the neurochemistry of DA neurons and the resulting accumulation of toxic by-products of abnormal DA metabolism could present a heightened risk for developing PD in individuals with loss of function mutation in the Aldh1a1 gene.
In conclusion, the present study examined the functioning of the DA system in young Aldh1a1−/− mice. Lack of Aldh1a1 expression in nigrostriatal DAergic neurons significantly increased striatal extracellular fluid levels of DA, significantly reduced striatal DA uptake, and significantly reduced terminal release of DA in response to KCl or amphetamine stimulation. Further characterization of these animals is necessary to more fully understand how alterations in Aldh1a1 gene expression may be involved in the development or perpetuation of PD. In particular it will be important to evaluate how aging and exposure to environmental toxicants may interact with abnormal Aldh1a1 gene function to influence the development of a Parkinsonian phenotype.
4. Experimental procedures
4.1. Animals
The method for production of Raldh1 (Aldh1a1) null mice on a C57Bl/6 background has been described previously (Fan et al., 2003). Aldh1a1−/− mice used to establish the current colony were provided by G. Duester (Sanford-Burnham Institute, CA). Mice were genotyped by PCR using custom primers; Aldh1a1 WT 5′ TAAAGACCTGGATAAGGCCATCA and 5′ ACGGTGCACAAAATAAACATCTG and Aldh1a1 neo 5′ AGGATCTCCTGTCATCTCACCTTGCTCCTG and 5′ AAGAACTCGTCAAGAAGGCGATAGAAGGCG (BioChem, Salt Lake City, UT). Wildtype male C57Bl/6 (Taconic, Germantown, NY) and age-matched Aldh1a1−/− mice, were housed in 12 h light/dark cycle with ad libitum food and water. Mice were 8–12 weeks old when studied. All procedures were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee, and carried out according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
4.2. Microdialysis
Animals were anesthetized with Ketamine/Xylazine (i.p., 8:1) and placed in a custom mouse stereotaxic apparatus. Using flat skull coordinates, a microdialysis probe cannula (Sci-Pro, Sanborn, NY) was affixed to the skull overlying the striatum at the following coordinates relative to Bregma: 0.4 mm rostral, 1.8 mm lateral and 2.0 mm below the skull surface. Animals recovered from surgery overnight and the following morning, a dialysis probe (2 mm membrane) (Sci-Pro, Sanborn, NY) was inserted into the striatum. The freely moving animal was tethered to a liquid swivel (Instech Laboratories, Inc., Plymouth Meeting, PA) attached to a syringe pump. Prior to insertion, probes were calibrated in vitro at 37 °C to verify functionality and to calculate relative recovery of DA and DOPAC. After insertion, probes were perfused with sterile artificial cerebrospinal fluid (aCSF, sodium chloride 145 mM; potassium chloride 2.8 mM; magnesium chloride 1.2 mM; and calcium chloride 1.2 mM) at a rate of 1.5 ml/min. Approximately 22 μl of dialysate was collected and analyzed every 15 min until a stable DA baseline (defined as 3 consecutive samples within approximately 10% variation) was achieved. The perfusate was then switched to aCSF containing 120 mM KCl for 30 min and then switched back to standard aCSF and samples were collected until DA levels re-stabilized. Following return to baseline, the probe was perfused with aCSF containing 50 μM d-amphetamine sulfate for 30 min. The perfusate was then switched back to standard aCSF and samples were collected until DA levels once again returned to a stable baseline level.
Dopamine and DOPAC levels were measured in dialysate using high-pressure liquid chromatography with electrochemical detection (HPLC-EC, ESA Coulochem III). Samples were separated using a C18 reverse-phase column and MD TM mobile phase (ESA, Chelmsford, MA) consisting of sodium dihydrogen phosphate 75 mM; 1-Octanesulfonic Acid 1.7 mM; .01% TEA; EDTA 25 μM; 10% Acetonitrile, pH 3.0 at a flow rate of 0.5 ml/min. Peak heights were analyzed using EZChrom Chromatography software (Scientific Software Inc., San Ramon, CA) and data processing was accomplished using Prism GraphPad (La Jolla, CA). Data were calculated as peak release.
At the conclusion of a microdialysis study, the animal was euthanized and the brain quickly removed. The striatum on the side opposite to the probe site was collected for postmortem analysis of DA and metabolite levels using methods previously described (Anderson et al., 2001). The remaining tissue including the striatum on the side used for microdialysis and ventral mesencephalon was post-fixed in 4% paraformaldehyde for verification of the probe insertion site and for cell counting studies (vide infra).
4.3. Tissue processing and cell counting
Serial coronal sections (30 μm thick) were cut frozen through the rostral–caudal extent of the SN and collected in 0.1 M phosphate-buffered saline (PBS). Every third section was processed for TH-IR. Adjacent sections were processed for ALDH1A1-immunoreactivity (ALDH1A1-IR) and the remaining section was stained for Nissl substance to detect total neuronal numbers. Immunohistochemistry was performed on free floating sections. Tissues were incubated in PBS containing 3% hydrogen peroxide and 0.3% Triton X-100 to inhibit endogenous peroxidase activity followed by 1 h block at room temperature in 5% (w/v) non-fat powdered milk (NFM) containing Triton X-100 (NFM-T). Sections were then incubated overnight at 4 °C with rabbit anti-TH (1:1,000; Pel-Freeze Inc., Rogers, AR) or rabbit anti-ALDH1A1 (1:10,000; Abcam Cambridge MA) in NFM-T. After washing in NFM-T, sections were incubated in biotinylated goat anti-rabbit antibody (1:1000 Jackson Immunoresearch, West Grove, PA) in NFM-T for 2 h at room temperature, washed in 0.1 M PBS and incubated in ABC reagent (Vector Labs, Burlingame, CA) for 1 h at room temperature. Tissue was washed again in 0.1 M PBS and visualized using Vector SG substrate, according to the manufacturer's protocol (Vector Labs, Burlingame, CA). Sections were mounted in sequential order, dried, cleared, and coverslipped. Slides were coded and analyzed blindly to estimate the number of TH-IR cells in the SNc using Stereo-Investigator software (MicroBrightField, Inc., Williston, VT) and an Olympus BX-60 microscope. The region of interest (ROI) was first outlined at 4× magnification and 100× magnification was used for counting. The system randomly overlaid grids of 195 μm×87 μm on the ROI. Counting frames (40.2 μm×40.2 μm) were used for counting neurons with clear cytoplasmic staining and a distinct nucleus contained within the counting frame. Cell counts were only accepted with a Gundersen CE of <0.1.
4.4. Protein analyses
Membrane and cytoplasmic proteins were extracted from the striatum of Aldh1a1−/− and wildtype C57Bl/6 controls (MEM-Per Extraction Kit, Pierce, Rockford, IL). Membrane protein fractions (for DAT and VMAT2) and cytosolic protein fractions (for TH and ALDH1A1) were separated on 4–12% polyacrylamide gels (Invitrogen, Inc.) and transferred to 0.2 μm nitrocellulose membranes (Biorad, Hercules, CA). A rat monoclonal antibody raised against the N-terminus of DAT (1:10,000, Millipore, Billerica, MA) and VMAT2 (1:500, Abcam, Cambridge, MA) were used to detect membrane proteins. Rabbit polyclonal antibodies were used to detect the cytoplasmic proteins; TH (1:10,000, Pel-Freeze Inc., Rogers, AR), and ALDH1A1 (1:10,000, Abcam, Cambridge, MA). Blots were blocked for 1 h in Tris-buffered saline (TBS) containing 5% non fat milk and 0.1% Tween 20 for 1 h at room temperature and then washed with TBS containing 0.1% Tween 20 (TTBS). Membranes were then incubated in appropriate secondary antibodies in TTBS for 1 h at room temperature (1:20,000, Pierce, Rockford, IL), washed in TTBS, and bands were visualized using chemiluminescence (Pierce Pico or Dura). Optical density of bands was quantified using MCID software (Cambridge, UK).
4.5. Dopamine uptake
Dopamine uptake was estimated in whole striatal homogenates using the protocol of Lau et al. (Lau, 2003). Total uptake was measured after adding 100 μl of fresh striatal homogenate to tubes containing assay buffer (50 mM Tris–HCl, pH 7.4; 120 mM NaCl; 5 mM KCl; and 0.32 M sucrose), 100 μM pargyline and 0.5 μM [3H]DA (specific activity=38.7 Ci/mmol, Perkin Elmer, Shelton, CT). Nonspecific uptake was assessed by the addition of 100 μM mazindol (incubated at 37 °C for 3 min) to the uptake assay buffer. Tubes containing samples were vortexed and incubated at 37 °C for 3 min. The reaction was terminated by placing the tubes in ice and then samples were vacuum filtered onto Whatman GF/B filters using a Brandel Cell Harvester. Following several rinses with cold assay buffer, filters were removed, dried, and added to scintillation vials containing 10 ml of scintillation fluid (ScintiSafe, Fisher Scientific, Pittsburgh, PA), and radioactivity was quantified by liquid scintillation spectrometry (Packard TriCarb 2200).
4.6. Data analyses
All data are reported as mean±standard error unless otherwise stated. Statistical analyses for each component study were performed using unpaired Student's t test, two-tailed (GraphPad, Prism, La Jolla, CA).
Acknowledgments
This research was supported by the American Parkinson's Disease Association and the F.M. Kirby Foundation, Inc.
REFERENCES
- Adams JD, Jr., Chang ML, Klaidman L. Parkinson's disease—redox mechanisms. Curr. Med. Chem. 2001;8:809–814. doi: 10.2174/0929867013372995. [DOI] [PubMed] [Google Scholar]
- Anderson DW, Neavin T, Smith JA, Schneider JS. Neuroprotective effects of pramipexole in young and aged MPTP-treated mice. Brain Res. 2001;905:44–53. doi: 10.1016/s0006-8993(01)02466-0. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson's disease pathogenesis. Brain Res. 2003;989:205–213. doi: 10.1016/s0006-8993(03)03354-7. [DOI] [PubMed] [Google Scholar]
- Erwin VG, Deitrich RA. Brain aldehyde dehydrogenase. Localization, purification, and properties. J. Biol. Chem. 1966;241:3533–3539. [PubMed] [Google Scholar]
- Fan X, Molotkov A, Manabe S-I, Donmoyer CM, Deltour L, Foglio MH, Cuenca AE, Blaner WS, Lipton SA, Duester G. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell. Biol. 2003;23:4637–4648. doi: 10.1128/MCB.23.13.4637-4648.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson's disease and in the ventral tegmental area in schizophrenia. Neurobiol. Dis. 2003;14:637–647. doi: 10.1016/j.nbd.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Grimm J, Mueller A, Hefti F, Rosenthal A. Molecular basis for catecholaminergic neuron diversity. Proc. Natl. Acad. Sci. U. S. A. 2004;101:13891–13896. doi: 10.1073/pnas.0405340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MB. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin–proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural Transm. 2004;111:1543–1573. doi: 10.1007/s00702-004-0212-1. [DOI] [PubMed] [Google Scholar]
- Lamensdorf I, Eisenhofer G, Harvey-White J, Nechustan A, Kirk K, Kopin IJ. 3,4-Dihydroxyphenylacetaldehyde potentiates the toxic effects of metabolic stress in PC12 cells. Brain Res. 2000;868:191–201. doi: 10.1016/s0006-8993(00)02309-x. [DOI] [PubMed] [Google Scholar]
- Lau YS. Measurement of dopamine uptake in neuronal cells and tissues. Methods Mol. Med. 2003;79:465–471. doi: 10.1385/1-59259-358-5:465. [DOI] [PubMed] [Google Scholar]
- Mandel S, Grunblatt E, Riederer P, Amariglio N, Jacob-Hirsch J, Rechavi G, Youdim MB. Gene expression profiling of sporadic Parkinson's disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann. N. Y. Acad. Sci. 2005;1053:356–375. doi: 10.1196/annals.1344.031. [DOI] [PubMed] [Google Scholar]
- Maring JA, Deitrich RA, Little R. Partial purification and properties of human brain aldehyde dehydrogenases. J. Neurochem. 1985;45:1903–1910. doi: 10.1111/j.1471-4159.1985.tb10550.x. [DOI] [PubMed] [Google Scholar]
- Meyer MJ, Mosely DE, Amarnath V, Picklo MJ., Sr. Metabolism of 4-hydroxy-trans-2-nonenal by central nervous system mitochondria is dependent on age and NAD+ availability. Chem. Res. Toxicol. 2004;17:1272–1279. doi: 10.1021/tx049843k. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu. Rev. Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Gu M, Taanman J-W, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM. Mitochondria in the etiology and pathogenesis of Parkinson's disease. Ann. Neurol. 1998;44:S89–S98. doi: 10.1002/ana.410440714. [DOI] [PubMed] [Google Scholar]
- Tank AW, Weiner H, Thurman JA. Enzymology and subcellular localization of aldehyde oxidation in rat liver. Oxidation of 3,4-dihydroxyphenylacetaldehyde derived from dopamine to 3,4-dihydroxyphenylacetic acid. Biochem. Pharmacol. 1981;30:3265–3275. doi: 10.1016/0006-2952(81)90598-0. [DOI] [PubMed] [Google Scholar]
- Westerlund M, Galter D, Carmine A, Olson L. Tissue- and species-specific expression patterns of class I, III, and IV Adh and Aldh 1 mRNAs in rodent embryos. Cell Tissue Res. 2005;322:227–236.. doi: 10.1007/s00441-005-0038-7. [DOI] [PubMed] [Google Scholar]