

Abstract
Soluble water-forming NAD(P)H oxidases constitute a promising NAD(P)+ regeneration method as they only need oxygen as cosubstrate and produce water as sole byproduct. Moreover, the thermodynamic equilibrium of O2 reduction is a valuable driving force for mostly energetically unfavorable biocatalytic oxidations. Here, we present the generation of an NAD(P)H oxidase with high activity for both cofactors, NADH and NADPH. Starting from the strictly NADH specific water-forming Streptococcus mutans NADH oxidase 2 several rationally designed cofactor binding site mutants were created and kinetic values for NADH and NADPH conversion were determined. Double mutant 193R194H showed comparable high rates and low K m values for NADPH (k cat 20 s-1, K m 6 µM) and NADH (k cat 25 s-1, K m 9 µM) with retention of 70% of wild type activity towards NADH. Moreover, by screening of a SeSaM library S. mutans NADH oxidase 2 variants showing predominantly NADPH activity were found, giving further insight into cofactor binding site architecture. Applicability for cofactor regeneration is shown for coupling with alcohol dehydrogenase from Sphyngobium yanoikuyae for 2-heptanone production.
Keywords: coenzyme selectivity, NADPH recycling, 2-heptanol oxidation, NADPH oxidase, site-directed mutagenesis, cofactor regeneration
Introduction
Enzyme catalyzed oxidation reactions have gained increasing interest in biocatalysis recently, reflected also by a number of excellent reviews on this topic published in the last years [1–3]. Oxidoreductases constitute an important group of biocatalysts as they facilitate not only the widely used stereoselective reduction of aldehydes and ketones but also the less well exploited oxidation of alcohols and amines. Oxidoreductases catalyzed oxidations are also used for production of chiral alcohols and amines by deracemization [1, 4–6].
Oxidoreductases, especially aldo-keto-reductases and dehydrogenases, act on the substrate by the transfer of electrons from or to a cofactor, mostly the nicotinamide-based nucleotides NAD(H) and NADP(H). As nicotinamide cofactors are expensive, regeneration of cofactors is necessary for economically feasible biocatalytic processes. While for the regeneration of the reduced cofactors NADH and NADPH several systems (engineered formate dehydrogenase [7, 8], phosphite dehydrogenase [9, 10], glucose dehydrogenase [11, 12] plus cosubstrate) are well established and widely used, universal regeneration systems for the oxidized forms NAD+ and NADP+ are less well developed.
Enzyme based, electrochemical, chemical, and photochemical regeneration methods are known. Coupled substrate or coupled enzyme systems [4, 13, 14] constitute two possibilities for enzymatic NAD(P)+ recycling. In these reaction set-ups the cofactor is regenerated via the reduction of a carbonyl group of a cosubstrate, catalyzed either by the production enzyme itself (coupled substrate) [15] or by an additionally added dehydrogenase (coupled enzyme; glutamate dehydrogenase [16, 17], lactate dehydrogenase [18]). Carbonyl cosubstrate reductions by dehydrogenases normally provide little thermodynamic driving force for mostly energetically unfavorable biocatalytic alcohol oxidations. Generally, it is therefore necessary to supply the cosubstrate in excess to achieve high substrate conversion rates. In recent studies several smart concepts have been introduced to reduce the need for cosubstrate. The use of one-way cosubstrates [19] or cofactor regeneration as an integral part of a redox neutral multi-enzyme network [20, 21] was reported.
Several cofactor regeneration systems benefit from the high driving force of molecular oxygen as hydrogen acceptor. One example therefore is a 9,10 phenantrenequinone/xylose reductase system where the quinone is auto-reoxidized by oxygen [22]. O2 reduction also drives cofactor regeneration via mediators as ABTS or Meldola's blue, which are reoxidized by a laccase under H2O formation [23–25]. Instead of using laccase the mediator reoxidation can also be achieved by electrochemical means, albeit at moderate turnover numbers. To overcome the still rather low productivity of electrochemical regeneration processes careful reaction and cell design is necessary [26–28]. In pure chemical regeneration processes the chemical agent directly reoxidizes the cofactor without biocatalyst. Often Ruthenium complexes are used as oxidants [14]. The direct regeneration of NAD(P)+ via FMN was found to be strongly accelerated by light-induced excitation of FMN [29].
A very promising NAD(P)+ regeneration method is the application of soluble NAD(P)H oxidases (EC 1.6.3.1 NOX) from bacteria or archaea which use molecular oxygen as oxidant. This regeneration method has the advantage of being cheap as no cosubstrate or mediator is needed. Straightforward downstream processing is possible as only hydrogen peroxide or water is formed as byproduct. Moreover, the high redox potential of oxygen results in a high thermodynamic driving force. The electron and hydrogen transfer from NADH to oxygen is catalyzed by known soluble NAD(P)H oxidases via a two electron transfer producing hydrogen peroxide (equation 1) or a four electron transfer producing water (equation 2) [30, 31].
| 1 |
| 2 |
The four electron transferring oxidases are the preferred choice for cofactor regeneration as they only form water as byproduct. In case of H2O2 production, catalase has to be added to the system to prevent enzyme damage by the peroxide. Water-forming NADH oxidases have been studied from several bacteria as Streptococcus [32–35], Lactobacillus [36–38], Lactococcus [39], Clostridium [40], Serpulina [41], Leuconostoc [42] and Bacillus [43]. NOX from P. furiosus [44] and T. kodakarensis [45] produce H2O and a significant level of H2O2 (72% for PfNOX, 25% for TkNOX). NAD(P)H oxidases belong to the pyridine nucleotide disulfide oxidoreductases (PNDOR) together with, among others, glutathione reductase and CoA-disulfide reductase [46]. NOX enzymes contain a single conserved redox-active cysteine that circulates between the thiol/thiolate and the sulfenic acid state during catalysis. Overoxidation of the cysteine leads to enzyme deactivation. Several NADH oxidases need FADH or DTT addition for optimal performance [36, 45, 47]. Enzymes with high specific activities (> 150 U/mg) were recently reported from L. sanfranciscensis, L. plantarum, L. rhamnosus and S. pyogenes [34, 37, 38, 48]. A drawback in using NADH oxidases for cofactor regeneration is that almost all water-forming NADH oxidases are specific for NADH. In wild type form only two water forming NOXs and one hydrogen peroxide forming NOX show activity with NADPH (around half / one third of activity with NADH depending on the NOX [38, 49, 50]. NOX 1299 from T. kodakarensis is the only NOX showing higher wild type activity with NADPH than with NADH but disadvantageously it produces high amounts of H2O2 [45]. NOX of L. plantarum was recently successfully mutated to accept NADPH but resulting in a simultaneous decrease in NADH activity [37]. The k cat value for NADH reaction of the variant with highest catalytic efficiency with NADPH was six-fold reduced compared to the wild type.
In this study we aimed at developing an NAD(P)H oxidase which is universally applicable for regeneration of NADH as well as NADPH. As starting point NADH specific water-forming S. mutans NADH oxidase 2 (SmNOX) was chosen. SmNOX was chosen as it was well characterized to be stable, highly active, not dependent on FADH or DTT addition [51], and had already been expressed in E. coli before [32]. Moreover, a crystal structure of a closely related enzyme from S. pyogenes was available which enabled us to model the SmNOX structure. In a thorough mutation study of the cofactor binding site a SmNOX mutant with matched activities with NADH and NADPH was generated. Mutants with increased NADPH/NADH activity ratios were identified by SmNOX library screening. The conversion of 2-heptanol to 2-heptanone with NADPH regeneration by engineered SmNOX was shown.
Methods
Strains and materials
E. coli TOP10F’ was originally bought from Invitrogen (Carlsbad, CA, USA), E. coli BL21-Gold (DE3) was from Stratagene (La Jolla, CA, USA). NAD(P)H was from Roche Diagnostics GmbH (Mannheim, Germany) or Roth (Karlsruhe, Germany). Materials for cloning were from Fermentas (St. Leon-Roth, Germany, now Thermo-Fisher Scientific) if not stated otherwise. All other chemicals were purchased from Sigma-Aldrich, Fluka (St. Luis, MO, USA) or Roth (Karlsruhe, Germany) if not stated otherwise.
Homology modeling of S. mutans NADH oxidase 2
Homology modeling for Streptococcus mutans NOX 2 was based on an X-ray structure of NADH oxidase from Streptococcus pyogenes (template: 2BC0A, 2.00 Å). The sequence identity between target and template was 77.5%. The homology model was created with the automated protein structure homology-modeling server SWISS-MODEL developed by the Protein Structure Bioinformatics group at the SIB - Swiss Institute of Bioinformatics and the Biozentrum University of Basel (version February 2008) [52].
Cloning and site directed mutagenesis
A synthetic S. mutans NOX 2 gene (protein sequence: GI:290580450) was ordered at DNA2.0 (Menlo Park, CA, USA) and ligated into a NdeI/HindIII cut pMS470Δ8 vector [53] downstream of the tac-promoter to give the vector pMSsN1Wt. Site directed mutagenesis of the S. mutans NOX 2 gene in vector pMSsN1Wt was performed following the Stratagene Quikchange Site-directed Mutagenesis Kit instruction (Stratagene, La Jolla, CA, USA). Primers used for site directed mutagenesis are listed in Table 1. PCR reaction mixtures (50 µL) contained template plasmid (28 pM), primers (0.2 µM each), dNTPs (200 µM each) and 10 x reaction buffer (5 µL) supplied with the polymerase. Pfu turbo polymerase (Stratagene, 2.5 units) was added to each tube. The amplification protocol comprised 30 seconds of initial denaturation at 95 °C, 18 cycles of denaturation (30 s, 95 °C), annealing (1 min, 55 °C) and extension (6 min, 68 °C), and a final 7 minutes extension period at 68 °C. After 1 h of DpnI digestion competent E. coli TOP10F’ cells were electrotransformed with the reaction mixture (2 µL). Successful incorporation of the desired mutations was verified by dideoxy sequencing.
Table 1.
Primers used in PCR for site directed mutagenesis of S. mutans NOX 2.
| Mutated amino acidsa) | Forward primerb) (mismatched bases are underlined) |
|---|---|
| Asp192→Ala | 5’-agaagttatcctgatcgccgttgttgacacctgcc-3’ |
| Asp192→Arg | 5’-aaagaagttatcctgatcaacgttgttgacacctgcc-3’ |
| Val193→Arg | 5’-gaagttatcctgatcgaccgtgttgacacctgcctggc-3’ |
| Val194→His | 5’-gttatcctgatcgacgttcatgacacctgcctggca-3’ |
| Ala199→Arg | 5’-gttgttgacacctgcctgcgtggttactacgaccaggac-3’ |
| Gly200→Lys | 5’-gttgacacctgcctggcaaaatactacgaccaggacctg-3’ |
| Asp192→Ala/Val193→Arg | 5’-taaagaagttatcctgatcgcccgtgttgacacctgcctggcag-3’ |
| Val194→His/Ala199→Argc) | 5’-gttcatgacacctgcctgcgtggttactacgaccaggac-3’ |
| Val194→His/Gly200→Lysc) | 5’-catgacacctgcctggcaaaatactacgaccaggacctg-3’ |
Numbering refers to S. mutans NOX sequence beginning with 1 for the starting methionine
reverse primers have reverse complementary sequence
for combination of mutations the plasmid carrying the Val194→His mutation was used as template
Preparation of cell free extracts
Electrocompetent E. coli BL21 (DE3) Gold cells were transformed with plasmid pMSsN1Wt or one of 15 variants thereof. Additionally, a plasmid pMSsN2 was transformed into an E. coli BL21 (DE3) giving a strain overexpressing Lactobacillus sanfranciscensis NOX (LsNOX). pMSsN2 is identical to pMSsN1 except that it carries the gene coding for LsNOX (synthetic variant, ordered at DNA2.0, protein sequence: GI:11862874) instead of the SmNOX. Precultures of all resulting E. coli strains were cultivated in LB media (50 mL) containing Ampicillin (100 mg/L) in baffled shake flasks (300 mL) at 37 °C and 130 rpm overnight. Main cultures were inoculated to an OD of 0.05 in the described medium (250 mL in 1 L baffled flasks) and cultivated at 37 °C and 130 rpm. NOX production was induced at OD 0.8-1 by addition of IPTG (1 mM). Cells were harvested after an overnight induction period (25 °C, 110 rpm) by centrifugation (15 minutes, 5000 rcf, Avanti J-20 XP centrifuge, Beckman Coulter, Krefeld, Germany, rotor JA-10). Cell pellets were diluted in potassium phosphate buffer (50 mM, pH 7.0) to a final volume of 25 mL. Cell breakage was achieved by ultrasonication with a Branson sonifier 250 (Branson ultrasonic corporation, Danbury, CT, USA) for 6 minutes at 50 W with continuous cooling, pulsed with one 700 ms pulse per second with a 1 cm diameter tip. Cell free lysates were prepared by collecting the supernatant of centrifugation at 36000 rcf (rotor JA-25.50) for 45 minutes and concentrating it to half the volume via Vivaspin 20 centrifugal concentration tubes with 30 kDa molecular weight cutoff (Sartorius, Göttingen, Germany). Cell free extracts from an E. coli strain expressing S. yanoikuyae ADH (SyADH) from the pEamTA based plasmid pEam_SyADH was prepared following the same protocol.
Protein content determination, SDS-PAGE
The protein content was determined with bichinonic acid protein assay (BCA) kit (Thermo Scientific, Waltham, MA, USA) using BSA as standard. For SDS-PAGE NuPAGE® 4-12% Bis-Tris Gels, 1.0 mm, from Invitrogen, (Carlsbad, CA, USA) were used with a NuPAGE MOPS SDS Running Buffer for Bis-Tris Gels. All strains were stored as glycerol stocks at -80 °C. Cell free extracts were stored in aliquots at -20 °C.
Enzyme activity assay
Initial rate data of NAD(P)H oxidation were acquired measuring the decrease in NAD(P)H absorption at 340 nm (ε= 6220 M-1 cm-1) in potassium phosphate buffer (50 mM, pH 7.0) at 25 °C. The tempered buffer was vortexed for saturation with oxygen before mixing with the enzyme and cofactor solution. Absorption measurements were performed on a Spectramax Plus 384 (Molecular Devices, Sunnyvale, CA, USA) or on a Synergy MX (Biotek, Winooski, VT, USA) in UV-star micro titer plates (Greiner, Kremsmünster, Austria). The total reaction volume was 200 µL, reactions were started by addition of NAD(P)H. Data collection was started after 5 seconds of mixing. For activity measurements with FAD, enzyme preparations first were pre-incubated in 10 fold concentration in 50 mM KPi containing 25 µM or 250 µM FAD and then were diluted 1:10 in the same buffer for initial rate detection after addition of NADH. In case of DTT addition the assay buffer contained 5 mM of DTT.
Determination of catalytic constants for SmNOX variants
Apparent kinetic parameters were obtained from initial rate measurements at air saturation oxygen level (250 µM) with eight cofactor concentrations varying over a concentration range of 5 to 10 times the apparent K m or to a maximum NAD(P)H concentration of 1 mM. Enzymes were applied as crude lysates in dilutions chosen to give rates between 0.001 and 0.05 ▵Abs/min and rates were constant for ≥ 1 minute. Appropriate controls containing crude lysate without overexpressed NOX verified that blank rates were insignificant for all conditions used. Results from initial rate measurements were fitted to a Michaelis-Menten type equation (equation 3) using unweighted least-squares regression analysis performed with Sigmaplot program version 11 (Systat Software Inc.).
| 3 |
v is the initial rate, v max is the apparent maximum rate (U/mg total protein in cell free extract), A the cofactor concentration and K m the apparent Michaelis constant for NAD(P)H at air saturation oxygen levels.
H2O2 determination
Enzyme assay reaction mixtures with 1 mM of NAD(P)H were fully converted with purified SmNOX wild type or variant 193R194H. The assay solution or glucose standard solutions were diluted 1 + 1 with an o-dianisidine / glucose oxidase / horse radish peroxidase mixture [54] and absorption was measured at 460 nm.
Recloning in pEHISTEV vector and enzyme purification
SmNOX wild type gene and variant 193R194H were recloned in pEHISTEV vector [55] via EcoRV/HindIII restriction sites. A pEHISTEV version was used in which a second EcoRV restriction site was eliminated by introduction of a silent mutation. Plasmid preparations checked for correct sequence were transformed into E. coli BL21 (DE3) Star. Expression and cell free extract preparation were done as described above but in LB/Kanamycin medium. The cells were harvested by centrifugation (5000 g, 15 minutes). The pellet was resuspended in 50 mM KPi pH 7.0 and disrupted by ultrasonication. The cell free extract was applied to a 5 mL Ni-Sepharose 6 Fast Flow column (GE Healthcare, Chalfont St Giles, UK). The tagged enzymes were obtained by a one-step purification using the buffers recommended in the manual. After purification, the enzyme buffer was exchanged to 50 mM KPi and enzymes were concentrated to protein concentrations above 5 mg/mL by Vivaspin 20 tubes with 10 kDa molecular weight cutoff (Sartorius) before storage at -20 °C. The 6xHis tag was cleaved off from 1 mg NOX by incubation with tobacco etch virus protease (TEV protease) in a reaction mixture containing 0.2 mM EDTA, and 1 mM DTT in 50 mM Tris/HCl buffer, pH 8.0 by overnight incubation at 4 °C. 10 µg TEV protease were used per mg NOX. The mixture was applied on the Ni-Sepharose 6 Fast Flow column and washed through with 15 mL of 30 mM sodium phosphate buffer containing 0.3 M NaCl and 20 mM imidazole, pH 7.5. The flow through was collected. Buffer exchange to 50 mM KPi samples and concentration to > 1 mg/mL protein was done in Vivaspin 20 centrifugal concentrators (Satorius AG, Göttingen, Germany), aliquots of concentrated NOX solutions were stored at -20 °C.
Library generation and cultivation
A sequence saturation mutagenesis library [56] of SmNOX gene with random mutations was bought at SeSaM Biotech (Aachen, Germany). The library was based on mutant SmNOX 194H200K. The library was cloned into pMS470 vector and transformed into E. coli TOP10F’. Transformants were picked into 60 µL LB/Ampicillin media in 384 well plates, grown overnight at 37 °C and 60% of humidity and stored as 15% glycerol stocks at -80 °C. Cultivation of the expression library was done in 96 well plate format. Preculture plates with 150 µL of LB/Ampicillin media per well were inoculated from glycerol stock plates and cultivated at 37 °C at 60% humidity for at least 12 hours. Main culture plates with V-shaped bottom contained 80 µL of LB/Ampicillin media and were inoculated from the preculture plates. After 8 hours of growth at 37 °C and 60% humidity SmNOX2 expression was induced by addition of 20 µL of a 0.5 mM IPTG solution in LB/Ampicillin media. The plates were kept at 28 °C and 60% humidity for 16 hours. Cells were harvested by 15 minutes of centrifugation at 2500 g. Supernatant was decanted and the cell pellets were frozen at -20 °C for at least two hours.
Screening of SmNOX variants from a random mutagenesis library for enhanced NADPH/NADH activity
Screening assays were carried out in 96 well plates. After thawing cell lysis was accomplished by addition of 100 µL lysis buffer (50 mM KPi, pH 7.0, 1 mg/mL lysozyme) and an 1 h incubation at 28 °C at 600 rpm. Cell debris was separated by centrifugation at 2500 g for 15 min at 4 °C. The supernatant was diluted 1 + 1 with 50 mM KPi, pH 7.0 and used for screening assays. 140 µL of 50 mM KPi, pH 7.0, were added to 10 µL of diluted supernatant in two plates in parallel. Reactions were started by addition of 50 µL of a 0.8 mM NADH or NADPH solution. Initial rates of NADH and NADPH conversion were measured by detection of decrease in absorption at 340 nm over three minutes. Activity with NADH and NADPH was compared for each well. 2800 clones were screened, 480 thereof were chosen for a re-screen and the best 40 thereof were measured in a re-re-screen. Best variants were finally cultivated in shake flasks. From best variants plasmid DNA was isolated with Gene JetTM Plasmid Miniprep Kit (Fermentas, St. Leon-Roth, Germany) and sent for sequencing.
Conversion experiments
Conversion experiments were set up in 1.5 mL reaction tubes. The reaction mixture contained NADP+ (100 µM) and 2-heptanol (10 mM) in potassium phosphate buffer (50 mM, pH 7.0) in a total volume of 500 µL. Sphingobium yanoikuyae ADH (SyADH) was applied as crude E. coli lysate and SmNOX 193R194H was applied as purified enzyme in amounts to give 1 U/mL. After 12 hours at 25 °C and 600 rpm 100 µL of n-butanol (50 mM) was added as internal standard for GC analysis and the mixture was extracted with ethyl acetate (500 µL). Substrate conversion was determined by GC-analysis on a Varian CP7503 gas chromatograph equipped with an FID detector (300 °C) and a Phenomenex ZB-FFAP column (30 m x 0.32 mm; 0.25 µM) with a Restek Hydroguard MT precolumn (5 m x 0.32 mm). H2 was used as carrier gas (2.7 mL/min). The following temperature program was used: 65 °C to 110 °C, 9 °C/min; 110 °C to 160 °C, 25 °C/min. The retention time for 2-heptanol was 2.48 min and for 2-heptanone 3.44 min.
Results and Discussion
Mutation Strategy
Water-forming Streptococcus mutans NADH oxidase 2 (SmNOX) is a monomeric 50 kDa enzyme which is NADH specific [51]. We intended to establish SmNOX as universal NAD(P)+ regeneration system by engineering the NADH specific wild type towards the effective usage of both cofactors, NADH and NADPH. Ideally, the created variant should have comparable characteristics for both cofactors to simplify application for cofactor regeneration in industrial processes with varying oxidizing enzymes. In NOX enzymes the nicotinamide cofactor is bound in the well described Rossmann fold manner [57]. In Rossmann fold enzymes often an acidic residue, typically an aspartate at the C-terminus of the second β-strand of the alternating βαβαβ-regions plays a key role in NAD(H) binding [58] by forming hydrogen bonds to the 2’-OH and 3’-OH of the adenine ribose. In contrast, NADP(H) specific Rossmann fold enzymes typically miss this acidic residue and instead carry a basic residue at the following amino acid position. Positive charges in the cofactor binding site facilitate the binding of the negatively charged phosphate group present in NADP(H) but not in NAD(H). The relevance of the described positions for cofactor specificity has first been shown in a mutation study of glutathione reductase by Scrutton et al. [58]. NADH oxidase from Lactobacillus sanfranciscensis (LsNOX) exhibits a limited NADPH activity in parallel to NADH activity [38]. An alignment of the SmNOX sequence with the glutathione reductase, B. anthracis coenzyme A-disulfide reductase and LsNOX sequence (Figure 1) indicates several positions of possible importance for NADPH activity. SmNOX and LsNOX show an aspartate at the expected position indicative for NADH activity (position 192 in SmNOX numbering including the starting methionine). The positive charge important for NADPH activity is missing at the following amino acid in both enzymes but in LsNOX at the +2 position counted from the aspartate a positively charged histidine is found. Position 196 and 200 are also occupied by positively charged residues. Like LsNOX, B. anthracis coenzyme A-disulfide reductase with dual cofactor specificity shows the negatively charged residue (here glutamate) in combination with a positively charged residue. The subtle side chain rearrangements and reformation of hydrogen bonds enabling the dual cofactor specificity of B. anthracis coenzyme A-disulfide reductase were recently described [59].
Figure 1.

Sequence alignment for the NAD(P)H-binding motifs of selected pyridine nucleotide disulfide oxidoreductases.
SmNOX: S. mutans NADH oxidase 2, LsNOX: L. sanfranciscensis NAD(P)H oxidase, BaCoADR: B. anthracis CoA-disulfide reductase, EcGR: E. coli Glutathione reductase, TkNOX 1299: T. kodakarensis NAD(P)H oxidase, cofactor specificities given in brackets, red: negatively charged residues typical for NADH converting enzymes, blue: positively charged residues typical for NADPH converting enzymes, conserved sequence motif GXGXXG/A for dinucleotide binding marked with *, alignment was done with ClustalW2.
For SmNOX we built a structural model based on the X-ray structure of NADH oxidase from S. pyogenes (77.5% identity, 2BC0A). A comparison of the cofactor binding site of SmNOX, LsNOX, and E. coli glutathione reductase (Figure 2) indicated that amino acid residues at SmNOX position 192-194 and 199-200 might possibly interact with the cofactor, while residues at position 195-198 are turned away from the cofactor. Positions 192-194, 199 and 200 were chosen for rational engineering of SmNOX with the main aim of introducing positively charged residues to enable NADPH activity. In addition, single mutations were combined to double, triple, and quadruple mutants.
Figure 2.

Cofactor binding sites of S. mutans NADH oxidase 2, L. sanfranciscensis NAD(P)H oxidase and E. coli Glutathione reductase.
Panel A: Swiss-model for S. mutans NOX 2, based on S. pyogenes NOX structure (77.5% identity, pdb 2BC0), ADP from aligned LsNOX structure pdb 2CDU, FAD from pdb 2BC0; panel B: L. sanfranciscensis NAD(P)H crystal structure pdb 2CDU with ADP bound [60]; panel C: E. coli Glutathione reductase crystal structure pdb 1GET [61], images were generated with PyMOL (Schrodinger Inc.)
Mutation and expression
For recombinant expression and engineering, a synthetic S. mutans NOX 2 gene was inserted into pMS470Δ8 vector to give the vector pMSsN1Wt. Site directed mutagenesis of the S. mutans NOX 2 gene was performed following the Stratagene Quikchange protocol. Wild type and mutants were expressed in E. coli BL 21(DE3) Gold cells. Moreover, LsNOX was expressed under the same conditions from vector pMSsN2, which is identical to pMSsN1Wt, except that it carries a synthetic gene for LsNOX instead of the SmNOX gene. SDS-PAGE gel analysis of cell free extracts indicated NOX over-expression for all variants with a strong band migrating to the expected position for 50 kDa. The band was not detected in the cell free extract of an E. coli BL21 strain without plasmid. In Figure 3 the SDS-PAGE analysis of cell free extracts for SmNOX wild type and single mutants are shown.
Figure 3.

4-12% Bis-Tris SDS-PAGE gel of cell free extracts of E. coli BL21 (DE3) Gold expressing SmNOX.
Apparent kinetic constants of cofactor specificity mutants
For SmNOX and LsNOX wild type and fourteen SmNOX variants initial rate data for the oxidation of NADH or NADPH in cell free extracts at air saturated oxygen levels were recorded. Maximal specific activities and apparent K m values were calculated by fitting velocities measured over a cofactor concentration range of up to 5-10 fold the K m value to equation 3. Results are listed in Table 2. All mutants were cultivated under identical conditions and SDS-PAGE analysis showed a comparable expression level for all variants (see Figure 3).
Table 2.
Apparent kinetic values and efficiencies for SmNOX mutants and LsNOX from cell free extracts.
| NADH | NADPH | ||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| v max a) (U/mg) | K m a) (µM) | v max/K m (U/mg mMol) | v max (U/mg) | K m (µM) | v max/K m (U/mg mMol) | ||
| SmNOX Wt | 3.5 ± 0.1 | 6 ± 1 | 580 | 0.09c) | |||
| LsNOX | 2.2 ± 0.1 | 9 ± 1 | 240 | 1.1 ± 0.1 | 5 ± 1 | 220 | |
| Mut1 | D192Ab) | 2.5 ± 0.1 | 18 ± 2 | 140 | 2.2 ± 0.1 | 140 ± 20 | 15 |
| Mut2 | D192N | 2.8 ± 0.1 | 19 ± 2 | 150 | 2.9 ± 0.1 | 190 ± 20 | 15 |
| Mut4 | V193R | 3.8 ± 0.4 | 9 ± 3 | 420 | 1.6 ± 0.1 | 140 ± 10 | 12 |
| Mut4 | V194H | 3.5 ± 0.1 | 11 ± 2 | 320 | 1.4 ± 0.1 | 150 ± 10 | 10 |
| Mut5 | A199R | 4.4 ± 0.1 | 9 ± 1 | 490 | 0.45c) | ||
| Mut6 | G200K | 3.4 ± 0.1 | 7 ±1 | 490 | 2.7 ± 0.2 | 120 ± 30 | 22 |
| Mut7 | 192A/193R | 2.9 ± 0.1 | 23 ± 2 | 130 | 4.8 ± 0.2 | 5 ± 1 | 960 |
| Mut8 | 194H/199R | 4.2 ± 0.1 | 12 ± 1 | 350 | 3.7 ± 0.1 | 71 ± 6 | 52 |
| Mut9 | 194H/200K | 3.1 ± 0.1 | 7 ± 1 | 440 | 2.6 ± 0.1 | 12 ± 1 | 217 |
| Mut10 | 193R/194H | 4.4 ± 0.1 | 7 ± 2 | 630 | 4.6 ± 0.1 | 7 ± 1 | 660 |
| Mut11 | 192A/193R/194H | 2.0 ± 0.1 | 16 ± 2 | 130 | 4.2 ± 0.2 | 3 ±1 | 1400 |
| Mut12 | 193R/194H/199R | 4.3 ± 0.1 | 7 ± 1 | 610 | 1.7 ± 0.1 | 4 ± 1 | 430 |
| Mut13 | 193R/194H/200K | 4.6 ± 0.2 | 4 ± 1 | 1200 | 3.3 ± 0.2 | 2 ± 0 | 1650 |
| Mut14 | 192A/193R/194H/199R | 2.3 ± 0.1 | 11 ± 0 | 210 | 6.3 ± 0 | 3 ± 0 | 2100 |
| Mut15 | 192A/193R/194H/200K | 3.1 ± 0.4 | 8 ± 1 | 390 | 4.0 ±.0.3 | 3 ± 1 | 1300 |
v max and apparent K m values were measured in cell free extracts at air-saturated oxygen levels
all indicated mutations confer to SmNOX numbering including the starting methionine
as saturation with NADPH could not be achieved v max/appK m was calculated from the initial linear section of the Michaelis-Menten curve
Ratios of NADPH and NADH activities and efficiencies measured from one cell free extract in parallel clearly indicated cofactor specificity changes between wild type and the variants. As expected, wild type SmNOX showed only marginal activity with NADPH, while LsNOX wild type showed NADPH oxidation, as reported [38]. In SmNOX Mut1 and Mut2 (D192A and D192N) the negatively charged aspartate, which is known to be a key residue for NADH binding, is missing. In absence of the negative charge the activity with NADH was slightly reduced and higher NADH K m values were detected. Remarkably, only by the exchange of the aspartate to non-polar or positively charged residues NADPH conversion rates increased to levels comparable to NADH rates, albeit with 10 fold higher K m values.
The introduction of a positively charged residue next to the aspartate in Mut3 and Mut4 (V193R, V193H) without removal of the aspartate also enabled activity with NADPH although to a lower extent than the aspartate removal. NADH activity was not reduced in Mut3 and Mut4 compared to wild type SmNOX containing extracts. The introduction of a positive charge was even more effective at position 200. NADH activity stayed unchanged compared to wild type for a Mut6 (G200K) variant, while NADPH activity already reached around 80% of the activity with NADH. A positively charged arginine at position 199 in Mut5 could not increase NADPH activity.
While single mutations already introduced quite high levels of NADPH activity, only the combination of mutations further decreased NADPH K m values to values lower than 10 µM. The best combination for creating a mutant with high and matched NADH and NADPH activities at low K m values is mutant Mut10 (V193R/V194H). Quadruple mutant Mut14 (192A/193R/194H/199R) showed the highest NADPH to NADH activity ratio of all mutants with a maximal NADPH activity three times as high as the NADH activity.
Sequence comparison of H2O2 and H2O forming NAD(P)H oxidase from T. kodakarensis (TkNOX) and SmNOX showed that TkNOX features an arginine at positions equivalent to SmNOX positions 194 and 199 and a lysine at the position equivalent to SmNOX position 200 [45] (Figure 1). The results from the SmNOX mutation study indicate that these positively charged residues might be responsible for making TkNOX the only known bacterial wild type NOX showing higher activity with NADPH than with NADH.
Since Scrutton et al. [58] reported the first NAD(P)H specificity engineering of an enzyme, a vast number of enzymes have been mutated to alter cofactor specificities. The outcome of these mutation studies provided evidence that the effects obtained by site directed mutagenesis of positions known to be relevant for cofactor specificity, especially the aspartate or glutamate at the end of the second β-sheet, vary tremendously. Only in very few cases the catalytic efficiency of reactions with the originally disfavored cofactor could be increased to values of the same order of magnitude as for reactions with the originally used cofactor in the unmutated enzyme [10, 62–64]. For mutants in which the conserved aspartate or glutamate was exchanged to smaller non-polar residues, the efficiency with NADP+ ranged from 1/4000 to 1/3 of the efficiency of the unmutated enzymes with NAD+ [10, 65, 66]. Especially rare are examples of increased efficiency with one cofactor while keeping also the efficiency with the other cofactor high [10, 63]. In the outstanding case of B. subtilis lactate dehydrogenase the mutation of valine, the residue equivalent to SmNOX V193, into arginine led to a 140 fold increased NADPH k cat value. The increase in NADPH k cat did not lead to a decrease but even to a four-fold increase in NADH k cat. K m values stayed at wild type NADH level for both cofactors for the variant. In NADH oxidase from L. plantarum [37] the introduction of positively charged residues next to the conserved aspartate enabled k cat values for NADPH of up to 69% of wild type level with NADH but decreased k cat for NADH to 58% in variant G178R and to 16% in G178V/L179R. Interestingly, the single mutation G178R led to a low NADH K m of 6 µM compared to 50 µM in the wild type form. NADPH K m of the same variant was 490 µM and could be decreased to 9 µM in the G178V/L179R double mutant.
In summary, also in comparison to other enzymes, SmNOX turned out to be an excellent choice for the generation of an NAD(P)H oxidase with comparable kinetic characteristics for both nicotinamide cofactors without drastic loss of activity or increase in K m compared to the wild type enzyme.
NADPH preferring SmNOX variants found in library screening
Around 3000 variants of a sequence saturation mutagenesis library (bought at SeSaM Biotech) built on SmNOX 194H200K were screened for increase in NADPH/NADH activity ratios. We chose a starting point for the library without the well-studied mutation 193R as we rather aimed at finding new promising mutation combinations that were so far unknown to give high activities with NADPH.
No variant with significantly increased NADPH activity without loss in NADH activity compared to SmNOX 194H200K could be detected in the library screening. However, three mutants were identified with clearly increased NADPH/NADH activity ratio albeit with concomitant decrease in NADH activity. Figure 4 shows NADH and NADPH activity levels of crude lysates of mutant 194H200K202N, 194H200K202C and 194H200K340V388T compared to variant SmNOX 194H200K. A 194H200K340V variant without 388T mutation was constructed to check for the influence of the mutation at position 340 without mutation at position 388. Variant 194H200K340V showed the same NADH/NADPH activity ratio as variant 194H200K340V388T. NADH activity was around 60% of NADPH activity in both cases. We conclude that mutation of position 340 has a high impact on cofactor specificity while mutation 388T probably has no influence on cofactor binding. Position 202 is located at the end of the loop which connects cofactor binding site strand βB and helix αB and starts after aspartate 192. The modeled SmNOX structure indicates that residue 202 is positioned too far away from bound NAD(P)H in order to allow direct interaction with the cofactor (Figure 2). Strikingly, position 340 is located at the beginning of a β-sheet in close vicinity to the before described loop end. The SmNOX wild type model indicates a potential hydrogen bond between Y202 and N340. In LsNOX the equivalent positions are occupied by a tyrosine and a valine, which cannot form the hydrogen bond. We speculate that a presumably higher flexibility of the loop caused by removal of the hydrogen bond hampers the activity with the more structurally demanding NADPH less than activity with NADH.
Figure 4.
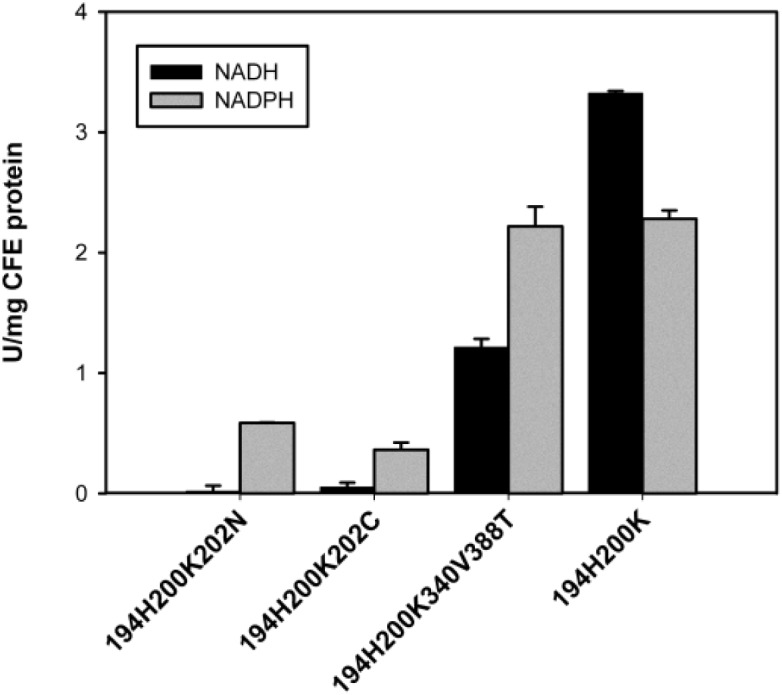
Cell free extract (CFE) activities of S. mutans NADH oxidase 2 variants found in a sequence saturation mutagenesis library by screening for improved NADPH/NADH activity ratios. The library was built on SmNOX 194H200K and initial rates were measured employing 300 µM NADH or NADPH.
Purification of SmNOX and determination of kinetic values
SmNOX wild type and variant 193R194H were recloned in vector pEHISTEV for expression with an N-terminal 6xHis tag. The enzymes were purified to apparent electrophoretic homogeneity by Ni-affinity chromatography as demonstrated in Figure 5. After purification the tag was cleaved off by treatment with TEV protease. Apparent kinetic values for NAD(P)H oxidation were determined in air saturated buffer as described in the methods section. k cat and K m values for SmNOX wild type and mutant 193R194H are shown in Table 3. The SmNOX wild type k cat value corresponded to a specific activity of 44 U/mg. Higuchi et al. [51] reported a specific activity of water-forming S. mutans NOX 2 of 100 U/mg. The lower specific activity measured here was not unexpected due to a lower assay temperature than in the previous study. However, this lower activity could also be caused by enzyme deactivation during the tag cleavage procedure which included an overnight incubation at 4 °C. With SmNOX mutant 193R194H now an efficient NADH oxidase is available which has very similar kinetic values for oxidation of NADH as well as NADPH.
Figure 5.
SDS-PAGE of purified S. mutans NADH oxidase 2.
Standard: SeeBlue® Plus2 Prestained Protein Standard; lane 1: wild-type; lane 2: 193R194H, SDS-PAGE was performed with an Invitrogen NuPAGE® system with a 4-12% Bis-Tris Gel and Coomassie staining
Table 3.
Apparent kinetic values and efficiencies for SmNOX wild type and variant 193R194H from purified protein.
| SmNOX variant | NADH | NADPH | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| k cat (s-1) | K m (µM) | k cat/K m (s-1µM-1) | k cat (s-1) | K m (µM) | k cat/K m (s-1µM-1) | |
| Wild type | 36.8±0.3 | 7±2 | 5±2 | n.d. | n.d. | n.d. |
| 193R194H | 25.5±0.9 | 9±2 | 3±1 | 20±2 | 6±2 | 3±1 |
n.d. not detected
H2O2 production, influence of FAD and DTT
Possible H2O2 formation was analyzed by detecting H2O2 values after SmNOX catalyzed total oxidation of up to 1 mM of NAD(P)H with o-dianisidine and HRP. For SmNOX wild type with NADH 2.7% of the catalytic conversions led to H2O2 formation, for SmNOX variant 193R194H with NADH in 3.3% of conversions and for NADPH in 4% of conversions H2O2 was formed. Other NOX enzymes were reported to lose FAD during purification [43]. Concentrated purified SmNOX was clearly yellow, indicating that FAD was still bound. Initial rate measurements after 30 min pre-incubation in 25 µm or 250 µM and with addition of the same concentration of FAD to the assay showed an insignificant increase of activity for SmNOX wild type and an insignificant decrease of activity for variant 193R194H. Several NADH oxidases have also been shown to be activated by addition of DTT [36, 47], probably by preventing the overoxidation of the catalytically active cysteine. SmNOX activity was only slightly increased by addition of 5 mM DTT to the initial rate measurements.
Conversion of 2-heptanol to 2-heptanone
Application of water-forming NADH oxidases as cofactor recycling system coupled to a biocatalytic oxidation has so far been demonstrated for L. sanfranciscensis NOX [47, 67, 68], L. brevis NOX [69, 70] and L. rhamnosus NOX [48]. Here, we demonstrate the NADP+ regeneration by cofactor specificity engineered SmNOX for 2-heptanol oxidation to 2-heptanone (Scheme 1). SmNOX was applied in a coupled enzyme system with alcohol dehydrogenase from Sphingobium yanoikuyae (SyADH). 10 mM of 2-heptanol were converted to 2-heptanone in the presence of 100 µM of NADP+ with and without SmNOX added to the assay. SyADH exhibits a low enantioselectivity for 2-heptanol [19] and therefore allows complete conversion if enough cofactor is supplied. NADP+ is cheaper than NADPH and therefore the preferred starting compound for cofactor recycling. Conversion rates are shown in Table 5. Nearly complete conversion could be achieved within 12 hours by addition of SmNOX. Without SmNOX, conversion rates around 1% as expected for a stoichiometric conversion of the 100 µM cofactor were found.
Scheme 1.
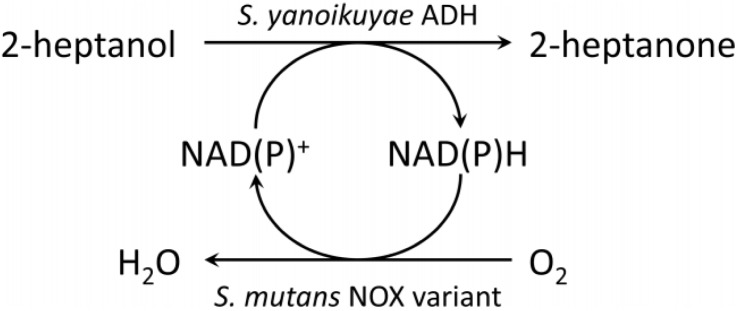
Cofactor regeneration by S. mutans NADH oxidase for 2-heptanol oxidation.
Table 5.
Enzymatic 2-heptanol to 2-heptanone conversion with S. yanoikuyae ADH with and without NADP+ regeneration within 12 hours.
| SmNOX variant | 2-heptanol | NADP+ | U/mL SyADH | U/mL SmNOX | % conversion | Cofactor turnovers |
|---|---|---|---|---|---|---|
| 193R194H | 10 mM | 100 µM | 1 | 2 | 97 | 97 |
| - | 10 mM | 100 µM | 1 | 0 | 1.2 | 1.6 |
Table 4.
Influence of FAD and DTT on SmNOX, initial rates for NADH activity in 50 mM KPi set to 100%.
| SmNOX variant | Buffer only | +25µM FAD | +250µM FAD | +5mM DTT |
|---|---|---|---|---|
| Wild type | 100 | 108 | 112 | 104 |
| 193R194H | 100 | 89 | 87 | 110 |
SD from at least triplicate measurements was always <16%
Conclusion
The NADH specific S. mutans NADH oxidase 2 belongs to the few NADH oxidases that produce water instead of hydrogen peroxide and is therefore well-suited to be used as cofactor recycling system. For general applicability SmNOX was engineered towards efficient usage of both cofactors, NADH and NADPH. The dual cofactor specificity was achieved by introducing positively charged amino acid residues for increased NADPH binding, while still retaining the aspartate which facilitates NADH binding. SmNOX variant V193R/V194H showed comparable high k cat and K m values for NADH and NADPH oxidation with only slight decrease in activity compared to SmNOX wild type. NADPH specificity was also found to be increased by the mutation of Y202 or N340. These two residues form a hydrogen bond between the end of the nucleotide binding loop and a near-by positioned β-sheet in SmNOX wild type, which is destroyed by the mutations. SmNOX variants were shown to be well active without a need to add FAD or DTT. Cofactor engineered S. mutans NAD(P)H oxidase 2 is therefore well suited for application as a versatile NAD(P)+ regeneration system, as was demonstrated for combination with S. yanoikuyae ADH for oxidation of 2-heptanol to 2-heptanone.
Acknowledgements
This work has been supported by the Federal Ministry of Economy, Family and Youth (BMWFJ), the Federal Ministry of Traffic, Innovation and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol and ZIT-Technology Agency of the City of Vienna through the COMET-Funding Programme managed by the Austrian Research Promotion Agency FFG.
We thank Prof. Wolfgang Kroutil for providing the S. yanoikuyae ADH expressing E. coli strain, Dr. Sigrid Egger for providing the TEV-protease and gratefully acknowledge the help of Monika Koch with expression and kinetic characterization of SmNOX double, triple and quadruple knock-outs.
Competing Interests
The authors have declared that no competing interests exist.
References
- 1.Hollmann F, Arends IWCE, Buehler K, Schallmey A, Bühler B (2011) Enzyme-mediated oxidations for the chemist. Green Chem 13: 226–265 [Google Scholar]
- 2.Monti D, Ottolina G, Carrea G, Riva S (2011) Redox reactions catalyzed by isolated enzymes. Chem Rev 111: 4111–4140 [DOI] [PubMed] [Google Scholar]
- 3.Romano D, Villa R, Molinari F (2012) Preparative biotransformations: Oxidation of alcohols. Chem Cat Chem 4: 739–749 [Google Scholar]
- 4.Kroutil W, Mang H, Edegger K, Faber K (2004) Biocatalytic oxidation of primary and secondary alcohols. Adv Synth Catal 346: 125–142 [Google Scholar]
- 5.Turner NJ (2011) Enantioselective oxidation of C-O and C-N bonds using oxidases. Chem Rev 111: 4073–4087 [DOI] [PubMed] [Google Scholar]
- 6.De Gonzalo G, Orden AA, Bisogno FR (2012) New trends in organic synthesis with oxidative enzymes. Curr Org Chem 16: 2598–2612 [Google Scholar]
- 7.Tishkov VI, Popov VO (2006) Protein engineering of formate dehydrogenase. Biomol Eng 23: 89–110 [DOI] [PubMed] [Google Scholar]
- 8.Hoelsch K, Sührer I, Heusel M, Weuster-Botz D (2013) Engineering of formate dehydrogenase: Synergistic effect of mutations affecting cofactor specificity and chemical stability. Appl Microbiol Biotechnol 97: 2473–2481 [DOI] [PubMed] [Google Scholar]
- 9.Johannes TW, Woodyer RD, Zhao H (2007) Efficient regeneration of NADPH using an engineered phosphite dehydrogenase. Biotechnol Bioeng 96: 18–26 [DOI] [PubMed] [Google Scholar]
- 10.Woodyer R, Van der Donk WA, Zhao H (2003) Relaxing the nicotinamide cofactor specificity of phosphite dehydrogenase by rational design. Biochemistry 42: 11604–11614 [DOI] [PubMed] [Google Scholar]
- 11.Wong C-H, Drueckhammer DG, Sweers HM (1985) Enzymatic vs. fermentative synthesis: Thermostable glucose dehydrogenase catalyzed regeneration of NAD(P)H for use in enzymatic synthesis. J Am Chem Soc 107: 4028–4031 [Google Scholar]
- 12.Kaswurm V, Hecke WV, Kulbe KD, Ludwig R (2013) Guidelines for the application of NAD(P)H regenerating glucose dehydrogenase in synthetic processes. Advanced Synthesis and Catalysis 355: 1709–1714 [Google Scholar]
- 13.Wichmann RA, Vasic-Racki DB (2005) Cofactor regeneration at the lab scale. Adv Biochem Engin/Biotechnol 92: 225–260 [DOI] [PubMed] [Google Scholar]
- 14.Rodríguez C, Lavandera I, Gotor V (2012) Recent advances in cofactor regeneration systems applied to biocatalyzed oxidative processes. Curr Org Chem 16: 2525–2541 [Google Scholar]
- 15.Bisogno FR, Lavandera I, Kroutil W, Gotor V (2009) Tandem concurrent processes: One-pot single-catalyst biohydrogen transfer for the simultaneous preparation of enantiopure secondary alcohols. J Org Chem 74: 1730–1732 [DOI] [PubMed] [Google Scholar]
- 16.Carrea G, Bovara R, Cremonesi P, Lodi R (1984) Enzymatic preparation of 12-ketochenodeoxycholic acid with NADP regeneration. Biotechnol Bioeng 26: 560–563 [DOI] [PubMed] [Google Scholar]
- 17.Lee LG, Whitesides GM (1986) Preparation of optically active 1,2-diols and (-hydroxy ketones using glycerol dehydrogenase as catalyst: Limits to enzyme-catalyzed synthesis due to noncompetitive and mixed inhibition by product. J Org Chem 51: 25–36 [Google Scholar]
- 18.Richter N, Zienert A, Hummel W (2011) A single-point mutation enables lactate dehydrogenase from Bacillus subtilis to utilize NAD+ and NADP+ as cofactor. Eng Life Sci 11: 26–36 [Google Scholar]
- 19.Lavandera I, Kern A, Resch V, Ferreira-Silva B, Glieder Aet al. (2008) One-way biohydrogen transfer for oxidation of sec-alcohols. Org Lett 10: 2155–2158 [DOI] [PubMed] [Google Scholar]
- 20.Müller CA, Akkapurathu B, Winkler T, Staudt S, Hummel Wet al. (2013) In vitro double oxidation of n-heptane with direct cofactor regeneration. Adv Synth Catal 355: 1787–1798 [Google Scholar]
- 21.Sattler JH, Fuchs M, Tauber K, Mutti FG, Faber K, Pfeffer J, Haas T, Kroutil W (2012) Redox self-sufficient biocatalyst network for the amination of primary alcohols. Angew Chem Int Ed 51: 9156–9159 [DOI] [PubMed] [Google Scholar]
- 22.Pival SL, Klimacek M, Nidetzky B (2008) Novel chemo-enzymatic mimic of hydrogen peroxide-forming NAD(P)H oxidase for efficient regeneration of NAD+ and NADP+ . Adv Synth Catal 350: 2305–2312 [Google Scholar]
- 23.Aksu S, Arends IWCE, Hollmann F (2009) A new regeneration system for oxidized nicotinamide cofactors. Adv Synth Catal 351: 1211–1216 [Google Scholar]
- 24.Ferrandi EE, Monti D, Patel I, Kittl R, Haltrich Det al. (2012) Exploitation of a laccase/meldola's blue system for NAD+ regeneration in preparative scale hydroxysteroid dehydrogenase-catalyzed oxidations. Adv Synth Catal 354: 2821–2828 [Google Scholar]
- 25.Könst P, Kara S, Kochius S, Holtmann D, Arends IWCEet al. (2013) Expanding the scope of laccase-mediator systems. Chem Cat Chem 5: 10, 3027–3032 [Google Scholar]
- 26.Kochius S,Park JB,Ley C,Könst P,Hollmann Fet al. (2013) Electrochemical regeneration of oxidised nicotinamide cofactors in a scalable reactor. J Mol Catal B: Enzym http://dx.doi.org/10.1016/j.molcatb.2013.07.006 [Google Scholar]
- 27.Kohlmann C, Märkle W, Lütz S (2008) Electroenzymatic synthesis. J Mol Catal B: Enzym 51: 57–72 [Google Scholar]
- 28.Hollmann F, Arends IWCE, Buehler K (2010) Biocatalytic redox reactions for organic synthesis: Nonconventional regeneration methods. Chem Cat Chem 2: 762–782 [Google Scholar]
- 29.Hollmann F, Hofstetter K, Schmid A (2006) Non-enzymatic regeneration of nicotinamide and flavin cofactors for monooxygenase catalysis. Trends Biotechnol 24: 163–171 [DOI] [PubMed] [Google Scholar]
- 30.Ashrafuddin Ahmed S, Claiborne A (1989) The streptococcal flavoprotein NADH oxidase. I. Evidence linking NADH oxidase and NADH peroxidase cysteinyl redox centers. J Biol Chem 264: 19856–19863 [PubMed] [Google Scholar]
- 31.Ashrafuddin Ahmed S, Claiborne A (1989) The streptococcal flavoprotein NADH oxidase. II. Interactions of pyridine nucleotides with reduced and oxidized enzyme forms. J Biol Chem 264: 19864–19870 [PubMed] [Google Scholar]
- 32.Matsumoto J, Higuchi M, Shimada M, Yamamoto Y, Kamio Y (1996) Molecular cloning and sequence analysis of the gene encoding the H2O-forming NADH oxidase from Streptococcus mutans. Biosci Biotechnol Biochem 60: 39–43 [DOI] [PubMed] [Google Scholar]
- 33.Hoskins DD, Whiteley HR, Mackler B (1962) The reduced diphosphopyridine nucleotide oxidase of Streptococcus faecalis: purification and properties. J Biol Chem 237: 2647–2651 [PubMed] [Google Scholar]
- 34.Gao H, Tiwari MK, Kang YC, Lee J-K (2012) Characterization of H2O-forming NADH oxidase from Streptococcus pyogenes and its application in l-rare sugar production. Bioorg Med Chem 22: 1931–1935 [DOI] [PubMed] [Google Scholar]
- 35.Yu J, Bryant AP, Marra A, Lonetto MA, Ingraham KAet al. (2001) Characterization of the Streptococcus pneumoniae NADH oxidase that is required for infection. Microbiology 147: 431–438 [DOI] [PubMed] [Google Scholar]
- 36.Hummel W, Riebel B (2003) Isolation and biochemical characterization of a new NADH oxidase from Lactobacillus brevis. Biotechnol Lett 25: 51–54 [DOI] [PubMed] [Google Scholar]
- 37.Lopez de Felipe F, Hugenholtz J (2001) Purification and characterisation of the water forming NADH-oxidase from Lactococcus lactis. Int Dairy J 11: 37–44 [Google Scholar]
- 38.Park JT, Hirano J-I, Thangavel V, Riebel BR, Bommarius AS (2011) NAD(P)H oxidase v from Lactobacillus plantarum (NoxV) displays enhanced operational stability even in absence of reducing agents. J Mol Catal B: Enzym 71: 159–165 [Google Scholar]
- 39.Riebel BR, Gibbs PR, Wellborn WB, Bommarius AS (2002) Cofactor Regeneration of NAD+ from NADH: Novel Water-Forming NADH Oxidases. Adv Synth Catal 344: 1156–1168 [Google Scholar]
- 40.Kawasaki S, Ishikura J, Chiba D, Nishino T, Niimura Y (2004) Purification and characterization of an H2O-forming NADH oxidase from Clostridium aminovalericum: Existence of an oxygen-detoxifying enzyme in an obligate anaerobic bacteria. Arch Microbiol 181: 324–330 [DOI] [PubMed] [Google Scholar]
- 41.Stanton TB, Sellwood R (1999) Cloning and characteristics of a gene encoding NADH oxidase, a major mechanism for oxygen metabolism by the anaerobic spirochete Brachyspira (Serpulina) hyodysenteriae. Anaerobe 5: 539–546 [Google Scholar]
- 42.Sakamoto M, Uchimura T, Komagata K (1996) Comparison of H2O-forming NADH oxidase from Leuconostoc mesenteroides subsp. mesenteroides NRIC 1541(T) and H2O2-forming NADH oxidase from Sporolactobacillus inulinus NRIC 1133(T). J Ferment Bioeng 82: 531–537 [Google Scholar]
- 43.Wang L, Chong H, Jiang R (2012) Comparison of alkyl hydroperoxide reductase and two water-forming NADH oxidases from Bacillus cereus ATCC 14579. Appl Microbiol Biotechnol 96: 1265–1273 [DOI] [PubMed] [Google Scholar]
- 44.Ward DE, Donnelly CJ, Mullendore ME, Van Der Oost J, De Vos WM, Crane EJ III (2001) The NADH oxidase from Pyrococcus furiosus: Implications for the protection of anaerobic hyperthermophiles against oxidative stress. Eur J Biochem 268: 5816–5823 [DOI] [PubMed] [Google Scholar]
- 45.Nisar MA, Rashid N, Bashir Q, Gardner QTAA, Shafiq MHet al. (2013) TK1299, a highly thermostable NAD(P)H oxidase from Thermococcus kodakaraensis exhibiting higher enzymatic activity with NADPH. J Biosci Bioeng 116: 39–44 [DOI] [PubMed] [Google Scholar]
- 46.Ojha S, Meng EC, Babbitt PC (2007) Evolution of function in the “two dinucleotide binding domains” flavoproteins. PLoS Compu Biol 3: 1268–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ödman P, Wellborn WB, Bommarius AS (2004) An enzymatic process to L-ketoglutarate from L-glutamate: The coupled system L-glutamate dehydrogenase/NADH oxidase. Tetrahedron: Asymmetry 15: 2933–2937 [Google Scholar]
- 48.Zhang Y-W, Tiwari MK, Gao H, Dhiman SS, Jeya Met al. (2012) Cloning and characterization of a thermostable H2O-forming NADH oxidase from Lactobacillus rhamnosus. Enzyme Microb Technol 50: 255–262 [DOI] [PubMed] [Google Scholar]
- 49.Jia B, Park S-C, Lee S, Pham BP, Yu Ret al. (2008) Hexameric ring structure of a thermophilic archaeon NADH oxidase that produces predominantly H2O. FEBS Journal 275: 5355–5366 [DOI] [PubMed] [Google Scholar]
- 50.Wu X, Kobori H, Orita I, Zhang C, Imanaka Tet al. (2012) Application of a novel thermostable NAD(P)H oxidase from hyperthermophilic archaeon for the regeneration of both NAD+ and NADP+ . Biotechnol Bioeng 109: 53–62 [DOI] [PubMed] [Google Scholar]
- 51.Higuchi M, Shimada M, Yamamoto Y, Hayashi T, Koga Tet al. (1993) Identification of two distinct NADH oxidases corresponding to H2O2-forming oxidase and H2O-forming oxidase induced in Streptococcus mutans. J Gen Microbiol 139: 2343–2351 [DOI] [PubMed] [Google Scholar]
- 52.Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22: 195–201 [DOI] [PubMed] [Google Scholar]
- 53.Balzer D, Ziegelin G, Pansegrau W, Kruft V, Lanka E (1992) KorB protein of promiscuous plasmid RP4 recognizes inverted sequence repetitions in regions essential for conjugative plasmid transfer. Nucleic Acids Res 20: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blaedel WJ, Uhl JM (1975) Nature of materials in serum that interfere in the glucose oxidase peroxidase o-dianisidine method for glucose, and their mode of action. Clin Chem 21: 119–124 [PubMed] [Google Scholar]
- 55.Liu H, Naismith JH (2009) A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr Purif 63: 102–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wong TS, Tee KL, Hauer B, Schwaneberg U (2004) Sequence saturation mutagenesis (SeSaM): a novel method for directed evolution. Nucleic Acids Res 32: e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rossmann MG, Moras D, Olsen KW (1974) Chemical and biological evolution of a nucleotide binding protein. Nature 250: 194–199 [DOI] [PubMed] [Google Scholar]
- 58.Scrutton NS, Berry A, Perham RN (1990) Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 343: 38–43 [DOI] [PubMed] [Google Scholar]
- 59.Wallen JR, Paige C, Mallett TC, Karplus PA, Claiborne A (2008) Pyridine nucleotide complexes with Bacillus anthracis coenzyme A-disulfide reductase: A structural analysis of dual NAD(P)H specificity. Biochemistry 47: 5182–5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lountos GT, Jiang R, Wellborn WB, Thaler TL, Bommarius ASet al. (2006) The crystal structure of NAD(P)H oxidase from Lactobacillus sanfranciscensis: Insights into the conversion of O2 into two water molecules by the flavoenzyme. Biochemistry 45: 9648–9659 [DOI] [PubMed] [Google Scholar]
- 61.Mittl PRE, Berry A, Scrutton NS, Perham RN, Schulz GE (1994) Anatomy of an engineered NAD-binding site. Protein Sci 3: 1504–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Antonio Bocanegra J, Scrutton NS, Perham RN (1993) Creation of an NADP-dependent pyruvate dehydrogenase multienzyme complex by protein engineering. Biochemistry 32: 2737–2740 [DOI] [PubMed] [Google Scholar]
- 63.Chen Z, Lee WR, Chang SH (1991) Role of aspartic acid 38 in the cofactor specificity of Drosophila alcohol dehydrogenase. Eur J Biochem 202: 263–267 [DOI] [PubMed] [Google Scholar]
- 64.Ehrensberger AH, Elling RA, Wilson DK (2006) Structure-guided engineering of xylitol dehydrogenase cosubstrate specificity. Structure 14: 567–575 [DOI] [PubMed] [Google Scholar]
- 65.Andreadeli A, Platis D, Tishkov V, Popov V, Labrou NE (2008) Structure-guided alteration of coenzyme specificity of formate dehydrogenase by saturation mutagenesis to enable efficient utilization of NADP+ . FEBS J 275: 3859–3869 [DOI] [PubMed] [Google Scholar]
- 66.Morina K, Schulte M, Hubrich F, Dörner K, Steimle Set al. (2011) Engineering the respiratory complex I to energy-converting NADPH:Ubiquinone oxidoreductase. J Biol Chem 286: 34627–34634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Könst P, Merkens H, Kara S, Kochius S, Vogel Aet al. (2012) Enantioselective oxidation of aldehydes catalyzed by alcohol dehydrogenase. Angew Chem Int Ed 51: 9914–9917 [DOI] [PubMed] [Google Scholar]
- 68.Riebel BR, Gibbs PR, Wellborn WB, Bommarius AS (2003) Cofactor regeneration of both NAD+ from NADH and NADP+ from NADPH: NADH oxidase from Lactobacillus sanfranciscensis. Adv Synth Catal 345: 707–712 [Google Scholar]
- 69.Findrik Z, Šimunovic I, Vasic-Racki D (2008) Coenzyme regeneration catalyzed by NADH oxidase from Lactobacillus brevis in the reaction of l-amino acid oxidation. Biochem Eng J 39: 319–327 [Google Scholar]
- 70.Hummel W, Kuzu M, Geueke B (2003) An efficient and selective enzymatic oxidation system for the synthesis of enantiomerically pure D-tert-Leucine. Org Lett 5: 3649–3650 [DOI] [PubMed] [Google Scholar]