

Abstract
Sexually dimorphic phenotypes are thought to largely result from sex differences in gene expression, and genes with sex-biased expression have been well characterized in adults of many species. Although most sexual dimorphisms manifest in adults, many result from sex-specific developmental trajectories, implying that juveniles may exhibit significant levels of sex-biased expression. However, it is unclear how much sex-biased expression occurs before reproductive maturity and whether preadult sex-biased genes should exhibit the same evolutionary dynamics observed for adult sex-biased genes. In order to understand the continuity, or lack thereof, and evolutionary dynamics of sex-biased expression throughout the life cycle, we examined sex-biased genes in pre-gonad tissue of two preadult stages and compared them with the adult gonad of Drosophila melanogaster. We found that the majority of the genome is sex-biased at some point in the life cycle, with some genes exhibiting conserved sex-biased expression and others displaying stage-specific sex bias. Our results also reveal a far more complex pattern of evolution for sex-biased genes throughout development. The most rapid evolutionary divergence occurred in genes expressed only in larvae within each sex, compared with continuously expressed genes. In females—but not males—this pattern appeared to be due to relaxed purifying selection in larva-limited genes. Furthermore, genes that retained male bias throughout life evolved more rapidly than stage-specific male-biased genes, due to stronger purifying selection in stage-specific genes. However, female-biased genes that were specific to larvae evolved most rapidly, a pattern that could not be definitively attributed to differences in adaptive evolution or purifying selection, suggesting that pleiotropic constraints on protein-coding sequences can arise when genes are broadly expressed across developmental stages. These results indicate that the signature of sex-specific selection can be detected well before reproductive maturity and is strongest during development.
Keywords: development, sex-specific selection, sexual conflict, sexual dimorphism
Introduction
Many sexual species exhibit considerable sexual dimorphism. These sex differences are often complex and therefore involve many genes. However, males and females share the majority of their genome, suggesting that much of the basis for this dimorphism must result from differences in expression of the same genes. Sex differences in gene expression have been well characterized in adult animals, particularly in classic model organisms, and several patterns have emerged that appear common to a wide range of taxa. In adults, more genes show a stronger bias toward male expression than toward female expression (reviewed by Ellegren and Parsch 2007; Zhang et al. 2007; Small et al. 2009; Graveley et al. 2011; Zhao et al. 2011; Martins et al. 2013). Furthermore, male-biased genes typically show more rapid gene turnover compared with female-biased and unbiased genes, and elevated sequence divergence, due at least in part to more rapid adaptive evolution, particularly for genes expressed in the germline. In contrast, female-biased genes often evolve at similar or slower rates compared with unbiased genes (Ellegren and Parsch 2007; Zhang et al. 2007; Parsch and Ellegren 2013). Together, these observations have led to the hypothesis that much of the evolution of sex-biased genes is driven by selection on males via sexual selection and conflict (Swanson and Vacquier 2002; Ranz et al. 2003; Connallon and Knowles 2005; Small et al. 2009; Artieri and Singh 2010).
Currently, studies of sex-biased gene expression focus almost exclusively on adults, and the ontogenetic complexity of sexual dimorphism and sex-biased genes remains largely unexplored. This trend reflects the expectation that sex-biased expression and sex-specific selection will be minimal in juveniles, which is in many ways reasonable, based on the outward apparent sexual monomorphism in juveniles of many species. Moreover, empirical studies suggest that the sexes are subject to similar selection as juveniles, with sex-specific selection and intralocus conflict only beginning in adults when the sexes have divergent reproductive interests (Chippindale et al. 2001; Rice and Chippindale 2001; Gibson et al. 2002). Thus, to the extent that sex-biased gene expression represents a history of sexual antagonism, minimal juvenile sex-biased expression should be expected, and those genes that show sex-biased expression only in juveniles should not display the same rapid evolution exhibited by adult sex-biased genes.
However, recent studies have challenged the generality both of juvenile sexually monomorphic gene expression and of uniform selection on juvenile males and females. A few studies have reported considerable sex-biased gene expression in juvenile stages (e.g., rainbow trout, Hale et al. 2011; silkworm, Zhao et al. 2011), and in some cases the pattern of sex-biased expression is quite distinct from the adult stage (Mank et al. 2010). Furthermore, sex-specific selection can act on juvenile fitness even without gross phenotypic differences between the sexes (Zikovitz and Agrawal 2013). These results are hard to reconcile with the view of juvenile sexual monomorphism. As a result, we currently lack a comprehensive understanding of the patterns and evolutionary dynamics of sexually dimorphic gene expression throughout development.
Here, we use high-throughput RNA sequencing to investigate the ontogenetic basis of sex-biased gene expression in Drosophila melanogaster and ask whether the expression and evolutionary dynamics of sex-biased genes in two preadult stages (third instar larvae and pre-pupae) reflect the adult pattern. In D. melanogaster, male-biased genes show a greater magnitude of bias than female-biased genes (Ranz et al. 2003; Ellegren and Parsch 2007) and exhibit more rapid protein divergence, due at least in part to more rapid adaptive evolution (Zhang et al. 2004; Jagadeeshan and Singh 2005; Zhang and Parsch 2005; Gnad and Parsch 2006; Pröschel et al. 2006; Haerty et al. 2007; Zhang et al. 2007; Baines et al. 2008; Artieri et al. 2009; Meisel 2011; Grath and Parsch 2012; Müller et al. 2012). These patterns are related to both sex differences in expression breadth across tissues, with sex-biased genes specific to reproductive tissues evolving more rapidly than broadly expressed genes (Grath and Parsch 2012; Parsch and Ellegren 2013), and chromosomal linkage, with male-biased X-linked genes evolving most rapidly (Baines et al. 2008; Grath and Parsch 2012; Kayserili et al. 2012; Müller et al. 2012; Parsch and Ellegren 2013; see also Meisel et al. 2012b).
To examine sex-biased expression, we focus on the most sexually dimorphic tissue of pre-gonad imaginal discs (in larvae and pre-pupae, stages at which sex differentiation is an ongoing process) and adult gonads. By isolating a single tissue, our study overcomes the problem of tissue allometry that has complicated analyses of sex-biased gene expression in studies of whole organisms, which can create an imprecise picture of overall sex bias (Chintapalli et al. 2007; Catalan et al. 2012; Perry and Mank forthcoming) and which very likely varies across larval, pre-pupal, and adult body plans. We ask three questions. 1) What is the pattern of sex-biased gene expression in pre-adults, including linkage, tissue specificity, and gene function? 2) What is the pattern of continuity of expression throughout development for male- and female-biased genes? 3) How does ontogeny interact with sex-specific selection to shape the divergence of sex-biased genes? Our results indicate that sex-biased gene expression in juvenile stages is both considerable and dynamic, and that the evolution of sex-biased genes throughout ontogeny is considerably more complex than previously realized.
Results
At both the third instar larval and the pre-pupal stages we sampled, pre-gonad tissue is more differentiated in males than females. In males, primary spermatocytes begin to dominate the pre-gonads of third instar larvae and are the dominant cell type in pre-pupae, with the first meiotic divisions occurring around the onset of pupation (Bodenstein 1950). Spermatocytes synthesize the RNA required for later spermatid development (Lindsley and Tokuyasu 1980). In females, pre-gonad tissue contains only oogonia with no oocytes yet present; the first meiotic divisions occur as the adult female emerges (King 1970; Mahowald and Kambysellis 1980). Ovarioles are well differentiated in pre-pupae, but differentiation has not yet commenced at the larval stage we sampled (Bodenstein 1950).
We found that gene expression measures among our biological replicates of each sex and life stage were strongly and positively correlated, with r ranging from 0.947 to 0.999 (P < 0.00005). Adult gene expression was positively correlated with a previous adult data set (rMALE = 0.865, n = 8,847, P < 0.0001; rFEMALE = 0.846, n = 7,474, P < 0.0001; Gan et al. 2010).
Sex-Biased Gene Expression in Larvae and Pre-pupae
In contrast to previous whole-body studies of pre-adult Drosophila (Lebo et al. 2009; Meisel et al. 2012a), we found that more than half of all expressed genes showed moderate to high levels of sex-biased expression (≥2-fold expression difference, Padj < 0.05). In both larvae and pre-pupae, more genes were female- than male-biased for all but the most extremely sex-biased genes (those with ≥10-fold expression difference, Padj < 0.05; fig. 1 and supplementary table S1, Supplementary Material online). However, the magnitude of differential expression between the sexes was greatest for male-biased genes (fig. 1 and supplementary table S1, Supplementary Material online). These patterns were broadly similar in larvae and pre-pupae.
Fig. 1.

Relative gene expression in males and females, for larval and pre-pupal autosomal and X-linked genes. Percentages within each panel indicate the percentage of all genes expressed at that stage that were at least 2-fold male-biased (MB; upper left, top number), male-limited (upper left, bottom number; i.e., genes with no expression in females), female-biased (FB; lower right, top number), and female-limited (lower right, bottom number), with all categories further defined by significantly different expression between the sexes (Padj < 0.05, FDR-corrected). The number of expressed genes is given in the lower left. Unbiased (UB) genes were defined as Padj > 0.05 and less than 2-fold difference between the sexes.
Compared with autosomes, a larger proportion of genes on the X chromosome was female-biased and a lower proportion was male-biased in both larvae and pre-pupae (fig. 1 and supplementary table S1, Supplementary Material online), consistent with both the feminization of the X chromosome (Meisel et al. 2012a) and incomplete or absent dosage compensation in the male germline (Meiklejohn et al. 2011; Meiklejohn and Presgraves 2012; but see also Hense et al. 2007; Vicoso and Charlesworth 2009). To further assess whether dosage compensation was absent in our preadult samples, we calculated 1) the ratio of male to female expression for X-linked genes at each stage (log2(mean male expression)/log2(mean female expression)), which should equal one with full dosage compensation, and 2) the ratio of X to autosomal expression in each sex and at each stage, which should equal one with full dosage compensation. We found, first, that the male:female expression ratio was 0.85 in larvae and 0.79 in pre-pupae. Second, the male X:autosome ratios were 0.90 and 0.88 in larvae and pre-pupae, respectively. The data therefore support a scenario of incomplete dosage compensation at these developmental stages.
Extreme sex bias (≥10-fold difference, Padj < 0.05) arises in different ways for male- and female-biased genes (fig. 2). Extremely male-biased genes are the product of both increased expression in males and decreased expression in females. In contrast, extremely female-biased genes are not upregulated in females but instead arise from downregulation in males. This pattern was more pronounced in pre-pupae than in larvae.
Fig. 2.

Mean relative gene expression (fragments per kilobase of transcript per million mapped reads) at different thresholds of sex-bias, for male- and female-biased genes expressed in males (blue) and females (red). For each threshold, sex-biased genes were categorized by a significant difference between the sexes (FDR-corrected). Error bars were too small to be meaningfully displayed.
Continuity of Sex-Biased Gene Expression
Relative gene expression was highly conserved between larvae and pre-pupae (supplementary fig. S1, Supplementary Material online). Within a sex, the developmental stages showed much greater similarity in gene expression than the sexes showed with each other within a developmental stage (r[FEMALE LARVAE-PREPUPAE] = 0.8831; r[MALE LARVAE-PREPUPAE] = 0.9553; r[LARVAL MALE-FEMALE] = 0.3984; r[PREPUPAL MALE-FEMALE] = 0.2737; all P < 0.0001).
To examine sex-bias conservation through to the adult stage, we examined the overlap in sex-bias categories among stages (fig. 3 and supplementary figs. S1 and S2, Supplementary Material online). For autosomal loci, male-biased genes more often retained their sex bias throughout life (58% vs. 46% for female-biased genes), whereas female-biased genes more often showed stage-specific sex bias (30% vs. 25%; fig. 3). This pattern was reversed for X-linked loci, potentially due to the overall reduction in male-biased genes on the D. melanogaster X. Adults showed higher stage specificity for sex bias compared with larvae and pupae. Altogether, 46–65% of sex-biased genes retained their sex bias throughout life, whereas 18–30% showed stage-specific sex bias.
Fig. 3.

Venn diagrams of the number and percentage of genes showing sex-biased gene expression in larvae, pre-pupae, and adults.
For the above analysis, we categorized adult sex bias using our adult data (see Materials and Methods). We compared the results with those obtained when adult sex bias was categorized using a meta-analysis of previous gene expression studies on mainly whole body flies (SEBIDA; Gnad and Parsch 2006; supplementary fig. S2, Supplementary Material online). We focused on the meta-analysis data as the best available summary across many studies. Previous individual studies that have examined adult gonad alone (as we have; e.g., Parisi et al. 2003, 2004) have been based on microarrays with less sensitivity to lowly expressed genes (Harrison, Wright et al. 2012), therefore resulting in relatively few genes called as sex-biased, especially for female-biased genes. In fact, by comparing data from five studies of adult D. melanogaster (including this one), we found that gene expression data are more similar between whole-body and gonad-only studies than between microarray and RNA-seq platforms (supplementary table S2, Supplementary Material online). Although our data were strongly correlated with those of the SEBIDA meta-analysis, our data yielded more genes categorized as sex-biased in adults, and therefore more genes with overlapping sex-biased expression among stages (compare fig. 3 and supplementary fig. S2, Supplementary Material online). This difference might result from the fact that the meta-analysis incorporates many studies with multiple sources of variation, or from our data being exclusively from the gonad, as some of the patterns of sex bias from the gonad can be diluted through the inclusion of somatic tissue in whole-body studies, or from differences in platforms among studies (Harrison, Wright et al. 2012).
Reversals in Sex-Specific Gene Expression
In order to identify genes that reversed the direction of sex-specific expression between developmental stages, we filtered our data for genes that were sex-biased (≥2-fold differential expression between the sexes and Padj < 0.05) or sex-limited (expressed in one sex only; Padj < 0.05) in larvae or pre-pupae, and sex-biased or sex-limited in the opposite direction in adults. Autosomal genes were more than three times as likely to switch from female-biased or female-limited expression to male-biased or male-limited expression as vice versa (7.5% [182/2,427] vs. 1.8% [37/2,026]; 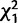 = 80.8, P < 0.000001; supplementary fig. S3, Supplementary Material online). X-linked genes showed a similar pattern, although the difference was smaller and not statistically significant (3.3% [25/743] vs. 1.5% [5/333];
= 80.8, P < 0.000001; supplementary fig. S3, Supplementary Material online). X-linked genes showed a similar pattern, although the difference was smaller and not statistically significant (3.3% [25/743] vs. 1.5% [5/333]; 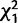 = 2.9, P = 0.08; supplementary fig. S4, Supplementary Material online). For these results, we classified adult sex bias using our own data, and we obtained similar results using the SEBIDA meta-analysis (supplementary text, Supplementary Material online; Gnad and Parsch 2006).
= 2.9, P = 0.08; supplementary fig. S4, Supplementary Material online). For these results, we classified adult sex bias using our own data, and we obtained similar results using the SEBIDA meta-analysis (supplementary text, Supplementary Material online; Gnad and Parsch 2006).
Evolutionary Divergence of Sex-Biased Genes
To test for differences in the rate of evolutionary divergence among categories of sex-biased and sex-limited genes, we compared the ratio of nonsynonymous to synonymous nucleotide substitutions (dN/dS) for sex-biased, sex-limited, and unbiased genes in larvae and pre-pupae along the D. melanogaster terminal lineage (table 1). The patterns we observed are broadly consistent with previous reports for adult flies; however, we identified several interesting deviations. Female-biased genes evolved more slowly than unbiased genes in both life stages (table 1). In contrast, male-biased and male-limited genes evolved more rapidly than unbiased genes, although this difference was not significant for all categories (table 1).
Table 1.
Divergence Estimates for Sex-Biased and Sex-Limited Gene Categories in Larvae and Pre-pupae.
| Stage | Location | Categorya | n Genes (kb/1000)b | dN (P)c | dS (P)c | dN/dS (P)c | DoS (P)d |
|---|---|---|---|---|---|---|---|
| Larva | Autosomes | UB | 659 (41.49) | 0.0062 | 0.0662 | 0.0930 | −0.14 |
| MB | 1202 (90.13) | 0.0082 (<0.001) | 0.0744 (<0.001) | 0.1107 (0.004) | −0.10 (<0.0001) | ||
| ML | 290 (24.83) | 0.0092 (<0.001) | 0.0780 (<0.001) | 0.1179 (0.004) | −0.12 (0.021) | ||
| FB | 1915 (167.31) | 0.0051 (0.001) | 0.0691 (0.013) | 0.0742 (<0.001) | −0.16 (0.116) | ||
| FL | 179 (12.23) | 0.0067 (0.410) | 0.0711 (0.023) | 0.0944 (0.892) | −0.17 (0.113) | ||
| X | UB | 58 (4.42) | 0.0091 | 0.0809 | 0.1123 | −0.10 | |
| MB | 111 (11.70) | 0.0149 (0.016) | 0.0975 (0.058) | 0.1524 (0.101) | −0.06 (0.244) | ||
| ML | 35 (3.85) | 0.0140 (0.044) | 0.1162 (0.004) | 0.1207 (0.811) | −0.09 (0.858) | ||
| FB | 442 (27.38) | 0.0050 (<0.001) | 0.0746 (0.170) | 0.0676 (0.004) | −0.14 (0.134) | ||
| FL | 36 (2.51) | 0.0056 (0.029) | 0.0794 (0.845) | 0.0703 (0.083) | −0.14 (0.458) | ||
| Pre-pupa | Autosomes | UB | 694 (44.31) | 0.0065 | 0.0674 | 0.0971 | −0.15 |
| MB | 1029 (75.35) | 0.0080 (<0.001) | 0.0738 (<0.001) | 0.1080 (0.076) | −0.11 (<0.0001) | ||
| ML | 440 (38.11) | 0.0098 (<0.001) | 0.0796 (<0.001) | 0.1225 (0.001) | −0.10 (<0.0001) | ||
| FB | 2080 (174.51) | 0.0050 (<0.001) | 0.0682 (0.424) | 0.0733 (<0.001) | −0.16 (0.214) | ||
| FL | 204 (14.39) | 0.0070 (0.455) | 0.0729 (0.006) | 0.0958 (0.881) | −0.17 (0.074) | ||
| X | UB | 62 (4.94) | 0.0088 | 0.0800 | 0.1098 | −0.13 | |
| MB | 95 (9.18) | 0.0120 (0.127) | 0.0961 (0.046) | 0.1246 (0.490) | −0.10 (0.109) | ||
| ML | 55 (6.54) | 0.0187 (<0.001) | 0.1135 (<0.001) | 0.1651 (0.071) | −0.03 (0.005) | ||
| FB | 459 (28.83) | 0.0052 (<0.001) | 0.0755 (0.345) | 0.0690 (0.003) | −0.14 (0.889) | ||
| FL | 30 (2.06) | 0.0048 (0.038) | 0.0762 (0.601) | 0.0633 (0.081) | −0.10 (0.314) | ||
aUnbiased (UB), male-biased (MB), female-biased (FB), male-limited (ML), or female-limited (FL).
bThe number of genes and total alignment length.
cP values from a permutation test comparing each set of genes with the unbiased genes for that category. Significant P values (<0.05) are italicized.
dMean values for the direction of selection statistic, calculated as the difference in the proportion of fixed differences that are nonsynonymous and the proportion of polymorphisms that are nonsynonymous (Stoletzki and Eyre-Walker 2011). Negative values indicate purifying selection; positive values indicate adaptive evolution. P values from a Wilcoxon nonparametric test.
To assess whether differences in divergence rates were due to increased positive selection or relaxed purifying selection, we used existing data on standing variation in protein-coding sequences (from the Drosophila Genome Reference Panel, DGRP; Mackay et al. 2012) to calculate the direction of selection (DoS) statistic, a measure of the difference in the proportions of nonsynonymous fixed differences and polymorphisms (Stoletzki and Eyre-Walker 2011). Positive DoS values indicate positive selection and adaptive evolution, whereas zero values indicate neutral evolution and negative values indicate purifying selection and the presence of segregating weakly deleterious mutations. Estimates of DoS were negative across all sex-bias categories (table 1). Comparisons among categories revealed evidence for relaxed purifying selection driving the rapid diversification of male-biased and male-limited autosomal genes (table 1). In contrast, female-biased genes, which evolved more slowly than unbiased genes, showed a nonsignificant trend toward stronger purifying selection compared with unbiased genes (table 1).
Evolutionary Divergence of Stage-Specific and Continuously Sex-Biased Genes
Given the extensive overlap in sex bias throughout life (fig. 3 and supplementary fig. S1, Supplementary Material online), the divergence of sex-biased genes in one stage may be driven by selection acting at another stage. To examine this possibility, we tested for differences in evolutionary divergence in genes that showed stage-specific or continuous sex-specific expression. We focused on autosomal genes because there were not enough X-linked stage-specific genes for a meaningful test.
We conducted two complementary tests. First, we examined divergence in genes with stage-specific expression in each sex—that is, expressed in a sex in one developmental stage and not expressed in that sex in the other stages—compared with genes continuously expressed in that sex throughout life. In both sexes, genes with stage-specific expression showed more rapid divergence than genes with continuous expression in that sex (fig. 4a and supplementary table S3, Supplementary Material online). Genes with expression limited to the larval stage evolved most rapidly within both sexes. We compared DoS estimates among these groups of genes to assess the contributions of positive and purifying selection to their evolutionary divergence. In females, the results are consistent with more rapid evolution due to relaxed purifying selection acting on larval-specific genes (fig. 4c). In contrast, we found similar DoS values for larval-specific and continuously expressed genes in males, and we were therefore unable to identify whether the more rapid evolution of larval-specific genes was due to relaxed purifying selection (fig. 4c).
Fig. 4.

Divergence rates (a, b; the ratio of nonsynonymous to synonymous substitutions, dN/dS) and the direction of selection (c, d) for autosomal genes with continuous or stage-limited expression within a sex (a, c), or continuous or stage-limited sex-bias in expression (b, d). Asterisks indicate a significant difference (P < 0.05, permutation test) between stage-specific genes and continuously expressed or sex-biased genes for that category. Bars indicate 95% bootstrapped confidence intervals; the bars for genes always expressed in (a) are present but very small due to the large sample sizes. The number of genes in each category is indicated.
Second, we examined genes for which sex bias in expression was stage-specific, compared with genes that were consistently male- or female-biased, or unbiased, throughout life. Male- and female-biased genes show distinct patterns of evolutionary divergence depending on the extent of sex-bias conservation throughout development (fig. 4b and supplementary table S4, Supplementary Material online). Genes that remained male-biased throughout life evolved more rapidly than genes that were male-biased in one stage only (fig. 4b). This pattern was similar to that of unbiased genes. In fact, when this ontogenetic pattern of sex bias was considered, male-biased genes no longer evolved more rapidly than unbiased genes; the dN/dS ratios of continuously male-biased and unbiased genes were statistically indistinguishable (permutation test, P = 0.605).
In contrast, female-biased genes with stage-specific sex bias evolved more rapidly than genes that maintained their female bias throughout development (fig. 4b). The most rapidly evolving female-biased genes—those with larva-specific female bias—had a dN/dS ratio that was statistically indistinguishable from the most rapidly evolving male-biased and unbiased genes (with continuous male-biased or unbiased expression; permutation tests, P = 0.434, P = 0.808, respectively), although the limited number of genes with larva-specific female bias would have reduced the statistical power of this test.
The DoS value for continuously male-biased genes is consistent with the action of relaxed purifying selection promoting their more rapid evolution (fig. 4d). However, DoS values for consistent and stage-specific genes with either unbiased or female-biased expression were statistically indistinguishable (fig. 4d), providing no support for differences in positive or purifying selection as an explanation for variation in the divergence rates of these genes.
We conducted the above analysis by categorizing adult sex-biased genes using our data. When we categorized adult sex bias using a meta-analysis of mostly whole body samples (SEBIDA, Gnad and Parsch 2006), the results showed similarities and differences (supplementary fig. S3 and table S5, Supplementary Material online). Again, the differences may be due to the greater sensitivity of RNA-seq compared with microarrays and our use of gonad tissue compared with mainly whole-body studies. Using the SEBIDA data, male-biased and female-biased genes were distinct from each other, with genes that were male-biased in all three life stages again evolving more rapidly than genes and with DoS values supporting relaxed purifying selection as the driving force. Genes that were continuously female-biased again evolved relatively slowly. However, here adult-specific female-biased genes evolved more rapidly than other female-biased genes, with DoS values supporting relaxed purifying selection driving this divergence, and unbiased genes showed no significant differences in divergence or DoS values between continuous and stage-specific expression.
Evolutionary Divergence of Tissue-Limited and Broadly Expressed Genes
In adult D. melanogaster, female-biased genes are more likely than male-biased genes to be expressed in multiple tissues (Grath and Parsch 2012). This may explain why female-biased genes in adults evolve more slowly, as the constraints of pleiotropic expression in multiple tissues can slow down protein evolution (Mank et al. 2008; Meisel 2011). Additionally, adult sex-biased genes with tissue-limited expression—particularly adult male-biased genes expressed only in reproductive tissues—tend to evolve more rapidly than those expressed broadly (Mank et al. 2008; Mank 2009; Meisel 2011; Grath and Parsch 2012). We tested whether these patterns are replicated in larvae by referencing gene expression data for seven larval somatic tissues (available in the FlyAtlas database, Chintapalli et al. 2007). We categorized sex-biased and sex-limited genes based on whether their expression was limited to larval pre-gonad only (i.e., expressed in our data set and with signal intensity <100 in each somatic tissue) or larval pre-gonad and at least one somatic tissue. Overall, 7,021 genes expressed in our larval data set were also reported in the FlyAtlas database. Compared with unbiased genes, female-biased genes were more likely to be expressed in both larval soma and pre-gonad, whereas female-limited, male-biased, and male-limited genes were less likely to be expressed broadly (table 2). Furthermore, autosomal genes were more likely to be expressed in multiple tissues than X-linked genes (61% vs. 57%; 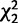 = 9.5, P = 0.009).
= 9.5, P = 0.009).
Table 2.
Breadth of Expression of Genes Expressed in Larval Pre-gonads, by Sex-Bias Category.
| Location | Categorya | n Genes Expressed in Both Larval Soma and Pre-gonad/Total (%) | 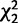 |
P Value |
|---|---|---|---|---|
| Autosomes | UB | 558/843 (66%) | ||
| MB | 728/1638 (44%) | 110.6 | <0.000001 | |
| ML | 78/437 (18%) | 275.1 | <0.000001 | |
| FB | 2070/2583 (80%) | 73.9 | <0.000001 | |
| FL | 128/297 (43%) | 53.3 | <0.000001 | |
| X | UB | 55/116 (47%) | ||
| MB | 61/247 (25%) | 22.2 | 0.000015 | |
| ML | 7/78 (9%) | 35.7 | <0.000001 | |
| FB | 554/717 (77%) | 49.6 | <0.000001 | |
| FL | 25/65 (38%) | 2.8 | 0.24 |
aGenes categorized by their expression in larval pre-gonad as unbiased (UB), male-biased (MB), female-biased (FB), male-limited (ML), or female-limited (FL). For each category, the frequency of broadly expressed genes is tested against unbiased genes for that chromosomal location.
To test whether expression breadth influences evolutionary divergence for preadult sex-biased genes, we compared genes with expression limited to larval pre-gonads or expression in pre-gonads and at least one somatic tissue. In larvae, autosomal genes specific to pre-gonad tissue evolved more rapidly than genes expressed more broadly (fig. 5a). For most sex-bias categories, DoS values did not differ significantly depending on expression breadth (fig. 5b). A curious exception was a higher DoS value for female-biased genes expressed in multiple tissues compared with pre-gonad specific genes, indicating relaxed purifying selection despite their slower divergence in protein-coding sequence (fig. 5). However, the magnitude of difference in DoS was not large between the categories. Like autosomal genes, male- and female-biased X-linked genes showed more rapid divergence when they were broadly expressed rather than limited to pre-gonad tissue (supplementary table S6, Supplementary Material online). There were too few X-linked unbiased and sex-limited genes with divergence data available to perform a meaningful test.
Fig. 5.

Divergence rates (a; the ratio of nonsynonymous to synonymous substitutions, dN/dS) and the direction of selection (b) for unbiased (UB), male-biased (MB), male-limited (ML), female-biased (FB), and female-limited (FL) autosomal genes expressed in multiple larval tissues (pre-gonad and at least one somatic tissue) or pre-gonad only. Asterisks indicate significant differences (P < 0.05, permutation test). Lines indicate 95% bootstrapped confidence intervals. The number of genes in each category is indicated.
Gene Ontology
Gene ontology (GO) analyses can be a useful exploratory tool, although it is difficult to draw firm conclusions about functional differences based on GO analyses alone (e.g., Pavlidis et al. 2012). To explore how gene function relates to developmental patterns of sex-biased expression, we tested for enriched GO terms in larvae and pre-pupae for 1) sex-biased, sex-limited, and unbiased genes, 2) genes with continuous or stage-specific sex bias, and 3) genes with consistent or reversed sex bias throughout development. We compared each subset of genes to all genes expressed at that stage.
In both larvae and pre-pupae, female-biased and female-limited genes were most enriched for metabolic, cellular, and developmental processes, as well as channel activity and compound binding functions, whereas male-biased genes were most enriched for male gamete generation and spermatogenesis (supplementary table S6, Supplementary Material online). This pattern is consistent with the hypothesis that female-biased genes function in more varied ways than male-biased genes, which is consistent with female-biased genes showing greater breadth of expression across tissues and developmental stages.
Genes that maintained their sex bias throughout development showed distinct enrichment from genes with stage-specific sex bias (supplementary table S7, Supplementary Material online), particularly for female-biased genes. Genes with female bias throughout life were most enriched for cellular and intracellular parts and cellular processes, whereas continuously male-biased genes were most enriched for male gamete generation and spermatogenesis. Genes that switched from female-biased to male-biased expression throughout development were most enriched for extracellular components, whereas genes that switched from male-biased to female-biased expression were not significantly enriched for any terms (supplementary table S8, Supplementary Material online).
Discussion
This study expands ongoing efforts to characterize the D. melanogaster developmental transcriptome (Arbeitman et al. 2002; Daines et al. 2011; Graveley et al. 2011; Mikhaylova and Nurminsky 2011) and to investigate the evolutionary genetics of potentially sexually antagonistic genes (Innocenti and Morrow 2010). Characterizing developmental patterns of sex differences in gene expression represents a major gap in understanding the evolutionary genetics of sex-biased genes (Artieri et al. 2009; Artieri and Singh 2010; Carreira et al. 2011; Meisel et al. 2012a; Parsch and Ellegren 2013). Our study redresses this gap by examining patterns of sex-biased gene expression and sex-specific selection in two juvenile stages of D. melanogaster. We find that investigating sex-biased genes from a developmental perspective yields novel insights. Many genes exhibited conserved sex-biased expression throughout development, whereas many others displayed stage-specific sex-biased expression. The pattern of continuity matters for the evolution of these genes: we detected more rapid evolution of genes expressed only in larvae within each sex compared with continuously expressed genes. Furthermore, genes with continuously male-biased expression evolved more rapidly than stage-specific male-biased genes by relaxed purifying selection, whereas female-biased genes showed the reverse pattern but with no evidence for differences in purifying or positive selection. Overall, when considering gene expression throughout development, differences in evolutionary divergence rates between male-biased and female-biased genes are minimized.
Sex-Biased Genes in Preadult Flies
Despite the lack of conspicuous phenotypic sexual dimorphism in preadult flies, we found that gene expression differences between the sexes were vast compared with the differences between developmental stages (fig. 3 and supplementary table S1 and fig. S1, Supplementary Material online). The frequency of extremely male-biased genes was higher in larvae than in pre-pupae (fig. 1 and supplementary table S1, Supplementary Material online), which may reflect a greater discrepancy between the sexes in gametogenesis in larvae before the onset of ovarian development. In contrast to our results, a previous microarray study reported more male- than female-biased expression in third instar larvae and did not detect decreased X-linked male-biased expression (Meisel et al. 2012a), potentially due to the differences between whole-body and tissue-specific microarray and RNA-seq analyses. Lebo et al. (2009) took a different approach by contrasting expression between wild-type flies and mutants lacking germline tissue, using microarrays, and report higher levels of sex-biased expression in pupae that are closer to what we observed in larvae and pre-pupae.
Continuity and Specificity in Sex Bias
In contrast to ontogenetic studies of sex bias in birds (Mank et al. 2010), where sex-biased expression is largely discontinuous among developmental stages, the D. melanogaster genome shows a complex mixture of genes with both continuous and stage-specific sex-biased expression; in fact, roughly 50% of genes fell into each group (fig. 3). Genes were more likely to retain male-biased than female-biased expression throughout development, which probably reflects the earlier onset of male gametogenesis (Bodenstein 1950). Consistent with this interpretation, continuously male-biased genes were enriched for GO terms associated with gametogenesis, whereas larval- and pre-pupal-specific male-biased genes were not enriched for any terms (supplemental table S6, Supplementary Material online).
Our finding that more genes switched from female to male expression than vice versa is consistent with the pattern in silkworm (Zhao et al. 2011). We found that adult male flies appeared to co-opt genes encoding extracellular components in larval females. Further functional characterization of these genes is needed to understand both the mechanistic basis of reversals in sex bias and the conservation of this pattern across taxa.
In contrast to our results, Gan et al. (2010) found higher stage specificity of male-biased genes compared with female-biased genes, despite the broad consistency between our data and theirs noted above. The difference might arise because the wild-type flies used in their study were less than 1 day posteclosion, such that oogenesis may not have been fully activated.
Evolutionary Divergence in Ontogenetic Context
The pattern of evolutionary divergence for sex-biased genes changed considerably when we accounted for developmental patterns. When we ignored the consistency of expression across developmental stages and considered divergence rates of larval and pre-pupal sex-biased genes, we obtained similar results to those reported for adult sex-biased genes (reviewed by Ellegren and Parsch 2007; Parsch and Ellegren 2013); namely, more rapid divergence for male-biased genes through relaxed purifying selection, slower divergence for female-biased genes with weak evidence for stronger purifying selection, and a fast-X effect (table 1).
However, when we incorporated developmental patterns, our analysis of genes with continuous or stage-specific sex bias in expression shows that patterns of evolutionary divergence of sex-biased genes are more nuanced than previously appreciated. Our results show that the well-known pattern of more rapid evolution of male-biased compared with female-biased and unbiased genes does not always hold when the developmental pattern is considered (fig. 4). Furthermore, male- and female-biased genes can evolve at similarly rapid rates, depending on their developmental pattern, but that stage limitation influences divergence rates in distinct ways in males and females (fig. 4b and d). This finding suggests that the sexes exhibit distinct patterns in how selection acts throughout development.
Our finding that stage-specific genes evolved more rapidly than continuously expressed genes within each sex supports the notion of developmental evolutionary constraints imposed by shared gene expression across multiple time points (Artieri et al. 2009; Mank et al. 2010), analogous to the constraints thought to be imposed by expression across multiple tissues (Haerty et al. 2007; Mank et al. 2008; Meisel 2011). Three further features of these results are notable. First, and surprisingly, the difference in divergence rates between sex-specific genes disappears when considering the continuity of expression across stages. Male- and female-expressed genes specific to larvae evolved equally rapidly, and genes continuously expressed in either sex also evolved at similar rates (fig. 4). Second, the more rapid divergence of female larval-specific genes was consistent with relaxed purifying selection acting on these genes, relative to genes continuously expressed in females. However, male larval-specific genes, though diverging relatively rapidly, did not show this pattern, suggesting that distinct evolutionary processes can shape the evolution of sex-specific gene expression even when divergence rates are similar. Finally, genes specific to third instar larvae within both sexes evolved most rapidly, with genes specific to adults evolving relatively slowly. This result contrasts with Artieri et al.’s (2009) finding that adult-specific genes evolved more rapidly than a combination of all larvae- and pupae-specific genes. Our study samples two precise developmental time points using a higher resolution assay of gene expression (RNA-seq as opposed to EST libraries), and we show that there is in fact not a continuous increase in rates of evolutionary divergence throughout development.
Conclusion
Overall, our results highlight that new aspects of the evolutionary dynamics of sex-biased genes and sex-specific selection can be uncovered by considering ontogenetic patterns of gene expression. Important next steps for progress in these areas include measuring the developmental patterns of sex-specific isoforms, which can be as frequent as sex-biased genes in adults (Gan et al. 2010; Chang et al. 2011; Daines et al. 2011), and characterizing early somatic sex differences in expression, which can be considerable in adults (Chang et al. 2011).
Materials and Methods
Experimental Animals and Developmental Stages
A fly stock was obtained in which the female-specific promoter Sxl was linked to a gene expressing GFP, such that female embryos express GFP and embryos can be sorted by sex (Graham et al. 2006). This construct was backcrossed into a genetically variable fly population (LHm, Chippindale et al. 2001; backcrossing performed by U. Friberg). Flies were maintained at 25 °C and a 12:12 light:dark schedule.
Gonad imaginal discs or (in adults) mature gonads were dissected from 1) wandering third instar larvae that had dark blue food visible in their guts (i.e., having recently stopped feeding and >12 h from pupation, Karim and Thummel 1992; equivalent to puff stage 1, Andres and Thummel 1994), 2) newly formed white prepupae, and 3) 24-h post-eclosion virgin adults.
Sample Preparation
The parents of flies used in this experiment were grown at a standardized low density (25/ml food) to minimize variation associated with parental rearing conditions. These parental flies were 3–5 days postemergence when we collected eggs for the flies to be sampled. The larvae and pre-pupae we sampled were grown at low density (25/ml food) on standard food media containing bromophenol blue (0.05%) to facilitate staging (Maroni and Stamey 1983). Dissections were performed from 2 to 7 h Zeitgeber time. Animals were sexed by fluorescence under a fluorescence microscope and by visual inspection of pre-gonads (Kerkis 1931). The gonadal discs were dissected on ice-cold phosphate buffered saline and immediately transferred to QIAzol lysis reagent (Qiagen, West Sussex, United Kingdom) on ice, followed by tissue rupture and storage at −80 °C. Although gonad tissue is comprised of somatic and germline cells, germline cells contribute the majority of mRNA (Gupta et al. 2006).
We sequenced three nonoverlapping pools (i.e., biological replicates) of flies per sex for both larvae and pre-pupae, along with one pool for each sex for adults to enable us to compare our results with existing data sets. We included more female than male pre-gonads within each pool for larvae and pre-pupae to reflect the higher RNA yield from male pre-gonads. The number of pairs of gonads ranged from 39 to 104 for larval and prepupal pools and was 10 for adult pools.
RNA was extracted using a lipid tissue kit (Qiagen RNeasy, West Sussex, United Kingdom), following manufacturer’s instructions and including the DNase digestion step. mRNA was prepared for sequencing using standard Illumina protocols.
Sequencing and Bioinformatics
Library preparation and RNA sequencing (Illumina HiSeq 2000) was performed by the Wellcome Trust Centre for Human Genetics, Oxford. Each biological replicate was barcoded and pooled on a single lane, yielding 37.17 Gb of 100 bp paired-end reads. Read quality and potential sequencing biases were assessed using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc, last accessed August 31, 2013). Trimmomatic (Lohse et al. 2012) was used to remove any residual Illumina adaptor sequence, trim leading, and trailing bases with a Phred quality score less than 4, and using a sliding window approach trim when the average Phred quality score over four bases dropped below 15. Any paired-end reads for which either read was shorter than 25 bp after trimming were removed from the data set. After filtering, there were on average 13 million reads per pool (biological replicate).
The genome of Drosophila melanogaster (Adams et al. 2000) was obtained from Ensembl (release 64; Flicek et al. 2012). Each pool’s filtered paired-end reads were mapped to the reference genome using RSEM (Li and Dewey 2011), which leverages the short-read aligner bowtie (Langmead et al. 2009). rRNA genes and rRNA pseudogenes were removed, as rRNA can bias expression estimates of mRNA genes. On average, 81.11% of reads mapped to the reference genome per pool and a total of 14,705 genes had mapped reads. A filter of four reads per million mappable reads (pmmr) was applied to remove nonexpressed and lowly expressed genes (Harrison, Mank et al. 2012; Moghadam et al. 2013). Genes were filtered if they did not pass the four-pmmr threshold in at least two out of three pools for either sex of either larvae or pupae pools, or did not pass the threshold for either adult pool, resulting in 10,587 genes that were expressed in at least one sex and life stage. The raw read count outputs from RSEM were quantile normalized. Chromosome location and GO terms (Ashburner et al. 2000) were obtained for each gene, if available, from the FlyBase gene associations (release FB2012_01; McQuilton et al. 2012).
Gene orthology information was obtained from FlyBase and one-to-one orthologs identified for the melanogaster subgroup (Drosophila 12 Genomes Consortium 2007; Larracuente et al. 2008), which includes D. melanogaster, D. sechellia, D. simulans, D. yakuba, D. erecta, and D. ananassae. Within the melanogaster subgroup, 9,162 orthologs were identified in FlyBase, and each orthologous group of six genes was aligned with PRANK (Löytynoja and Goldman 2005). Nine of these orthologous groups were removed from the data set due to a member’s codons not being in multiples of three. A further 27 orthologous groups had erroneous FlyBase entries and were missing one or more of the melanogaster group species genes. Repeatmasker (http://www.repeatmasker.org, last accessed August 31, 2013) identified 882 genes as containing retrotransposons, including long interspersed nuclear elements (LINEs) and long terminal repeats (LTRs), or DNA transposons, and these groups were removed from the data set. In-frame stop codons were found in 124 orthologous groups genes and a further 13 orthologous groups were excluded because genes were less than 100 bp in aligned gapless length. These filtering steps left 8,112 one-to-one orthologs of the six melanogaster group species.
These 8,112 genes were analyzed with PAML (version 4.4b; Yang 2007), using the melanogaster subgroup phylogeny (Drosophila 12 Genomes Consortium 2007; Larracuente et al. 2008). Along the D. melanogaster branch, the number of nonsynonymous substitutions (Dn), the number of potential nonsynonymous sites (N), the number of nonsynonymous substitutions per nonsynonymous site (dN), the number of synonymous substitutions (Ds), the number of potential synonymous sites (S), the number of synonymous substitutions per synonymous site (dS), and their ratio (dN/dS) were extracted for each orthologous group. These values were verified against dN/dS results from several previous studies (Parisi et al. 2003; Ranz et al. 2003; Gibson et al. 2004; Parisi et al. 2004; Stolc et al. 2004; McIntyre et al. 2006; Goldman and Arbeitman 2007; Wayne et al. 2007; Mikhaylova et al. 2008; Ayroles et al. 2009; Gan et al. 2010; Innocenti and Morrow 2010; Wasbrough et al. 2010; Wyman et al. 2010; Graveley et al. 2011) obtained from the sex bias database SEBIDA (version 3.0; Gnad and Parsch 2006).
Data on standing variation in protein-coding sequences were obtained from the Drosophila Genome Reference Panel (DGRP; Database file date July 18, 2012; Mackay et al. 2012) to calculate the numbers of synonymous (Ps) and nonsynonymous polymorphisms (Pn). For the 8,112 genes used for the dN/dS analysis, SNPs were extracted from the DGRP database coded as either synonymous or nonsynonymous, with an additional filter requiring the minor allele to be present in at least 10% of lines (17/168). These data were used to calculate the direction of selection statistic, DoS = Dn/(Dn + Ds) − Pn/(Pn + Ps) (Stoletzki and Eyre-Walker 2011).
Analyses
Within a developmental stage, we defined sex-biased genes as having at least 2-fold differential expression and sex-limited genes as having zero expression in one sex. For both categories, we required a significant P value from a t-test after multiple testing correction (Padj < 0.05, Benjamini Hochberg correction [Benjamini and Hochberg 1995], false discovery rate [FDR] 5%). This approach to categorizing sex-biased genes was consistent with two alternative approaches (EdgeR and DESeq): our categorization showed an 87–95% overlap with these methods, which compares well with the overlap observed between them (79–93%). We also explored patterns of sex-biased gene expression using different threshold of differential expression.
To examine the continuity of sex-biased gene expression throughout development, we used two methods to categorize adult genes. First, we used our own adult data, following the above procedure, requiring Padj < 0.05 in a genome-wide χ2 test. Second, we used categories defined by the SEBIDA meta-analysis with a 2-fold threshold (Gnad and Parsch 2006).
In all analyses, we treated autosomes and X chromosomes separately because they display different patterns of sex-biased expression and rates of evolutionary divergence in D. melanogaster (e.g., Meisel et al. 2012a).
To test for differences in dN/dS among groups of genes, we used permutation tests with 1,000 iterations. To test for differences in DoS, we used Wilcoxon nonparametric tests. We present bootstrapped confidence intervals (based on 1,000 replicates) with dN/dS and DoS values. t-tests, correlations, χ2 tests, and Wilcoxon tests were calculated using JMP v. 10 (SAS Institute Inc., Cary, NC, USA). Permutation tests and bootstrapping were conducted in Excel 2010 (Microsoft, Redmond, Washington, USA).
We used GOrilla (Eden et al. 2009) to test for GO term enrichment in groups of sex-biased genes.
Supplementary Material
Supplementary analyses, supplementary figures S1–S4, and supplementary tables S1–S8 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank the Wellcome Trust Centre for Human Genetics in Oxford for sequencing, U. Friberg and R. Dean for fly stocks, M. Pointer and L. Rowe for discussion, and S. Montgomery, A. Wright, F. Zimmer, and two anonymous reviewers for constructive comments that improved the manuscript. The authors acknowledge the use of the UCL Legion High Performance Computing Facility (Legion@UCL), and associated support services, in the completion of this work. This work was supported by the Elizabeth Hannah Jenkinson Fund and the John Fell Oxford University Press Research Fund (grants to J.C.P.) and the European Research Council (grant 260233 to J.E.M.).
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Andres AJ, Thummel CS. Methods for quantitative analysis of transcription in larvae and prepupae. Methods Cell Biol. 1994;44:565–573. doi: 10.1016/s0091-679x(08)60932-2. [DOI] [PubMed] [Google Scholar]
- Arbeitman MN, Furlong EEM, Imam F, Johnson E, Null BH, Baker BS, Krasnow MA, Scott MP, Davis RW, White KP. Gene expression during the life cycle of Drosophila melanogaster. Science. 2002;297:2270–2275. doi: 10.1126/science.1072152. [DOI] [PubMed] [Google Scholar]
- Artieri CG, Haerty W, Singh RS. Ontogeny and phylogeny: molecular signatures of selection, constraint, and temporal pleiotropy in the development of Drosophila. BMC Biol. 2009;7:42. doi: 10.1186/1741-7007-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artieri CG, Singh RS. Demystifying phenotypes: the comparative genomics of evo-devo. Fly. 2010;4:18–20. doi: 10.4161/fly.4.1.10509. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroles JF, Carbone MA, Stone EA, Jordan KW, Lyman RF, Magwire MM, Rollmann SM, Duncan LH, Lawrence F, Anholt RR, et al. Systems genetics of complex traits in Drosophila melanogaster. Nat Genet. 2009;41:299–307. doi: 10.1038/ng.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines JF, Sawyer SA, Hartl DL, Parsch J. Effects of X-linkage and sex-biased gene expression on the rate of adaptive protein evolution in Drosophila. Mol Biol Evol. 2008;25:1639–1650. doi: 10.1093/molbev/msn111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. 1995;57:289–300. [Google Scholar]
- Bodenstein D. The postembryonic development of Drosophila. In: Demerec M, editor. Biology of Drosophila. New York: John Wiley and Sons; 1950. pp. 275–367. [Google Scholar]
- Carreira VP, Soto IM, Mensch J, Fanara JJ. Genetic basis of wing morphogenesis in Drosophila: sexual dimorphism and non-allometric effects of shape variation. BMC Dev. Biol. 2011;11:32. doi: 10.1186/1471-213X-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalan A, Hutter S, Parsch J. Population and sex differences in Drosophila melanogaster brain gene expression. BMC Genomics. 2012;13:654. doi: 10.1186/1471-2164-13-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PL, Dunham JP, Nuzhdin SV, Arbeitman MN. Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC Genomics. 2011;12:364. doi: 10.1186/1471-2164-12-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JAT. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- Chippindale AK, Gibson JR, Rice WR. Negative genetic correlation for adult fitness between sexes reveals ontogenetic conflict in Drosophila. Proc Natl Acad Sci U S A. 2001;98:1671–1675. doi: 10.1073/pnas.041378098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connallon T, Knowles LL. Intergenomic conflict revealed by patterns of sex-biased gene expression. Trends Genet. 2005;21:495–499. doi: 10.1016/j.tig.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Daines B, Wang H, Wang L, Li Y, Han Y, Emmert D, Gelbart W, Wang X, Li W, Gibbs R, et al. The Drosophila melanogaster transcriptome by paired-end RNA sequencing. Genome Res. 2011;21:315–324. doi: 10.1101/gr.107854.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Q, Chepelev I, Wei G, Tarayrah L, Cui K, Zhao K, Chen X. Dynamic regulation of alternative splicing and chromatin structure in Drosophila gonads revealed by RNA-seq. Cell Res. 2010;20:763–783. doi: 10.1038/cr.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, Riley-Berger R, Harshman L, Kopp A, Vacha S, Nuzhdin S, Wayne M. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics. 2004;167:1791–1799. doi: 10.1534/genetics.104.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Chippindale AK, Rice WR. The X chromosome is a hot spot for sexually antagonistic fitness variation. Proc R Soc Lond B Biol Sci. 2002;269:499–505. doi: 10.1098/rspb.2001.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad F, Parsch J. Sebida: a database for the functional and evolutionary analysis of genes with sex-biased expression. Bioinformatics. 2006;22:2577–2579. doi: 10.1093/bioinformatics/btl422. [DOI] [PubMed] [Google Scholar]
- Goldman TD, Arbeitman MN. Genomic and functional studies of Drosophila sex hierarchy regulated gene expression in adult head and nervous system tissues. PLoS Genet. 2007;3:e216. doi: 10.1371/journal.pgen.0030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham P, Thompson J, Griswold K, Schedl P, Pulak R. Sorting and collecting females from males at high speed. Drosoph Inf Serv. 2006;89:123–128. [Google Scholar]
- Grath S, Parsch J. Rate of amino acid substitution is influenced by the degree and conservation of male-biased transcription over 50 myr of Drosophila evolution. Genome Biol Evol. 2012;4:346–359. doi: 10.1093/gbe/evs012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Parisi M, Sturgill D, Nuttall R, Doctolero M, Dudko OK, Malley JD, Eastman PS, Oliver B. Global analysis of X-chromosome dosage compensation. J Biol. 2006;5:3. doi: 10.1186/jbiol30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerty W, Jagadeeshan S, Kulathinal RJ, Wong A, Ravi Ram K, Sirot LK, Levesque L, Artieri CG, Wolfner MF, Civetta A, et al. Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics. 2007;177:1321–1335. doi: 10.1534/genetics.107.078865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale MC, Xu P, Scardina J, Wheeler PA, Thorgaard GH, Nichols KM. Differential gene expression in male and female rainbow trout embryos prior to the onset of gross morphological differentiation of the gonads. BMC Genomics. 2011;12:404. doi: 10.1186/1471-2164-12-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PW, Mank JE, Wedell N. Incomplete sex chromosome dosage compensation in the Indian meal moth, Plodia interpunctella, based on de novo transcriptome assembly. Genome Biol Evol. 2012;4:1118–1126. doi: 10.1093/gbe/evs086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PW, Wright AE, Mank JE. The evolution of gene expression and the transcriptome-phenotype relationship. Semin Cell Dev Biol. 2012;23:222–229. doi: 10.1016/j.semcdb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hense W, Baines JF, Parsch J. X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol. 2007;5:e273. doi: 10.1371/journal.pbio.0050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti P, Morrow EH. The sexually antagonistic genes of Drosophila melanogaster. PLoS Biol. 2010;8:e1000335. doi: 10.1371/journal.pbio.1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeeshan S, Singh RS. Rapidly evolving genes of Drosophila: Differing levels of selective pressure in testis, ovary, and head tissues between sibling. Mol Biol Evol. 2005;22:1793–1801. doi: 10.1093/molbev/msi175. [DOI] [PubMed] [Google Scholar]
- Karim FD, Thummel CS. Temporal coordination of regulatory gene expression by the steroid hormone ecdysone. The EMBO Journal. 1992;11:4083–4093. doi: 10.1002/j.1460-2075.1992.tb05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayserili MA, Gerrard DT, Tomancak P, Kalinka AT. An excess of gene expression divergence on the X chromosome in Drosophila embryos: implications for the faster-X hypothesis. PLoS Genet. 2012;8:e1003200. doi: 10.1371/journal.pgen.1003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkis J. The growth of the gonads in Drosophila melanogaster. Genetics. 1931;16:212–224. doi: 10.1093/genetics/16.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RC. Ovarian development in Drosophila melanogaster. New York: Academic Press; 1970. [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larracuente AM, Sackton TB, Greenberg AJ, Wong A, Singh ND, Sturgill D, Zhang Y, Oliver B, Clark AG. Evolution of protein-coding genes in Drosophila. Trends Genet. 2008;24:114–123. doi: 10.1016/j.tig.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Lebo MS, Sanders LE, Sun F, Arbeitman MN. Somatic, germline and sex hierarchy regulated gene expression during Drosophila metamorphosis. BMC Genomics. 2009;10:80. doi: 10.1186/1471-2164-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey C. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley DL, Tokuyasu KT. Spermatogenesis. In: Ashburner M, Wright TRF, editors. The Genetics and Biology of Drosophila. London: Academic Press; 1980. pp. 226–294. [Google Scholar]
- Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, Usadel B. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012;40:W622–W627. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 2005;102:10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TF, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald AP, Kambysellis MP. Oogenesis. In: Ashburner M, Wright TRF, editors. The genetics and biology of Drosophila. New York: Academic Press; 1980. pp. 141–225. [Google Scholar]
- Mank JE. Sex chromosomes and the evolution of sexual dimorphism: lessons from the genome. Am Nat. 2009;173:141–150. doi: 10.1086/595754. [DOI] [PubMed] [Google Scholar]
- Mank JE, Hultin-Rosenberg L, Zwahlen M, Ellegren H. Pleiotropic constraint hampers the resolution of sexual antagonism in vertebrate gene expression. Am Nat. 2008;171:35–43. doi: 10.1086/523954. [DOI] [PubMed] [Google Scholar]
- Mank JE, Nam K, Brunström B, Ellegren H. Ontogenetic complexity of sexual dimorphism and sex-specific selection. Mol Biol Evol. 2010;27:1570–1578. doi: 10.1093/molbev/msq042. [DOI] [PubMed] [Google Scholar]
- Maroni G, Stamey SC. Use of blue food to select synchronous, late third instar larvae. Drosoph Inf Serv. 1983;59:142–143. [Google Scholar]
- Martins MJ, Mota CF, Pearson GA. Sex-biased gene expression in the brown alga Fucus vesiculosus. BMC Genomics. 2013;14:294. doi: 10.1186/1471-2164-14-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre L, Bono L, Genissel A, Westerman R, Junk D, Telonis-Scott M, Harshman L, Wayne M, Kopp A, Nuzhdin S. Sex-specific expression of alternative transcripts in Drosophila. Genome Biol. 2006;7:R79. doi: 10.1186/gb-2006-7-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuilton P, St. Pierre SE, Thurmond J, The FlyBase Consortium FlyBase 101— the basics of navigating FlyBase. Nucleic Acids Res. 2012;40:D706–D714. doi: 10.1093/nar/gkr1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Landeen EL, Cook JM, Kingan SB, Presgraves DC. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol. 2011;9:e1001126. doi: 10.1371/journal.pbio.1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Presgraves DC. Little evidence for demasculinization of the Drosophila X chromosome among genes expressed in the male germline. Genome Biol Evol. 2012;4:1007–1016. doi: 10.1093/gbe/evs077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP. Towards a more nuanced understanding of the relationship between sex-biased gene expression and rates of protein-coding sequence evolution. Mol Biol Evol. 2011;28:1893–1900. doi: 10.1093/molbev/msr010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG. Disentangling the relationship between sex-biased gene expression and X-linkage. Genome Res. 2012a;22:1255–1265. doi: 10.1101/gr.132100.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG. Faster-X evolution of gene expression in Drosophila. PLoS Genet. 2012b;8:e1003013. doi: 10.1371/journal.pgen.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova LM, Nguyen K, Nurminsky DI. Analysis of the Drosophila melanogaster testes transcriptome reveals coordinate regulation of paralogous genes. Genetics. 2008;179:305–315. doi: 10.1534/genetics.107.080267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova LM, Nurminsky DI. Lack of global meiotic sex chromosome inactivation, and paucity of tissue-specific gene expression on the Drosophila X chromosome. BMC Biol. 2011;9:29. doi: 10.1186/1741-7007-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadam HK, Harrison PW, Zachar G, Szekely T, Mank JE. The plover neurotranscriptome assembly: transcriptomic analysis in an ecological model species without a reference genome. Mol Ecol Resour. 2013;13:696–705. doi: 10.1111/1755-0998.12096. [DOI] [PubMed] [Google Scholar]
- Müller L, Grath S, von Heckel K, Parsch J. Inter- and intraspecific variation in Drosophila genes with sex-biased expression. Int J Evol Biol. 2012;2012:963976. doi: 10.1155/2012/963976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Edwards P, Minor J, Naiman D, Lü J, Doctolero M, Vainer M, Chan C, Malley J, et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol. 2004;5:1–18. doi: 10.1186/gb-2004-5-6-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, Eastman S, Oliver B. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003;299:697–700. doi: 10.1126/science.1079190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsch J, Ellegren H. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 2013;14:83–87. doi: 10.1038/nrg3376. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Jensen JD, Stephan W, Stamatakis A. A critical assessment of storytelling: gene ontology categories and the importance of validating genomic scans. Mol Biol Evol. 2012;29:3237–3248. doi: 10.1093/molbev/mss136. [DOI] [PubMed] [Google Scholar]
- Perry JC, Mank JE. From genotype × environment to transcriptome × environment: identifying and understanding the environmental influences in the gene expression underlying sexually selected traits. In: Hunt J, Hosken D, editors. Genotype-by-Environment Interactions and Sexual Selection. Chichester (UK): Wiley-Blackwell; Forthcoming. [Google Scholar]
- Pröschel M, Zhang Z, Parsch J. Widespread adaptive evolution of Drosophila genes with sex-biased expression. Genetics. 2006;174:893–900. doi: 10.1534/genetics.106.058008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science. 2003;300:1742–1745. doi: 10.1126/science.1085881. [DOI] [PubMed] [Google Scholar]
- Rice WR, Chippindale AK. Intersexual ontogenetic conflict. J Evol Biol. 2001;14:685–693. [Google Scholar]
- Small CM, Carney GE, Mo QX, Vannucci M, Jones AG. A microarray analysis of sex- and gonad-biased gene expression in the zebrafish: evidence for masculinization of the transcriptome. BMC Genomics. 2009;10:579. doi: 10.1186/1471-2164-10-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolc V, Gauhar Z, Mason C, Halasz G, van Batenburg MF, Rifkin SA, Hua S, Herreman T, Tongprasit W, Barbano PE, et al. A gene expression map for the euchromatic genome of Drosophila melanogaster. Science. 2004;306:655–660. doi: 10.1126/science.1101312. [DOI] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. Estimation of the neutrality index. Mol Biol Evol. 2011;28:63–70. doi: 10.1093/molbev/msq249. [DOI] [PubMed] [Google Scholar]
- Swanson WJ, Vacquier VD. The rapid evolution of reproductive proteins. Nat Rev Genet. 2002;3:137–144. doi: 10.1038/nrg733. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. The deficit of male-biased genes on the D. melanogaster X chromosome is expression-dependent: a consequence of dosage compensation? J Mol Evol. 2009;68:576–583. doi: 10.1007/s00239-009-9235-4. [DOI] [PubMed] [Google Scholar]
- Wasbrough ER, Dorus S, Hester S, Howard-Murkin J, Lilley K, Wilkin E, Polpitiya A, Petritis K, Karr TL. The Drosophila melanogaster sperm proteome-II (DmSP-II) J Proteomics. 2010;73:2171–2185. doi: 10.1016/j.jprot.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Wayne ML, Telonis-Scott M, Bono LM, Harshman L, Kopp A, Nuzhdin SV, McIntyre LM. Simpler mode of inheritance of transcriptional variation in male Drosophila melanogaster. Proc Natl Acad Sci U S A. 2007;104:18577–18582. doi: 10.1073/pnas.0705441104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman M, Agrawal A, Rowe L. Condition-dependence of the sexually dimorphic transcriptome in Drosophila melanogaster. Evolution. 2010;64:1836–1848. doi: 10.1111/j.1558-5646.2009.00938.x. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007;450:233–238. doi: 10.1038/nature06323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Hambuch TM, Parsch J. Molecular evolution of sex-biased genes in Drosophila. Mol Biol Evol. 2004;21:2130–2139. doi: 10.1093/molbev/msh223. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Parsch J. Positive correlation between evolutionary rate and recombination rate in Drosophila genes with male-biased expression. Mol Biol Evol. 2005;22:1945–1947. doi: 10.1093/molbev/msi189. [DOI] [PubMed] [Google Scholar]
- Zhao M, Zha X-F, Liu J, Zhang W-J, He N-J, Cheng D-J, Dai Y, Xiang Z-H, Xia Q-Y. Global expression profile of silkworm genes from larval to pupal stages: toward a comprehensive understanding of sexual differences. Insect Sci. 2011;18:607–618. [Google Scholar]
- Zikovitz AE, Agrawal AF. The condition dependency of fitness in males and females: the fitness consequences of juvenile diet assessed in environments differing in key adult resources. Evolution. 2013;67:2849–2860. doi: 10.1111/evo.12170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.