

SUMMARY
Autism spectrum disorder (ASD) is a complex developmental syndrome of unknown etiology. Recent studies employing exome- and genome-wide sequencing have identified nine high-confidence ASD (hcASD) genes. Working from the hypothesis that ASD-associated mutations in these biologically pleiotropic genes will disrupt intersecting developmental processes to contribute to a common phenotype, we have attempted to identify time periods, brain regions, and cell types in which these genes converge. We have constructed coexpression networks based on the hcASD “seed” genes, leveraging a rich expression data set encompassing multiple human brain regions across human development and into adulthood. By assessing enrichment of an independent set of probable ASD (pASD) genes, derived from the same sequencing studies, we demonstrate a key point of convergence in midfetal layer 5/6 cortical projection neurons. This approach informs when, where, and in what cell types mutations in these specific genes may be productively studied to clarify ASD pathophysiology.
INTRODUCTION
Autism spectrum disorders (ASDs) are defined by impairments in reciprocal social interaction, often accompanied by abnormalities in language development as well as repetitive behaviors and/or restricted interests. Considerable genetic and phenotypic heterogeneity has complicated efforts to establish the biological substrates of the syndrome. However, a sea change is currently underway in the genetics and genomics of ASD. Although genome-wide efforts to identify common genetic variation contributing to the syndrome have not yet led to reproducible results (State and Levitt, 2011), the identification of the important contribution of rare de novo mutations (Jamain et al., 2003; Sanders et al., 2011; Sebat et al., 2007) combined with high-throughput sequencing technology has recently led to the systematic discovery of loss of function (LoF) de novo coding mutations carrying comparatively large biological effects in ASD (Iossifov et al., 2012; Kong et al., 2012; Neale et al., 2012; O’Roak et al., 2011, 2012a, 2012b; Sanders et al., 2012). As a result, the set of associated genes has increased markedly during the past 18 months, and this number will continue to grow steadily and predictably as additional cohorts of ASD families are sequenced (Buxbaum et al., 2012). Moreover, recent advances are further clarifying the genomic architecture of ASD. While de novo point mutations have so far been estimated to play a contributory role in approximately 15% of affected individuals, estimates of locus heterogeneity imparted by these mutations alone already range from several hundred to more than 1,000 genes (He et al., 2013; Iossifov et al., 2012; Sanders et al., 2012).
The increasing number of genes carrying rare coding mutations with strong association to the human phenotype presents unprecedented opportunities for translational neuroscience. At the same time, the combination of extraordinary locus heterogeneity and biological pleiotropy poses considerable obstacles to the dissection of the pathophysiology of ASD, including the challenge of designing productive functional studies for a given gene in the absence of knowing when and where in the brain to investigate the identified risk mutations. This issue is particularly relevant given the fact that many of the genes discovered to date are involved in multiple biological processes at multiple points during development. Moreover, identical mutations in the same gene can lead to widely disparate psychiatric and neurological syndromes (Malhotra and Sebat, 2012). Consequently, a determination of spatiotemporal convergence among groups of disease-related mutations, all known to lead to ASD, may be particularly helpful as a first step toward identifying the functional perturbations specifically relevant for this phenotype.
With this in mind, we have set out to address the key question of if and when, in what brain regions, and in which cell types specific groups of ASD-related mutations converge during human brain development. To pursue this question, we have taken a “bottom-up” approach to gene coexpression network analysis, focusing initially on only nine “seed” genes carrying multiple de novo LoF mutations and thereby showing the strongest evidence for association with ASD. By focusing on these nine “high confidence” (hcASD) genes, we have sought to minimize the noise that can accompany network analyses based on inputs with widely varying evidence for association. Moreover, we have restricted input genes to those identified only via “hypothesis-naïve” exome- and genome- wide sequencing and have set a consistent statistical threshold for inclusion, minimizing the confounds that may accompany attempts to clarify mechanism using inputs that may have been identified, in part, based on their biological plausibility.
To evaluate these nine seed genes, we have used spatially and temporally rich mRNA expression data from developing human brain as the substrate for constructing networks. This choice is based on several key considerations: first, that an analysis of the expression trajectories of ASD-associated genes in typically-developing human brain can provide insight into normal biological mechanisms that go awry in ASD (State and Sestan 2012); second, that highly correlated gene expression is likely to reflect shared function and/or regulation; and third, and perhaps most importantly, that recent work in characterizing the human brain transcriptome underscores the spatial and temporal dynamism that occurs during development and provides the ability to exploit this dimensionality (Kang et al., 2011). These types of data are not yet available for protein-protein interaction and gene-ontology databases.
To search for points of convergence among spatially and temporally defined coexpression networks, we relied on a second, nonoverlapping set of probable ASD (pASD) genes found to carry a single de novo LoF mutation and derived from the same studies as those yielding the hcASD seed genes. This inclusion criterion was likewise designed to minimize selection bias and batch effects and to provide relatively uniform independent evidence for association with ASD across this set of inputs.
Our analysis identifies robust, statistically significant evidence for convergence of the input set of hcASD and pASD risk genes in glutamatergic projection neurons in layers 5 and 6 of human midfetal prefrontal and primary motor-somatosensory cortex (PFC-MSC). Given the extensive genetic and phenotypic heterogeneity underlying ASD and the small fraction of risk genes that we have examined in this study, this likely represents only one of several such points of convergence. Nonetheless, the analytic approach presented here clarifies key variables relevant for productive functional studies of specific ASD genes carrying LoF mutations, providing an important step in moving from gene discovery to an actionable understanding of ASD biology.
RESULTS
Identification of hcASD Seed Genes
To identify input genes for this analysis, we have consolidated a cohort of 1,043 families (599 quartets, 444 trios) from a combination of published data (987 families; 543 quartets, 444 trios) (Iossifov et al., 2012; Kong et al., 2012; Neale et al., 2012; O’Roak et al., 2012b) and exome sequencing of an additional 56 quartets from the Simons Simplex Collection (SSC), a well-characterized ASD cohort consisting of families with one affected proband, two unaffected parents, and, in the majority of pedigrees, at least one unaffected sibling (Fischbach and Lord, 2010). Among the 1,043 probands, 144 de novo LoF mutations were identified (Table S1 available online), with LoF defined as a premature stop codon, canonical splice-site disruption, or frameshift insertion/deletion. Within the 599 quartets from the SSC (Iossifov et al., 2012; Sanders et al., 2012), we observed 75 de novo LoF mutations in 72 of the affected probands compared to 34 in 32 of the unaffected matched sibling controls (OR 2.21 95%CI: 1.45–3.36; p=5×10−5, binomial exact test; Figure S1 and Table S1).
Based on this data set, the observation of three de novo LoF mutations in the same gene in unrelated individuals identifies an ASD gene (false discovery rate [FDR] q = 0.0002; >99.9% chance of being a true ASD gene). Moreover, genes with two or more de novo LoF mutations are also very likely to be true ASD genes (q=0.02; 97.8% chance of being a true ASD gene). Thus, we refer to genes with two or more de novo LoF mutations in unrelated individuals as hcASD genes. Finally, we find that genes carrying a single de novo LoF mutation are more likely than not to be true ASD genes (q=0.45; 54.7% chance of being a true ASD gene). We subsequently refer to these as probable ASD (pASD) genes. Overall, our analysis of the 1,043 families identifies one previously unreported hcASD gene with two de novo LoF mutations: Ankyrin 2, neuronal (ANK2); confirms eight other hcASD genes (Figure 1A); and identifies 122 pASD genes (Table S1).
Figure 1. Overview of Coexpression Analysis Workflow and Associated Data Sets.

A) Nine hcASD genes derived from our data and additional published data sets. The number of independent de novo loss of function (LoF) mutations identified is indicated in parentheses. ANK2 is a novel ASD-associated gene. B) A comprehensive data set of spatiotemporal gene expression spanning human brain development (Kang et al., 2011) was used to perform coexpression analysis. This data set spans 12 brain regions during periods 1 and 2 (embryonic and early fetal development) and 16 regions from period 3 to period 15 (early fetal to late adulthood). Neocortical regions are in red. C) Periods of human brain development as defined by Kang et al. (2012). PCW, postconceptual weeks; M, postnatal months; Y, postnatal years. D) The hcASD genes (black) are used as “seeds” to build coexpression networks along spatial and temporal dimensions. E) Mean expression levels for two genes are plotted as a function of period of development (temporal axis) and region of the brain (spatial axis). The images illustrate that highly correlated genes have similar expression profiles across these dimensions. The Pearson’s correlation value quantifies the similarity between these profiles. F) Networks are interrogated for enrichment of an independent set of 122 probable ASD (pASD) genes (gray). G) Networks are tested for enrichment of layer and cell-type specific genes. FC, frontal cerebral wall; PC, parietal cerebral wall; TC, temporal cerebral wall; OC, occipital cerebral wall; HIP, hippocampal anlage (periods 1–2), hippocampus (periods 3–15); VF, ventral forebrain; MGE, medial ganglionic eminence; LGE, lateral ganglionic eminence; CGE, caudal ganglionic eminence; DIE, diencephalon; DTH, dorsal thalamus; URL, upper (rostral) rhombic lip; OFC, orbital prefrontal cortex; DFC, dorsal prefrontal cortex; VFC, ventral prefrontal cortex; MFC, medial prefrontal cortex; M1C, primary motor cortex; S1C, primary somatosensory cortex; IPC, posterior inferior parietal cortex; A1C, primary auditory cortex; STC, superior temporal cortex; ITC, inferior temporal cortex; V1C, primary visual cortex; AMY, amygdala; STR, striatum; MD, mediodorsal nucleus of the thalamus; CBC, cerebellar cortex. See also Figure S1 and Table S1.
Constructing Spatiotemporal Coexpression Networks
To interrogate coexpression dynamics of the nine hcASD genes in the context of human brain development, we utilized a previously published spatially and temporally rich transcriptome data set (Kang et al., 2011) generated as part of the BrainSpan project. These exon-level expression data originate from 16 regions of the human brain sampled in 57 clinically unremarkable postmortem brains of diverse ancestry (31 males, 26 females) that span 15 consecutive periods of neurodevelopment and adulthood from 5.7 postconception weeks (PCW) to 82 years (Figures 1B and 1C).
To identify specific time points and regions of brain development relevant to ASD, we partitioned the data into developmental periods and subsets of brain regions. Temporally, we used a sliding window of three consecutive time periods across the 15 total periods, producing 13 windows. Spatially, our preliminary analysis (Figure S2A) suggested that clustering the brain regions based on transcriptome data from fetal development (period 3–7) would optimize resolution. Therefore, brain regions with high transcriptional similarity were identified using hierarchical clustering of fetal transcriptome data. Four main clusters were observed (Figure 2A): (1) V1C, ITC, IPC, A1C, and STC (V1C-STC cluster); (2) M1C, S1C, VFC, MFC, DFC, and OFC (prefrontal and primary motor-somatosensory cortex or PFC-MSC cluster); (3) STR, HIP, and AMY; and (4) MD and CBC (MD-CBC cluster). In total, we generated 52 networks (13 time windows by four anatomical clusters) corresponding to 52 spatiotemporal windows (Figure 1D). These networks were created by calculating pairwise Pearson correlation coefficients between each of the nine hcASD genes and 16,947 genes from the exon array data set. To focus on the most relevant coexpression partners, we identified the 20 coexpressed genes best correlated to each hcASD gene (Figure 1E) and having a correlation coefficient R 0.7.
Figure 2. Convergence in Prefrontal and Primary Motor-Somatosensory Cortex Regions During Midfetal Development.

A) Hierarchical clustering of brain regions based on transcriptional similarity during fetal development (periods 3–7) divides the brain regions into four groups, demarcated by color. These clusters also reflect actual topographical proximity and functional segregation. B) To achieve spatiotemporal resolution, coexpression networks were formed from 52 subsets of the expression data based on 13 developmental stages (in three period windows) and four sets of brain regions (clusters shown in A). Each of the networks was tested for enrichment of 122 pASD genes. This heatmap shows the negative log10(p-value) (hypergeometric test) for enrichment in each network with developmental stages on the x-axis and brain regions on the y-axis. Networks that are not significant are in white; nominally significant networks are in light red; networks that are significant after correction for multiple comparisons are in red with the negative log10(p-value) noted. C) pASD gene enrichment is statistically significant, after correction for multiple comparisons, by hypergeometric test (gray line) in the given regions and time points. Black lines show p values estimated from the permutation test based on the number of pASD genes within the networks (corrected for multiple comparisons); the vertical red line shows the number of pASD genes observed for that specific network. pASD enrichment in period 3–5 (p=0.003), 4–6 (p-0.05), and 8–10 (p=0.04) networks remains significant, whereas the period 7–9 network (p=0.8) does not.
PFC-MSC, prefrontal cortex and primary motor-somatosensory cortex; V1C-STC, V1C, ITC, IPC, A1C, STC. See also Figure S2 and Table S2.
pASD Gene Enrichment Is Localized to Specific Spatiotemporal Networks
We reasoned that if a particular coexpression network captures ASD-related biology, then we would expect to see additional ASD genes mapping within that network. Consequently, to determine whether any of the 52 spatiotemporal networks showed convergence of ASD gene expression, we first asked whether any showed a statistically significant enrichment of pASD genes (Figure 1F) based on a hypergeometric test.
We identified enrichment of pASD genes in just 4 of the 52 networks (Figure 2B), after Bonferroni correction for multiple testing: PFC-MSC in period 3–5 (10–19 PCW; p=9.9×10−6); PFC-MSC in period 4–6 (13–24 PCW; p=1.2×10−3); V1C-STC in period 7–9 (24 PCW to 12 months of age; p=0.02); and MD-CBC in period 8–10 (birth to six years of age; p=3.5×10−4).
Permutation Testing Highlights Networks Derived from Midfetal Prefrontal and Primary Motor-Somatosensory Cortex
Although the hypergeometric test is a computationally efficient approach to an initial evaluation of enrichment, it assumes that all genes have an equal chance of being found in the network; however, the rate of de novo LoF mutations varies with gene size and GC content, violating this assumption. Moreover, the correlation-based network construction could potentially lead to inflated p values. Thus, we conducted separate permutation tests with 100,000 iterations for each of the four putatively enriched networks (Figure 2C). In each iteration, we randomly selected nine hcASD seed genes, with the probability of seed-gene selection based on gene size and GC content, and then constructed the corresponding coexpression network using the identical criteria noted above. We then quantified the presence of pASD genes within each network and assessed the significance of the observed enrichment, correcting for multiple comparisons. Using this more stringent permutation test (Figure 2C), pASD enrichment remained in the PFC-MSC networks at periods 3–5 (p=0.003) and 4–6 (p=0.05), and in the MD-CBC network at period 8–10 (p=0.04). Enrichment was no longer observed in the fourth (period 7–9) network. Because the MD-CBC network had relatively few tissue samples available compared with the PFC-MSC networks at periods 3–5 and 4–6 (26, 107, and 140 samples, respectively; Figure S2F), we elected to focus our subsequent analyses on the two PFC-MSC networks, hereafter referred to as the ‘midfetal’ networks. Additional permutation tests either varying the number of top coexpressed genes chosen or permuting the set of pASD genes instead of the hcASD genes, as well as a cross-validation experiment and a temporal analysis of individual periods, confirmed the robustness of our findings (Figures S2C–S2E and Table S2). The two midfetal networks are displayed in Figures 3A and 3B, the period 8–10 network is displayed in Figure S3, and network genes are summarized in Table S3.
Figure 3. The Period 3–5 and Period 4–6 PFC-MSC Networks.

(A and B) The period 3–5 (A) and period 4–6 (B) coexpression networks are displayed with the force-directed layout function of Cytoscape using correlation as the edge weight (Cline et al., 2007). Gene coexpression analysis included the 20 genes best correlated with each hcASD gene. The hcASD seed genes are in black; pASD genes identified within the network are in gray; and top 20 co-expressed genes that are not pASD genes are in white. The lines (edges) reflect coexpression correlations ≥0.7 and the shade represents the strength of the correlation; positive correlations are in in red; negative correlations are in blue. (C and D) The pASD genes enriched within the networks in (A) and (B) represent those with the highest probability of being true ASD genes. The TADA score combines de novo mutation data with inherited variant data from trios, rare variant case-control data, and estimates of mutation rate in order to estimate the probability of ASD association for each gene (He et al., 2013). The histograms show the results of permutation tests (100,000 iterations each) assessing the combined TADA score in the period 3–5 (C) and period 4–6 (D) networks; the observed scores, shown by the vertical red lines, are highly significant (p=6.0×10−5 and p=2.7×10−4, respectively). See also Figure S3 and Tables S3 and S4.
The Midfetal Networks Capture Biologically Meaningful Data
Given the hypothesis that strongly correlated genes share regulation and/or function, we assessed whether the genes in the two midfetal networks had greater than expected similarity in their spatiotemporal expression patterns. As predicted, both the period 3–5 and 4–6 networks had particularly high correlation coefficients, reflecting networks more connected than expected by chance (p=0.03 and p=0.02, respectively; Figures 3A and 3B), suggesting that the genes within these networks are biologically related.
The most connected hcASD gene within the period showing the strongest evidence for convergence (period 3–5 network in PFC-MSC) was T-box, brain, 1 (TBR1), a transcription factor known to be involved in forebrain development. Consequently, we assessed the relationship between TBR1 and the other genes in this network. First, we investigated whether its perturbation altered expression of other network genes. Using whole mouse cortex isolated from Tbr1−/− and Tbr1+/+ littermates at postnatal day 0 (P0, equivalent to human early midfetal development; Workman et al., 2013), we conducted RNA sequencing (RNAseq) and identified differentially expressed (DEX) genes (Table S4). Four of these DEX genes were observed in the network; NR4A2 and SV2B were both downregulated, whereas FEZF2 and NEFM were both upregulated. Furthermore, all four DEX genes have previously been identified as TBR1 targets by chromatin immunoprecipitation sequencing (ChIP-seq) analysis in N2A cells (Han et al., 2011). Given the regulatory function of TBR1 and its prominence in the midfetal networks, we evaluated if our results were primarily a result of inclusion of this gene. We found that this was not the case: removal of TBR1 from the coexpression analysis did not alter any of our findings regarding spatiotemporal convergence (Figure S2B).
pASD Genes Within Midfetal Networks Are More Likely To Be Associated with ASD
As noted, we estimate that over half of the 122 pASD genes represent true ASD risk genes. We consequently hypothesized that if the midfetal networks effectively capture ASD biology, then pASD genes within these networks would show greater evidence of association with ASD compared to those present in the 100,000 permuted networks. To test this, we turned to TADA, a newly developed statistical approach that integrates de novo and inherited variant data with estimates of gene mutability to yield gene-specific p values (He et al., 2013). TADA p values for each gene were calculated based on the whole-exome data in our study as well as case-control data (935 cases and 870 controls) from the ARRA Autism Sequencing Consortium study (Liu et al., 2013). Because all the pASD genes were defined by the presence of a single de novo LoF mutation, any variability in gene-specific p values derived from the TADA analysis represents independent evidence for association. Based on 100,000 permutations, we observed that the TADA p values for pASD genes within both midfetal networks were significantly lower than expected (Figures 3C and 3D).
Human Midfetal Laminar-Specific Expression Data Implicate Inner Cortical Plate in ASD
Given the well-established variability in gene expression by cortical layer, we reasoned that spatiotemporal networks identified in the PFC-MSC would reflect a heterogeneous signal from multiple layers within each dissection. Consequently, to increase the resolution of our analysis, we investigated whether the set of genes present in each midfetal network were more highly correlated within a particular cortical layer than expected by chance. Therefore, we utilized layer-specific microarray data that were obtained from laser microdissected (LMD) prenatal human brain by the BrainSpan Consortium (http://www.brainspan.org). Samples were derived from frontal neocortex of four brains, corresponding to periods 4, 5, or 6, and each region was dissected into nine layers, spanning from cortical surface to ventricular surface and including the outer (CPo) and inner (CPi) cortical plate.
To evaluate for layer specificity, we recreated both the midfetal periods 3–5 and 4–6 coexpression networks within each layer and assessed each layer-specific network for increased connectivity. This approach preserves the overall structure of the hcASD seed gene networks, including their temporal properties. Because many of the hcASD and pASD genes show expression throughout developing brain, we hypothesized that the expression dynamics of the ASD-related networks would be more informative than, for example, evaluating differential expression of individual genes. In practice, for each LMD layer, we calculated the Pearson’s correlation coefficients for all gene pairs that were found to be present within the midfetal networks and then summed the coefficients within the layer to quantify network connectivity. The observed value was assessed using a permutation test and 100,000 iterations. We identified increased connectivity in a single layer, CPi, for the period 3–5 midfetal network (corrected p=2.7×10−4; Figures 4A and 4B; Figure S4).
Figure 4. The PFC-MSC Networks Show Enrichment for Markers of Deep Layer Projection Neurons.

(A and B) To improve the spatial resolution of the coexpression analysis, the midfetal networks were assessed in an independent prenatal transcriptome (http://www.brainspan.org) data set comprised of microarray-based gene expression profiles of laser microdissected (LMD) human midfetal (period 4–6) brains. Each hcASD seed gene network was recreated within each layer from this LCM data and the significance of the observed connectivity (sum of correlations along network connections) for each layer was assessed by permutation test. The period 3–5 network (A) shows significant connectivity in the CPi region (inner cortical plate) corresponding to neocortical layers 5–6 (corrected p=2.7×10−4), whereas the the period 4–6 network (B) is not significantly connected in any layer. C) The period 3–5 PFC-MSC network is enriched for markers of deep layer cells in mouse cortex. At each postnatal day (P) indicated, differential gene expression analysis of RNA-seq data was performed on three cortical layers of two mouse cortices (1 male, 1 female) to identify genes exclusively differentially expressed in a particular layer (markers). The “superficial” layer corresponds to the human marginal zone and CPo (human layer 2 [L2] and L3), layer “four” corresponds to human CPo (human L4), and the “deep” layer corresponds to human CPi and subplate (human L5/L6 and subplate). From P4 to P10 (mid- to late fetal development), human orthologues of murine deep layer marker genes are significantly enriched in the period 3–5 network (hypergeometric test). From P14 to P180 (adulthood), there is insufficient resolution to differentiate layers and none are significantly enriched. (D and E) Enrichment of a set of previously published cell-type specific marker genes (Kang et al., 2011) is specific to cortical glutamatergic projection neurons (CPNs) in both midfetal networks (p=0.02 for period 3–5; p=0.01 for period 4–6). Markers of deep layer CPNs (L5/L6) are significantly enriched in both networks (p=0.02 for period 3–5; p=0.01 for period 4–6) whereas markers of superficial layer CPNs (L1 to L4) are not. All p values were assessed by permutation test with 100,000 iterations unless otherwise noted.
See also Figure S4 and Tables S5 and S6.
Mouse Laminar-Specific Expression Data Implicate Deep Cortical Layers in ASD
Given the role of neuronal migration in early brain development, we considered whether the observed localization to CPi for the period 3–5 PFC-MSC network might change over time. Because the LMD data were limited to a narrow developmental interval, we turned to a time series of neocortical layer-derived RNA-seq data for mouse brain development. These data were composed of two mouse brains, one male and one female, for each of six time points from P4 to P180 (corresponding in humans to late midfetal development to adulthood; Workman et al., 2013). At each time point, we sampled three cortical zones: superficial layers (SL; L1 to 3), layer 4 (L4), and deep layers (DL; L5/L6) and identified genes that were upregulated in only one of these three zones (Table S5). We then assessed whether a greater than expected enrichment of these zone-specific genes was observed in the period 3–5 PFC-MSC network. Consistent with our findings in human fetal cortex, only deep-layer (L5/L6) markers showed enrichment (p<0.04, hypergeometric test) and this enrichment was constrained to the earlier developmental time points P4 to P10 (equivalent to human late midfetal to late fetal; Figure 4C). This suggests that our finding of localization to CPi in human fetal brain for this hcASD-derived network was not a consequence of neurons that ultimately migrate to superficial cortical layers.
Gene-Marker-Based Analysis Implicates Layer 5/6 Glutamatergic Projection Neurons in ASD
Both the human fetal and mouse data localize the period 3–5 midfetal network to the deep layers of developing cortex. We next considered layer- and cell- specific gene markers to add a third, independent verification of this result and to increase our resolution to a specific cell type within the cortex. We utilized a previously published set of 40 marker genes representing five cell types and superficial and deep cortical layers in the developing human brain (Kang et al., 2011; Table S6). We assessed enrichment for these markers in the midfetal networks using permutation testing with 100,000 iterations. Significant enrichment was observed for cortical glutamatergic projection neurons (CPNs) in the period 3–5 and 4–6 networks (3 out of 17 markers in both; p=0.02 and p=0.01 for 3–5 and 4–6, respectively). Further inspection revealed that these three markers are specific for deep cortical layer (layers L5 and L6) CPNs (3 out of 11 in both; p=0.02 and p=0.01 for 3–5 and 4–6, respectively, Figures 4D and 4E).
Expression Profiling Confirms the Expression of hcASD Genes in Midfetal Cortical Projection Neurons
Our analysis of the ASD-associated midfetal networks predicts that the majority of the genes within the networks should be expressed in the CPi in midfetal prefrontal cortex. We tested this prediction by conducting immunostaining or in situ hybridization in period 5–6 coronal sections from human frontal cortex for five representative hcASD genes: TBR1, POGZ, CHD8, and DYRK1A (immunostaining), and SCN2A (in situ hybridization). As predicted, all five genes show robust expression in CPi projection neurons of this tissue (Figure 5). CHD8, DYRK1A, and SCN2A were observed in the majority of cortical plate neurons, including projection neurons in CPi and CPo. Consistent with our findings, TBR1, the most connected gene in our period 3–5 network, is exclusively expressed in CPi projection neurons in these developmental periods (Kwan et al., 2012). However, we note the expression of most other genes in the networks is not expected to be restricted to the CPi, as the association with this layer was observed at the network level.
Figure 5. hcASD Genes are Expressed in Midfetal Deep Layer Projection Neurons.
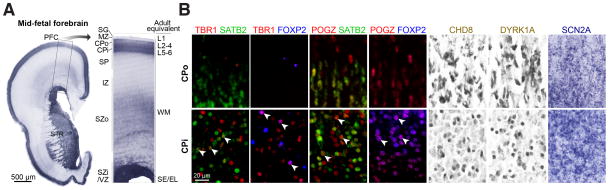
A) A coronal tissue section through the prefrontal cortex (PFC) and striatum (STR) of a midfetal forebrain was Nissl stained to visualize cells in distinct developmental zones. The darkest labeled zones are denser with cells, namely the VZ (ventricular zone) and SZi (inner subventricular zone), CPi (inner cortical plate), and CPo (outer cortical plate). The corresponding adult zones are labeled on the right of the higher magnified boxed area. B) Tissue sections of PFC areas at approximately equivalent ages (18–21 PCW) were stained with either antibodies (as labeled on top of the first six columns; fluorescence in red, green, or blue; DAB in brown) or in situ hybridization probes (last column, SCN2A). Images in the top row were taken at the boundary between CPo and MZ (marginal zone), and images in the bottom row at the boundary between CPi and SP (subplate). Arrows indicate cells colabeled for an hcASD gene and a marker gene (SATB2 or FOXP2).
SG, subpial granular zone; IZ, intermediate zone; SZo, outer subventricular zone; L, layer; WM, white matter; SE/EL, subependymal/ependymal layer.
To determine localization in different types of CPNs, we performed double immunofluorescent labeling using antibodies against the proteins encoded by the hcASD genes TBR1 and POGZ, along with two well-known markers for subtypes of cortical projection neurons, FOXP2 and SATB2 (Kwan et al., 2012). Both TBR1 and POGZ colocalized with FOXP2, a marker of deep-layer subcortical projection neurons that is exclusively present in the CPi. In addition, in some CPi neurons, we observed TBR1 and POGZ colocalizing with SATB2, a marker of intracortical (corticocortical) projection neurons, indicating that both subgroups of CPi projection neurons express TBR1 and POGZ.
DISCUSSION
The complexity of the genetic contribution to common neuropsychiatric conditions, including ASD, poses ample challenges for translational neuroscience. We sought to address some of these challenges by focusing on key questions that other analytical approaches have been hard pressed to tackle: specifically, when, where, and in what cell type should a specific ASD-related mutation or group of mutations be studied to begin to identify relevant pathophysiological mechanisms?
Given recent studies suggesting that as many as 1,000 genes or more could contribute to ASD (He et al., 2013; Iossifov et al., 2012; Sanders et al., 2012), our analysis has uncovered a surprising degree of developmental convergence. Despite starting with only nine hcASD seed genes, we have identified highly significant and robust evidence for the contribution of coexpression networks relevant to L5 and L6 CPNs in two overlapping periods of midfetal human development (3–5 and 4–6) corresponding to 10–24 PCW. These results strongly support the hypothesis that the marked locus heterogeneity underlying ASD will point to a much smaller set of underlying pathophysiological mechanisms.
Although there is clear evidence for the role of synaptic proteins in the pathogenesis of ASD, these findings point to the contribution of mechanisms extending beyond the synapse (State and Sestan, 2012). The identification of a functionally diverse set of risk genes within ASD networks, encoding proteins that are found in distinct cell compartments, is consistent with the hypothesis that alterations in multiple distinct pathways or processes may lead to the ASD phenotype by virtue of their spatial and temporal properties. For example, the CPi projection neurons of the midfetal PFC-MSC are among the first cortical neurons to form synaptic connections, and these early neural circuits may be particularly vulnerable to a variety of genetic perturbations and related functional disturbances that may all ultimately increase the risk for ASD.
To maximize the accuracy of our analysis, we focused considerable attention on the selection of input data. As noted, we restricted seed genes to those carrying multiple de novo LoF mutations and pASD genes to those carrying a single de novo LoF mutation—all derived from the same set of sequencing studies. Although this excluded several previously identified and well-established ASD genes, it established statistically based and consistent inclusion criteria, provided input data with relatively uniform evidence for involvement in ASD, and removed any bias introduced by the selection of genes that may have been originally identified, in part, by mechanistic hypotheses.
Similarly, although the evidence for vulnerability in some aspects of development of midfetal CPNs in ASD is quite robust, there is also support for the notion that additional points of convergent biology will ultimately be found in ASD. For example, in addition to midfetal cortical development, we have identified preliminary evidence for pASD enrichment in thalamus and cerebellum during postnatal development. Notably, two previously well-established ASD genes, NRXN1 and NLGN4X, also are found within this coexpression network (Supplemental Figure S3). In a similar vein, we note that none of the well-established syndromic ASD genes, such as FMR1, TSC1, TSC2, or PTEN, are found in the networks highlighted by our analyses. Taken together, these findings suggest that additional time points and brain regions are likely to be identified as both the number of bona fide risk genes grows and the depth of data on the molecular landscape of human brain development increases.
In this regard, we note the overall similarities between our findings and those reported in the accompanying study by Parikshak et al. (2013) in this issue of Cell, which uses an alternative approach to identify spatial and temporal convergence in genes involved in neurodevelopmental disorders. They also identify a potential role of CPNs in ASD, including in midfetal development and in L5/L6, yet their results also implicate L2/L3 in ASD. This difference is unsurprising, given differences in the input genes selected, in their approach to network/module construction and in their selection of expression data sets. By using a select small set of hcASD genes, our approach prioritized specificity over sensitivity, and we fully anticipated there would be undetected points of temporal and spatial convergence in ASD risk.
Nonetheless, the findings presented here immediately constrain important variables in the study of specific ASD-related mutations and provide the basis to pursue well-informed hypotheses regarding which mutations in which genes would be most likely to show overlapping molecular, cellular, or circuitlevel phenotypes. Given the increasing pool of ASD genes and mutations and the wide range of biological processes the encoded proteins perform, this ability to focus future in vitro and in vivo studies on subsets of genes based on their spatial and temporal properties and network relationships promises to have considerable value in the pursuit of a deeper understanding of pathophysiological mechanisms as well as in the identification of treatment targets.
EXPERIMENTAL PROCEDURES
For more details on any of these sections, please see the Extended Experimental Procedures. All experiments involving animals were performed in accordance with a protocol approved by Yale University’s Committee on Animal Research.
Exome Data and Mutations
Whole-blood derived DNA for 56 families chosen at random from the Simons Simplex Collection was subjected to exon capture using the Nimblegen EZExomeV2.0 array and sequenced with 74bp paired-end reads on the Illumina HiSeq 2000. Sequence reads were aligned to hg19 using BWA (Li and Durbin, 2009). Single-nucleotide variants were predicted using SAMtools (Li et al., 2009), whereas insertion-deletions were predicted using local realignment with Dindel (Albers et al., 2011). All de novo variants were confirmed using PCR and Sanger sequencing. Additional mutation data were obtained from the supplemental material from published papers (Iossifov et al., 2012; Kong et al., 2012; Neale et al., 2012; O’Roak et al., 2012a, 2012b; Sanders et al., 2012).
Construction of Spatiotemporal Coexpression Networks
Gene-level expression data (Platform GPL5175; Affymetrix GeneChip Human Exon 1.0 ST Array) were downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO accession number GSE25219) (Kang et al., 2011). Expression data from the core probe set were used in coexpression analysis (exceptions: for hcASD gene CHD8 and the pASD genes FLG, FREM3, FRG2C, LMTK3, THSD7A, UBN2, and ZNF594, data from the extended probe set were utilized). Within each postmortem brain, gene expression values were determined per region, resulting in a vector of expression values (expression profile) for each gene by brain region and brain sample. Within each spatiotemporal window, the expression profile was trimmed to relevant regions and brain samples only, and the Pearson correlation coefficient for the trimmed expression profile was calculated for each pairwise combination of genes. For each seed gene, the top 20 best-correlated genes with an absolute correlation coefficient with the seed gene of R0.7 were selected; each network is composed of the hcASD genes and their top correlated genes. Edges are then formed between any network genes with absolute correlation coefficient R 0.7. Any hcASD genes without edges were removed.
Temporal windows were determined solely by creating overlapping sets of three consecutive periods of development, starting with periods 1–3 and ending at periods 13–15. To determine spatial windows, brain regions were grouped according to transcriptional similarity during fetal development (periods 3–7): based on pairwise Spearman correlations, brain regions were hierarchically clustered in R using the “hclust” function (1 − corr2 as the distance, clustering using Ward’s method).
Permutation Tests
The permutation tests used throughout are based on selecting nine pseudo-hcASD seed genes weighted on the likelihood of observing multiple de novo LoF mutations by chance. A total of 100,000 sets of nine pseudo-hcASD genes were identified and separate coexpression networks were built for each of the four networks assessed. These 100,000 networks were used as the basis for the following permutation tests: pASD gene enrichment, connectivity, TADA p values, layer-specific marker genes, and cell-type-specific marker genes.
TBR1 KO mRNA-Seq and Data Analysis
Total RNA was isolated from freshly dissected P0 neocortices of Tbr1−/− and Tbr1+/+ littermates using a QIAGEN RNeasy Mini Kit. Libraries were prepared using an Illumina mRNA-Seq Sample Prep Kit. Amplified cDNA was size-selected at 250 bp and validated using the Agilent Bioanalyzer DNA 1000 system. The final product was subjected to cluster generation using an Illumina Standard Cluster Generation Kit v4. Libraries were sequenced to generate 74 bp single-end reads using the Illumina Genome Analyzer pipeline. Differentially expressed genes were identified based on a |log2(fold change)| ≥0.5 and adjusted P ≤0.05 based on Fisher’s exact test.
Network Analysis with Human Laminar-Specific Expression Data
Region- and layer-specific gene expression data were obtained from the BrainSpan Prenatal LMD Microarray project. This data set profiles gene expression in four brains spanning periods 4–6 of development (15–21 PCW; see technical white paper at http://www.brainspan.org). To assess the relevance of the two networks to each of the nine fetal layers or compartments, we determined normed Pearson’s correlation coefficients between network genes connected in the original midfetal networks, using expression data from each layer separately. By summing these correlations, we were able to estimate the overall connectivity of the networks layer-by-layer and then to assess the significance of this connectedness using a permutation test with 100,000 iterations.
Enrichment Analysis in Mouse Neocortical Layers Across Development
Different layers of the mouse cerebral neocortex were microdissected from live tissue sections of the Dcdc2a-Gfp transgenic reporter mouse (obtained from the GENSAT project; Schmidt et al., 2013), which expresses GFP selectively in L4 pyramidal and stellate glutamatergic excitatory neurons. Tissue samples were collected from the primary somatosensory area (equivalent to human S1C) of mouse brains at P4, P6, P8, P10, P14, and P180. Three laminar zones were isolated at each time point: superficial layers (SL; L2/l3 including marginal zone or L1, and pia), L4, and deep layers (DL; L5/L6 including transient subplate zone or adult white matter). At each time point, genes exclusively upregulated in only one laminar zone (marker genes) were identified.
Immunostaining and In Situ Hybridization
Coronal sections from 17–20 PCW human frontal cortex were used to validate the cellular resolution of ASD gene expression. For immunohistochemistry, sections were pretreated with 0.3% H2O2 followed by incubation in blocking buffer at room temperature and then incubation for 24–48 hr at 4°C in primary antibodies. Tissue was then incubated with biotin-labeled secondary antibodies, conjugated with avidin-biotin-peroxidase complex (Vector Laboratories), and visualized with DAB (Vector Laboratories). Images were taken using a digital scanner (Aperio ScanScope).
Immunofluorescent staining was performed similarly as aforementioned, without peroxide pretreatment, and with longer incubations in blocking buffer, primary and secondary antibodies, and intermittent washes. DAPI was included during incubation with secondary antibodies to counterstain nuclei.
For in situ hybridization, brain sections were first mounted on charged slides, followed by postfixation in 4% paraformaldehyde-PBS and then a 0.1 M triethanolamine with 0.25% acetic anhydride pretreatment. Sections were then incubated overnight at 65°C with 500 ng/ml of digoxigenin (DIG)-labeled cRNA probes corresponding to human SCN2A nucleotides 1,817–2,954 (NM_001040143). Sections were washed and incubated overnight at 4°C with alkaline phosphatase-conjugated anti-DIG antibodies, followed by signal detection using NBT/BCIP chromogen (Roche) diluted in a polyvinyl alcohol (PVA) buffer.
Accession Numbers
The whole-exome sequence data from the additional 56 quartets reported in this paper are available from the Sequence Read Archive (SRA) of the NCBI under BioProject PRJNA224099. The Tbr1−/− and Tbr1+/+ RNA-seq data reported in this paper are available from the SRA under BioProject PRJNA224108. The longitudinal neocortical layer-derived RNA-seq data for mouse brain development reported in this paper are available from the SRA under BioProject PRJNA224095.
Supplemental Information
Supplemental Information includes Extended Experimental Procedures, four figures, and six tables and can be found with this article online at http://dx. doi.org/10.1016/j.cell.2013.10.020.
Acknowledgments
We are grateful to the families participating in the Simons Foundation Autism Research Initiative (SFARI) Simplex Collection (SSC). This work was supported by a gift from the Overlook International Foundation (to M.W.S., N.S., B.D., K.R., and J.N.), as well as grants from the Simons Foundation (to M.W.S., N.S., K.R., and J.N.), the Kavli Foundation (to N.S.), the National Institute of Mental Health (RC2 MH089956 to M.W.S., U01 MH081896 to N.S., and R37 MH057881 to B.D. and K.R.), the Foster-Davis Foundation Inc. (NARSAD DI to N.S.), the Howard Hughes Medical Institute (International Student Research Fellowship to both S.J.S. and W.H.), and the Canadian Institutes of Health Research (Doctoral Foreign Study Award to A.J.W.). We would like to thank the SSC principal investigators (A.L. Beaudet, R. Bernier, J. Constantino, E.H. Cook Jr, E. Fombonne, D. Geschwind, D.E. Grice, A. Klin, D.H. Ledbetter, C. Lord, C.L. Martin, D.M. Martin, R. Maxim, J. Miles, O. Ousley, B. Peterson, J. Piggot, C. Saulnier, M.W. State, W. Stone, J.S. Sutcliffe, C.A. Walsh and E. Wijsman) and the coordinators and staff at the SSC clinical sites; the SFARI staff, in particular M. Benedetti; the Rutgers University Cell and DNA repository for accessing biomaterials; the Yale Center of Genomic Analysis, in particular J. Overton, S. Umlauf, I. Tikhonova and A. Lopez, for generating sequencing data; T. Brooks-Boone, N. Wright-Davis and M. Wojciechowski for their help in administering the project at Yale; and H. Rankin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R. Dindel: accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Daly MJ, Devlin B, Lehner T, Roeder K, State MW, Consortium TAS. The Autism Sequencing Consortium: Large-Scale, High-Throughput Sequencing in Autism Spectrum Disorders. Neuron. 2012;76:1052–1056. doi: 10.1016/j.neuron.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protocols. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Han W, Kwan KY, Shim S, Lam MM, Shin Y, Xu X, Zhu Y, Li M, Sestan N. TBR1 directly represses Fezf2 to control the laminar origin and development of the corticospinal tract. Proc Natl Acad Sci U S A. 2011;108:3041–3046. doi: 10.1073/pnas.1016723108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD, et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013;9:e1003671. doi: 10.1371/journal.pgen.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y-h, Narzisi G, Leotta A, et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AMM, Pletikos M, Meyer KA, Sedmak G, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Wong WS, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Šestan N, Anton ES. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development. 2012;139:1535–1546. doi: 10.1242/dev.069963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Sabo A, Neale BM, Nagaswamy U, Stevens C, Lim E, Bodea CA, Muzny D, Reid JG, Banks E, et al. Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet. 2013;9:e1003443. doi: 10.1371/journal.pgen.1003443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature genetics. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012a;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012b;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple Recurrent De Novo CNVs, Including Duplications of the 7q11.23 Williams Syndrome Region, Are Strongly Associated with Autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EF, Kus L, Gong S, Heintz N. BAC transgenic mice and the GENSAT database of engineered mouse strains. Cold Spring Harb Protoc. 2013;2013 doi: 10.1101/pdb.top073692. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci. 2011;14:1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, Sestan N. Neuroscience. The emerging biology of autism spectrum disorders. Science. 2012;337:1301–1303. doi: 10.1126/science.1224989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman AD, Charvet CJ, Clancy B, Darlington RB, Finlay BL. Modeling Transformations of Neurodevelopmental Sequences across Mammalian Species. The Journal of Neuroscience. 2013;33:7368–7383. doi: 10.1523/JNEUROSCI.5746-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information includes Extended Experimental Procedures, four figures, and six tables and can be found with this article online at http://dx. doi.org/10.1016/j.cell.2013.10.020.