

Abstract
Angiopoietin-like 4 (Angptl4) is a glucocorticoid receptor (GR) primary target gene in hepatocytes and adipocytes. It encodes a secreted protein that inhibits extracellular LPL and promotes adipocyte lipolysis. In Angptl4 null mice, glucocorticoid-induced adipocyte lipolysis and hepatic steatosis are compromised. Markedly, insulin suppressed glucocorticoid-induced Angptl4 transcription. To unravel the mechanism, we utilized small molecules to inhibit insulin signaling components and found that phosphatidylinositol 3-kinase and Akt were vital for the suppression in H4IIE cells. A forkhead box transcription factor response element (FRE) was found near the 15 bp Angptl4 glucocorticoid response element (GRE). Mutating the Angptl4 FRE significantly reduced glucocorticoid-induced reporter gene expression in cells. Moreover, chromatin immunoprecipitation revealed that GR and FoxO1 were recruited to Angptl4 GRE and FRE in a glucocorticoid-dependent manner, and cotreatment with insulin abolished both recruitments. Furthermore, in 24 h fasted mice, significant occupancy of GR and FoxO1 at the Angptl4 GRE and FRE was found in the liver. In contrast, both occupancies were diminished after 24 h refeeding. Finally, overexpression of dominant negative FoxO1 mutant abolished glucocorticoid-induced Angptl4 expression, mimicking the insulin suppression. Overall, we demonstrate that both GR and FoxO1 are required for Angptl4 transcription activation, and that FoxO1 negatively mediates the suppressive effect of insulin.
Keywords: glucocorticoid receptor, forkhead box protein O1, chromatin immunoprecipitation, glucocorticoid response element
Angiopoietin-like 4 (Angptl4), also known as the fasting-induced adipose factor, or Fiaf, encodes a secreted protein that inhibits LPL activity (1–3). LPL hydrolyzes TGs in lipoproteins, such as chylomicrons and VLDLs, into monoacylglycerol and FFAs, which can then be taken up into the cells. Angptl4 can also induce lipolysis in adipocytes (1–4). For adipocytes, the net metabolic effect of Angptl4 is to mobilize lipids to plasma. The importance of Angptl4 in the regulation of lipid homeostasis is supported by a series of physiological studies (2, 5). First, injecting ANGPTL4 recombinant proteins rapidly increases plasma TG and FFA levels (5, 6). Mice overexpressing Angptl4 in muscle and adipose tissue have increased plasma TG levels (7), and adenoviral-mediated overexpression of Angptl4 in liver causes hyperlipidemia and hepatic steatosis (8). Second, compared with WT mice, those lacking Angptl4 gene (Angptl4−/−) are hypolipidemic and have lower plasma FFA levels (9, 10). Furthermore, treating mice with Angptl4 antibody reduces plasma TG levels (11). When fed with high-fat diet, Angptl4−/− mice become obese faster than WT mice (12). However, these mice eventually develop fibrinopurulent peritonitis, ascites, intestinal fibrosis, and cachexia (9). Finally, genetic studies also support the critical role of Angptl4 in the regulation of lipid homeostasis. Population-based sequencing of human ANGPTL4 gene uncovered genetic variations that contribute to a reduced level of plasma TG (13). Also, serum ANGPTL4 levels and white adipose tissue (WAT) ANGPTL4 expression are inversely correlated in monozygotic twins.
The expression of Angptl4 gene is modulated by various signals. Thiazolidinedione, fibrate, and FFA induce Angptl4 transcription through members of the PPAR family, PPARγ, α, and β/δ, respectively (14, 15). Hypoxia and transforming growth factor β also activate Angptl4 transcription (16–18). Our group previously showed that glucocorticoids stimulate Angptl4 transcription in adipocytes and hepatocytes (19). A glucocorticoid response element (GRE) was identified in 3′ untranslated region of rat Angptl4 gene and is located between +6,267 and +6,241 [relative to transcription start site (TSS)] (19). The Angptl4 GRE sequence is conserved within rat, mouse, and human. Notably, Angptl4 expression is highly induced upon fasting, and glucocorticoid signaling is required for this fasting response (20). Physiological studies further confirmed that Angptl4 is involved in glucocorticoid-regulated lipid metabolism. Excess glucocorticoid-induced fatty liver and hyperlipidemia are protected in Angptl4−/− mice (19). Furthermore, glucocorticoid-induced lipolysis in WAT is reduced in Angptl4−/− mice (20).
Previous studies have shown that serum ANGPTL4 levels and the expression of ANGPTL4 are inversely correlated with insulin sensitivity (21–23). In 3T3-L1 adipocytes, insulin suppresses Angptl4 gene expression (24, 25). Notably, in adipocytes, glucocorticoids promote lipolysis, whereas insulin inhibits this process. Moreover, insulin resistance could lead to dyslipidemia and hepatic steatosis, which both can be a result of excess or prolonged glucocorticoid exposure. Based on these data, we propose that insulin suppresses glucocorticoid-induced Angptl4 transcription to antagonize glucocorticoid-modulated lipid metabolism. In this report, we investigated the effect of insulin on glucocorticoid-stimulated Angptl4 gene expression and unraveled the transcriptional mechanism underlying insulin-suppressed glucocorticoid-induced Angptl4 transcription.
MATERIALS AND METHODS
Cell culture
H4IIE rat hepatoma cells were cultured in DMEM (Mediatech) with 5% FBS (Tissue Culture Biologicals) and incubated at 37°C with 5% CO2. For all cell culture experiments, H4IIE cells were grown to 95% confluence, and treatments were diluted in DMEM only and applied to cultured cells. Rat primary hepatocytes were provided by the Cell Biology Core of University of California, San Francisco Liver Center. Treatments were applied as follows: 0.5 μM dexamethasone (Dex; Sigma D4902), 1 nM insulin (Sigma I9278), 10 μM phosphatidylinositol 3-kinase (PI3K) inhibitor GDC-09410 (Selleck S1064), 5 μM Akt inhibitor API-2 (Tocris 2151), 0.5 nM mammalian target of rapamycin (mTOR) inhibitor MK-8669 (also known as deforolimus, Selleck S1022), 200 nM S6 kinase (S6K) inhibitor rapamycin (Cayman 13346), and 5 μM glycogen synthase kinase (GSK) inhibitor SB-216763 (Tocris 1616).
Animals
Male 8-week-old C57BL/6 mice were purchased from Charles River. The control group of mice was continuously fed, while the experimental group of mice was fasted for 24 h starting at 10 AM, or fasted and refed the next morning at 10 AM for 24 h. Then, the mice were euthanized, and their liver tissues were collected at the same time. The Office of Laboratory Animal Care at the University of California, Berkeley (#R306-0111) approved all animal experiments conducted.
RNA isolation and quantitative PCR
Total RNA from H4IIE cells was isolated using Nucleospin RNA II kit (Macherey-Nagel 740955), and total RNA from mouse liver was prepared with TRI Reagent® RT (Molecular Research Center Inc.). To synthesize randomly primed cDNA, 0.5 μg of total RNA, 4 μl of 2.5 mM 2’-deoxynucleoside 5’-triphosphate, and 2 μl of 15 μM random primers (New England Biolabs) were mixed at a volume of 16 μl and incubated at 70°C for 10 min. Then, a 4 μl cocktail containing 25 units of Moloney Murine Leukemia Virus Reverse Transcriptase (New England Biolabs), 10 units of RNasin (Promega) and 2 μl of 10× reaction buffer (New England Biolabs) was added and incubated at 42°C for 1 h and then 95°C for 5 min. The cDNA was diluted appropriately for real-time quantitative PCR (qPCR) using the EVA QPCR SuperMix Kit (Biochain) following the manufacturer's protocol. qPCR was performed in a StepOne PCR System (Applied Biosystems) and analyzed with the ΔΔ-Ct method, as supplied by the manufacturer. Rpl19 gene expression was used for internal normalization. Mouse primers used were mRpl19_cDNA_Fo: ATGGAGCACATCCACAAGC; mRpl19_cDNA_Re: TCCTTGGTCTTAGACCTGCG; mAngptl4_cDNA_Fo: AAGATGCACAGCATCACAGG; and mAngptl4_cDNA_Re: ATGGATGGGAAATTGGAGC. Rat primers used were rRPL19_cDNA_Fo: ACAAGCGGATTCTCATGGAG; rRPL19_cDNA_Re: TCCTTGGTCTTAGACCTGCG; rAngptl4_cDNA_Fo: AGACCCGAAGGATAGAGTCCC; and rAngptl4_cDNA_Re: CCTTCTGGAACAGTTGCTGG.
Plasmids, transfection, and luciferase reporter assay
Reporter plasmids harboring different GR binding regions (GBRs) were cotransfected with pcDNA3-hGR (150 ng) and pRL Renilla (100 ng) into H4IIE cells in 12-well plates. pGL4.10-E4TATA reporter plasmid was generated by insertion of a 50 bp minimal E4 TATA promoter sequence into the BglII to HindIII sites of vector pGL4.10 to drive luciferase expression. Different lengths of the Angptl4 GBR regions were then inserted into the upstream of KpnI and XhoI sites of E4 TATA sequences. The QuikChange Lightning mutagenesis kit (Stratagene) was used to make site-directed mutations per the manufacturer's instructions. Lipofectamine 2000 (Invitrogen) was used to transfect H4IIE cells according to the technical manual. Twenty-four hours posttransfection, cells were treated with control ethanol, 0.5 μM Dex, 1 nM insulin in DMEM only for 16–20 h. Cells were then harvested, and their luciferase activities were measured with the Dual-Luciferase Reporter Assay kit (Promega) according to the technical manual. Cloning primers used were Luc_rAngptl4_KpnI_+6529bp: gctgcaGGTACCgctcttgttacctgctatgt; Luc_rAngptl4_XhoI_+6030bp: cgctctCTCGAGtggagatgcagagggacca; Luc_rAngptl4_KpnI_+6376bp: gctgcaGGTACCggaagctgaaatcactggga; Luc_rAngptl4_XhoI_+6181bp: cgctctCTCGAGggttccaaggcacagctca; and Luc_rAngptl4_KpnI_+6244bp: gctgcaGGTACCcagagaacaaaatgttctgagg. Mutagenesis primers used were Luc_rAngptl4_mtFOX_sense: CAAAGTTGGAGTAAAGATGTTCCTCGGGTGGAG and Luc_rAngptl4_mtFOX_antisense: CTCCACCCGAGGAACATCTTTACTCCACACTTTG. Human expression vector for dominant negative Akt was described previously (26) and was provided by Dr. Gary Firestone (University of California Berkeley).
Chromatin immunoprecipitation assay
H4IIE cells (1 × 108 to 2 × 108 cells) were treated with control ethanol, 0.5 µM Dex, 1 nM insulin, or a combination of Dex and insulin for 30 min, followed by cross-linking with formaldehyde at a final concentration of 1% at room temperature for 5 min. The reactions were quenched with 0.125 M glycine. The cells were then washed twice with 1× PBS and scraped and lysed in cell lysis buffer (50 mM HEPES-KOH at pH 7.4, 1 mM EDTA, 150 mM NaCl, 10% glycerol, 0.5% Triton X-100), supplemented with protease inhibitor (PI) cocktails (Calbiochem). The cell lysate was incubated for 15 min at 4°C, and the crude nuclear extract was collected by centrifugation at 600 g for 5 min at 4°C. The nuclei were resuspended in 1 ml of ice-cold RIPA buffer (10 mM Tris-HCL at pH 8.0, 1 mM EDTA, 150 mM NaCl, 5% glycerol, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, supplemented with PI). The chromatin was fragmented on ice with Branson Sonifier 250 sonicator (4 min total, 20 s pulse at 30% power followed by 40 s pause). To remove insoluble components, the samples were centrifuged at 13,000 rpm for 15 min at 4°C, and the supernatant was collected. Fifty to 100 μl supernatant was aliquoted as input, and the rest of supernatant was divided equally for antibody addition. One and half micrograms of normal rabbit IgG antibody (sc-2027, Santa Cruz Biotechnology), 2.5 μg of rabbit polyclonal anti-GR antibody (IA-1), 4 μg of FoxO1 antibody (sc-11350×, Santa Cruz Biotechnology), 4 μg of FoxO3 antibody (07-702, Millipore), or 4 μg of hepatic nuclear factor-3β/FoxA2 antibody (sc-9187×, Santa Cruz Biotechnology) was added to the supernatant and nutated at 4°C overnight. Our GR antibody, IA-1, was raised in a rabbit against a synthetic peptide comprising residues 75–103 of human GR. The IA-1 antibody was purified from the serum by binding to human GR fragment 27–506 immobilized to agarose beads (Sterogene Actigel), then eluted at low pH. The eluted antibody was neutralized, concentrated to 1 µg/µl, and stored at −80°C. We found IA-1 to be at least as specific for GR by western and chromatin immunoprecipitation (ChIP) sequencing compared with the published N499 and H-300 (SC-8992, Santa Cruz Biotechnology).
The next day, 100 μl of 50% protein A/G plus-agarose bead slurry (Santa Cruz Biotechnology) was added into each immunoprecipitated sample and nutated for 2 h at 4°C. These washes followed: twice with RIPA buffer, twice with RIPA buffer containing 500 mM NaCl, twice with LiCl buffer (20 mM Tris at pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% Tergitol-type NP-40 (NP-40, nonyl phenoxypolyethoxylethanol), 0.5% sodiumdeoxycholate), and one last time with RIPA buffer. After removing the remaining wash buffer, 75 μl of proteinase K solution (Tris-EDTA (TE) at pH 8.0, 0.7% SDS, 200 μg/ml proteinase K) was added to each immunoprecipitation reaction, followed by incubation at 55°C for 3 h and 65°C overnight to reverse formaldehyde cross-linking. ChIP DNA fragments were purified with QIAquick PCR purification kit (Qiagen), eluting in 60–120 μl of Qiagen Elution Buffer. ChIP DNA samples were then subjected to real-time qPCR analysis with the following primers: rAngptl4_ChIP_+6093bp_Fo: TTGACCGACTGGAGATAGGG and rAngptl4_ChIP_+6205bp_Re: ATGTTGTGAGCTGTGCCTTG.
Mouse liver ChIP assay
Liver tissues were harvested from control and experimental groups of C57BL/6 mice, minced with a razor blade, and collected in 1× SSC buffer (150 mM NaCl and 15 mM sodium citrate). Samples were then washed on a nutator, followed by centrifugation at 4,000 rpm for 3 min at 4°C to remove supernatant. The liver pellets were resuspended in PBS and cross-linked with 1% formaldehyde for 15 min at room temperature with gentle shaking, and the reaction was quenched with the addition of glycine. Centrifugation followed to remove supernatant, and the cell pellets were washed with ice-cold PBS plus PI with shaking. Once PBS was removed, the cells were resuspended in hypotonic buffer (10 mM HEPES at pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2% NP-40, 1 mM EDTA, 5% sucrose) with PI, 1.5 mM spermine, and 0.5 mM spermidine added right before use. Cells were homogenized using Polytron PT 2100 homogenizer for four pulses of 10 s each. Homogenized cells in hypotonic buffer are mounted onto cushion buffer (10 mM Tris-HCl at pH 7.5, 15 mM NaCl, 60 mM KCl, 1 mM EDTA, 10% sucrose, with PI, 1.5 mM spermine, and 0.5 mM spermidine added right before use) and centrifuged at 4,000 rpm at 4°C for 5 min, followed by the removal of supernatant. SDS-sonication buffer (50 mM Tris-HCl at pH 8.0, 2 mM EDTA, and 1% SDS added right before use) was applied to resuspend the nuclei pellet for sonication at 60% output for five times, 10 s each. To remove insoluble components, the samples were centrifuged at 13,000 rpm for 15 min at 4°C, and the supernatant was collected. Then, 3 vol of dilution buffer (20 mM Tris-HCl at pH 8.0, 2 mM EDTA, 200 mM NaCl, 1% Triton X-100, and 0.1% Na-deoxycholate) was added to the supernatant, and 100 μl of 50% protein A/G plus-agarose bead slurry with 5 μg of IgG antibody was added to each sample for preclearing at 4°C for 1 h with gentle shaking. Then, the sample was centrifuged at 4,000 rpm at 4°C for 3 min, 100 μl of the supernantant was saved for input, and the rest was divided accordingly for immunoprecipitation overnight. The next day, 60 μl of 50% protein A/G plus-agarose bead slurry was applied, and the samples were nutated for 2 h at 4°C. The following 500 μl washes were done, all supplemented with PI: once with TSE I (20 mM Tris-HCl at pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, and 0.1% SDS); once with TSE II (20 mM Tris-HCl at pH 8.0, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, and 0.1% SDS); once with TSE III (10 mM Tris-HCl at pH 8.0, 1 mM EDTA, 0.25 M LiCl, 1% NP-40, and 1% Na-deoxycholate), and twice with TE (10 mM Tris-HCl at pH 8.0 and 1 mM EDTA) buffer. Centrifugation at 8,000 rpm at 4°C for 1 min was used to remove supernatant between washes. Freshly prepared 400 μl elution buffer (100 mM NaHCO3 and 1% SDS) was added to each sample, and elution buffer (up to 400 μl in total) was added for input. The samples were nutated for 1 h at room temperature. To reverse the cross-linking, after centrifugation, supernatant was transferred to new Eppendorf tubes, and a final concentration of 200 mM NaCl was added to each sample, followed by 65°C water bath incubation for >6 h. The next day, 8 μl of 0.5 M EDTA, 16 μl of 1 M Tris-HCL at pH 6.5, and 1.5 μl of 25 mM proteinase K were added to each sample, followed by 65°C water bath incubation for 1 h. To purify DNA, chloroform extraction was done for each sample, and real-time qPCR was carried out for data analysis.
Adenovirus infection and Western blotting
FoxO1Δ256 adenoviruses were provided from Dr. Mimmo Accili (Columbia University) (27), which express human influenza hemagglutinin (HA) epitope tag-containing proteins. H4IIE cells were infected with adenoviruses for 48 h, followed by 5–6 h treatment of control ethanol, 0.5 μM Dex, or Dex plus 1 nM insulin. Antibody against HA tag (Roche 11583816001) was used to detect the overexpression level of FoxO1, and GAPDH (abcam ab9483) was included as internal control.
RESULTS
Insulin suppressed glucocorticoid-induced Angptl4 gene expression
Mouse primary hepatocytes were treated with 0.5 µM of Dex (a synthetic glucocorticoid), 1 nM insulin, a combination of insulin and Dex, or ethanol (vehicle control) for 5 h. At the end of the treatment, RNA was isolated from these cells, and reverse transcription was performed. Real-time PCR (qPCR) was then used to monitor the expression of Angptl4. We found that Angptl4 gene expression was ∼3.4-fold higher in Dex-treated cells than ethanol-treated cells (Fig. 1A). Although insulin treatment did not significantly decrease Angptl4 expression, insulin treatment abolished Dex-induced Angptl4 expression (Fig. 1A).
Fig. 1.

Gene expression of Angptl4. A: Mouse primary hepatocytes and H4IIE hepatoma cells were treated with control ethanol (EtOH), 0.5 µM Dex, 1 nM insulin, or Dex plus insulin, and in combination with one of the following inhibitors: 10 µM PI3K inhibitor GDC-0941, 5 µM Akt inhibitor API-2, 0.5 nM MK-8669 and 200 nM rapamycin, two TORC1 inhibitors, or 5 µM GSK inhibitor SB-216763. RNA isolation and RT-qPCR followed. Primers specific to Angptl4 and Rpl19 (internal control) gene were used in qPCR. Fold induction was calculated by normalizing to Rpl19 and taking the ratio of treatment over ethanol. Error bars indicate standard error of the mean. B: Schematic presentation of insulin signaling pathway in hepatocytes. Small-molecule inhibitors used to inhibit insulin signaling components are shown. C: Mouse primary adipocytes were treated with control ethanol (EtOH), 0.5 µM Dex, 1 nM insulin, or Dex plus insulin for 5 h. The expression of Angptl4 was assessed by RT-qPCR. Error bars indicate standard error of the mean. * P < 0.05 and n ≥ 3.
We previously showed that Dex markedly augmented the expression of Angptl4 in rat H4IIE hepatoma cells. We thus examined whether insulin inhibited Angptl4 expression in H4IIE cells. H4IIE cells were treated with 0.5 µM of Dex, 1 nM insulin, a combination of insulin and Dex, or ethanol for 5 h. Gene expression of Angptl4 was then performed. Indeed, we found that Dex increased Angptl4 expression ∼18.9-fold (Fig. 1A). Insulin treatment abolished this induction (Fig. 1A), whereas insulin alone did not significantly affect Angptl4 expression. Overall, these results support that the H4IIE cell line is a viable cell culture model for studying the mechanism of insulin effect on Angptl4 expression.
Identification of signaling molecules in insulin signaling pathway required to inhibit glucocorticoid-induced Angptl4 gene expression
To learn how insulin inhibits Dex-induced Angptl4 expression, various small molecules that inhibit the activity of each signaling protein in the insulin signaling pathway were applied (Fig. 1B). In detail, GDC-0941 inhibits class I PI3K (28), whereas API-2 inhibits Akt (29). MK-8669 and rapamycin inhibited mTORC1 (30, 31), which results in a reduction of p70 S6K activity. In addition, the GSK-3β inhibitor SB-216763 was used to mimic insulin effect (32), as Akt inhibits GSK-3β. We found that MK-8669 and rapamycin treatment did not affect insulin response, whereas GDC-0941 and API-2 significantly reduced the ability of insulin to inhibit Dex-induced Angptl4 gene expression (from 1.1-fold to 2.7- and 28.7-fold, respectively, Fig. 1A). Most likely, PI3K and Akt participate in the observed insulin repression, whereas mTORC1 does not. Moreover, cotreatment of Dex and SB-216763 reduced glucocorticoid-induced Angptl4 from 18.9- to 6.5-fold (Fig. 1A), suggesting that inhibiting GSK-3β mimicked the repressive effect of insulin on glucocorticoid-induced Angptl4 expression.
We also examined whether insulin suppresses Dex-induced Angptl4 expression in mouse primary adipocytes. We treated mouse primary adipocytes isolated from epididymal fat depots with ethanol, Dex, insulin, and Dex plus insulin for 5 h. RNA was isolated from these cells, and qPCR was used to monitor Angptl4 expression. We found that Dex increased Angptl4 expression ∼1.5-fold (Fig. 1C). This induction was abolished by insulin treatment (Fig. 1C). Insulin treatment alone did not reduce Angptl4 expression statistically; however, the trend of Angptl4 expression was lower in insulin-treated cells than in ethanol-treated ones.
FOX response element is required for maximal glucocorticoid-induced Angptl4 transcription
The 500 bp GBR of rat Angptl4, located from nt position +6,030 to +6,529 relative to its TSS, was inserted upstream of a luciferase reporter plasmid pGL4.10-E4TATA (33, 34)to create pWT (Fig. 2A). Dex treatment increased the activity of this reporter gene 12.7-fold (Fig. 2B), and a consensus GRE was previously identified at nt +6,228 to +6,242 (19). Here, we showed that insulin was able to suppress Dex-induced reporter gene activity from 12.7-fold to 4.8-fold (Fig. 2B). This indicates that pWT contains cis-acting elements that mediate Dex and insulin effects.
Fig. 2.

Mutagenesis and reporter assay reveal the importance of Fox binding site to glucocorticoid response. The 15 bp GRE of Angptl4 is located at chr17: 33,911,836 to 33,911,850, equivalent to nt position +6,229 to +6,243 relative to Angptl4 TSS. All nt positions shown are relative to rat Angptl4 TSS. A: WT GBR (+6,030 to +6,529) is inserted upstream of a luciferase gene in reporter plasmid pGL4.10-E4TATA (denoted as pWT). Deletions of WT GBR give the following reporter plasmids: pDel-1 (nt +6,030 to +6,376), pDel-2 (nt +6,181 to +6,529), pDel-3 (nt +6,181 to +6,376), and pDel-4 (nt +6,181 to +6,244). B: Reporter plasmids (250 ng) described in A were cotransfected with a human GR expression vector (150 ng) and a Renilla internal control plasmid (50 ng) into H4IIE cells. Twenty-four hours posttransfection, cells were treated with control ethanol (EtOH), 0.5 µM Dex, 1 nM insulin, or Dex plus insulin for 20–24 h. Fold induction is calculated by taking the ratio of treatment over ethanol. C: GRE and FRE of rat Angptl4 are shown. The nt +6,324 of FRE is mutated from C to G for mutant FRE (pMut-FRE). D: Reporter assays described in B were carried out for pWT and pMut-FRE. Ethanol-treated pWT luciferase activity was set as 0%, while Dex-treated pWT was 100%. E: pWT (250 ng) was cotransfected with a human GR expression vector (150 ng), a Renilla internal control plasmid (50 ng), and a human expression vector for dominant negative Akt (DN Akt; 300 or 600 ng) into H4IIE cells. Twenty-four hours posttransfection, cells were treated with control ethanol, 0.5 µM Dex, 1 nM insulin, or Dex plus insulin for 20–24 h. The results are presented as relative activities to Dex-induced pWT reporter activity. Error bars represent standard error of the mean. ** P < 0.05 for relative activity with no DN Akt cotransfection versus relative activity with DN Akt cotransfection; N.D. indicates no difference. Error bars represent standard error of the mean. * P < 0.05.
To locate the cis-acting elements responsible for insulin suppression, we deleted portions of pWT to get pDel-1, pDel-2, and pDel-3, which contain the genomic region of rat Angptl4 nt +6,030 to +6,376; nt +6,181 to +6,529; and nt +6,181 to +6,376 relative to its TSS, respectively (Fig. 2A). Markedly, insulin was able to suppress Dex response in all three reporter genes. These results suggest that cis-acting elements mediating Dex and insulin effects are located within nt +6,181 and +6,376.
We used TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html) to look for binding motif(s) for other transcription factors close to the Angptl4 GRE. Binding motifs for Sex determining region Y protein (SRY), Acute myeloid leukemia 1 protein (AML1), TATA, GR, FoxA2 (also known as HNF-3β), Ikaros-1 (IK-1), Ik-2, and Ik-3 were found when we set the threshold at 85 (Fig. 2C). Among them, FoxA2 binding motif and its potential binding proteins, FoxA2, FoxO1, and FoxO3, have a direct link to insulin signaling. Akt has been reported to phosphorylate and export FoxA2, FoxO1, and FoxO3 from the nucleus, thereby inactivating these transcription factor proteins (35, 36). For simplicity, this FoxA2 binding site will be referred to as the Forkhead box (FOX) response element (FRE) from now on.
To examine whether the Angptl4 FRE relayed the suppression of insulin on glucocorticoid-induced Angptl4 expression, we constructed the pDel-4 reporter, in which the FRE region was deleted from the pDel-3 reporter gene (Fig. 2A). Dex treatment markedly increased the activity of this pDel-4 reporter gene (Fig. 2B). However, insulin was unable to repress Dex response (Fig. 2B). These results suggested that the FRE plays a critical role in the repressive function of insulin on Dex-induced Angptl4.
Previous studies have demonstrated that FRE can cooperate with GRE to confer a maximal glucocorticoid response on gluconeogenic genes, such as phosphoenolpyruvate carboxykinase (Pepck) and glucose-6-phosphatase (G6Pase). Notably, the role of the FRE in Dex-activated Angptl4 transcription cannot be answered by comparing the Dex response on pDel-3 and pDel-4 reporters, because the Angptl4 GRE in pDel-4 is located 132 bp closer to the luciferase TSS than pDel-3 (Fig. 2A). The distance between the GRE and the basal transcription machinery could affect the magnitude of hormone response. Therefore, to learn whether the FRE is required for maximal Dex response in Angptl4, we mutated one nucleotide in the FRE in pWT to obtain pMut-FRE (Fig. 2C). With the mutated FRE, Dex-induced luciferase activity was significantly reduced 66% (Fig. 2D, from 100% to 34%), which indicates that this FRE is essential for conferring glucocorticoid response. For pWT reporter, insulin treatment reduced 80% of Dex response (Fig. 2D). For pMut-FRE, the ability of insulin to suppress Dex-induced luciferase activity was greatly reduced (Fig. 2D). These results again confirm the important role of this FRE in mediating the repressive effect of insulin. The reason that pMut-FRE still responded to insulin is likely because a single nucleotide mutation in FRE did not entirely eliminate the binding of FoxA2, FoxO1, and/or FoxO3 to this element. This is in contrast to pDel-4, in which the deletion of the entire FRE results in a complete loss of insulin response.
In Fig. 1A, we showed that Akt was involved in insulin-repressed glucocorticoid-induced Angptl4 expression. To test whether Akt directly regulates Angptl4 transcription through the GBR, we cotransfected an expression vector harboring a dominant negative form of Akt (DN Akt) with pWT into H4IIE cells. As shown in Fig. 2E, insulin suppressed ∼80% of Dex-induced pWT activity. With cotransfection of 0.3 μg DN Akt, insulin only repressed ∼50% of Dex-induced pWT activity. Furthermore, cotransfection of 0.6 μg DN Akt completely abolished insulin suppression (Fig. 2E). These results suggest that Akt is involved in the repressive effect of insulin on the enhancer activity of the Angptl4 GBR.
The recruitment of GR and FoxO1 to the rat Angptl4 GBR
We performed ChIP with specific antibodies against GR, FoxA2, FoxO1, and FoxO3 to determine which of them were recruited to the Angptl4 GBR. H4IIE cells were treated with ethanol, Dex, insulin, or a combination of insulin and Dex for 30 min, followed by ChIP assay. At basal condition under ethanol treatment, there was no significant GR recruitment at the Angptl4 GBR (Fig. 3). With Dex treatment, we observed significant GR occupancy (Fig. 3). Moreover, FoxO1 was recruited to the Angptl4 GBR with Dex treatment (Fig. 3). Interestingly, insulin treatment abolished Dex-induced recruitment of both GR and FoxO1 to the Angptl4 GBR (Fig. 3). The recruitment of FoxA2 and FoxO3 with Dex treatment did not reach statistical significance, with P values of 0.07 and 0.79, respectively (FoxO3 data was not shown).
Fig. 3.
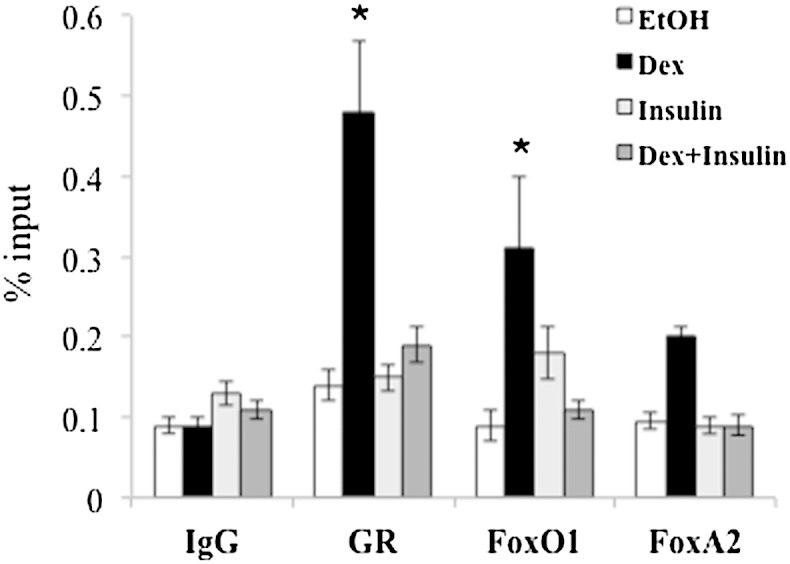
ChIP shows that insulin treatment abolished Dex-induced GR and FoxO1 recruitment. H4IIE cells were treated with control ethanol, 0.5 µM Dex, 1 nM insulin, or a combination of Dex and insulin for 30 min, followed by ChIP with antibody against IgG, GR, FoxO1, and FoxA2. Primer used here flanks nt +6,093 and +6,205 relative to Angptl4 TSS. Error bars indicate SEM. * P < 0.05.
The recruitment of GR and FoxO1 to the mouse Angptl4 GBR in vivo
To examine whether GR and FoxO1 are recruited to the mouse Angptl4 GBR in a similar pattern in vivo, we monitored the occupancy of these transcription factors upon fasting and refeeding cycle. Fasting elevates circulating corticosterone levels, while refeeding increases plasma insulin levels. Mice were fasted for 24 h from 10 AM. The next morning at 10 AM, food was replenished along with 5% glucose included in the drinking water. After 24 h, liver tissues were collected for gene expression and ChIP assays. The three groups were 1) continuous feeding (ad libitum), 2) 24 h fasting (fasted), and 3) 24 h fasting followed by 24 h refeeding (fasted/refed). Liver tissues were collected at the same time. We found that Angptl4 expression in 24 h fasted animals was 4-fold higher than that of ad libitum mice (Fig. 4A). After refeeding for 24 h, Angptl4 expression returned to levels the same as the ad libitum mice (Fig. 4A). With ChIP assay, in fasted mice both GR and FoxO1 occupied the Angptl4 GBR (Fig. 4B). In contrast, in fasted/refed mice, the occupancy of these two transcription factors diminished greatly (Fig. 4B). These results were reminiscent of our observations in H4IIE cells. In ad libitum mice, we detected significant GR occupancy on the GRE (Fig. 4B). However, FoxO1 was absent from the FRE (Fig. 4B). It is important to note that there is no induction of Angptl4 expression in ad libitum mice. These results further support the notion that the FRE is required for glucocorticoid-activated Angptl4. In summary, high glucocorticoid levels result in the recruitment of GR and FoxO1 to the Angptl4 GBR, whereas high insulin levels disrupt this recruitment.
Fig. 4.

In vivo (A) Angptl4 gene expression and (B) ChIP assay during fasting and refeeding cycle. C57BL/6 mice were treated as follows: 1) ad libitum (Ad Lib), 2) fasted for 24 h (Fast), and 3) fasted for 24 h then refed for 24 h (Fast/Re-fed). Error bars indicate standard error of the mean. * P < 0.05.
Overexpression of FoxO1 mutant lacking transactivation domain abolished the effects of glucocorticoids and insulin on Angptl4 gene
To further test the importance of the Angptl4 FRE in glucocorticoid- and insulin-regulated Angptl4 transcription, we overexpressed a mutant form of FoxO1 protein with adenoviral system in H4IIE cells. The FoxO1 mutant FoxO1Δ256 lacks the transactivation domain. FoxO1Δ256 is believed to act dominant negatively, as it can bind to its cognate cis-acting element but cannot synergize with GR to activate gene transcription. Indeed, compared with the overexpression of control LacZ (Ad-LacZ), FoxO1Δ256 overexpression (Ad-FoxO1Δ256) markedly reduced the ability of Dex to increase Angptl4 expression in H4IIE cells (Fig. 5). Furthermore, when FoxO1Δ256 was overexpressed, insulin treatment did not significantly suppress Dex-induced Angptl4 expression. These results indicate that FoxO1 mediates the suppressive effect of insulin on Dex-induced Angptl4 expression. Under basal conditions, Angptl4 expression increased ∼2-fold in FoxO1Δ256-overexpressing cells (Fig. 5). Notably, FoxO1 did not occupy Angptl4 FRE when H4IIE cells were treated with ethanol (Fig. 3). Therefore, it is unlikely that FoxO1Δ256 directly regulates basal Angptl4 expression. The increased basal Angptl4 expression is likely due to indirect effect of FoxO1Δ256 in H4IIE cells.
Fig. 5.
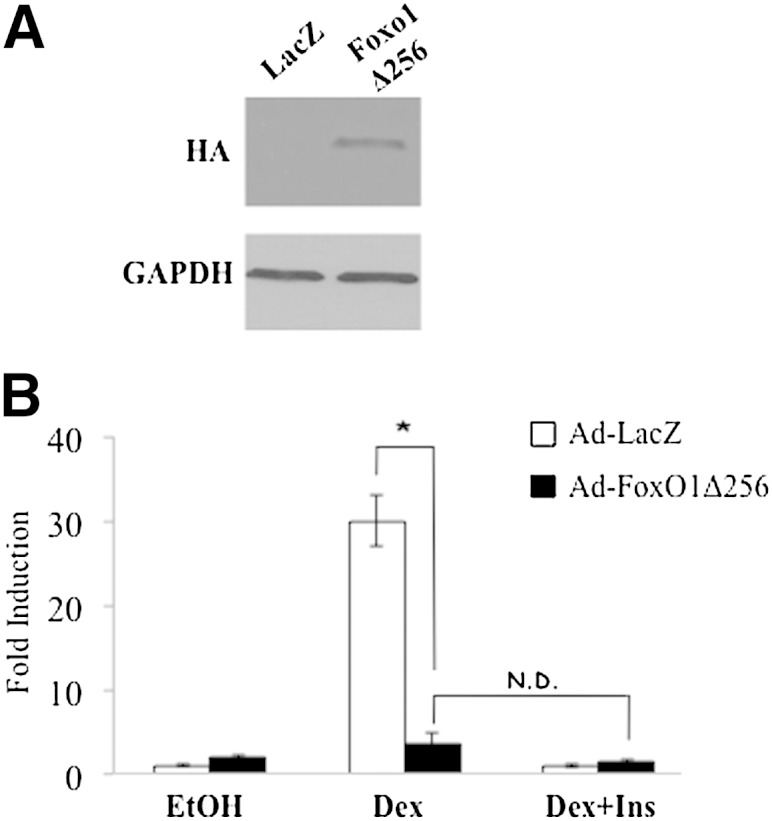
FoxO1 mediates insulin response on Dex-induced Angptl4 gene expression. H4IIE cells were infected with adenovirus carrying control LacZ (Ad-LacZ) or mutant FoxO1 lacking transactivation domain (Ad-FoxO1Δ256). A: Immunoblotting shows overexpression levels. B: After 48 h of infection, cells were treated with control ethanol, Dex, or a combination of Dex plus insulin for 6 h, followed by RT-qPCR for Angptl4 gene expression. Fold induction was calculated by normalizing to Rpl19 and taking the ratio of treatment over ethanol. Error bars indicate standard error of the mean. * P < 0.05; N.D. indicates no difference.
DISCUSSION
In this report, we have identified several novel aspects of the mechanism governing the transcriptional regulation of Angptl4 gene by glucocorticoids (GCs) and insulin. We found that an Angptl4 FRE, located close to the Angptl4 GRE, is vital for a maximal GC response in Angptl4 transcription (Fig. 6A). FoxO1 is recruited to this FRE in a GC-dependent manner both in vitro and in vivo, and the transactivation domain of FoxO1 is required for the GC response. Moreover, insulin represses GC-induced Angptl4 transcription through the activation of PI3K and Akt (Fig. 6B). In contrast, mTORC1 and its downstream effectors are not involved in this process. Furthermore, the Angptl4 FRE is also required for the repressive effect of insulin on GC-induced Angptl4 transcription. With the presence of insulin, the recruitment of FoxO1 and GR to their respective response elements is markedly reduced (Fig. 6B), which results in decreased GC response. Such a phenomenon is also confirmed in vivo. Upon fasting, which elevates circulating corticosterone levels and keeps insulin levels low, GR and FoxO1 occupy their respective response elements. However, after refeeding, circulating insulin levels are increased, and the occupancies of both GR and FoxO1 on their respective response elements are markedly reduced. Notably, ChIP sequencing results from the Encyclopedia of DNA Elements (ENCODE) Consortium found that there is a binding site for FoxO1 adjacent to the GRE of 3′ untranslated region of human ANGPTL4 gene (University of California, Santa Cruz genome browser). Likely, this mechanism of FoxO1 and GR-coregulated Angptl4 transcription is conserved in humans.
Fig. 6.

Proposed mechanism of Angptl4 transcription. A: When glucocorticoids are present without insulin, FoxO1 binds to the Angptl4 FRE and helps GR transcribe Angptl4. B: When both glucocorticoid and insulin are present, Akt inactivates FoxO1 by phosphorylation, which results in the export of FoxO1 from the nucleus. Without FoxO1 protein, glucocorticoid-induced Angptl4 gene expression is greatly reduced.
The mechanism of insulin-repressed GC-induced Angptl4 transcription is reminiscent of the mechanism of the transcriptional regulation of two important gluconeogenic genes: Pepck and G6Pase. For all three genes, their FREs are required for both GC and insulin response (37–39). Moreover, the insulin repression acts through PI3K and Akt, whereas mTORC1 is not involved (40, 41). Insulin indirectly activates PI3K, which in turn increases the activity of Akt for FoxO1 phosphorylation and leads to the exclusion of FoxO1 from the nucleus to cytosol. Gsk3, which is inactivated by Akt, has been reported to potentiate FoxO1 activity for Pepck and G6Pase expression (42, 43). Our data showed that inhibiting Gsk3 activity mimicked insulin repression on GC-induced Angptl4 expression. However, these results did not confirm that Gsk3 is required for the insulin repression, as Akt can directly inactivate FoxO1. In fact, a previous study suggested that Gsk3 is not involved in insulin-repressed Pepck and G6Pase expression in vivo, despite the pharmacological inhibition of Gsk3 mimicking the repressive effect of insulin (42).
There is one key feature that is distinct between the regulation of Angptl4 and the two previously mentioned gluconeogenic genes. FoxO1 binds to its respective FREs in the Pepck and the G6Pase promoters prior to GC treatment (39, 44). In contrast, FoxO1 is recruited to the Angptl4 FRE only upon GC treatment. This difference may explain the absence of insulin repression on basal Angptl4 expression in H4IIE cells from our experiments. In mouse 3T3-L1 adipocytes, previous studies reported that basal Angptl4 expression was suppressed by insulin (24, 25). These findings suggest that insulin exerts tissue-specific effects on basal Angptl4 expression. Alternatively, insulin may suppress Angptl4 expression in mouse primary hepatocytes and rat H4IIE hepatoma cells at time point(s) that we did not examine. FoxO1 is implicated in insulin-repressed basal Angptl4 expression in 3T3-L1 cells (24). It would be interesting to test whether the Angptl4 FRE we identified here associates with FoxO1 in 3T3-L1 adipocytes under basal conditions.
So far, we know that FoxO1 plays a critical role in GC-activated Angptl4 transcription. ChIP experiments in mouse liver showed that GR occupied the Angptl4 GRE during ad libitum and fasted states. However, the expression of Angptl4 was markedly lower in the ad libitum state than in the fasted state. This is likely due to the absence of FoxO1 on the Angptl4 FRE during the ad libitum state. It appears that the transactivation domain of FoxO1 is required for GC response to Angptl4, as overexpression of FoxO1Δ256 hindered Dex-induced Angptl4 transcription in H4IIE cells. Most likely, the assembly of the transcriptional regulatory complex, one that includes GR, FoxO1, and their associated coregulatory proteins, is required to confer maximal GC response to Angptl4 transcription. The fact that insulin treatment, which leads to FoxO1 exclusion from the nucleus, significantly reduced the recruitment of GR to the Angptl4 GRE suggests that the association of FoxO1 with the Angptl4 FRE is required for the stable interaction between GR and the Angptl4 GRE. Similar mechanisms were found in the regulation of Pepck gene. The transactivation domain of FoxA2 was required for a complete GC response on rat Pepck gene transcription (45), and the binding of FoxA2 and/or FoxO1 to its Pepck FRE (also known as the insulin response sequence or accessory element 2) potentiates the association of GR to the Pepck GRE (46). Notably, GR by itself does not bind to Pepck GRE well (47), whereas GR binds the Angptl4 GRE greatly as this GRE is a perfect palindromic consensus GRE (AGAACAnnnTGTTCT) (19). This notion was further supported by our results, in which in the ad libitum state significant numbers of GRs occupied the GRE even though FoxO1 was absent from the FRE.
One of major metabolic functions of Angptl4 is to mobilize fatty acids from adipocytes to circulating plasma by inhibiting extracellular LPL and promoting intracellular lipolysis in adipocytes. This aspect is physiologically important during fasting and starvation and is consistent with the physiological function of GCs. Increased Angptl4 proteins induced by GCs in liver can also enter circulation to promote lipid mobilization as mentioned. In contrast, insulin suppresses lipolysis and therefore antagonizes the effect of GCs on Angptl4 transcription. Moreover, insulin also inhibits the transcription of another GC-induced lipolytic gene, adipocyte TG lipase (Atgl, also known as desnutrin) (48). The Atgl GRE has not been identified; however, FoxO1 has been shown to activate Atgl gene transcription (49). The expression of FoxO1 is induced by GCs in adipocytes (50, 51). Thus, FoxO1 might mediate the GC response on the Atgl gene as well. Intriguingly, FoxO1 appears to be essential for a maximal GC response of genes that are repressed by insulin, including Angptl4, Pepck, and G6Pase. In opposition to their antagonist effects on lipolysis, GCs have been reported to synergize with insulin to potentiate lipogenesis (52–55). However, FoxO1 has been reported to suppress lipogenic screbp-1c gene expression (56). Furthermore, FoxO1 can reduce hepatic lipogenic Chrebp protein stability (57).
Therefore, we speculate that insulin-controlled FoxO1 participates not only in certain aspects of GC functions but also plays a pivotal role in determining the effects of GCs on lipid metabolism. We have previously shown that GCs activate lipolysis and lipogenesis concurrently in WATs (51). We hypothesize with results from the current studies that in adipocytes high FoxO1 activity aids the lipolytic effects of GCs. On the contrary, with low FoxO1 activity, as in the presence of insulin, GC-induced lipogenesis is favored. Future studies will test the proposed model and therefore provide insights into the fundamental understanding of insulin and glucocorticoid actions.
Footnotes
Abbreviations:
- Angptl4
- angiopoietin-like 4
- ChIP
- chromatin immunoprecipitation
- Dex
- dexamethasone
- DN Akt
- dominant negative form of Akt
- FOX
- forkhead box
- FRE
- Fox response element
- GR
- glucocorticoid receptor
- GBR
- GR binding region
- GRE
- glucocorticoid response element
- G6Pase
- glucose-6-phosphatase
- GSK
- glycogen synthase kinase
- Pepck
- phosphoenolpyruvate carboxykinase
- PI
- protease inhibitor
- PI3K
- phosphatidylinositol 3-kinase
- pMut-FRE
- mutant FRE
- pWT
- pGL4.10-E4TATA containing the Angptl4 GBR
- qPCR
- quantitative PCR
- TSS
- transcription start site
- WAT
- white adipose tissue
This work is supported by National Institutes of Health Grant R01 DK083591, a Dissertation Award Fellowship from the University of California Tobacco-Related Diseases Research Program (T.K.), and the Dr. and Mrs. James C. Y. Soong Fellowship of University of California at Berkeley (T-C.C.).
REFERENCES
- 1.Hato T., Tabata M., Oike Y. 2008. The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc. Med. 18: 6–14. [DOI] [PubMed] [Google Scholar]
- 2.Kersten S. 2005. Regulation of lipid metabolism via angiopoietin-like proteins. Biochem. Soc. Trans. 33: 1059–1062. [DOI] [PubMed] [Google Scholar]
- 3.Li C. 2006. Genetics and regulation of angiopoietin-like proteins 3 and 4. Curr. Opin. Lipidol. 17: 152–156. [DOI] [PubMed] [Google Scholar]
- 4.Gray N. E., Lam L. N., Yang K., Zhou A. Y., Koliwad S., Wang J. C. 2012. Angiopoietin-like 4 (Angptl4) protein is a physiological mediator of intracellular lipolysis in murine adipocytes. J. Biol. Chem. 287: 8444–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida K., Shimizugawa T., Ono M., Furukawa H. 2002. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J. Lipid Res. 43: 1770–1772. [DOI] [PubMed] [Google Scholar]
- 6.Akiyama T. E., Lambert G., Nicol C. J., Matsusue K., Peters J. M., Brewer H. B., Jr, Gonzalez F. J. 2004. Peroxisome proliferator-activated receptor beta/delta regulates very low density lipoprotein production and catabolism in mice on a Western diet. J. Biol. Chem. 279: 20874–20881. [DOI] [PubMed] [Google Scholar]
- 7.Mandard S., Zandbergen F., van Straten E., Wahli W., Kuipers F., Muller M., Kersten S. 2006. The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J. Biol. Chem. 281: 934–944. [DOI] [PubMed] [Google Scholar]
- 8.Xu A., Lam M. C., Chan K. W., Wang Y., Zhang J., Hoo R. L., Xu J. Y., Chen B., Chow W. S., Tso A. W., et al. 2005. Angiopoietin-like protein 4 decreases blood glucose and improves glucose tolerance but induces hyperlipidemia and hepatic steatosis in mice. Proc. Natl. Acad. Sci. USA. 102: 6086–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bäckhed F., Ding H., Wang T., Hooper L. V., Koh G. Y., Nagy A., Semenkovich C. F., Gordon J. I. 2004. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA. 101: 15718–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Köster A., Chao Y. B., Mosior M., Ford A., Gonzalez-DeWhitt P. A., Hale J. E., Li D., Qiu Y., Fraser C. C., Yang D. D., et al. 2005. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology. 146: 4943–4950. [DOI] [PubMed] [Google Scholar]
- 11.Desai U., Lee E. C., Chung K., Gao C., Gay J., Key B., Hansen G., Machajewski D., Platt K. A., Sands A. T., et al. 2007. Lipid-lowering effects of anti-angiopoietin-like 4 antibody recapitulate the lipid phenotype found in angiopoietin-like 4 knockout mice. Proc. Natl. Acad. Sci. USA. 104: 11766–11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bäckhed F., Manchester J. K., Semenkovich C. F., Gordon J. I. 2007. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA. 104: 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romeo S., Pennacchio L. A., Fu Y., Boerwinkle E., Tybjaerg-Hansen A., Hobbs H. H., Cohen J. C. 2007. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 39: 513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Georgiadi A., Lichtenstein L., Degenhardt T., Boekschoten M. V., van Bilsen M., Desvergne B., Muller M., Kersten S. 2010. Induction of cardiac Angptl4 by dietary fatty acids is mediated by peroxisome proliferator-activated receptor beta/delta and protects against fatty acid-induced oxidative stress. Circ. Res. 106: 1712–1721. [DOI] [PubMed] [Google Scholar]
- 15.Yoon J. C., Chickering T. W., Rosen E. D., Dussault B., Qin Y., Soukas A., Friedman J. M., Holmes W. E., Spiegelman B. M. 2000. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol. Cell. Biol. 20: 5343–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Jan S., Amy C., Cazes A., Monnot C., Lamande N., Favier J., Philippe J., Sibony M., Gasc J. M., Corvol P., et al. 2003. Angiopoietin-like 4 is a proangiogenic factor produced during ischemia and in conventional renal cell carcinoma. Am. J. Pathol. 162: 1521–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H., Ge C., Zhao F., Yan M., Hu C., Jia D., Tian H., Zhu M., Chen T., Jiang G., et al. 2011. Hypoxia-inducible factor 1 alpha-activated angiopoietin-like protein 4 contributes to tumor metastasis via vascular cell adhesion molecule-1/integrin beta1 signaling in human hepatocellular carcinoma. Hepatology. 54: 910–919. [DOI] [PubMed] [Google Scholar]
- 18.Padua D., Zhang X. H., Wang Q., Nadal C., Gerald W. L., Gomis R. R., Massague J. 2008. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 133: 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koliwad S. K., Kuo T., Shipp L. E., Gray N. E., Backhed F., So A. Y., Farese R. V., Jr, Wang J. C. 2009. Angiopoietin-like 4 (ANGPTL4, fasting-induced adipose factor) is a direct glucocorticoid receptor target and participates in glucocorticoid-regulated triglyceride metabolism. J. Biol. Chem. 284: 25593–25601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray N. E., Lam L. N., Yang K., Zhou A. Y., Koliwad S., Wang J. C. 2012. Angiopoietin-like 4 (Angptl4) is a physiological mediator of intracellular lipolysis in murine adipocytes. J. Biol. Chem. 287: 8444-8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Raalte D. H., Brands M., Serlie M. J., Mudde K., Stienstra R., Sauerwein H. P., Kersten S., Diamant M. 2012. Angiopoietin-like protein 4 is differentially regulated by glucocorticoids and insulin in vitro and in vivo in healthy humans. Exp. Clin. Endocrinol. Diabetes. 120: 598–603. [DOI] [PubMed] [Google Scholar]
- 22.Soronen J., Laurila P. P., Naukkarinen J., Surakka I., Ripatti S., Jauhiainen M., Olkkonen V. M., Yki-Jarvinen H. 2012. Adipose tissue gene expression analysis reveals changes in inflammatory, mitochondrial respiratory and lipid metabolic pathways in obese insulin-resistant subjects. BMC Med. Genomics. 5: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizutani N., Ozaki N., Seino Y., Fukami A., Sakamoto E., Fukuyama T., Sugimura Y., Nagasaki H., Arima H., Oiso Y. 2012. Reduction of insulin signaling upregulates angiopoietin-like protein 4 through elevated free fatty acids in diabetic mice. Exp. Clin. Endocrinol. Diabetes. 120: 139–144. [DOI] [PubMed] [Google Scholar]
- 24.Yamada T., Ozaki N., Kato Y., Miura Y., Oiso Y. 2006. Insulin downregulates angiopoietin-like protein 4 mRNA in 3T3–L1 adipocytes. Biochem. Biophys. Res. Commun. 347: 1138–1144. [DOI] [PubMed] [Google Scholar]
- 25.Dutton S., Trayhurn P. 2008. Regulation of angiopoietin-like protein 4/fasting-induced adipose factor (Angptl4/FIAF) expression in mouse white adipose tissue and 3T3–L1 adipocytes. Br. J. Nutr. 100: 18–26. [DOI] [PubMed] [Google Scholar]
- 26.Failor K. L., Desyatnikov Y., Finger L. A., Firestone G. L. 2007. Glucocorticoid-induced degradation of glycogen synthase kinase-3 protein is triggered by serum- and glucocorticoid-induced protein kinase and Akt signaling and controls beta-catenin dynamics and tight junction formation in mammary epithelial tumor cells. Mol. Endocrinol. 21: 2403–2415. [DOI] [PubMed] [Google Scholar]
- 27.Nakae J., Kitamura T., Silver D. L., Accili D. 2001. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Invest. 108: 1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Folkes A. J., Ahmadi K., Alderton W. K., Alix S., Baker S. J., Box G., Chuckowree I. S., Clarke P. A., Depledge P., Eccles S. A., et al. 2008. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 51: 5522–5532. [DOI] [PubMed] [Google Scholar]
- 29.Yang L., Dan H. C., Sun M., Liu Q., Sun X. M., Feldman R. I., Hamilton A. D., Polokoff M., Nicosia S. V., Herlyn M., et al. 2004. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 64: 4394–4399. [DOI] [PubMed] [Google Scholar]
- 30.Mahalingam D., Sankhala K., Mita A., Giles F. J., Mita M. M. 2009. Targeting the mTOR pathway using deforolimus in cancer therapy. Future Oncol. 5: 291–303. [DOI] [PubMed] [Google Scholar]
- 31.Loewith R., Jacinto E., Wullschleger S., Lorberg A., Crespo J. L., Bonenfant D., Oppliger W., Jenoe P., Hall M. N. 2002. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell. 10: 457–468. [DOI] [PubMed] [Google Scholar]
- 32.Coghlan M. P., Culbert A. A., Cross D. A., Corcoran S. L., Yates J. W., Pearce N. J., Rausch O. L., Murphy G. J., Carter P. S., Roxbee Cox L., et al. 2000. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7: 793–803. [DOI] [PubMed] [Google Scholar]
- 33.Lin Y. S., Carey M. F., Ptashne M., Green M. R. 1988. GAL4 derivatives function alone and synergistically with mammalian activators in vitro. Cell. 54: 659–664. [DOI] [PubMed] [Google Scholar]
- 34.Bolton E. C., So A. Y., Chaivorapol C., Haqq C. M., Li H., Yamamoto K. R. 2007. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 21: 2005–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calnan D. R., Brunet A. 2008. The FoxO code. Oncogene. 27: 2276–2288. [DOI] [PubMed] [Google Scholar]
- 36.Wolfrum C., Asilmaz E., Luca E., Friedman J. M., Stoffel M. 2004. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 432: 1027–1032. [DOI] [PubMed] [Google Scholar]
- 37.Onuma H., Vander Kooi B. T., Boustead J. N., Oeser J. K., O'Brien R. M. 2006. Correlation between FOXO1a (FKHR) and FOXO3a (FKHRL1) binding and the inhibition of basal glucose-6-phosphatase catalytic subunit gene transcription by insulin. Mol. Endocrinol. 20: 2831–2847. [DOI] [PubMed] [Google Scholar]
- 38.Wang J. C., Stromstedt P. E., O'Brien R. M., Granner D. K. 1996. Hepatic nuclear factor 3 is an accessory factor required for the stimulation of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Mol. Endocrinol. 10: 794–800. [DOI] [PubMed] [Google Scholar]
- 39.Hall R. K., Wang X. L., George L., Koch S. R., Granner D. K. 2007. Insulin represses phosphoenolpyruvate carboxykinase gene transcription by causing the rapid disruption of an active transcription complex: a potential epigenetic effect. Mol. Endocrinol. 21: 550–563. [DOI] [PubMed] [Google Scholar]
- 40.Dickens M., Svitek C. A., Culbert A. A., O'Brien R. M., Tavare J. M. 1998. Central role for phosphatidylinositide 3-kinase in the repression of glucose-6-phosphatase gene transcription by insulin. J. Biol. Chem. 273: 20144–20149. [DOI] [PubMed] [Google Scholar]
- 41.Sutherland C., O'Brien R. M., Granner D. K. 1995. Phosphatidylinositol 3-kinase, but not p70/p85 ribosomal S6 protein kinase, is required for the regulation of phosphoenolpyruvate carboxykinase (PEPCK) gene expression by insulin. Dissociation of signaling pathways for insulin and phorbol ester regulation of PEPCK gene expression. J. Biol. Chem. 270: 15501–15506. [DOI] [PubMed] [Google Scholar]
- 42.Lipina C., Huang X., Finlay D., McManus E. J., Alessi D. R., Sutherland C. 2005. Analysis of hepatic gene transcription in mice expressing insulin-insensitive GSK3. Biochem. J. 392: 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakamaki J., Daitoku H., Kaneko Y., Hagiwara A., Ueno K., Fukamizu A. 2012. GSK3beta regulates gluconeogenic gene expression through HNF4alpha and FOXO1. J. Recept. Signal Transduct. Res. 32: 96–101. [DOI] [PubMed] [Google Scholar]
- 44.Vander Kooi B. T., Streeper R. S., Svitek C. A., Oeser J. K., Powell D. R., O'Brien R. M. 2003. The three insulin response sequences in the glucose-6-phosphatase catalytic subunit gene promoter are functionally distinct. J. Biol. Chem. 278: 11782–11793. [DOI] [PubMed] [Google Scholar]
- 45.Wang J. C., Stromstedt P. E., Sugiyama T., Granner D. K. 1999. The phosphoenolpyruvate carboxykinase gene glucocorticoid response unit: identification of the functional domains of accessory factors HNF3 beta (hepatic nuclear factor-3 beta) and HNF4 and the necessity of proper alignment of their cognate binding sites. Mol. Endocrinol. 13: 604–618. [DOI] [PubMed] [Google Scholar]
- 46.Stafford J. M., Wilkinson J. C., Beechem J. M., Granner D. K. 2001. Accessory factors facilitate the binding of glucocorticoid receptor to the phosphoenolpyruvate carboxykinase gene promoter. J. Biol. Chem. 276: 39885–39891. [DOI] [PubMed] [Google Scholar]
- 47.Scott D. K., Stromstedt P. E., Wang J. C., Granner D. K. 1998. Further characterization of the glucocorticoid response unit in the phosphoenolpyruvate carboxykinase gene. The role of the glucocorticoid receptor-binding sites. Mol. Endocrinol. 12: 482–491. [DOI] [PubMed] [Google Scholar]
- 48.Villena J. A., Roy S., Sarkadi-Nagy E., Kim K. H., Sul H. S. 2004. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 279: 47066–47075. [DOI] [PubMed] [Google Scholar]
- 49.Chakrabarti P., Kandror K. V. 2009. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J. Biol. Chem. 284: 13296–13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee M. J., Gong D. W., Burkey B. F., Fried S. K. 2011. Pathways regulated by glucocorticoids in omental and subcutaneous human adipose tissues: a microarray study. Am. J. Physiol. Endocrinol. Metab. 300: E571–E580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu C. Y., Mayba O., Lee J. V., Tran J., Harris C., Speed T. P., Wang J. C. 2010. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS ONE. 5: e15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y., Jones Voy B., Urs S., Kim S., Soltani-Bejnood M., Quigley N., Heo Y. R., Standridge M., Andersen B., Dhar M., et al. 2004. The human fatty acid synthase gene and de novo lipogenesis are coordinately regulated in human adipose tissue. J. Nutr. 134: 1032–1038. [DOI] [PubMed] [Google Scholar]
- 53.Amatruda J. M., Danahy S. A., Chang C. L. 1983. The effects of glucocorticoids on insulin-stimulated lipogenesis in primary cultures of rat hepatocytes. Biochem. J. 212: 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai Y., Song Z., Wang X., Jiao H., Lin H. 2011. Dexamethasone-induced hepatic lipogenesis is insulin dependent in chickens (Gallus gallus domesticus). Stress. 14: 273–281. [DOI] [PubMed] [Google Scholar]
- 55.Wang J. C., Gray N. E., Kuo T., Harris C. 2012. Regulation of triglyceride metabolism by glucocorticoid receptor. Cell Biosci. 2: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng X., Zhang W., O-Sullivan I., Williams J. B., Dong Q., Park E. A., Raghow R., Unterman T. G., Elam M. B. 2012. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J. Biol. Chem. 287: 20132–20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ido-Kitamura Y., Sasaki T., Kobayashi M., Kim H. J., Lee Y. S., Kikuchi O., Yokota-Hashimoto H., Iizuka K., Accili D., Kitamura T. 2012. Hepatic FoxO1 integrates glucose utilization and lipid synthesis through regulation of Chrebp O-glycosylation. PLoS ONE. 7: e47231. [DOI] [PMC free article] [PubMed] [Google Scholar]