

Abstract
Rationale
The bile acid receptor Farnesoid-X-Receptor (FXR) regulates many aspects of lipid metabolism by various complex and not fully understood molecular mechanisms. We set out to investigate the molecular mechanisms for FXR-dependent regulation of lipid and lipoprotein metabolism.
Objective
To identify FXR-regulated microRNAs that were subsequently involved in regulating lipid metabolism.
Methods and Results
ATP binding cassette transporter A1 (ABCA1) is a major determinant of plasma High Density Lipoprotein (HDL)-cholesterol levels. Here we show that activation of the nuclear receptor FXR in vivo increases hepatic levels of miR-144, which in turn lower hepatic ABCA1 and plasma HDL levels. We identified two complementary sequences to miR-144 in the 3′ untranslated region (UTR) of ABCA1 mRNA that are necessary for miR-144-dependent regulation. Overexpression of miR-144 in vitro decreased both cellular ABCA1 protein and cholesterol efflux to lipid-poor apolipoprotein A-I (ApoA-I) protein, whilst overexpression in vivo reduced hepatic ABCA1 protein and plasma HDL-cholesterol. Conversely, silencing miR-144 in mice increased hepatic ABCA1 protein and HDL-cholesterol. In addition, we utilized tissue-specific FXR deficient mice to show that induction of miR-144 and FXR-dependent hypolipidemia requires hepatic, but not intestinal FXR. Finally, we identified functional FXR response elements (FXREs) upstream of the miR-144 locus, consistent with direct FXR regulation.
Conclusion
We have identified a novel pathway involving FXR, miR-144 and ABCA1 that together regulate plasma HDL cholesterol.
Keywords: FXR, HDL cholesterol, miRNA
INTRODUCTION
High plasma concentrations of low density lipoprotein (LDL) are a known risk factor for the development of cardiovascular disease 1. Conversely, high plasma levels of high density lipoprotein (HDL) are considered to be cardio-protective 2. The putative protective mechanisms include the anti-inflammatory properties of HDL and its role in reverse cholesterol transport, a process involving transport of cholesterol from atherosclerotic lesions back to the liver for subsequent excretion in bile 3.
The importance of ATP binding cassette transporter A1 (ABCA1) in HDL synthesis was first realized when inactivating mutations in the ABCA1 gene were shown to be causative for Tangier disease. Patients with Tangier’s disease have very low HDL levels, (less than 5% of normal levels) 4. Although ABCA1 is expressed in numerous cell types, tissue-specific deletion studies in mice demonstrated that hepatic, intestinal and adipose tissue ABCA1 contribute 70–80%, 15–20% and 15%, respectively, of the total plasma HDL 5, 6. Although bone marrow transplantation studies in mice have identified a critical role for ABCA1 in mediating cholesterol efflux from macrophages to lipid-poor apoproteins 7, there is no evidence that macrophage ABCA1 affects plasma HDL.
ABCA1 mRNA and protein levels are highly regulated by both transcriptional and post-transcriptional mechanisms. Activation of the Liver X Receptor (LXR, Nr1h3) results in increased transcription of the ABCA1 gene and increased levels of functional protein 8, 9. More recent studies demonstrated that ABCA1 is also regulated post-transcriptionally by two microRNAs, miR-33a and miR-33b, derived from intronic regions of the sterol-response-element-binding protein genes SREBP-2 and SREBP-1, respectively 10–14. Importantly, miR-33a is highly conserved across all species whereas miR-33b is absent from rodents although present in primates. Studies with mice 10–13 and non-human primates 15 demonstrated that modest changes in miR-33 levels (less than 2-fold) were sufficient to alter hepatic ABCA1 protein and plasma HDL levels (15–20%). Low cellular sterol levels result in activation of SREBP-2 and subsequent induction of genes involved in cholesterol synthesis and uptake 16. Simultaneous induction of miR-33 reduces ABCA1-dependent cholesterol efflux, thus providing a complementary mechanism to maintain cellular cholesterol levels. Taken together, these studies show that SREBP-2 and miR-33 function together to maintain cellular cholesterol homeostasis.
High expression of the Farnesoid X Receptor (FXR, Nr1h4) is limited to liver, intestine, kidney and the adrenal gland 17. Activation of FXR regulates genes controlling synthesis, secretion and resorption of bile acids, detoxification of hepatotoxins 18, maintenance of the intestinal barrier, and lipoprotein metabolism 17, 19, 20. Although bile acids were originally identified as the endogenous ligands for FXR 21, 22, subsequent studies demonstrated that specific bile acids also activate other nuclear receptors and the G-protein coupled receptor, TGR5 19, 20. Thus, a number of synthetic FXR-specific agonists, including GW4064 and WAY-362450 have been developed 23, 24. In the current study we utilized a recently described potent FXR agonist (GSK2324) 25 to identify hepatic microRNAs that are differentially regulated in vivo. Using a combination of gain- and loss-of-function studies, we show that one of these microRNAs, miR-144, is a direct FXR target that regulates hepatic ABCA1 protein levels and plasma HDL cholesterol levels.
METHODS
Mice, Diets, and Treatments
C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Fxr−/− mice were backcrossed to C57BL/6J mice for 10 generations at UCLA. Floxed Fxr−/− mice were previously described 26, 27 and were backcrossed for 12 generations to a C57BL/6J background at UCLA, before being crossed to Vilin-Cre or Albumin-Cre mice (Jackson Laboratories). Mice were fed a standard chow diet (NIH31 modified mouse/rat diet, catalogue no. 7013, Harlan Teklad) ad libitum. GSK2324 was a kind gift from Drs. David Deaton and Tim Willson (GlaxoSmithKline). In the original publication by Bass et al. 25 the authors used the parent FXR agonist GW4064 to generate GSK8062. A quinolone derivative of GSK8062 was termed 1c, but was also called GSK2324 25, the term used in the current paper. Importantly, the activation of FXR with GSK2324 is over 100-fold greater than the effect on other nuclear receptors 25. Where indicated, 10–12 week old mice were treated with either vehicle (water) or GSK2324 (30mpk, once a day) for 3 days unless otherwise stated in figure legends. Comparison between GSK2324 and GW4064 were carried out after 3 doses of either vehicle (0.5% Tween 80) and either of the solubilized FXR agonists via I.P. injections. For anti-miR treatments, mice were fed Western Diet (Research Diets, D12079B) for a total of 44 days. Anti-miRs were designed and synthesized by Regulus Therapeutics and delivered via I.P injections at 5mpk. In all experiments unless otherwise stated, mice were fasted for 4–6 hours and sacrificed in the early afternoon. All animal experiments were approved by the Office of Animal Research Oversight (OARO) at UCLA.
microRNA Expression Profiling
Total RNA was extracted from mouse livers treated for 3 days with vehicle or GSK2324 using Qiagen miRNeasy kit according to manufacturer’s instructions. Samples were prepared by pooling RNA from 2–3 livers and triplicate samples per condition were analyzed. Microarray hybridization and scanning was performed at UCLA Department of Pathology and Laboratory Medicine Clinical Microarray Core using Exiqon miRCURY LNA microRNA microarrays. Data analysis was carried out using the limma and vsn packages in R (v2.12.0). Calculated p-values from the moderated t-statistics were adjusted using Benjamini & Hochberg method for multiple testing. miRNAs with adjusted p-value <0.05 and fold change greater than 25% were considered differentially expressed.
Primary Hepatocytes
Mouse primary hepatocytes were isolated and cultured as described previously 18. Hepatocytes were infected with an MOI of 5 for each adenovirus either 6 or 12 hours after isolation, and cultured for an additional 48h with or without specific treatments, as described in the figure legends. Cholesterol efflux was carried out as previously described 9.
Cell Culture and Luciferase Reporter Assay
Hep3B (ATCC) cells were cultured according to ATCC recommendations. Cells were seeded in 6- or 12-well plates and when 70–80% confluent, infected with Ad-pre-miRNA for 24–48 hours before harvesting either total RNA using the miRNeasy kit (QIAGEN), or protein in RIPA buffer. For reporter assays, the mouse 3′ UTR of the ABCA1 mRNA was cloned downstream of the luciferase reporter gene in the pGL3 Promoter plasmid. The regulation of the ABCA1 3′UTR by miR-144 was determined by co-transfecting increasing concentrations of pSico pre-miR-144 (or pre-miR-451 or pSico GFP) plasmid with the luciferase-ABCA1 3′UTR reporter plasmid into HEK293Ad cells (Agilent). Cells were harvested 48 hours after transfection. Alternatively, HEK293Ad cells were co-transfected with the luciferase-ABCA1 3′UTR reporter with different concentrations of miR-144 (miR-144-3p) or miR-144* (miR-144-5p) mimics (Dharmacon) and harvested 48 hours after transfection. For miR-144 promoter studies, the 3kb region upstream of human pre-miR-144 was cloned into pGL4.10 plasmid (Promega) and transfection was carried out as described previously 18 in Hep3B cells. Luciferase reporter values were normalized to β-galactosidase activity to correct for transfection efficiency.
Plasma Lipid and Lipoprotein Analysis
All plasma lipid samples were analyzed by the Atherosclerosis Research Unit (ARU) Lipid Core Facility, which is certified by the Centers for Disease Control (CDC) lipid standardization program (Lab ID number LSP-251). Plasma triacylglyceride, total and HDL cholesterol were determined as previously described 26. Plasma lipoprotein profiles were obtained by modified Column Lipoprotein Profile (CLiP) method 28. Briefly, 15ml of plasma were diluted with 60ml of saline, and 10ml injected into a Superose-6 column (GE Healthcare) using elution buffer [saline/2mM EDTA/0.01% sodium azide (pH = 7.4)] at a flow rate of 0.6 mL/min at 40°C. The FPLC eluate was immediately mixed with cholesterol reagent (Thermo Scientific) at a flow rate of 0.3ml/min, and incubated at 40°C in a 5m KOT coiled reactor. The final mixture entered a spectrophotometric detector set at 500 nm, and the profiles were collected in real time using the LC solution software (Shimadzu).
Adenovirus Preparation
The generation of Ad-Control, Ad-pre-miR-144 and Ad-pre-miR-451 was carried out as described previously 10 except that the sequences amplified were those of miR-144 or miR-451 including approximately 100bp at the 5′ and 3′ ends. Adenovirus particles were prepared using the AdEasy system (Agilent) and purified by CsCl gradient centrifugation. The virus was dialyzed for 48 hours and stored at −80°C. Particles were quantified by serial dilution methods and detection of GFP positive plaques in HEK293Ad cells (Agilent). To overexpress miRNAs in mice, 109 plaque forming units (PFU) of adenovirus were transfused into 8–10 week old C57/BL6 mice via tail vein injection. Livers and plasma were collected 5 days post-infection after a 4–5 hour fast.
Real Time PCR
Total RNA was isolated with the miRNAeasy kit (QIAGEN) according to the manufacturer’s instructions. Gene expression was determined from cDNA synthesized using Reagents for Taqman kit (Applied Biosystems) from 500ng of total RNA and using a Lighcycler480 Real-time qPCR machine and Lightcycler 480 Mastermix (Roche). Relative gene expression was determined using an efficiency corrected method and efficiency was determined from a 3-log serial dilutions standard curve made from cDNA pooled from all samples. Primers were designed across exon-exon boundaries using Roche UPL guidelines. Primer sequences are shown in Online Table IV. Results were normalized to Tbp, 36B4 or β-actin mRNA. Relative miRNA expression was determined by Taqman RT-qPCR using pre-designed miR-144 (for miR-144-3p), miR-144* (for miR-144-5p) or miR-451 primer probe sets (Applied Biosystems) from cDNA synthesized from 100 ng total RNA (Applied Biosciences), and normalized to SnoRNA 202.
Western Blot Assay
Whole liver lysates were homogenized, or cell lysates were prepared in RIPA buffer (1x PBS with 1% SDS, 5g/L sodium deoxycholate, 1% NP-40) supplemented with protease inhibitor cocktail (Roche) fortified with additional PMSF, Leupeptin, Aprotinin and ALLN (Sigma) and quantified using the BCA assay (Pierce). Equal amounts of protein were separated on 4–12% acrylamide gels (Invitrogen) and transferred to a PVDF membrane (Millipore). Membranes were blocked for 1–16 hours in 5% non-fat milk solution in Tris-Buffered Saline containing 0.5% Tween 20 and probed with antibodies to ABCA1 (Novus Biologicals) or mouse monoclonal β-Actin (Sigma) for 1–16 hours. HRP detection was carried out using ECL plus reagent (GE Healthcare) according to the manufacturer’s instructions. Densitometric analysis was carried out using Quantity One software (BioRad) and relative ABCA1 protein levels expressed as fold changes after normalization to β-actin protein levels.
Statistical Analysis
Statistical analysis was performed using Prism Graphpad software (V5.0) or Microsoft Excel. Comparison between control and treatment group(s) was carried out using either a student t-test or One-way ANOVA and statistical significance is shown as described in the figure legends.
RESULTS
Activation of FXR in vivo Induces Hepatic miR-144 and miR-451
To identify new pathways regulated by FXR, we utilized GSK2324, a novel, water-soluble FXR agonist 25. As shown in Online Figure IA, treatment of wild-type, but not Fxr−/− mice with GSK2324 increased the hepatic expression of the bile acid export pump (Bsep/Abcb11), a well characterized FXR target gene 20. Further, induction of Bsep, and other known FXR target genes, was more pronounced in GSK2324-treated compared to GW4064-treated mice, suggesting GSK2324 is a more potent FXR agonist than GW4064 (Online Figure IB and data not shown).
Although activated FXR functions to directly induce target genes, FXR can also result in reduced expressions of many genes via indirect mechanisms. The latter is best exemplified by the FXR-dependent induction of SHP and FGF15/19 that together repress genes involved in bile acid synthesis 19. We hypothesized that FXR may also induce miRNAs that would act in turn to negatively regulate specific genes. To test this hypothesis, we isolated total hepatic RNA from wild-type mice treated with vehicle or GSK2324 25 and performed miRNA expression profiling. Differential miRNA expression analysis was carried out as described in the Methods section. This approach identified five miRNAs that were differentially expressed in the livers of GSK2324-treated C57BL/6 mice (Figure 1A, Online Figure IC (Volcano plot) and Online Table I). Of these five miRNAs, three were significantly induced (miR-144, -451 and -691; Online Table I) but only miR-144 and -451 are highly conserved across many phyla 29. In addition, miR-144 and miR-451, were previously reported to have high basal hepatic expression 30. The finding that miR-144 and miR-451 are co-transcribed as a bicistronic cluster (Figure 1B) from an intergenic region on mouse chromosome 11 29, 31, 32 is also consistent with the co-ordinate induction of both miRNAs. Further, two mature miRNAs, miR-144 (also known as miR-144-3p) and miR-144* (also known as miR-144-5p) are generated from one of the stem-loops (Figure 1B). Taqman-based RT-qPCR confirmed the induction of miR-451, miR-144 and miR-144* in wild-type, but not Fxr−/− mice treated with GSK2324 (Figure 1C and Online Figures ID and IE). Importantly, all three miRNAs were also induced following treatment with the more commonly used FXR agonist GW4064, when used at an equivalent dose (Figure 1D and Online Figure IE). The finding that miR-144, miR-144* and miR-451 were induced to similar levels by GSK2324 (Figure 1 and Online Figure I) is in agreement with the bicistronic organization and common regulation of these microRNAs.
Figure 1. In vivo GW4064 and GSK2324 Treatment Induces Hepatic Expression of miR-144 and miR-451.

(A) Total hepatic RNA was isolated from wild-type C57BL/6 mice treated for 3 days with vehicle or GSK2324 (30mpk/day) (n=9–10 mice/group). MicroRNA expression profiling was carried out using Exiqon microarrays in triplicate for each condition from samples pooled from 2–3 livers. Mean expression indicates the average of normalized hepatic expression values of vehicle- and GSK2324-treated mice. Log2 fold change indicates the change in miRNA expression after comparing GSK2324 to vehicle treatment. Open circles represent the significantly altered miRNAs (adjusted p<0.05 and fold change greater than 25%). (B) Diagram of the miR-144/-451 cluster, with sequences of miR-144* (miR-144-5p), miR-144 (miR-144-3p) and miR-451 identified. (C and D) Fxr+/+ (WT) and Fxr−/−(KO) mice (n=9/10 mice/group) were treated for 3 days with vehicle or GSK2324 (30mpk/day) (C), or vehicle, GW4064 (60mpk/d) or GSK2324 (60mpk/d) (D). Hepatic miRNAs were quantified using Taqman-based RT-qPCR and normalized to SnoRNA 202. Data are presented as mean ± SEM. Significance was measured with one-way ANOVA or student t-test. * indicates p<0.05, ** indicates p<0.01.
ABCA1 Is Regulated by miR-144
Using bioinformatic prediction tools 33 we compiled a list of mRNAs targeted by either miR-144 (Online Table II), or -451 using 4 different prediction softwares, and focused on the 117 putative targets that were common to all 4 prediction programs for miR-144 (Online Table II). Surprisingly, miR-451 had very few conserved target genes (not shown). Given the known role of FXR in the regulation of bile acid metabolism, we first searched for predicted miR-144 (or miR-451) target genes that would affect bile acid genes, but we failed to identify such targets (not shown). However, among the top predicted target genes for miR-144 was ABCA1. Analysis of the 3′UTR of ABCA1 mRNAs from numerous mammals identified two conserved putative miR-144 binding sites (Figure 2A). In addition, the mouse Abca1 3′UTR contains one additional predicted miR-144 binding site not present in other species (data not shown).
Figure 2. The 3′ UTR of the Mouse Abca1 mRNA Contains Two Complementary Sequences that Are Necessary for miR-144-Dependent Silencing.

(A) Schematic representation of the 3′UTR (3156 nucleotides) of mouse Abca1 mRNA with the putative complementary sequences to miR-144 and conservation shown for multiple species. (B–C) HEK293Ad cells (n=6 wells/condition) were transfected with a β-galactosidase expression plasmid, and plasmids encoding a luciferase ABCA1 3′UTR (3.1kb) for wild-type (B–D) or mutant 3′UTR lacking the miR-144 binding sites (C). Overexpression of miRNAs was carried out following co-transfection of pSicoR GFP plasmid expressing either pre-miR-144 or pre-miR-451 (B and C). Alternatively, cells were treated with mature miR-144 (miR-144-3p) or miR-144* (miR-144-5p), or control mimics (D). Luciferase activity was determined 48 hours after transfection and normalized to β–galactosidase activity. The activity of WT or mutant reporter plasmids in the absence of a miRNA overexpression plasmid or control mimic were independently set to 100%. Results are representative of at least 3 independent experiments. Data are presented as mean ± SEM. Significance was measured with one-way ANOVA and *** indicates p <0.001.
To examine whether miR-144 directly targets ABCA1, we utilized a luciferase reporter plasmid containing the 3′UTR from mouse Abca1 mRNA. Overexpression of miR-144, but not miR-451, reduced luciferase activity in a dose-dependent manner (Figure 2B). Deletion of the two predicted miR-144 binding sites rendered the ABCA1 reporter gene unresponsive to overexpression of miR-144 (Figure 2C). Individual mutations to the miR-144 binding site were not sufficient to abolish the response (data not shown). Consequently, we conclude that the two conserved miR-144 binding sites in the 3′UTR of ABCA1 are sufficient and necessary for miR-144-dependent repression of ABCA1.
These overexpression studies utilized a pre-miR-144 expression plasmid that produces endogenously processed mature miR-144 and miR-144*. To investigate whether only one or both mature forms of miR-144 could regulate ABCA1, we transfected HEK293Ad cells with mimics of miR-144, or miR-144* or a control mimic together with the luciferase ABCA1 3′UTR reporter plasmid. As expected, only the miR-144 mimic, and not the miR-144* or control mimic repressed the luciferase reporter plasmid (Figure 2D). The finding that the miR-144* mimic did not affect luciferase activity was expected as the ABCA1 3′UTR does not contain predicted miR-144* binding sites. We conclude that mature miR-144 (miR-144-3p), but not miR-144* (miR-144-5p) is responsible for the regulation of ABCA1.
To investigate whether miR-144 overexpression would regulate endogenous ABCA1 mRNA and/or protein levels, we treated primary mouse hepatocytes with an adenovirus expressing pre-miR-144 or pre-miR451, thus allowing for endogenous processing and generation of mature miR-144 and miR-451, respectively. Overexpression of miR-144 in primary mouse hepatocytes led to decreased ABCA1 protein levels, although unexpectedly Abca1 mRNA levels were unchanged (Figure 3A and B). Additionally, overexpression of miR-144 also resulted in decreased efflux of cellular cholesterol to lipid-poor apoA-I (Figure 3C), consistent with the decrease in ABCA1 protein levels (Figure 3B). These effects were specific for miR-144 since overexpression of miR-451 had no effect on ABCA1 mRNA or protein levels or cholesterol efflux to apoA-I (Figure 3A–C). Overexpression of miR-144 in a human hepatoma cell line (Hep3B) also resulted in a decrease in both ABCA1 protein and efflux of cholesterol to apoA-I, in the absence of a change in ABCA1 mRNA (Figure 3D–F). These results demonstrate that overexpression of miR-144 results in decreased ABCA1 protein levels in both mouse and human hepatocytes, suggesting that the mechanism may be conserved across these species.
Figure 3. Overexpression of miR-144 in Cultured Cells Reduces ABCA1 Protein Levels and Cholesterol Efflux to ApoA-I.

(A–C) Primary mouse hepatocytes and (D–F) human hepatoma Hep3B cells were infected with control adenovirus, Ad-pre-miR-144 or Ad-pre-miR-451 and cells analyzed after 24–48 hours for ABCA1 mRNA (A, D), ABCA1 protein (B, E) and the ability of the cells to efflux radiolabelled cholesterol to lipid-poor ApoA-I (C, F). Data are representative of at least 3 independent experiments (n=4–6 wells/condition) and Western blotting quantification values are average densitometry values from 3 wells/condition. Data are presented as mean ± SEM and significance was measured with one-way ANOVA or student t-test. *p<0.05, **p<0.01.
Overexpression of miR-144 in Mice Decreases Hepatic ABCA1 Protein and Plasma HDL Cholesterol
To evaluate the in vivo consequences of hepatic overexpression of miR-144, we injected wild-type mice on a chow diet with either a control adenovirus or an adenovirus expressing pre-miR-144 driven by a U6 promoter. This pre-miR-144 requires endogenous processing to produce mature, active miR-144. After 5 days, hepatic mature miR-144 levels increased 60% (Figure 4A), an increase similar to that observed after treatment of mice with the FXR agonist GSK2324 (compare Figure 4A and Figure 1C). Importantly, the increase in miR-144 was associated with a significant decrease in hepatic ABCA1 protein without a change in Abca1 mRNA (Figure 4B). The expression of other genes involved in lipoprotein and HDL biogenesis and metabolism (SR-BI, ApoAI, ApoAII, ApoE) were unchanged (Online Figure IIA). This latter result was expected since none of these genes were present in the list of predicted miR-144 target genes generated by four independent algorithms (Online Table II). In contrast, the expression of a known miR-144 target, nuclear factor-erythroid 2-related factor 2 (Nfe2l2, also known as NRF2) 34 and a novel target predicted from our in silico analysis (Rarβ, in Online Table II) were modestly decreased (Online Figure IIA). Importantly, total plasma and HDL cholesterol (Figure 4C and D) levels decreased in response to hepatic miR-144 overexpression in the absence of changes in plasma triacylglycerides (Online Figure IIB), consistent with a specific effect of miR-144 overexpression on plasma HDL cholesterol. Taken together, these data demonstrate that a modest increase in hepatic miR-144 levels in vivo is sufficient to reduce both hepatic ABCA1 protein and plasma HDL cholesterol.
Figure 4. Overexpression of miR-144 in vivo Reduces both Hepatic ABCA1 Protein and Plasma HDL Cholesterol Levels.
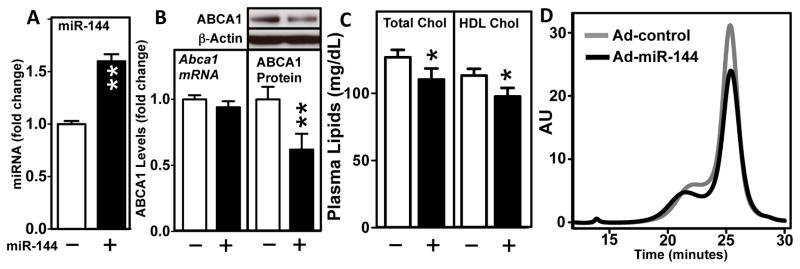
(A–D) Wild-type C57BL/6 mice (8–10/group) were infected with Ad-control or Ad-pre-miR-144. After 5 days, (A) Hepatic mature miR-144 was determined by Taqman-based RT-qPCR. In addition, (B) Abca1 mRNA and protein were determined SYBR green RT-qPCR and Western blotting respectively. Insert shows representative Western blot and protein changes were quantified by densitometry after normalization to β-actin (n=5/group). (C) Plasma total and HDL cholesterol, (D) plasma lipoprotein profiles determined by FPLC were determined as described in the Methods section. Data are shown as mean + SEM. Significance was determined using student t-test. *p<0.05, **p<0.01.
In vivo Silencing of miR-144 Increases Hepatic ABCA1 Protein and Plasma HDL
Having used gain-of-function approaches to show the effect of overexpression of hepatic miR-144, we next investigated whether loss-of-function would raise hepatic ABCA1 and plasma HDL cholesterol. Prior studies in mice and monkeys have successfully used chimeric 2′-fluoro/2′-methoxyethyl, phosphorothioate backbone modified anti-miRs to reduce the expression of a number of miRNAs, including miR-122 and miR-33 15, 35. Consequently, we generated antisense oligonucleotides that specifically target miR-144 (also known as miR-144-3p) and administered them to C57BL/6 mice fed a Western diet for 6 weeks. Bi-weekly anti-miR treatments were initiated after 2 weeks on the Western diet and continued for an additional 4 weeks before termination of the experiment. Anti-miR-144 or control anti-miR treatments did not affect body weight (Online Figure IIIA) or increase plasma ALT and AST levels compared to vehicle-treated mice (Online Figure IIIB). Plasma lipids were quantified after 2 weeks (Online Figure IIIC) and after 4 weeks (Figure 5) of anti-miR treatments at the termination of the experiment.
Figure 5. Silencing of miR-144 with anti-miR Increases Hepatic ABCA1 Protein and Plasma HDL Cholesterol.

(A–D) Wild-type C57BL/6 mice (n=10/group) were fed a Western diet for 6 weeks. Treatment with either a control anti-miR, anti-miR-144 or vehicle (phosphate buffered saline) was performed twice weekly (I.P.) at 5mpk for the final 4 weeks. (A) Hepatic miR-144 levels were measured by Taqman-based RT-qPCR analysis, normalized to SnoRNA 202. (B) Hepatic ABCA1 protein and mRNA levels were determined by Western blotting (representative) and quantified by densitometry analysis (n=4–5/group) or RT-qPCR (n=10/group) respectively and normalized to β-actin or 36B4. (C) Plasma total cholesterol and HDL-cholesterol levels and (D) plasma cholesterol lipoprotein profiles were determined as described in the Methods section. Data are shown as mean ± SEM. Significance was measured with one-way ANOVA or student t-test. * p<0.05; ***p<0.001.
Anti-miR-144 treatment significantly reduced hepatic miR-144 levels (Figure 5A). However, analysis of absolute changes in the levels of silenced microRNAs may be confounded by the formation of stable intracellular miRNA:anti-miR complexes 35. Nonetheless, the fact that reduced miR-144 levels were detected, suggests that at the very least, the anti-miR-144 accumulated in the livers of treated mice, and likely reduced miR-144 availability. Importantly, anti-miR-144 treatment for 4 weeks resulted in increased levels of hepatic ABCA1 protein, total plasma and HDL cholesterol, without significantly affecting Abca1 mRNA (Figure 5B–D) or plasma triacylglyceride levels (data not shown). The increase in plasma HDL cholesterol was also observed after only 2 weeks of anti miR-144 treatment (Online Figure IIIC). Silencing of miR-144 for 4 weeks had no effect on hepatic scavenger receptor B1 (Scarb1, also known as SR-B1) and ApoAI mRNA, and, as expected, induced the expression of both Nfe2l2 and Rarβ (Online Figure IIID). Surprisingly, anti-miR-144 treatment also increased the expression of hepatic ApoAII and ApoE mRNAs (Online Figure IIID). This latter result may be due to indirect regulation since the 3′UTRs of ApoAII and ApoE do not contain putative miR-144 binding sites, and neither ApoAII or ApoE mRNAs were altered by miR-144 overexpression (Online Figure IIA). Taken together, these results demonstrate that silencing of miR-144 results in increased hepatic ABCA1 protein levels and plasma HDL cholesterol.
Next, we also determined whether acute silencing of miR-144 in mice fed a chow diet also affected plasma lipid levels. We treated male, chow-fed C57BL/6 mice with 2 treatments of anti-miR-144 (5mpk) on days 1 and 4 and sacrificed the mice on day 7. Analysis of hepatic miR-144 levels indicated a significant reduction in miR-144 levels (Online Figure IVA), suggesting that the anti-miR-144 was likely present in the liver. Moreover, miR-144 silencing caused a modest increase in ABCA1 protein, without affecting mRNA (Online Figure IVB), consistent with previous experiments. Importantly, silencing miR-144 for 7 days was sufficient to elevate plasma total and HDL levels (Online Figure IVC and D), although these changes were more modest than silencing miR-144 for 28 days in the presence of a Western diet. None-the-less, these results demonstrate that silencing miR-144 both acutely and chronically in chow-fed or Western diet-fed mice results in increased plasma HDL levels.
miR-144 Is a Direct FXR Target
FXR activation in the liver directly induces FXR target genes, which in turn, act as effectors for the FXR-dependent changes in bile acid and lipid metabolism. In addition, activation of intestinal FXR induces FGF15/19 and this secreted protein then modulates hepatic gene expression 19, 20. To identify whether hepatic or intestinal FXR is required for induction of miR-144 and -451 in the liver, we generated tissue-specific FXR knockout mice by crossing mice containing floxed FXR alleles to mice that express either villin- or albumin-driven Cre recombinase. This approach generated mice that do not express FXR in the intestine (I-Fxr−/−) or liver (L-Fxr−/−), respectively. Treatment of the wild-type (floxed) Fxr mice or I-Fxr−/− mice with GSK2324, resulted in normal induction of both miR-144 and -451 (Figure 6A). In contrast, miR-144, -451 (Figure 6A) and miR-144* (Online Figure IF) were not induced in the livers of L-Fxr−/− mice, demonstrating the requirement for hepatic FXR for induction of miR-144/-451. Consistent with this result, we show that GSK2324 treatment of wild type or I-Fxr−/− mice for 3 days resulted in decreased total plasma and HDL cholesterol levels, whereas, plasma lipid levels were not decreased following treatment of L-Fxr−/− mice with GSK2324 (Figure 6B). Moreover, the hyperlipidemic effect previously described in whole-body Fxr−/− mice 36 was also observed in untreated L-Fxr−/−, but not I-Fxr−/− mice (Figure 6B). Together, these studies identify hepatic FXR as being critical for induction of miR-144 and for the GSK2324-dependent decrease in plasma cholesterol levels. The change in plasma cholesterol levels is likely a result of multiple mechanisms since FXR also induces the expression of hepatic Scarb1, the HDL receptor (Online Figure VA) 26.
Figure 6. Hepatic FXR Is Required for the Induction of miR-144 and the Hypocholesterolemic Effects of GSK2324. The miR-144/-451 Cluster Is a Direct FXR Target.

Liver-specific (L-KO) and Intestine-specific (I-KO) Fxr−/− mice and Fxr+/+ (floxed) littermate (WT) mice were treated with vehicle or GSK2324 for 3 days at 30mpk/day. (A) Hepatic miR-144 and miR-451 levels were determined by Taqman RT-qPCR and (B) Plasma total and HDL cholesterol levels were determined as described in the Methods section. (C) The human miR-144/-451 promoter cloned into a luciferase reporter plasmid was transfected together with a β-galactosidase expression plasmid and increasing amounts of pcDNA human FXRα2 expression plasmid into Hep3B cells in the presence or absence of GSK2324 (1μM). Promoter activity was normalized to β-galactosidase activity shown as fold changes. (D) Hepatic Abca1 mRNA and (E) ABCA1 protein levels were determined by RT-qPCR or Western blotting respectively in Fxr+/+ (WT) and Fxr−/− (KO) mice treated with vehicle or GSK2324 (30mpk/day, 3d) (n=5–9/group). Data are shown as mean ± SEM. Significance was measured with one-way ANOVA or student t-test. *p<0.05; ***p<0.001.
To determine whether miR-144/-451 is a direct target of FXR, we analyzed data from a genome-wide hepatic FXR response element (FXRE) ChIP-Seq study 37. This analysis identified an FXRE located approximately 12kb upstream of the mouse miR-144/-451 locus (Online Figure VB), whilst no FXRE was identified in the proximal promoter. Importantly, this FXRE was both conserved in rats, and was flanked by an additional nuclear receptor half-site (TGGCACT) (Online Figure VB), consistent with previous findings for many, but not all FXREs 37, 38. To investigate whether the distal putative FXRE functions as a bona fide FXRE, we generated a reporter plasmid containing 3 copies of the FXRE upstream of a minimal thymidine kinase promoter driving a luciferase reporter gene. FXR overexpression and activation with GSK2324 caused a dose-dependent induction in the miR-144 FXRE reporter plasmid (Online Figure VC).
In contrast, luciferase activity was not induced when the 3 copies of the FXRE were mutated (Online Figure VC). In contrast to mice and rats, analysis of the human genomic region corresponding to the 3kb region upstream of the human miR-144/-451 locus identified a putative FXRE. This response element is also conserved in non-human primates (Online Figure VB). A luciferase reporter gene under the control of the human miR-144/-451 promoter was induced following co-expression of FXR and treatment with GSK2324 (Figure 6C). The FXR-dependent activation of the human promoter was abolished when the predicted single FXRE was mutated (Figure 6C). Thus, we conclude that both primates and rodents, contain distinct functional FXREs in the miR-144/-451 loci that are important for the FXR-mediated induction of both miR-144 and -451.
The data we have described thus far suggests that treatment of mice with GSK2324 will result in a decrease in hepatic ABCA1 protein. To test this hypothesis, we treated wild-type and Fxr−/− mice with GSK2324 for 3 days. FXR activation resulted in a modest decrease in Abca1 mRNA, but a more pronounced decrease in ABCA1 protein (Figure 6D and E). This effect was specific, since treatment of Fxr−/− mice did not change either ABCA1 mRNA or protein levels (Figure 6D and E). These results are consistent with an FXR-dependent, post-transcriptional regulation of ABCA1.
DISCUSSION
Previous studies have demonstrated that treatment of mice and rats with FXR agonists, such as GW4064 or WAY-362450, results in decreased plasma triacylglyceride and cholesterol levels 24, 26, 39. FXR agonists were also shown to induce the hepatic expression of Scarb1 mRNA and protein that could explain, at least in part, the FXR-dependent reduction in plasma HDL 26. The results presented here identify a complementary, previously unrecognized pathway that links hepatic FXR activation to induction of miR-144 and subsequent reduction in hepatic ABCA1 protein. We used both gain-of-function and loss-of-function studies to show that changes in the levels of hepatic miR-144 are sufficient to regulate hepatic ABCA1 protein and plasma HDL-cholesterol. This effect was specific as plasma triacylglyceride levels were unaffected by changes in miR-144 levels. The physiological importance of this FXR-dependent dual regulation of plasma HDL levels through Scarb1 and miR-144 remains to be clearly elucidated but may be related to efficient channeling/partitioning of intrahepatic cholesterol into bile.
A very recent report showed that enhanced HDL uptake via overexpression of Scarb1 increased hepatic cellular cholesterol, which was then preferentially transported into bile, but the effect required blunted ABCA1 activity which was achieved with probucol 40. These latter results suggest that induction of Scarb1 alone is not sufficient to prevent the re-circulation of cholesterol, which may then be re-secreted into nascent HDL via ABCA1. Thus, our current data suggest that not only does FXR promote the clearance of circulating HDL (via Scarb1), but it also minimizes the re-secretion of intracellular sterols into new HDL (as illustrated in Figure 7). Together, these data suggest that FXR activation induces uptake of plasma HDL cholesterol into the liver, and subsequently facilitates the excretion of cholesterol and cholesterol-derived bile acids into the bile. Indeed, we have previously demonstrated that FXR activation with GW4064 increases reverse cholesterol transport 26, consistent with the altered sterol flux. The findings reported in the current study showing that FXR reduces hepatic ABCA1 protein via a post-transcriptional mechanism provides important evidence supporting this complex directional movement of cholesterol (Figure 7). FXR activation in vivo with GSK2324 or GW4064 (solubilized in 0.5% Tween 80) did not induce the expression of the transporters ABCG5/G8 (not shown), which are involved in the transport of cholesterol into the bile 41. Importantly, in the sterol flux model proposed by Annema and colleagues 40, an increase in Abcg5/Abcg8 expression was not required, suggesting that endogenous levels of ABCG5/G8 protein are sufficient to mediate cholesterol movement into the bile.
Figure 7. A Model Linking FXR Activation to Hepatic Cholesterol Transport.

Activated FXR results in induction of SCARB1 resulting in increased uptake of plasma HDL cholesterol. Activated FXR also induces the expression of miR-144 leading to a reduction in ABCA1 protein levels and cholesterol efflux to lipid-poor ApoAI. Consequently, internalized cholesterol may be preferentially channeled towards biliary excretion via ABCG5/ABCG8 rather than re-secretion via ABCA1 to produce pre-β-HDL, a precursor of HDL.
Hepatic miR-144 levels were induced to a similar extent in GSK2324-treated wild-type and I-Fxr−/− mice (Figure 6A) demonstrating that intestinal FXR has no role in regulating hepatic miR-144 levels or the hypolipidemic effects of FXR activation. These data, together with the observation that intestinal miR-144 levels were low/undetectable 30, suggest that the effects noted in the current report are dependent upon hepatic miR-144 but independent of intestinal miR-144.
Recent studies demonstrated that miR-33 represses the expression of ABCA1 mRNA and protein 10–12. Importantly, the complementary sequences in the mouse or human ABCA1 3′UTR that bind miR-33 are quite distinct from the sites that bind miR-144. Thus, miR-33 and miR-144 presumably regulate ABCA1 mRNA independently and in response to different signals that result from either changes in cellular sterol levels and subsequent SREBP-2 transcription or to changes in hepatic FXR activation, respectively. It is also interesting to note that miR-144 regulates ABCA1 largely at the protein level. The precise mechanism by which miRNAs regulate the levels of their targets is currently not well understood and it will be important to determine precisely how miR-144 regulates its various targets, including ABCA1. Some studies have suggested that the region outside the seed sequence may play a role in determining whether a miRNA modulates translational arrest (reduced complementarity) or mRNA destabilization (with more complementary base pairing) 42. Interestingly, the very first miRNA that was discovered (lin-4) was indeed shown to largely affect translation of its target, lin-14 43, 44. Further studies are required to determine the precise mechanism for how miR-144 regulates ABCA1.
The observation that hepatic miR-144 regulates plasma lipid levels was unexpected. Indeed, miR-144 and miR-451 were initially discovered as microRNAs processed independent of Dicer 29, 31, 32. Mice lacking both miR-144 and miR-451 45, 46 or lacking miR-451 alone 45 exhibited impaired late erythroblast maturation, a phenotype attributed solely to miR-451. Thus, the function of miR-144 remained an enigma. However, since the liver is not involved in erythroblast maturation in adulthood, it suggested that miR-144 or miR-451 had additional functions in adult mammals. The current report provides insight into one function of hepatic miR-144 in adult mammals.
The current study demonstrates that the mouse and human miR-144/-451 loci are direct targets for FXR. ChIP-Seq data had previously identified a putative FXRE 12kb upstream of the transcriptional start site (TSS) of the mouse miR-144-451 locus 37. In the current study we demonstrated that this FXRE functions to drive transcription of a luciferase reporter gene in an FXR- and GSK2324-dependent manner (Online Figure VC). In addition, we identified a functional FXRE in the proximal promoter of human miR-144/-451 (Figure 6C). These data demonstrate that the human and mouse miR-144/-451 cluster is a direct target of FXR, although the relative position of the functional FXREs upstream of the miR-144/-451 cluster is not strictly conserved. This is not without precedent, since the precise location of binding sites for many transcription factors, including other nuclear receptors in the liver, were previously shown not to be strictly conserved although regulation of the target genes themselves was maintained 47. Curiously, the mouse FXRE is also conserved in rats and the human FXRE is also conserved in the macaque (Online Figure VB) suggesting an evolutionary divergence in higher mammals, but nonetheless, a conserved response.
In summary, the current studies identify a new pathway that links FXR activation to induction of hepatic miR-144 and subsequent repression of ABCA1 and plasma HDL cholesterol. In mice, FXR activation has been shown to increase miR-144 (in the current study), lower plasma HDL, increase RCT 20 and lower atherosclerosis 39. Interestingly, FXR agonists are currently in Phase III clinical trials 20. However it is not known if treatment of humans with FXR agonists will produce similar effects on HDL levels and/or properties, RCT and atherosclerosis.
Recent studies in which miR-33, that also targets ABCA1, was silenced were reported to promote regression of pre-existing atherosclerotic lesions in Ldlr−/− mice 48 but have no effect on lesion development 49. Genetic loss of miR-33 in an atherosclerosis mouse model (Apoe−/− mice) showed increased HDL and reduced lesions 50. In contrast, bone marrow transplantation from miR-33 knockout mice into Apoe−/− mice also showed decreased atherosclerotic lesions, despite the absence of a change in plasma HDL levels 50, suggesting that macrophage miR-33 may be particularly important in lesion development. Importantly, the regulation of miR-144 in macrophages has not yet been determined. However, since FXR is not expressed in macrophages, the regulation of miR-144 in macrophages is likely independent of FXR. Moreover, the long-term effect of altering miR-144 levels in the liver or other tissues on atherosclerosis is also unknown and remains to be determined.
Supplementary Material
Acknowledgments
SOURCES OF FUNDING
This work was supported in part by United States Public Health Service, grants HL30568 and HL68445 (to P.A.E.), HL107794 (to Á.B.) the Laubish fund at UCLA (P.A.E.). T.Q. de A.V. (11POST7240070), E.J.T. (11POST7300060) and M.C. (10POST3660048) were partially supported by post-doctoral fellowships from the American Heart Association Western States Affiliate.
ABBREVIATIONS
- ABCA1
ATP binding cassette transporter A1
- ABCG5/G8
ATP binding cassette transporter G5/G8
- Bsep
Bile acid export pump
- ChIP
Chromatin immunoprecipitation
- FXR
Farnesoid X Receptor
- FXRE
FXR response element
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- LXR
Liver X Receptor
- miR
microRNA
- Nfe2l2
Nuclear factor (erythroid-derived 2)-like 2
- PFU
Plaque forming units
- RARβ
Retinoic acid receptor beta
- Scarb1
Scavenger receptor B1
- SREBP
Sterol response element binding protein
- TSS
Transcriptional start site
- UTR
Untranslated region
Footnotes
DISCLOSURES
Christy Esau is an employee of Regulus Therapeutics, all other authors have nothing to disclose.
References
- 1.Goldstein JL, Brown MS. The LDL receptor. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. The American journal of medicine. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 3.Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006;113:2548–2555. doi: 10.1161/CIRCULATIONAHA.104.475715. [DOI] [PubMed] [Google Scholar]
- 4.Brunham LR, Singaraja RR, Hayden MR. Variations on a gene: rare and common variants in ABCA1 and their impact on HDL cholesterol levels and atherosclerosis. Annual review of nutrition. 2006;26:105–129. doi: 10.1146/annurev.nutr.26.061505.111214. [DOI] [PubMed] [Google Scholar]
- 5.Chung S, Sawyer JK, Gebre AK, Maeda N, Parks JS. Adipose tissue ATP binding cassette transporter A1 contributes to high-density lipoprotein biogenesis in vivo. Circulation. 2011;124:1663–1672. doi: 10.1161/CIRCULATIONAHA.111.025445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. The Journal of clinical investigation. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. The Journal of biological chemistry. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- 9.Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marquart TJ, Allen RM, Ory DS, Baldan A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y, Hasegawa K, Yokode M, Kimura T, Kita T. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, MacDougald OA, Bommer GT. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem. 2010;285:33652–33661. doi: 10.1074/jbc.M110.152090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it’s been. Genes & development. 2009;23:2578–2591. doi: 10.1101/gad.1854309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Edwards PA. FXR signaling in metabolic disease. FEBS letters. 2008;582:10–18. doi: 10.1016/j.febslet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 18.Lee FY, de Aguiar Vallim TQ, Chong HK, Zhang Y, Liu Y, Jones SA, Osborne TF, Edwards PA. Activation of the farnesoid X receptor provides protection against acetaminophen-induced hepatic toxicity. Mol Endocrinol. 2010;24:1626–1636. doi: 10.1210/me.2010-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiological reviews. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 20.Vallim TQdA, Tarling EJ, Edwards PA. Pleiotropic Roles of Bile Acids in Metabolism. Cell metabolism. 2013 doi: 10.1016/j.cmet.2013.03.013. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 22.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmer JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 23.Evans MJ, Mahaney PE, Borges-Marcucci L, Lai K, Wang S, Krueger JA, Gardell SJ, Huard C, Martinez R, Vlasuk GP, Harnish DC. A synthetic farnesoid X receptor (FXR) agonist promotes cholesterol lowering in models of dyslipidemia. American journal of physiology. Gastrointestinal and liver physiology. 2009;296:G543–552. doi: 10.1152/ajpgi.90585.2008. [DOI] [PubMed] [Google Scholar]
- 24.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, Moore LB, Wilson JG, Lewis MC, Jones SA, Willson TM. Identification of a chemical tool for the orphan nuclear receptor FXR. Journal of medicinal chemistry. 2000;43:2971–2974. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 25.Bass JY, Caravella JA, Chen L, Creech KL, Deaton DN, Madauss KP, Marr HB, McFadyen RB, Miller AB, Mills WY, Navas F, 3rd, Parks DJ, Smalley TL, Jr, Spearing PK, Todd D, Williams SP, Wisely GB. Conformationally constrained farnesoid X receptor (FXR) agonists: heteroaryl replacements of the naphthalene. Bioorganic & medicinal chemistry letters. 2011;21:1206–1213. doi: 10.1016/j.bmcl.2010.12.089. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Yin L, Anderson J, Ma H, Gonzalez FJ, Willson TM, Edwards PA. Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. The Journal of biological chemistry. 2010;285:3035–3043. doi: 10.1074/jbc.M109.083899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. Journal of lipid research. 2007;48:2664–2672. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Garber DW, Kulkarni KR, Anantharamaiah GM. A sensitive and convenient method for lipoprotein profile analysis of individual mouse plasma samples. Journal of lipid research. 2000;41:1020–1026. [PubMed] [Google Scholar]
- 29.Yang JS, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, Lai EC. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15163–15168. doi: 10.1073/pnas.1006432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y, Schug J, McKenna LB, Le Lay J, Kaestner KH, Greenbaum LE. Tissue-specific regulation of mouse microRNA genes in endoderm-derived tissues. Nucleic acids research. 2011;39:454–463. doi: 10.1093/nar/gkq782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, Wolfe SA, Giraldez AJ. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116:4338–4348. doi: 10.1182/blood-2009-04-214817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis S, Propp S, Freier SM, Jones LE, Serra MJ, Kinberger G, Bhat B, Swayze EE, Bennett CF, Esau C. Potent inhibition of microRNA in vivo without degradation. Nucleic acids research. 2009;37:70–77. doi: 10.1093/nar/gkn904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 37.Chong HK, Infante AM, Seo YK, Jeon TI, Zhang Y, Edwards PA, Xie X, Osborne TF. Genome-wide interrogation of hepatic FXR reveals an asymmetric IR-1 motif and synergy with LRH-1. Nucleic acids research. 2010;38:6007–6017. doi: 10.1093/nar/gkq397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, Guo GL. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology. 2010;51:1410–1419. doi: 10.1002/hep.23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartman HB, Gardell SJ, Petucci CJ, Wang S, Krueger JA, Evans MJ. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR−/− and apoE−/− mice. Journal of lipid research. 2009;50:1090–1100. doi: 10.1194/jlr.M800619-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Annema W, Dikkers A, Freark de Boer J, Gautier T, Rensen PC, Rader DJ, Tietge UJ. ApoE promotes hepatic selective uptake but not RCT due to increased ABCA1-mediated cholesterol efflux to plasma. Journal of lipid research. 2012;53:929–940. doi: 10.1194/jlr.M020743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adajacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 42.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annual review of biochemistry. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 43.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 44.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 45.Rasmussen KD, Simmini S, Abreu-Goodger C, Bartonicek N, Di Giacomo M, Bilbao-Cortes D, Horos R, Von Lindern M, Enright AJ, O’Carroll D. The miR-144/451 locus is required for erythroid homeostasis. The Journal of experimental medicine. 2010;207:1351–1358. doi: 10.1084/jem.20100458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patrick DM, Zhang CC, Tao Y, Yao H, Qi X, Schwartz RJ, Jun-Shen Huang L, Olson EN. Defective erythroid differentiation in miR-451 mutant mice mediated by 14-3-3zeta. Genes & development. 2010;24:1614–1619. doi: 10.1101/gad.1942810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, Talianidis I, Flicek P, Odom DT. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. The Journal of clinical investigation. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marquart TJ, Wu J, Lusis AJ, Baldan A. Anti-miR-33 Therapy Does Not Alter the Progression of Atherosclerosis in Low-Density Lipoprotein Receptor-Deficient Mice. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:455–458. doi: 10.1161/ATVBAHA.112.300639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horie T, Baba O, Kuwabara Y, Chujo Y, Watanabe S, Kinoshita M, Horiguchi M, Nakamura T, Chonabayashi K, Hishizawa M, Hasegawa K, Kume N, Yokode M, Kita T, Kimura T, Ono K. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE−/− mice. Journal of the American Heart Association. 2012;1:e003376. doi: 10.1161/JAHA.112.003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.