

Abstract
Liposomes constitute one of the most popular nanocarriers for the delivery of cancer therapeutics. However, since their potency is limited by incomplete drug release and inherent instability in the presence of serum components, their poor delivery occurs in certain circumstances. In this study, we address these shortcomings and demonstrate an alternative liposomal formulation, termed crosslinked multilamellar liposome (CML). With its properties of improved sustainable drug release kinetics and enhanced vesicle stability, CML can achieve controlled delivery of cancer therapeutics. CML stably encapsulated the anticancer drug doxorubicin (Dox) in the vesicle and exhibited a remarkably controlled rate of release compared to that of the unilamellar liposome (UL) with the same lipid composition or Doxil-like liposome (DLL). Our imaging study demonstrated that the CMLs were mainly internalized through a caveolin-dependent pathway and were further trafficked through the endosome-lysosome compartments. Furthermore, in vivo experiments showed that the CML-Dox formulation reduced systemic toxicity and significantly improved therapeutic activity in inhibiting tumor growth compared to that of UL-Dox or DLL-Dox. This drug packaging technology may therefore provide a new treatment option to better manage cancer and other diseases.
Keywords: crosslinked multilamellar liposome, cancer therapy, doxorubicin, intracellular trafficking, nanomedicine, positron emission tomography (PET)
1. INTRODUCTION
Optimal treatment by many drugs often requires maintenance of the drug level for a prolonged time in order to achieve the therapeutic goals. While anticancer treatments require prolonged retention of highly concentrated cytotoxic drug levels to maximize antitumor effect, such requirement also poses the risk of systemic toxicity. Consequently, nanoparticle-based drug delivery systems, which can modulate the toxicity profile of anticancer drugs and improve drug circulation, have been widely viewed as a new treatment option for cancer therapeutics [1-3]. Many studies have demonstrated the accumulation of nanocarriers in the abnormal tumor microenvironment through the enhanced permeability and retention (EPR) effect as an advantage of nanoparticle-based drugs [4-6]. To date, approximately 20 therapeutic nanoparticles have received FDA approval for clinical use [2, 3].
Liposomes are one of the most popular nanocarriers for delivering many biologically active substances [7]. Based on their ability to encapsulate both hydrophilic and hydrophobic drugs, liposomal formulations of anticancer drugs have been extensively evaluated for treating cancers [6, 8, 9]. Among the many benefits of liposomal delivery of anthracyclin, e.g., doxorubicin or daunorubicin, compared with the administration of the free drug, is reduced cardiac toxicity with remaining therapeutic efficacy to tumors. However, poor stability and limited drug loading have remained prohibitive obstacles for practical applications. Furthermore, inherent instability of lipid-based carriers in the presence of serum components results in the fast-burst release of the chemotherapeutic drugs, which has limited their utility for the delivery of anticancer agents [9]. To overcome these challenges, a PEGylated liposomal drug formulation was developed by using an ammonium sulfate gradient loading procedure, enabling stable drug entrapment into liposomes and their extended blood-circulation time [10]. Notably, the liposome encapsulating doxorubicin (Dox), Doxil/Caelyx, exhibited an improved safety profile by reducing cardiac toxicity and enhancing penetration and accumulation in solid tumors. Consequently, it has been used in the treatment of a wide range of cancers [11-13]. Indeed, such liposomal drug formulations do appear to improve accumulation of liposomes at the tumor site. However, slow and incomplete drug release could still lead to low drug bioavailability within tumor tissue, limiting, in turn, therapeutic activity [14-17]. Furthermore, a lack of controlled-release properties of encapsulated drug may lead to toxic side effects, such as palmar–plantar erythrodysesthesia that is thought to result from unwanted drug distribution to skin during prolonged circulation of liposomal Dox [18], thus requiring further development or improvement in liposomal drug formulation. Attempts were made to improve the drug release rates of Dox by altering liposomal lipid compositions, but this method led to uncontrolled and rapid drug release kinetics, which also lowered therapeutic efficacy [19]. Therefore, a strategy to improve liposome-based anticancer drugs should involve the development of a stable liposomal formulation with improved drug release from the carrier in a controlled and sustained manner, thereby enhancing bioavailability.
In this study, we investigated a previously reported liposomal formulation [20] as an anticancer drug nanocarrier and examined whether it could offer improved drug release, along with stable and sustainable delivery of cancer therapeutics. This liposomal formulation involves the creation of a robust multilamellar structure of the liposome by covalently crosslinking interlipid bilayers. As a nanocarrier platform for chemotherapy drug delivery applications, our study demonstrates that these crosslinked multilamellar liposomes (CMLs) can lower systemic toxicity and enhance therapeutic efficacy. In addition, the intracellular trafficking of CMLs was monitored and visualized to provide a better understanding of their delivery mechanisms.
2. MATERIALS AND METHODS
2.1. Cell lines, Antibodies, Reagents, and Mice
B16 tumor cells (B16-F10, ATCC number: CRL-6475) and HeLa cells were maintained in a 5% CO2 environment with Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Inc., Manassas, VA) supplemented with 10% FBS (Sigma-Aldrich, St. Louis, MO) and 2 mM of L-glutamine (Hyclone Laboratories, Inc., Omaha, NE). The mouse monoclonal antibodies against clathrin, caveolin-1, EEA1, and the rabbit polyclonal antibody specific to trans-Golgi network (TGN38) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The mouse monoclonal antibody to Lamp-1 was purchased from Abcam (Cambridge, MA). Alexa488-goat anti-mouse immunoglobulin G (IgG) and Alexa594-goat anti-rabbit IgG antibodies were purchased from Invitrogen (Carlsbad, CA). Chloropromazine, Nystatin, and MβCD were obtained from Sigma-Aldrich.
All lipids were obtained from NOF Corporation (Japan): 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DOPG), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl) butyramide (maleimide-headgroup lipid, MPB-PE). 64Cu was obtained from Washington University (St. Louis, MO) and the University of Wisconsin (Madison, WI). 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (SNHS) were purchased from Thermo Scientific (Rockford, IL).
Female C57BL/6 mice, 6-10 weeks old, were purchased from Charles River Breeding Laboratories (Wilmington, MA). All mice were held under specific pathogen-reduced conditions in the animal facility of the University of Southern California (USA). All experiments were performed in accordance with the guidelines set by the National Institutes of Health and the University of Southern California on the Care and Use of Animals.
2.2. Synthesis of CMLs, ULs, and DLLs
Liposomes were prepared based on the conventional dehydration-rehydration method. 1.5 μmol of lipids of DOPC, DOPG, and MPB-PE at the molar ratio of the lipid composition of DOPC:DOPG:MPB-PE = 40:10:50, were mixed in chloroform, and the organic solvent in the lipid mixture was evaporated under argon gas and dried under vacuum overnight to form dried thin lipid films. The resultant dried film was hydrated in 10 mM Bis-Tris propane at pH 7.0 containing doxorubicin at a molar ratio of 0.5:1 (drugs:lipids), with vigorous vortexing every 10 min for 1 h, and then applied with 4 cycles of 15-s sonication (Misonix Microson XL2000, Farmingdale, NY) on ice at 1 min intervals for each cycle. To induce divalent-triggered vesicle fusion, MgCl2 was added to make a final concentration of 10 mM. The resulting multilamellar vesicles were further crosslinked by addition of Dithiothreitol (DTT, Sigma-Aldrich) at a final concentration of 1.5 mM for 1 h at 37°C. The resulting vesicles were collected by centrifugation at 14,000 g (12,300 RPM) for 4 min and then washed twice with PBS. For pegylation of CMLs, the liposomes were further incubated with 1 μmol of 2 kDa mPEG-SH (Laysan Bio Inc., Arab, AL) for 1 h at 37°C. The particles were then centrifuged and washed twice with PBS. Nonencapsulated doxorubicin was removed by a PD-10 Sephadex gel filtration column, and then the final products were stored in PBS at 4°C. Similarly, unilamellar liposomes (ULs) were prepared with the same lipid composition through rehydration, vortexing and sonication, as described above, except divalent-induced vesicle fusion and DTT crosslinking processes. The ULs were collected by centrifugation at 250,000 g for 90 min and then washed twice with PBS. Pegylation of ULs was carried out by incubation with 1 μmol of 2 kDa PEG-SH. Doxil-like liposomes (DLLs) were prepared using an ammonium sulfate pH gradient method as described [10]. Briefly, lipid film (HSPC:Cholesterol:DSPE-PEG2000=56:38:6) was rehydrated with 240 mM of ammonium sulfate buffer pH 5.4 with vigorous vortexing. Small unilamellar vesicles were prepared using sonication and extrusion at 60°C through 100 nm polycarbonate filters 20 times using a mini-Extruder (Avanti Polar Lipids, Alabaster, AL). The DLLs were collected by centrifugation at 250,000 g (45,400 RPM) for 90 min, washed twice with PBS, and then resuspended with HBS pH 7.4 (20 mM HEPES, 150 mM NaCl) containing doxorubicin hydrochloride. The particles were then centrifuged and washed twice with PBS. Nonencapsulated doxorubicin was removed by a PD-10 Sephadex gel filtration column, and then the final products were stored in PBS at 4°C.
2.3. Characterization of physical properties
The hydrodynamic size and size distribution of CMLs, ULs and DLLs were measured by dynamic light scattering (Wyatt Technology, Santa Barbara, CA). For cryo-electron microscopy imaging, the liposome samples were applied to the grid and plunge-frozen in liquid ethane using the FEI Mark III Vitrobot. CryoEM images were collected using a Tecnai T12 electron microscope (FEI Company) equipped with a Gatan Ultrascan 2k by 2k CCD camera.
2.4. In vitro drug encapsulation, release kinetics, and cytotoxicity
To study the loading capacity of Dox, Dox-loaded CMLs, ULs, or DLLs were collected and then washed twice with PBS, followed by lipid extraction of vesicles with 1% Triton X-100 treatment. Lipid concentrations of liposome suspensions were determined by phosphate assay [21]. Dox fluorescence (excitation 480 nm, emission 590 nm) was then measured by a Shimadzu RF-5301PC spectrofluorometer (Japan). To determine a half-time t1/2 whereby 50% of entrapped Dox is released from liposomes, CMLs or ULs were incubated at 37°C in 10% fetal bovine serum (FBS)-containing media, and the releasing media were collected to measure Dox fluorescence at regular time intervals. To obtain the release kinetics of Dox from liposomes, Dox-loaded CMLs, ULs or DLLs were incubated at 37°C in 10% fetal bovine serum (FBS)-containing media, the releasing media were removed from CMLs, ULs, or DLLs incubated at 37°C for quantification of Dox fluorescence every day, and fresh media were replaced for continuous monitoring of drug release.
B16 or HeLa cells were plated at a density of 5 × 103 cells per well in D10 media in 96-well plates and grown for 6 h. The cells were then exposed to a series of concentrations of Dox-loaded CMLs, ULs or DLLs for 48 h, and the cytotoxicity of Dox-liposomes was assessed using the Cell Proliferation Kit II (XTT assay) from Roche Applied Science, according to the manufacturer’s instructions.
2.5. Confocal Imaging
To label liposome particles with DiD lipophilic dyes, DiD dyes were added to the lipid mixture in chloroform at a ratio of 0.01:1 (DiD:lipids), and the organic solvent in the lipid mixture was evaporated under argon gas to incorporate DiD dyes into a lipid bilayer of vesicles. For the colocalization study with endocytic markers, HeLa cells were seeded on glass bottom dishes (MatTek Corporation, Ashland, MA) and grown at 37°C overnight. The cells were then incubated with DiD-labeled CML particles for 30 min at 4°C to synchronize internalization. After washing with PBS, the treated cells were then warmed to 37°C to initiate particle internalization for the indicated time periods. The cells were fixed, permeabilized with 0.1% Triton X-100, and then immunostained with the corresponding antibodies specific to clathrin, caveolin-1, EEA1, TGN38, or Lamp-1 and counterstained with DAPI (Invitrogen).
Fluorescence images were acquired on a Yokogawa spinning-disk confocal scanner system (Solamere Technology Group, Salt Lake City, UT) using a Nikon eclipse Ti-E microscope equipped with a 60×/1.49 Apo TIRF oil objective and a Cascade II: 512 EMCCD camera (Photometrics, Tucson, AZ, USA). Image processing and data analysis were carried out using the Nikon NIS-Elements software. To quantify the extent of colocalization, the Manders’ overlap coefficients (MOC) were generated using the Nikon NIS-Elements software by viewing more than 50 cells at each time point.
2.6. Uptake inhibition assay
HeLa cells (1 × 105 cells) were pretreated with chlorpromazine (CPZ, 25 μg/ml), nystatin (50 μg/ml), or methyl-β-cyclodextrin (MβCD, 15 mM) to disrupt clathrin- or caveolin-mediated entry pathway. The cells were then incubated with DiD-labeled CMLs for 1 h at 37°C in the presence of CPZ and nystatin or in the absence of MβCD. The cells were then washed twice with PBS. To disrupt energy-dependent internalization of CMLs, HeLa cells were incubated with DiD-labeled CMLs at 4°C for 1 h and then washed twice with cold PBS. The cellular uptake of particles was determined by measuring DiD fluorescence using the spectrofluorometer and was normalized based on the fluorescent intensity acquired upon incubation at 37°C for 1 h.
2.7. Maximum tolerated dose study
C57BL/6 female mice (6-10 weeks old) were administered by a single intravenous injection through tail vein with CML-Dox or free Dox at doses of 0, 20 and 40 mg/kg Dox equivalents. The mice were weighed and monitored daily for 8 days after injection.
2.8. In vivo tumor challenge
C57BL/6 female mice (6-10 weeks old) were inoculated subcutaneously with 1 × 106 B16 melanoma tumor cells. The tumors were allowed to grow for 6 days to a volume of 50~100 mm3 before treatment. On day 6, the mice were injected intravenously through tail vein with CML-Dox, UL-Dox, or DLL-Dox at a dose of 1 or 4 mg/kg Dox equivalent every other day (six mice per group), and tumor growth and body weight were then monitored for an additional 10 days by the end of the experiment. The length and width of the tumor masses were measured with a fine caliper every other day after Dox-liposome injection. Tumor volume was expressed as 1/2 × (length × width2).
2.9. In vivo PET imaging and biodistribution
For radiolabeling liposomes, amine-terminated PEG-SH was used for PEGylation of ULs and CMLs, while DSPE-PEG-NH2 was used for PEGylation of DLLs, in order to introduce amine groups onto liposomes for further reaction. Unless noted otherwise, all chemicals were analytic grade from Sigma-Aldrich (St. Louis, MO). 64Cu was produced using the 64Ni(p,n)64Cu nuclear reaction and supplied in high specific activity as 64CuCl2 in 0.1 N HCl. The bifunctional chelator AmBaSar was synthesized as reported [22]. AmBaSar was activated by EDC and SNHS. Typically, 5 mg of AmBaSar (11.1 μmol) in 100 μL water and 1.9 mg of EDC (10 μmol) in 100 μL water were mixed together, and 0.1 N NaOH (150 μL) was added to adjust the pH to 4.0. SNHS (1.9 mg, 8.8 μmol) was then added to the stirring mixture on ice-bath, and 0.1 N NaOH was added to finalize the pH to 4.0. The reaction remained at 4 °C for 30 min. The theoretical concentration of active ester AmBaSar-OSSu was calculated to be 8.8 μmol. Then, 5–20 times AmBaSar-OSSu (based on molar ratios) were loaded to the liposomes of interest. The pH was adjusted to 8.5 using borate buffer (1M, pH 8.5). The reaction remained at 4 °C overnight, after which the size-exclusion PD-10 column was employed to afford the AmBaSar-conjugated liposomes in PBS buffer. AmBaSar-liposome was labeled with 64Cu by addition of 1–5 mCi of 64Cu (50–100 μg AmBaSar-liposome per mCi 64Cu) in 0.1 N phosphate buffer (pH 7.5), followed by 45 min incubation at 40 °C. 64Cu-AmBaSar-liposome was purified on a size exclusion PD-10 column using PBS as the elution solvent. Positron emission tomography (PET) imaging of the mice was performed using a microPET R4 rodent model scanner (Concorde Microsystems, Knoxville, TN). The B16-F10 tumor-bearing C57/BL6 mice were imaged in the prone position in the microPET scanner. The mice were injected with approximately 100 μCi 64Cu-AmBaSar-liposome via the tail vein. For imaging, the mice were anaesthetized with 2% isoflurane and placed near the center of the field of view (FOV), where the highest resolution and sensitivity are obtained. Static scans were obtained at 1, 3, and 24h post-injection. The images were reconstructed by a two-dimensional ordered subsets expectation maximum (2D-OSEM) algorithm. Time activity curves (TAC) of selected tissues were obtained by drawing regions of interest (ROI) over the tissue area. The counts per pixel/min obtained from the ROI were converted to counts per ml/min by using a calibration constant obtained from scanning a cylinder phantom in the microPET scanner. The ROI counts per ml/min were converted to counts per g/min, assuming a tissue density of 1 g/ml, and divided by the injected dose to obtain an image based on ROI-derived percent injected dose of 64Cu tracer retained per gram (%ID/g). For biodistribution, animals were sacrificed 24h post-injection; tissues and organs of interest were harvested and weighed. Radioactivity in each organ was measured using a gamma counter, and radioactivity uptake was expressed as percent injected dose per gram (%ID/g). Mean uptake (%ID/g) for each group of animals was calculated.
2.10. Pharmacokinetics and quantification of Dox in tumors
C57/BL6 mice bearing B16 tumors (diameter 0.5~1 cm) were injected with free Dox, UL-Dox, DLL-Dox, or CML-Dox at a dose of 10 mg/kg Dox equivalent. To examine pharmacokinetics, blood was collected by retro-orbital bleeding at the indicated time points, and then plasma was obtained by centrifuging the samples at 14,000 g for 10 min. To detect Dox in tumors, the tumors were collected at 24 h after injection and were then homogenized. Dox was extracted by adding methanol to the homogenized samples, followed by vortexing and freeze/thaw cycles. After the extraction of Dox with further purification using an Amicon Ultra 10,000 MWCO centrifugal filter, Dox concentration was quantified by reverse phase HPLC using a C18 column.
3. RESULTS
3.1. Preparation and characterization of crosslinked multilamellar liposomal doxorubicin
Our goal was to generate a liposomal formulation with improved bioavailability of liposomal drugs and enhanced vesicle stability. To accomplish this, multilamellar vesicles were formed through covalently crosslinking functionalized headgroups of adjacent lipid bilayers, as illustrated in Figure 1A. This design was adapted from a recently reported multistep procedure based on the conventional dehydration-rehydration method [20]: (1) the incorporation of a thiol-reactive maleimide headgroup lipid (N-(3-Maleimide-1-oxopropyl)-L-α-phosphatidylethanolamine, MPB-PE) onto the surface of unilamellar liposome (UL); (2) divalent cation-triggered vesicle fusion that yields a multilamellar structure; and (3) interbilayer crosslinking across the opposing sides of lipid bilayers through the reactive headgroups with dithiothreitol (DTT) to generate robust and stable vesicles. As a final step, the surface of the crosslinked multilamellar liposome (CML) was PEGylated with thiol-terminated PEG, which could further improve vesicle stability and blood circulation half-life [7, 23]. Additionally, liposomes of approximately the same size and composition as Doxil, termed Doxil-like liposome (DLL), were also prepared for comparison (Figure 1E). First, we characterized the physical properties of CML compared to those of non-crosslinked UL with the same lipid composition or conventional liposome formulation DLL. The hydrodynamic size of the particles was measured by dynamic light scattering (DLS), and the result showed a slight increase in the mean diameter of CML (~220 nm) compared to that of the UL (~200 nm) (Figure 1B and 1C), whereas the size of DLL was much smaller (~129 nm), as expected. It also indicated that CML particles exhibited a narrow size distribution (polydispersity: 0.101±0.0082, Figure 1D), suggesting no significant aggregation of particles during the crosslinking process. In addition, the CML particles are remarkably stable and can be stored in PBS over two weeks at 4°C without significant change in size or size distribution (data not shown).
Figure 1.
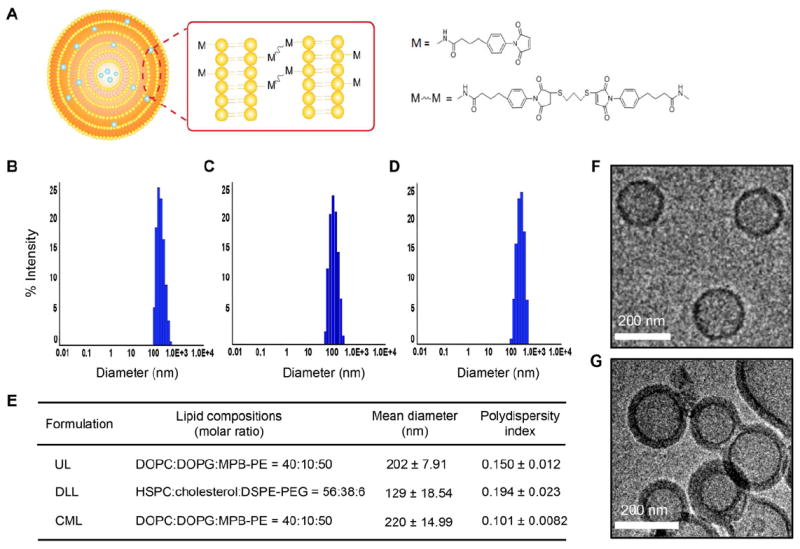
Stable liposomal formulation and physical characterization of CMLs. (A) Schematic representation of the crosslinked multilamellar liposome. (B, C) The hydrodynamic size distribution of PEGylated ULs (B), DLLs (C), and CMLs (D), as measured by dynamic light scattering. (E) The mean diameter and polydispersity of ULs, DLLs and CMLs. (F, G) Visualization of unilamellar and multilamellar structure of vesicles. Cryo-electron microscopy images of ULs (E) and CMLs (F).
To further confirm the multilamellar structure of liposomes, CML particles were imaged by cryo-electron microscopy. ULs were utilized as a control. The images demonstrated that the CML exhibited multilayered vesicle formation with thick walls (Figure 1F and Supplemetary Figure S1), while only a single thin-layer of lipid membrane was found in ULs (Figure 1E), suggesting that the covalent linkage between adjacent bilayers could lead to a stable multilamellar structure of vesicles. In addition, the mean diameter of CML estimated by cryo-electron microscopy was ~160 nm.
3.2. In vitro drug encapsulation, release kinetics, and cytotoxicity
Next, we examined whether the multilamellar structure of CML could improve the loading capacity of the anticancer agent doxorubicin (Dox) into vesicles compared to that of the unilamellar liposomal formulation (Figure 2A). To test its ability to encapsulate drugs, the lipid film was rehydrated in Dox-containing buffer to form Dox-loaded UL or CML. The result showed that the CML formulation could achieve higher Dox-encapsulation efficiency (~85%) than that of the UL (~39.7%) and that the amount of Dox loaded in CML (~250 μg per mg of lipids) was increased by ~4-fold compared with UL (Figure 2B), which was also higher loading efficiency than that of DLL (~160 μg per mg of lipids) [24]. Taken together, these results suggest that the formation of multilamellar structure via vesicle fusion apparently fosters the entrapment of extra drug payload into liposomes.
Figure 2.
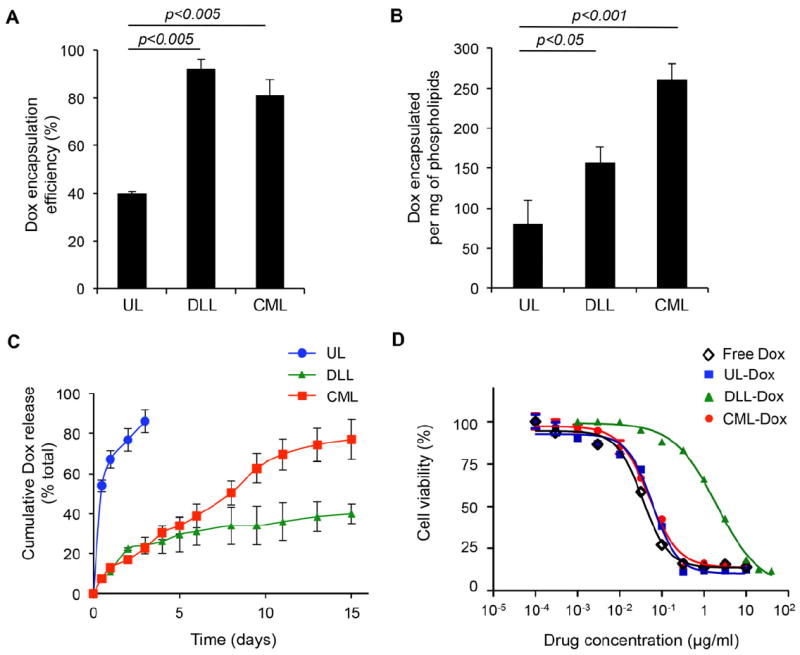
Enhanced vesicle stability and sustainable release kinetics of CMLs. (A) Encapsulation efficiency of doxorubicin (Dox) into the UL, DLL, or CML. (B) Total Dox loading per phospholipids (μg/mg). (C) In vitro release kinetics of doxorubicin from ULs, DLLS, and CMLs. (D) In vitro cytotoxicity of free Dox, UL-Dox, DLL-Dox, and CML-Dox in B16 melanoma tumor. The cytotoxicity was measured by a standard XTT assay. Error bars represent the standard deviation of the mean from triplicate experiments.
It is well known that lipid vesicles are exceedingly unstable in the presence of serum, thus limiting their utility as a drug carrier. Serum components disrupt liposome membranes, which causes leakage of their aqueous contents. As an anticancer drug carrier, the stability of liposomal formulations has been intrinsically linked to both toxicity level and therapeutic activity of the drug payload [25-27]. Therefore, we investigated the vesicle stability of CML in vitro upon exposure to a serum environment relative to the controlled release of its contents. Doxloaded ULs, DLLs, and CMLs were stored at 37°C in 10% FBS-containing media, and in vitro drug release rates were measured. As shown in Figure 2C, ULs had the expected burst release (most released within 2 days), whereas slower and linearly sustained release kinetics (up to two weeks) was seen for CMLs, indicating that the CML formulation could improve vesicle stability in the presence of serum components by forming a crosslinked multilamellar structure. Although significantly slower release kinetics was also observed in DLLs, less than 40% of encapsulated Dox was released from DLLs at 37°C in two weeks, while CMLs could release ~80% of encapsulated Dox in two weeks, suggesting that CML formulation could remarkably improve drug release compared with DLLs.
Next, we determined whether this sustained and improved drug release profile of CML could affect cytotoxicity in cells as compared to that of the UL and DLL. Free Dox or Dox-loaded ULs, DLLs, or CMLs were incubated with B16 cells for 48h, and the cytotoxicity of Dox-liposomes was then measured by a standard XTT assay. In vitro cytotoxicity data revealed that the half-maximal response (EC50) for CMLs was ~0.05 μg/ml for B16 cells, similar to that of free Dox and ULs (Figure 2D), suggesting that CMLs were able to maintain Dox cytotoxicity in cells, notwithstanding sustained drug release of the CML formulation. A similar result was also observed in HeLa cells (Supplementary Figure S2). Furthermore, DLLs exhibited higher EC50, 2.3 μg/ml (Figure 2D), which is consistent with previous reports indicating that Doxil has an EC50 about two orders of magnitude higher (lower cytotoxic activity) than free Dox [18], suggesting that improved drug release of CML formulation could augment cytotoxicity of liposomal drug, which is likely a result of enhanced uptake and intracellular delivery of Dox to the cells.
3.3. Cellular uptake, internalization pathway, and intracellular trafficking of CML
Endocytosis is generally considered one of the main entry mechanisms for various drug nanocarriers [28, 29]. Several endocytic pathways, including clathrin- and caveolin-mediated endocytosis, have been characterized as major routes for cell internalization [30-32]. Since the intracellular fate of nanocarriers is determined by the endocytic pathways utilized upon their entry to cells, elucidating the fundamental basis of intracellular processing of drug carriers can provide crucial insights for improving the efficiency of drug delivery and developing designs of drug carriers. Hence, to accomplish this, we focused on unraveling the entry mechanism and the subsequent intracellular trafficking of CMLs by visualizing fluorescent 1,1-dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine (DiD)-labeled CML particles and endocytic structures, i.e., clathrin and caveolin, in HeLa cells after 15 min incubation at 37°C. As shown in Figure 3A, a significant colocalization of CML particles with discrete caveolin-1 signals was observed, whereas, no remarkable colocalization between CMLs and clathrin was detected after 15 min incubation, even though some particles were overlaid with clathrin structures. The quantification of CML particles colocalized with caveolin-1 or clathrin structures by analyzing more than 50 cells suggested that the caveolin-mediated pathway might be involved in the entry of CMLs (Figure 3B). Involvement of the caveolin pathway was further confirmed by drug inhibition assays (Figure 3C). Pretreatment of cells with nystatin or methyl-β-cyclodextrin (MβCD), either of which is known to disrupt caveolin-dependent internalization [33, 34], significantly decreased the uptake of CML particles in HeLa cells. However, no inhibitory effect on their uptake was observed when cells were pretreated with chlorpromazine (CPZ), a drug known to inhibit clathrin-dependent internalization by blocking clathrin polymerization [35]. Results from the inhibition assay further verified that the entry of CMLs is mediated by caveolin-dependent endocytosis. It also appeared that incubation of CMLs with cells at 4°C for 1 h significantly diminished the cellular uptake of CMLs by ~66.7% compared to their internalization upon incubation at 37°C for 1 h, verifying that CMLs enter cells via energy-dependent endocytosis.
Figure 3.
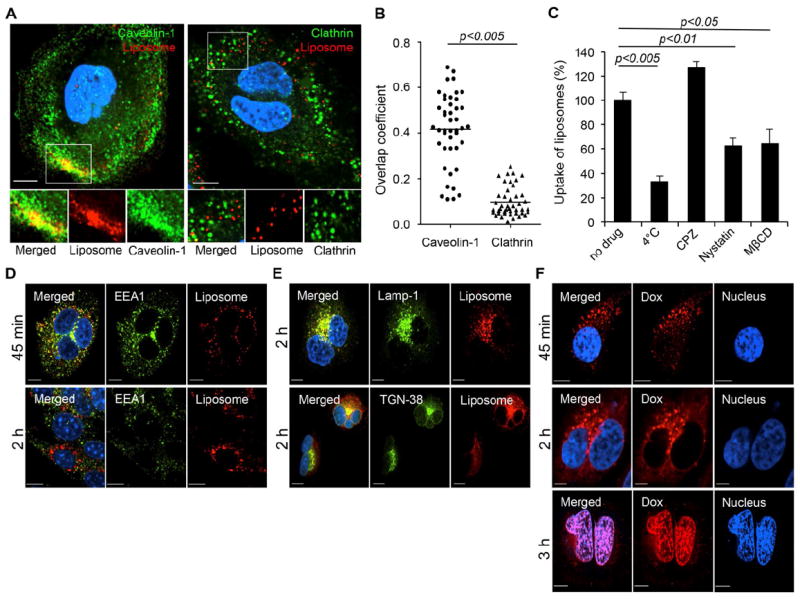
Caveolin-mediated internalization of CMLs and subsequent intracellular processing. (A) HeLa cells were incubated with DiD-labeled CML particles (red) for 30 min at 4 °C to synchronize internalization. The cells were then shifted to 37°C for 15 min, fixed, permeabilized, and immunostained with anti-caveolin-1 (green) or anti-clathrin antibody (green). The boxed regions are enlarged in the bottom panels. (B) Quantification of CML particles colocalized with caveolin-1 or clathrin signals after 15 min of incubation. Overlap coefficients were calculated with Manders’ overlap coefficients (MOC) by viewing more than 50 cells of each sample using the Nikon NIS-Elements software. Error bars represent the standard deviation of the mean from analysis of multiple images. (C) Inhibition of energy-dependent internalization by incubation at 4°C, clathrin-dependent internalization by chlorpromazine (CPZ, 25 μg/ml), and caveolin-dependent internalization by nystatin (50 μg/ml) and methyl-β-cyclodextrin (MβCD, 15mM). The uptake of DiD-labeled CML particles was determined by measuring DiD fluorescence. (D) Intracellular trafficking of CMLs through early endosomes. HeLa cells were incubated with DiD-labeled CML particles (red) for 30 min at 4 °C to synchronize internalization. The cells were then shifted to 37°C for 45 min or 2 h and immunostained with anti-EEA1 antibody (green). (E) Involvement of lysosomes and trans-Golgi network in trafficking of CMLs. Lysosomes and trans-Golgi network were visualized with anti-Lamp1 (upper panel) and anti-TGN38 (lower panel) antibody, respectively. (F) Confocal images of Dox-loaded CML particles. HeLa cells were incubated with Dox-loaded CMLs for 30 min at 4 °C and then shifted to 37°C for 45 min, 2 h or 3 h. For all images, the nucleus of cells was counterstained with DAPI. Scale bar represents 10 μm. Error bars represent the standard deviation of the mean from triplicate experiments.
Although caveolin-mediated entry and the subsequent intracellular processing remain poorly understood, cargos endocytosed through caveolae are believed to be transported to an organelle called “caveosome” [36, 37]. Cargo that traffics through the caveosome is thought to be further transported to the Golgi apparatus and/or endoplasmic reticulum (ER) [32]. It is also proposed that caveosomes may fuse directly to the early endosomes in a GTPase Rb5-dependent manner and may also proceed through the conventional endocytic pathway (endosomes/lysosomes) [37, 38]. To further demonstrate the subsequent intracellular fate of CMLs, DiD-labeled CML particles were evaluated for their colocalization with the early endosome (EEA-1)[39], lysosome (Lamp-1)[40], and trans-Golgi (TGN-38)[41] markers at different incubation times at 37°C. After incubation of 45 min, most CML particles were found in the EEA1+ early endosomes (Figure 3D, upper), whereas much less colocalization was detected between CMLs and EEA1 after 2 h incubation (Figure 3D, lower). Rather, at 2 h incubation, CML particles were mainly found in lysosomes (Figure 3E, upper), and some colocalization of CMLs with trans-Golgi was also observed (Figure 3E, lower). These imaging results demonstrated that CML particles could be primarily trafficked from caveosomes to the early endosome-lysosome compartments and could also traffic to the trans-Golgi network, possibly through the early endosomes.
In addition, imaging of drug release with Dox-loaded CMLs showed that a large amount of dot-shaped Dox fluorescence could be observed in the cytoplasm after 45 min incubation in HeLa cells (Figure 3F, upper), which suggests that Dox-liposome complexes were still located in the endosomes without significant release of Dox. After 2 h incubation, Dox fluorescent signals had diffused out of liposomes into the cytoplasm, but some dot-shaped Dox fluorescence could still be observed (Figure 3F, middle). At 3 h incubation, Dox fluorescence was mainly detected in the nucleus of cells with no clear observation of dot-shaped Dox in the cytoplasm (Figure 3F, lower), indicating that CML-Dox particles were most likely transported up to lysosomes where the encapsulated Dox was released into the cytoplasm prior to lysosomal degradation.
3.4. In vivo toxicity, cardiac toxicity, and tolerability
Despite recent advances in chemotherapeutic agents for cancer, their clinical applications are often limited by systemic toxicity. Therefore, various formulations of the drugs have been evaluated to achieve reduction in systemic toxicity. To determine the toxicity and tolerability of CML-Dox, we estimated the maximum tolerated dose by a single intravenous administration to C57/BL6 mice. The weights and general health of the mice were monitored for 8 days after injection of CML-Dox or free Dox at doses of 0, 20 and 40 mg/kg Dox equivalents (Figure 4A). As expected, a significant loss of body weight was observed at both 20 and 40 mg/kg of free Dox. Especially, mice receiving 40 mg/kg of free Dox exhibited obvious signs of toxicity. However, mice in the groups receiving CML-Dox appeared healthy. Mice receiving 20 mg/kg of CML-Dox showed no loss of weight throughout the experiment. Some loss of weight was observed in mice receiving 40 mg/kg of CML-Dox, but body weights were recovered 4 days post-injection. The results indicated that CML-Dox was much less toxic to mice (maximum tolerated dose > 40 mg/kg) than free Dox (maximum tolerated dose < 20 mg/kg). Furthermore, histopathologic analysis indicated that free Dox (20 mg/kg) caused severe damage of cardiac tissue such as myofibrillary loss and disarray, whereas no significant histopathologic changes in cardiac tissue from mice treated with CML-Dox (20 mg/kg Dox equivalent) or no drug (Figure 4B).
Figure 4.
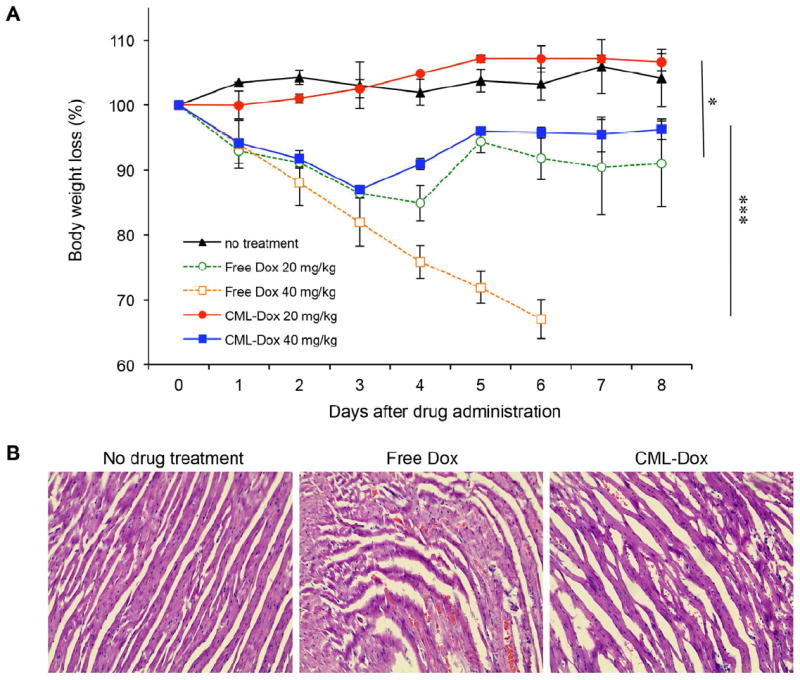
In vivo toxicity and tolerability. C57/BL6 mice were administered a single intravenous injection with CML-Dox or free Dox. (A) Average mouse weight loss over time. Data are presented as the percentage of the initial weights for mice treated with no injection (black line), CML-Dox (20 mg/kg Dox equivalent, red line; 40 mg/kg Dox equivalent, blue line), or free Dox (20 mg/kg Dox equivalent, green line; 40 mg/kg Dox equivalent, orange line). Error bars represent the standard deviation of the mean; n = 2 for each treatment group (*p<0.05, ***p<0.005). (B) Histologic appearance of cardiac tissues obtained from C57/BL6 mice with no drug treatment or administered a single intravenous injection with free Dox or CML-Dox at 20 mg/kg Dox equivalent.
3.5. In vivo therapeutic antitumor efficacy
Next, a mouse tumor model was used to validate the therapeutic efficacy of the CML-Dox formulation, compared with that of UL-Dox or DLL-Dox. At day 0, C57/BL6 mice were inoculated subcutaneously with B16 melanoma tumor cells. On day 6, mice were injected intravenously with UL-Dox, DLL-Dox, or CML-Dox at doses of 1 or 4 mg/kg Dox equivalents every other day, and tumor growth and body weights were then monitored for an additional 10 days. Mice in the group receiving 1 mg/kg CML-Dox showed significant tumor inhibition, whereas the treatment of mice with the equivalent Dox concentration of UL-Dox exhibited no inhibition at all (Figure 5A). At the higher dose of CML-Dox (4 mg/kg Dox), a dramatic suppression of tumor growth was observed in the group (Figure 5A and 5B), representing a significantly augmented therapeutic efficacy compared to that of UL-Dox. It also showed that CML-Dox exhibited slightly better antitumor effect compared with the conventional liposome formulation, DLL-Dox. No weight loss was seen for the duration of the experiment, even at the high dose of 4 mg/kg (Figure 5C), indicating the absence of systemic toxicity from this CML formulation.
Figure 5.
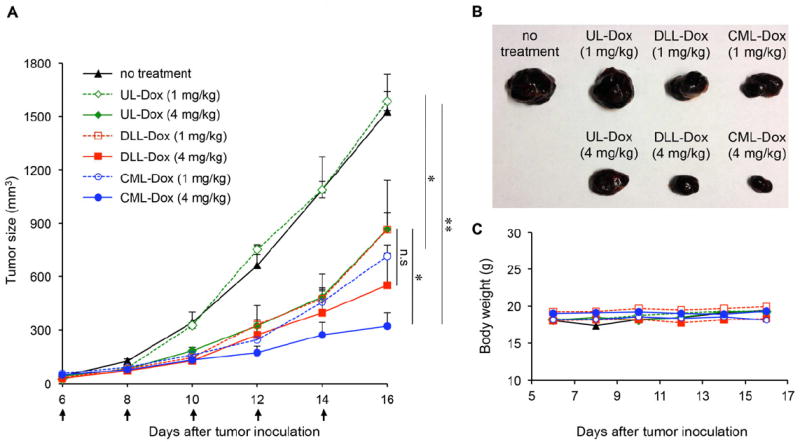
Antitumor effect of Dox-loaded CMLs, ULs, and DLLs in the B16 melanoma tumor model. (A) Tumor growth was measured after treatment with no injection (black line), CML-Dox (1 mg/kg DOX equivalent, blue dotted line; 4 mg/kg DOX equivalent, blue solid line), UL-Dox (1 mg/kg DOX equivalent, green dotted line; 4 mg/kg DOX equivalent, green solid line), or DLL-Dox (1 mg/kg DOX equivalent, red dotted line; 4 mg/kg DOX equivalent, red solid line). Error bars represent the standard error of the mean; n = 6 for each treatment group (*p<0.05, **p<0.01). (B) Excised tumors from each treatment group at 16 days after tumor inoculation. (C) Average mouse weight loss over the duration of the experiment.
3.6. Positron emission tomography (PET) imaging, pharmacokinetics, and biodistribution
To further investigate the basis of the enhanced therapeutic effectiveness of CML-Dox compared to that of UL-Dox or DLL-Dox, biodistribution patterns of liposomes were evaluated in mice bearing B16 tumors with in vivo PET imaging. The process for preparing radiolabeled liposomes is shown in Figure 6A. The bifunctional chelator AmBaSar was used in the 64Cu labeling due to the superior in vivo stability of 64Cu-AmBaSar over other 64Cu-chelator, such as 64Cu-DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) [42, 43]. As shown in Figure 6B, the PET images were obtained at several time points (1, 3, 24 h) after intravenous injection of 64Cu-AmBaSar-labeled ULs, DLLs, or CMLs. After 1 h of administration, radioactivity was present mainly in well-perfused organs, and accumulation in tumors was detected in DLLs and CMLs compared with ULs. Furthermore, the accumulation of DLLs and CMLs in tumors significantly increased after 3 and 24 h of injection, whereas accumulation of ULs in the bladder was observed after 3 h of administration as a consequence of rapid degradation.
Figure 6.
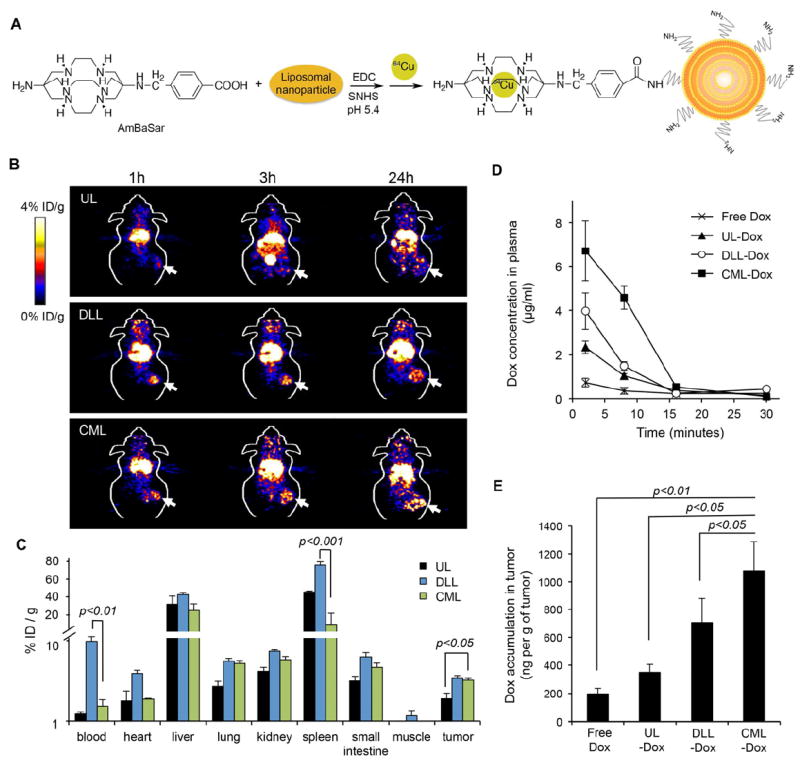
Biodistribution of drug carriers and accumulation of Dox in tumors. (A) Preparation of 64Cu-AmBaSar-labeled liposomes. (B) In vivo PET images of C57/BL6 mice bearing B16 tumors at 1, 3, and 24 h post-injection of 64Cu-AmBaSar-labeled UL, DLL, or CML. (C) Biodistribution of liposomes in different tissues at 24 h after injection with 64Cu-AmBaSar-labeled UL, DLL, or CML shown as percentage of injection dose per g of tissues (% ID/g). (D) The pharmacokinetics of Dox in plasma of C57/BL6 mice bearing B16 tumors injected with free Dox, UL-Dox, DLL-Dox, or CML-Dox at a dose of 10 mg/kg Dox equivalent. At given time intervals, blood was collected by retro-orbital bleeding. (E) Accumulation of Dox in tumors. C57/BL6 mice bearing B16 tumors were intravenously injected with free Dox, UL-Dox, DLL-Dox, or CML-Dox at a dose of 10 mg/kg Dox equivalent. The mean Dox concentrations in tumors were shown. Error bars represent the standard deviation of the mean; n = 3 for each treatment group.
In addition, the tumors and tissues of interest were then excised at 24 h post-injection and weighed, and accumulation levels of particles in the tumors and tissues were determined by measuring radioactivity (Figure 6C). This biodistribution assay revealed significantly higher accumulation of CMLs in tumors than that of ULs with the same lipid composition, suggesting that CMLs with improved vesicle stability could indeed enhance accumulation of drug carriers at the tumor site. Although no significant difference was observed between CMLs and DLLs relative to the accumulation of liposomes in the tumors, it is noteworthy that remarkably higher accumulation of DLLs in blood, heart, and spleen was detected compared to that of CMLs. This result indicated that DLLs could exhibit longer blood circulation compared to that of CMLs, but it could also lead to unwanted drug distribution to heart and spleen, which could induce severe toxic side effects, including cardiac toxicity.
The pharmacokinetic analysis showed that Dox levels of CML-Dox in serum were higher than those of free Dox and UL-Dox and, interestingly, even higher than Dox levels of DLL-Dox in serum at early time points (within 15 min) after injection (Figure 6D). This observation may suggest that DLL particles were preferentially absorbed by tissues, such as spleen and heart, quickly after administration. However, after having accumulated in tissues, some particles were slowly returned to blood circulation, thus explaining the longer blood retention of DLLs compared to that of CMLs at later times. In addition, Dox levels accumulated into tumors were further examined by measuring Dox concentration in collected tumors at 24 h after administration. As shown in Figure 6E, a significantly higher Dox level was detected in the tumors treated with CML-Dox, compared to that of free Dox, UL-Dox, or DLL-Dox. This result confirmed that the CML formulation could enhance penetration of Dox into the tumors. Moreover, even though both CMLs and DLLs exhibited a similar level of liposome accumulation in tumors, CMLs could achieve a higher Dox concentration in tumors based on their improved drug release (i.e., enhanced drug bioavailability), which corresponded well with the improved antitumor activity of CML-Dox in the mouse tumor model.
4. DISCUSSION
The overarching aim of this study was to evaluate a crosslinked multilamellar liposomal formulation of the anticancer agent doxorubicin for cancer therapeutics. We have demonstrated that crosslinked multilamellar structures of the CMLs not only offer controllable and sustainable drug release kinetics with increased vesicle stability, but also provide enhanced drug bioavailability, compared to the conventional unilamellar liposomes. It was also demonstrated that CMLs stably entrapped Dox in the vesicle and that the remarkable stability of CMLs allowed for long-term storage without a significant change in their size properties. Although some studies reported toxicity of some thiol-containing compounds including DTT in various cell lines, which is mainly caused by thiol-induced apoptosis [44], all thiol groups of DTT used as the cross-linker during the CML synthesis are quantitatively reacted with the maleimide headgroup of the lipid, and additionally, unreacted DTT was removed from CMLs after crosslinking. Thus, the thiol-induced toxicity should be minimized in this CML formulation.
In the present study, we demonstrated that the enhanced delivery of CML-Dox to tumor cells in vitro and in vivo improved anticancer activity and led to better tumor reduction and inhibition of tumor progression, when compared with the antitumor activity of non-crosslinked unilamellar liposome with the same lipid composition (UL) or Doxil-like liposome (DLL). Generally, the vesicle stability and drug release rate of liposomes are determined by liposome size, structure, and lipid composition. Although a higher drug release rate could augment drug bioavailability in tumors, the rapid release of drug from liposomes usually causes an equally rapid drug clearance in plasma, resulting in lower therapeutic efficacy to tumors. However, the crosslinked multilamellar structure of CML could achieve a controlled and sustained drug release profile, even though CML was composed of low-Tm (transition temperature) phospholipids, thus resulting in enhanced and sustained drug release kinetics. Consequently, the increased bioavailability of CML-Dox, together with improved vesicle stability, could allow for higher therapeutic activity, both in vitro and in vivo.
Furthermore, the entry mechanism and subsequent intracellular trafficking of CMLs were determined by direct visualization of interactions between CMLs and cellular endocytic structures. Our imaging study suggested that CML particles enter cells via caveolin-dependent endocytosis. Several studies have demonstrated that the caveolin-mediated pathway is a main route for uptake of liposomes [45, 46]. Moreover, efforts have been made in recent years to elucidate the detailed molecular mechanism underlying caveolae-mediated endocytosis and the subsequent intracellular fate of cargos. For example, simian virus 40 (SV40) is known to utilize the caveolae-mediated pathway for its entry and to be transported from the caveosome to endoplasmic reticulum (ER) to mediate infection [47]. Cholera toxin binding subunit (CTxB) is also believed to enter cells in a caveolin-dependent manner and to traffic to the Golgi complex, possibly through early endosomes [48]. Another recent study demonstrated that caveolin-dependent infectious entry of human papillomavirus type 31 (HPV31) could proceed to the endolysosomal compartments for successful infection [49]. These prior studies indicate that cargos utilizing caveolin-mediated endocytosis can traffic to multiple distinct intracellular destinations. Similarly, our imaging results with intracellular organelles suggested that CML particles internalized through caveolae might be predominantly transported further through the conventional endocytic pathway (i.e., early endosome to lysosome) and also traffic from the early endosomes to the trans-Golgi network, indicating that intracellular trafficking of CMLs might involve multiple distinct intracellular pathways. Additionally, it also appeared that the encapsulated Dox was released to cytoplasm of the cell prior to lysosomal degradation. The process for releasing the drug from the liposomes presumably involves the disruption of the integrity of the liposome bilayer in the presence of phospholipases, i.e., enzymes that hydrolyze phospholipids into fatty acids and other lipophilic substances present within endolysosomal compartments [50, 51].
Cholesterol is well known to rigidify and stabilize the liposomal membranes and has been widely used for current liposomal formulations [18, 50]. Addition of cholesterol to CML formulation may also provide rigid bilayers, promoting drug retention. However, incorporation of cholesterol in lipid bilayers could also hamper the process of crosslinking inter-lipid bilayers, which leads to vesicle instability in CML formulation. In addition, it was previously reported that the presence of cholesterol in the liposome membrane dramatically inhibits phospholipase activity [52], which suggests that cholesterol might disrupt cellular drug release from endolysosomal compartments and then decrease cytotoxic activity in tumor cells.
A previous report showed that the interbilayer-crosslinked multilamellar vesicle carrying protein antigens and adjuvants could steadily generate potent humoral and cellular immune responses for vaccine delivery [20]. In our study, we further extended this liposomal formulation to anticancer therapeutics and demonstrated many promising features of CMLs as a new nanocarrier platform for chemotherapy drug delivery applications. The CML formulation of Dox could reduce systemic toxicity, most likely by the controlled drug release. Furthermore, enhanced vesicle stability with higher Dox bioavailability enabled the improved in vivo therapeutic activity to tumors. It is noteworthy that CML-Dox treatment of B16 tumors, which is known as one of the most aggressive types of tumors, exhibited significant inhibition of tumor growth compared to that treated with the conventional liposomes UL-Dox and DLL-Dox. Our biodistribution study revealed that the enhanced therapeutic efficacy of CMLs resulted from the augmented accumulation of drugs at tumor sites and also showed lower accumulation of CMLs in heart and spleen compared to that of DLLs, which could improve the effectiveness and safety of drugs by minimizing the unwanted side effects.
5. CONCLUSION
This study suggests that the CML formulation potentially provides a versatile platform for cancer therapy. Moreover, this method can be readily applied to a combinatorial therapy, especially for codelivery of hydrophobic and hydrophilic drugs, which can offer several potential advantages, including synergistic effects and suppressed drug resistance. This liposomal formulation is expected to greatly advance current nanomedicine research and offer many new options in the treatment of a wide range of diseases.
Supplementary Material
Acknowledgments
We thank the USC NanoBiophysics Core Facility. This work was supported by National Institutes of Health grants (R01AI068978 and P01CA132681), a translational acceleration grant from the Joint Center for Translational Medicine, the USC Department of Radiology, the National Cancer Institute (P30CA014089), and a grant from the Ming Hsieh Institute for Research on Engineering Medicine for Cancer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 2.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–64. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–82. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 4.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–92. [PubMed] [Google Scholar]
- 5.Cho KJ, Wang X, Nie SM, Chen Z, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14:1310–6. doi: 10.1158/1078-0432.CCR-07-1441. [DOI] [PubMed] [Google Scholar]
- 6.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–71. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 7.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–60. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 8.Park JW. Liposome-based drug delivery in breast cancer treatment. Breast Cancer Res. 2002;4:93–7. doi: 10.1186/bcr432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J Pharm Sci. 2008;97:4696–740. doi: 10.1002/jps.21358. [DOI] [PubMed] [Google Scholar]
- 10.Haran G, Cohen R, Bar L, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151:201–15. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 11.Gabizon AA. Pegylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest. 2001;19:424–36. doi: 10.1081/cnv-100103136. [DOI] [PubMed] [Google Scholar]
- 12.O’Shaughnessy JA. Pegylated liposomal doxorubicin in the treatment of breast cancer. Clin Breast Cancer. 2003;4:318–28. doi: 10.3816/cbc.2003.n.037. [DOI] [PubMed] [Google Scholar]
- 13.Gabizon AA. Stealth liposomes and tumor targeting: One step further in the quest for the magic bullet. Clin Cancer Res. 2001;7:223–5. [PubMed] [Google Scholar]
- 14.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 15.Laginha KM, Verwoert S, Charrois GJR, Allen TM. Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Clin Cancer Res. 2005;11:6944–9. doi: 10.1158/1078-0432.CCR-05-0343. [DOI] [PubMed] [Google Scholar]
- 16.Al-Jamal WT, Al-Ahmady ZS, Kostarelos K. Pharmacokinetics & tissue distribution of temperature-sensitive liposomal doxorubicin in tumor-bearing mice triggered with mild hyperthermia. Biomaterials. 2012;33:4608–17. doi: 10.1016/j.biomaterials.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 17.van Lummel M, van Blitterswijk WJ, Vink SR, Veldman RJ, van der Valk MA, Schipper D, et al. Enriching lipid nanovesicles with short-chain glucosylceramide improves doxorubicin delivery and efficacy in solid tumors. FASEB J. 2011;25:280–9. doi: 10.1096/fj.10-163709. [DOI] [PubMed] [Google Scholar]
- 18.Barenholz Y. Doxil - The first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–34. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Charrois GJR, Allen TM. Drug release rate influences the pharmacokinetics, biodistribution, therapeutic activity, and toxicity of pegylated liposomal doxorubicin formulations in murine breast cancer. Biochim Biophys Acta. 2004;1663:167–77. doi: 10.1016/j.bbamem.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu HP, Huang B, et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat Mater. 2011;10:243–51. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itaya K, Ui M. A new micromethod for the colorimetric determination of inorganic phosphate. Clin Chim Acta. 1966;14:361–6. doi: 10.1016/0009-8981(66)90114-8. [DOI] [PubMed] [Google Scholar]
- 22.Cai H, Li Z, Huang CW, Park R, Shahinian AH, Conti PS. An improved synthesis and biological evaluation of a new cage-like bifunctional chelator, 4-((8-amino-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosane-1-ylamino)methyl) benzoic acid, for 64Cu radiopharmaceuticals. Nucl Med Biol. 2010;37:57–65. doi: 10.1016/j.nucmedbio.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Dos Santos N, Allen C, Doppen AM, Anantha M, Cox KAK, Gallagher RC, et al. Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: Relating plasma circulation lifetimes to protein binding. Biochim Biophys Acta. 2007;1768:1367–77. doi: 10.1016/j.bbamem.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 24.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–744. [PubMed] [Google Scholar]
- 25.Allen TM, Mumbengegwi DR, Charrois GJR. Anti-CD19-targeted liposomal doxorubicin improves the therapeutic efficacy in murine B-cell lymphoma and ameliorates the toxicity of liposomes with varying drug release rates. Clin Cancer Res. 2005;11:3567–73. doi: 10.1158/1078-0432.CCR-04-2517. [DOI] [PubMed] [Google Scholar]
- 26.Charrois GJR, Allen TM. Multiple injections of pegylated liposomal doxorubicin: pharmacokinetics and therapeutic activity. J Pharmacol Exp Ther. 2003;306:1058–67. doi: 10.1124/jpet.103.053413. [DOI] [PubMed] [Google Scholar]
- 27.Charrois GJR, Allen TM. Rate of biodistribution of stealth liposomes to tumor and skin: influence of liposome diameter and implications for toxicity and therapeutic activity. Biochim Biophys Acta. 2003;1609:102–8. doi: 10.1016/s0005-2736(02)00661-2. [DOI] [PubMed] [Google Scholar]
- 28.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–27. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 29.Dobson PD, Kell DB. Carrier-mediated cellular uptake of pharmaceutical drugs: an exception or the rule? Nat Rev Drug Discov. 2008;7:205–20. doi: 10.1038/nrd2438. [DOI] [PubMed] [Google Scholar]
- 30.Le Roy C, Wrana JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol. 2005;6:112–26. doi: 10.1038/nrm1571. [DOI] [PubMed] [Google Scholar]
- 31.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–12. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pelkmans L, Helenius A. Endocytosis Via Caveolae. Traffic. 2002;3:311–20. doi: 10.1034/j.1600-0854.2002.30501.x. [DOI] [PubMed] [Google Scholar]
- 33.Neufeld EB, Cooney AM, Pitha J, Dawidowicz EA, Dwyer NK, Pentchev PG, et al. Intracellular trafficking of cholesterol monitored with a cyclodextrin. J Biol Chem. 1996;271:21604–13. doi: 10.1074/jbc.271.35.21604. [DOI] [PubMed] [Google Scholar]
- 34.Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RGW. Caveolin, a protein-component of caveolae membrane coats. Cell. 1992;68:673–82. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- 35.Wang LH, Rothberg KG, Anderson RGW. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123:1107–17. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelkmans L, Burli T, Zerial M, Helenius A. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell. 2004;118:767–80. doi: 10.1016/j.cell.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–94. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 38.Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J Cell Biol. 2010;191:615–29. doi: 10.1083/jcb.201003086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999;397:621–5. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- 40.Carlsson SR, Roth J, Piller F, Fukuda M. Isolation and characterization of human lysosomal membrane-glycoproteins, h-lamp-1 and h-lamp-2 - major sialoglycoproteins carrying polylactosaminoglycan. J Biol Chem. 1988;263:18911–9. [PubMed] [Google Scholar]
- 41.Luzio JP, Brake B, Banting G, Howell KE, Braghetta P, Stanley KK. Identification, sequencing and expression of an integral membrane-protein of the trans-golgi network (TGN38) Biochem J. 1990;270:97–102. doi: 10.1042/bj2700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Jin Q, Huang C, Dasa S, Chen L, Yap L-P, et al. Trackable and targeted phage as positron emission tomography (PET) agent for cancer imaging. Theranostics. 2011;1:371–80. doi: 10.7150/thno/v01p0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai H, Fissekis J, Conti PS. Synthesis of a novel bifunctional chelator AmBaSar based on sarcophagine for peptide conjugation and 64Cu radiolabelling. Dalton Trans. 2009:5395–400. doi: 10.1039/b902210d. [DOI] [PubMed] [Google Scholar]
- 44.Held K, Sylvester F, Hopcia K, Biaglow J. Role of Fenton chemistry in thiol-induced toxicity and apoptosis. Radiat Res. 1996;145:542–53. [PubMed] [Google Scholar]
- 45.Huth US, Schubert R, Peschka-Suss R. Investigating the uptake and intracellular fate of pH-sensitive liposomes by flow cytometry and spectral bio-imaging. J Control Release. 2006;110:490–504. doi: 10.1016/j.jconrel.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 46.Pollock S, Antrobus R, Newton L, Kampa B, Rossa J, Latham S, et al. Uptake and trafficking of liposomes to the endoplasmic reticulum. FASEB J. 2010;24:1866–78. doi: 10.1096/fj.09-145755. [DOI] [PubMed] [Google Scholar]
- 47.Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol. 2001;3:473–83. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 48.Torgersen ML, Skretting G, van Deurs B, Sandvig K. Internalization of cholera toxin by different endocytic mechanisms. J Cell Sci. 2001;114:3737–47. doi: 10.1242/jcs.114.20.3737. [DOI] [PubMed] [Google Scholar]
- 49.Smith JL, Campos SK, Wandinger-Ness A, Ozbun MA. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J Virol. 2008;82:9505–12. doi: 10.1128/JVI.01014-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42:419–36. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 51.Andresen TL, Jensen SS, Jorgensen K. Advanced strategies in liposomal cancer therapy: problems and prospects of active and tumor specific drug release. Prog Lipid Res. 2005;44:68–97. doi: 10.1016/j.plipres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 52.Mouritsen OG, Jorgensen K. A new look at lipid–membrane structure in relation to drug research. Pharm Res. 1998;15:1507–19. doi: 10.1023/a:1011986613392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.