

Abstract
Simulations were performed to understand the relative contributions of molecular parameters to longitudinal (r1) and transverse (r2) relaxivity as a function of field, and to obtain theoretical relaxivity maxima over a range of fields to appreciate what relaxivities can be achieved experimentally. The field dependent relaxivity of a panel of gadolinium and manganese complexes with different molecular parameters: water exchange rates, rotational correlation times, hydration state, etc were measured to confirm that measured relaxivities were consistent with theory. The design tenets previously stressed for optimizing r1 at low fields (very slow rotational motion; chelate immobilized by protein binding; optimized water exchange rate) do not apply at higher fields. At 1.5T and higher fields, an intermediate rotational correlation time is desired (0.5 – 4 ns), while water exchange rate is not as critical to achieving a high r1. For targeted applications it is recommended to tether a multimer of metal chelates to a protein-targeting group via a long flexible linker to decouple the slow motion of the protein from the water(s) bound to the metal ions. Per ion relaxivities of 80, 45, and 18 mM−1s−1 at 1.5, 3, and 9.4T respectively are feasible for Gd3+ and Mn2+ complexes.
Introduction
MR imaging is increasingly moving to higher fields. There is now a large installed base of 3T scanners that are used clinically. 7T whole body scanners are available from the major MRI equipment manufacturers and there are a number installed worldwide. Recently human imaging at 9.4T has been reported (1). Higher fields bring greater signal:noise (SNR) and its attendant benefits of higher spatial resolution and/or reduced acquisition times.
Contrast agents are widely used in clinical MRI, with about a third of clinical scans being contrast enhanced (2,3). Clinical contrast agent usage is dominated by gadolinium-based T1-contrast agents which increase signal intensity on T1-weighted images, shorten acquisition times, and improve diagnostic confidence. Contrast agents are characterized by their relaxivity (r1 or r2) which is defined as the change in relaxation rate (R1,2 = 1/T1,2 in units of s−1) of solvent water protons upon addition of contrast agent, normalized to the concentration of contrast agent ([CA] in units of mM), equation 1:
| [1] |
The longitudinal relaxation times (T1) of tissue and blood increase with increasing field (4). For a contrast agent with equivalent relaxivity at two fields, the T1 change would be greater at the higher field because the inherent tissue R1 is slower at higher field. This is true of commercial extracellular fluid agents like Gd-DTPA whose relaxivity is fairly independent of field (5). However relaxivity is not a constant, but depends on external parameters such as applied field and temperature, and on molecular parameters such as the hydration state of the molecule and the molecular size. These molecular parameters can be tuned and optimized to create contrast agents with much higher relaxivities than conventional Gd-DTPA (6). High relaxivity contrast agents can be used at lower doses, or can be used in molecular imaging to detect low concentration targets.
There is an ongoing need to create new contrast agents. Molecular imaging agents bring additional specificity as a result of targeting (7-9). Recently, the incidence of nephrogenic systemic fibrosis (NSF) has highlighted the need for safe agents where the metal ion is completely eliminated from the body (10-13). While there are other contrast mechanisms available (T2*, chemical exchange saturation transfer, hyperpolarized agents), T1 relaxation has many salutary effects. For instance, T1 agents provide positive image contrast, can shorten acquisition times, are shelf stable, and have a proven track record in the clinic.
It is well known that T1 relaxivity typically decreases with increasing at high fields. Small, fast tumbling molecules like Gd-DTPA show a modest decrease in r1 with field (5), while slow tumbling molecules like MS-325 bound to serum albumin have high relaxivity that peaks between 0.5-1.0T and then shows a sharp drop in r1 with field (5,6). The strategy of markedly slowing rotational diffusion results in large gains in r1 at 1.5T but at high fields this strategy is much less effective. As new T1 agents are prepared, it would be valuable to design agents that have excellent relaxation properties over a broad range of imaging field strengths in order to take advantage of current 1.5T scanners and the increasing number of higher field scanners. This has been recognized in the recent work by Livramento et al. who have described new compounds with improved r1 at high fields (14-18).
Transverse relaxivity, r2, is static or increases with increasing field strength resulting in an increasing r2/r1 ratio with increasing field strength. Like r1, transverse relaxivity is dependent on a range of molecular parameters. In addition, for paramagnetic contrast agents like those based on gadolinium or manganese, the magnetization of the complex increases linearly with field and this results in greater susceptibility contrast at higher fields. Since r1 is decreasing with field but r2 is not, increasing the dose to offset lower r1 may not be effective at high field because of T2* effects at high doses. In molecular imaging, high doses may also result in target saturation where the amount of contrast achieved at the targeted is limited. New T1 reagents therefore should work effectively in high field imagers and provide high relaxivity over a range of field strengths and not be limited to 1.5T and lower fields.
The aims of this work are twofold. The first is to understand the relative contributions of the various molecular parameters to r1 and r2 as a function of field to determine what molecular factors are key in the design of high field, high relaxivity contrast agents. The second aim is to obtain theoretical relaxivity maxima over a range of fields to appreciate what can and cannot be achieved experimentally. Relaxivities greater than 100 mM−1s−1 per ion have been posited in the literature (2,19) and may be achieved at low fields (0.5T) but are such high values theoretically feasible at 3T and higher? If not, what represents “high relaxivity” at high fields? Here, we simulated the effect of different molecular parameters, including internal and anisotropic rotation, on the relaxivity of gadolinium and manganese complexes. We also measured field dependent relaxivities of a panel of gadolinium and manganese complexes with different water exchange rates, rotational correlation times, etc to ascertain if the measurements at high fields were consistent with theory.
Results
Theory
Solvation spheres
The metal complex can be viewed as having separate coordination spheres. The inner-sphere (IS) consists of ligands directly bonded to the metal ion. For contrast agents like GdDOTA, Figure 1, the inner-sphere is the DOTA ligand and a water molecule. The second coordination sphere (SS) is less well defined and consists of water molecules hydrogen bonded to the metal complex and possibly counterions. Beyond the second-sphere there is less organized structure and water freely diffuses, and this is referred to as the outer-sphere (OS). We define the 2nd sphere in terms of water residency time, τm’, which must be longer than the diffusion correlation time of water. Relaxivity can be factored into contributions arising from water in each coordination sphere, eqn 2.
| [2] |
Figure 1.

Factors influencing solvent water relaxation. The metal complex has an inner-sphere (IS) of nitrogen and oxygen atoms from the DOTA ligand and a coordinated water molecule. There is a distinct 2nd hydration sphere (SS) with Gd-H distance r’, and water molecules from both spheres undergo exchange with bulk water at rates 1/τm and 1/τm’ for 1st and 2nd sphere exchange
Inner-sphere relaxivity
The effect of chemical exchange on relaxation has been dealt with in terms of the Bloch equations by McConnell (20) and applied to the problem of water exchange at paramagnetic metal ions by Swift and Connick (21). Relaxation of bulk water arising from exchange of water molecules associated with a paramagnetic ion is given by:
| [3] |
| [4] |
where Ri is the observed relaxation rate, Ri0 is the rate in the absence of paramagnetic species, Rim is the relaxation rate of the water bound to the metal ion (i=1,2), τm is the lifetime of the water coordinated to the metal ion (its reciprocal is the water exchange rate kex = 1/τm), Δωm is the chemical shift of the coordinated water, and Pm is the mole fraction of water coordinated to the metal ion. This can be expressed in terms of the hydration number, q, which is the number of water molecules bound to the metal ion (M):
| [5] |
where square brackets denote concentration. For ions such as Gd3+ and Mn2+, the chemical shift of the bound water is small at most fields compared to its transverse relaxation rate and the Δωm term can be neglected. Combining equations 1-4 and neglecting Δωm, one arrives at expressions for two site exchange in terms of relaxivity, where the superscript “IS” is used to denote the relaxivity due to the inner-sphere (metal bound) water molecules:
| [6] |
where [H2O] is the water concentration in millimolar (55,600 mM).
Paramagnetic relaxation is fairly well understood. There are three mechanisms that contribute: dipole-dipole (DD) coupling between the paramagnetic ion and the hydrogen of water, scalar (SC) or contact relaxation, and Curie spin (CS) relaxation. The first two mechanisms were described by Solomon (22) and Bloembergen (23). Curie spin relaxation, first described by Gueron (24), results from modulation of the permanent magnetic moment and can become important at high fields as the complex becomes increasingly magnetized. At the fields that we are concerned with (1.5T and higher), only dipolar relaxation contributes appreciably to T1m for Gd(III) or Mn(II) induced nuclear relaxation whereas all three mechanisms can contribute to T2m, i.e. 1/T2m = 1/T2DD +1/T2SC+1/T2CS. The relevant expressions for 1.5T and above are:
| [7] |
| [8] |
| [9] |
| [10] |
Here, S is the spin quantum number, rMH is the ion-proton distance, ωH is the Larmor frequency of the proton (rad/s), ωS is the Larmor frequency of the electron, γH is the proton magnetogyric ratio, ge is the electronic g-factor (ge = 2 for Gd(III) and Mn(II)), μB is the Bohr magneton, μ0 is the permittivity of vacuum, A/ħ is the hyperfine coupling constant between the ion and the hydrogen nucleus, kB is the Boltzmann constant, and T is the absolute temperature.
The correlation time for each relaxation mechanism can have contributions from different processes: rotational motion (1/τR), longitudinal relaxation of the unpaired electrons (1/T1e), and water exchange (1/τm). Dipole-dipole relaxation can be modulated by either of these three, but at high fields electronic relaxation is slow relative to rotational motion and T1e does not contribute as a correlation time. By definition, scalar relaxation is independent of rotation, and Curie spin relaxation is independent of electronic relaxation:
| [11] |
| [12] |
| [13] |
These simplified expressions differ from the more general forms that cover low fields. There are several dispersive terms with 1/ωS2τc2 dependence that are vanishingly small at 1.5T and these have been omitted for clarity. In addition electronic relaxation is slow at high fields. Values of T1e directly measured for Gd3+ complexes at 0.34T were in the 1 – 5 ns range (25,26) and these times increase with increasing field. At very low fields T1e can be quite short and under conditions where 1/T1e is fast relative to rotational reorientation (slow motion regime) the description of electronic relaxation is complex and the simple analytical expressions of Bloembergen and Morgan do not hold (23). However at the fields we are considering (≥1.5T), the static zero field splitting of the complexes (<0.05T) is much less than the Zeeman energy and T1e >> τR. Although in principle 1/T1e is multiexponential, Belorizky and Fries showed that 1/T1e could be well approximated by a simple analytical expression (27). As a result, the usual modified Solomon-Bloembergen-Morgan (MSBM) equations are applied here. In a full analysis of the nuclear magnetic relaxation dispersion (NMRD) profile of MS-325 bound to albumin we found that high field relaxation was adequately described by MSBM while the description of the entire curve required application of the slow motion theory.
Here we describe the field dependence of electronic relaxation for Mn2+ and Gd3+ by:
| [14] |
Electronic relaxation for Mn2+ and Gd3+ in solution at high fields is a result of transient distortions of the compound that result in a modulation of the crystal field interaction parameters (23), also referred to as zero-field splitting (ZFS). The magnitude of this transient ZFS is given by Δ2 and this is related to crystal field terms D and E by Δ2 = (2/3)D2 + 2E2. The correlation time for this modulation is denoted τv and is on the order of picoseconds.
Second- and outer-sphere relaxivity
For the water in the second-sphere, relaxivity can be described by the same mechanisms as for inner-sphere water. We denote second-sphere parameters with a prime:
| [15] |
Strictly, each water should have its own relaxation time and residency time, but we assume here that all waters in a given coordination sphere behave the same. The ion-hydrogen distance r’ is longer and the residency time τm’ is shorter for waters in the second-sphere.
The outer-sphere contribution to relaxivity has been described by Freed (28,29). This is dominated by the diffusion time of water and a distance of closest approach. This is not something that can be readily changed by modifying the contrast agent. For the range of fields considered here, r1OS should be constant. We assume a value of 2 mM−1s−1 for r1OS based on literature values (30).
Effect of internal motion
For multimeric contrast agents or protein-targeted agents, rotation about the ion-hydrogen vector may not be well approximated by an isotropic model. Most targeted agents are bifunctional molecules that contain a moiety that binds to a protein linked to a relaxation enhancing moiety. If there is flexibility between the targeting moiety and the relaxation moiety then the motion that the coordinated water proton “sees” will be a function of the slow global motion of the protein and the faster internal motion of relaxation moiety (metal part) (31). Similarly for metal complexes attached to the surface of a dendrimer, the rotational correlation time at a given field will have contributions from the slow overall motion of the dendrimer as well as from faster local motions because of internal flexibility (32,33). To look at the effects of internal motion on relaxivity we used a model-free approach originally described by Lipari and Szabo (34). They showed that for many complicated models of motion, the spectral density function could be approximated as:
| [16] |
for dipolar longitudinal relaxation, where F represents an order parameter (usually denoted S but here F to not to confuse with spin number) that can vary between 0 and 1, τg is the global correlation time (which is the same as τc above for DD relaxation or τcs for CS relaxtion) and τl is a local correlation time that takes into account fast local motion (τf). When F2 = 1, motion is isotropic and the system is governed by the global relaxation time. When F2 = 0, relaxation is governed by the fast local motion. This approach has been successfully applied to explain the field dependent relaxivity of certain contrast agents (31,35-38).
Simulations
Relaxivity due to inner-sphere water
We simulated relaxivity as a function of external field (B0 = ωH/γH), rotational correlation time, and water residency time for a water in the inner-sphere, i.e. directly bound to the metal ion, of a Gd3+ complex and a Mn2+ complex. For Gd3+, the metal ion – water hydrogen distance was set to 3.1Å and the isotropic hyperfine constant was set to 0.1 MHz based on previous EPR studies (39-43). For Mn we use a distance of 2.9Å based on the Mn-Owater bond length determined from X-ray crystallography (44) and assuming a similar orientation of the bound water as in the Gd3+ case (41). A/h for Mn2+ was set to 1.0 MHz, the value reported for the Mn2+ aqua ion (23). We used previously determined values of Δ2 and τv for MS-325 and MnL1 for the simulations (35,38,45). Electronic relaxation was slow with respect to rotational diffusion for the field strengths that we are concerned with here (1.5 to 15T).
Effect of rotational dynamics on relaxivity due to inner-sphere water
The field dependent relaxivity for the inner-sphere water of a Gd complex with a water exchange rate similar to GdDTPA or GdDOTA (τm ~100 ns at 37 °C) is plotted in Figure 2 for 3 different isotropic rotational correlation times: 0.1, 1, and 10 ns. The short correlation time would be similar to a small metal complex like GdDTPA, while the long correlation time would reflect the protein-bound case like MS-325 bound to albumin. Protein binding (slow rotation, long τR) is an effective strategy at low field for increasing r1. However by 3T, this strategy offers diminishing returns and the r2/r1 ratio balloons with increasing field strength. This suggests that increasing the dose of a slow rotation contrast agent to decrease T1 has less effect at high field because of the large r2 effect. The fast tumbling complex exhibits a relatively field independent, but low r1 relaxivity. However by 7T, the r1 relaxivity of the fast tumbling case is now higher than the slow tumbling case. The intermediate motion regime (1 ns) also shows a fall off in r1 with field but this is less dramatic. The intermediate case offers the best combination of high r1 and low r2/r1 over a wide range of fields. For r2 the field dependence is rather flat and is proportional to the rotational correlation time. At very high fields, r2 starts to increase and this is due to an increasing contribution from the Δωm term in equation 3. Although this shift is relatively small, above 12 T it starts to approach the magnitude of R2m.
Figure 2.

Effect of rotation on r1 (A), r2 (B), or the ratio r2/r1 (C) as a function of field for the inner-sphere water of a Gd3+ complex with a water residency time 100 ns. τR = 0.1 ns (•••), 1.0 ns (---), or 10 ns (—). Inset in A is a magnified view.
For Mn2+, we observe a similar effect for r1 as a function of field and rotational correlation time, Figure 3a. Again, slow tumbling is a potent strategy at low fields, but not at high fields. The intermediate correlation time provides better high field relaxivity. For r2 there are some differences. Because the hyperfine interaction between the metal ion and the water proton is about an order of magnitude higher for Mn2+ than for Gd3+, there is a significant scalar contribution to r2 for Mn2+ complexes. This contribution is modulated by a correlation time that has contributions from τm and T1e. T1e increases with the square of the magnetic field and does not contribute significantly at high fields. In Figure 3b and 3c, the effect of the water residency time is shown for τm = 5 ns or 100 ns. For Mn2+ compounds, we see that r2 increases with increasing τR like for Gd3+ but r2 also increases with increasing τm.
Figure 3.

Effect of rotation on r1 (A), r2 (B and C) as a function of field for the inner-sphere water of a Mn2+ complex. Water residency times (τm) of 5 or 100 ns do not affect the r1 result, but r2 depends on τm because of a scalar contribution to r2: τm = 5 ns (B) or 100 ns (C). τR = 0.1 ns (•••), 1.0 ns (---), or 10 ns (—). Inset in A is a magnified view.
Effect of water exchange dynamics on relaxivity due to inner-sphere water
Relaxivity depends inversely on the water residency time, τm, and the relaxation time of the bound water, T1,2m. Obviously if τm becomes too long, there is little/no exchange of the relaxed water with the bulk and relaxivity will be limited. If τm becomes less than the rotational correlation time, then it starts to modulate the interaction energy, i.e. it becomes τc. A very short τm results in an increased T1m (relaxation of the bound water is less efficient) and relaxivity suffers. There is some optimum range, then for τm.
It is apparent that there is an optimum correlation time for r1 at each field. At high fields τc should be about 1/ωH. We simulated the effect of water exchange (kexchange = 1/τm) on the Gd3+ relaxivity at 3 field strengths: 0.47T, 3T, and 9.4T. We chose τR values that would give very high relaxivities for each field strength, 20, 1.5, and 0.5 ns respectively. The effect of water exchange on relaxivity is shown in Figure 4. Figure 4 demonstrates again, that maximum T1 relaxivity falls off with increasing field strength and that there is an ideal range for water exchange kinetics. However this ideal range of water exchange rates is much broader at higher fields. Similar results are obtained for Mn2+ with the additional effect of τm on r2 as shown in Figure 3.
Figure 4.

Effect of water residency time τm (ns) for an inner-sphere water on r1 (—) and r2 (---) under optimal rotational conditions at 0.47T (A, τR = 20 ns), 3T (B, τR = 1.5 ns), and 9.4T (C, τR = 0.5 ns). The optimal τm range increases as field increases.
In order to better appreciate the interplay between field strength, water exchange and rotational dynamics, we looked at τm vs τR for T1 relaxivity at 4 different field strengths. In Figure 5, relaxivity is presented as a contour plot with each color representing 20% increments of the maximum relaxivity at that given field strength. Figure 5 reveals that while it is theoretically possible to achieve very high relaxivity at low fields, this is difficult to achieve because it requires a narrow combination of correlation times. At 0.47T the largest area is represented by the lowest quintile of relaxivity. Conversely, at higher field strengths the highest quintile of relaxivity can be achieved with a wider range of water exchange rates and rotational dynamics. At higher fields the values of τR giving rise to the highest relaxivity become increasingly shorter.
Figure 5.
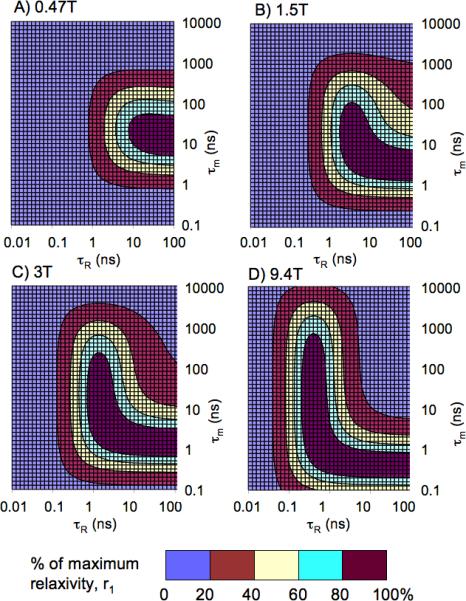
Interplay between water residency time (τm) and rotational motion (τR) on maximum achievable r1 or an inner-sphere water. Higher relaxivities can be achieved at lower fields, but at higher fields there is much larger dynamics space to achieve the top 20% of attainable r1.
Effect of internal motion on relaxivity due to inner-sphere water
It is apparent from equation 16 that internal motion will reduce relaxivity. The best scenario is either when there is no internal motion and rotation is isotropic, or when the local motion is completely decoupled from the slow motion (F2 = 1 or 0). This is illustrated in Figure 6 for relaxivity at 3T. At 3T, isotropic rotation with τR = 1.5 ns would lead to a high relaxivity. For a slow tumbling system (τglobal = 10 ns) with fast internal motion (τlocal = 0.1 ns), relaxivity is poor at all combinations of motion; slow and fast motion do not equal intermediate motion. When the internal motion is slower (τlocal = 1 ns, Figure 6b), then relaxivity is higher, but the relaxivity is highest when this internal motion is completely decoupled (F2 = 0) from the slower global motion.
Figure 6.

Effect of internal motion on relaxivity due to one inner-sphere water molecule at 3T, τm =100 ns. A) Simulation for global correlation time, τglobal = 10 ns and local correlation time, τlocal = 0.1 ns. B) τglobal = 10 ns, τlocal =1 ns. r1 is always highest at either F2 = 0 or 1.
Effect of second-sphere hydration on relaxivity
Waters in the second-sphere typically have very short residency times (τm’) which limits their probability of being relaxed. However there is good evidence that τm’ can be increased through hydrogen bonding. For instance GdDOTP (tetraphosponate analog of DOTA) has similar relaxivity to GdDOTA even though the former has no inner-sphere water (46). We showed that a q=0 GdDOTP-fatty acid derivative could have very high relaxivity (40 mM−1s−1 at 0.47T) when bound to serum albumin (47). This is likely due to strong hydrogen bonding of 2nd-sphere water to the anionic phosphonate oxygens. Relaxometry showed that there were 2nd-sphere exchangeable proton(s) with a lifetime of 1 ns (47). This seems to be general to phosphonate complexes (30,48-50). For example replacing one acetate on DOTA with a phosphonate still results in a q=1 complex, but the phosphonate has a 28% higher relaxivity which can only be attributed to a 2nd-sphere effect.
Using the q’/r’GdH value obtained previously, we simulated 2nd-sphere relaxivity for Gd3+ complexes using different correlation times for rotation and for the lifetime of the water in the 2nd-sphere. Some of these are shown in Figure 7. As may be expected, the relaxivity effect from protons in the 2nd-sphere can be significant if the residency time is relatively long, and if rotational motion is relatively slow. The effect is quite meaningful at high fields where shorter correlation times are preferred.
Figure 7.

Second-sphere relaxivities, r1 (A) and r2 (B) calculated for one water with r’GdH = 3.5 Å at different water residency times and rotational correlation times. τR = 10 ns, τm’ = 0.1 ns (•••); τR = 1 ns, τm’ = 1 ns (---); or τR = 10 ns, τm’ = 1 ns (—).
Effect of hydration on relaxivity
The inner-sphere contribution to relaxivity scales linearly with the number of water molecules bound. The simulations in Figures 2-4 would result in double the relaxivity if there were two inner-sphere waters. Similarly for the 2nd-sphere effect, more long-lived protons at an equivalent distance to the ion will increase the relaxivity.
Experimental Relaxivities
Field dependence on inner-sphere relaxivity
The compounds studied in this work are shown in Figure 8. These compounds provide a range of rotational correlation times and the correlation time can be altered further by protein binding. Relaxivities for these compounds determined at ambient temperature (21°C) and 3 field strengths are listed in Table 1.
Figure 8.

Compounds studied in this work.
Table 1.
Relaxivities (r1, r2) per metal ion determined at 1.4, 4.7, and 9.4T at 21 °C with units mM−1s−1 in either phosphate (10 mM) buffered saline (PBS) pH 7.4 or PBS+4.5% w/v human serum albumin (HSA), or 10 mg/mL human fibrin. Error estimated at ±10%.
| Compound | r1 1.4T | r1 4.7T | r1 9.4T | r2 1.4T | r2 4.7T | r2 9.4T |
|---|---|---|---|---|---|---|
| MS-325 | 8.30 | 7.21 | 5.14 | 9.27 | 14.0 | 15.7 |
| MS-325+HSA | 24.3 | 11.20 | 7.16 | 55.8 | 93.6 | 105 |
| GdDTPA-BSA | 10.4 | 5.99 | 3.79 | 13.6 | 13.6 | 13.9 |
| EP-2104R | 15.6 | 9.43 | 6.39 | 19.3 | 14.2 | 13.0 |
| EP-2104R+fibrin | 16.3 | 10.0 | 5.94 | 23.5 | 32.8 | 36.7 |
| MnL1 | 5.32 | 4.46 | 4.05 | 8.37 | 7.3 | 7.00 |
| MnL1+HSA | 27.2 | 5.78 | 3.48 | 96.7 | 80.6 | 94.6 |
| MnCl2 | 6.63 | 5.92 | 5.14 | 60.2 | 110 | 117 |
Discussion
Comparison of measured relaxivities with simulations
The measured relaxivities and their field dependencies are in the range one would expect on the basis of what is known about the correlation times of these molecules. For MS-325 bound to the protein serum albumin, it was previously shown (45) that r1 reaches a maximum between 0.5 and 1T and then declines, while r2 increases above 1T. At much higher fields (4.7 and 9.4T), r1 continues to decrease while r2 is large. In the absence of protein, MS-325 has a τR of ~0.18 ns at room temperature. Its relaxivity, both r1 and r2, is rather field independent as predicted by theory. At 9.4T the T1 relaxivity of MS-325 is about the same with or without albumin, but r2 is an order of magnitude higher in the presence of albumin. The protein-binding strategy which works well at low field offers no benefit for improving r1 at 9.4T.
We also looked at Gd-DTPA-BSA which has the Gd chelated by a diethylenetriaminetetraacetate-monoamide that is covalently attached to albumin. The monoamide complex is known to have about 3-fold slower water exchange rate than the diethylenetriaminepentaacetate (DTPA) chelator that is used in MS-325 (51). This slower water exchange rate mutes the r2 effect. The slow exchange also limits r1 but this is more important at low fields, see Figure 5. At 1.4T r1 for Gd-DTPA-BSA is much lower than MS-325/HSA but this difference is reduced as the field strength is increased.
The complex MnL1 is a manganese analog of MS-325 (38). Analogous field dependent relaxivity is observed for MnL1 in the presence and absence of albumin, as was seen for MS-325. At low field, there is a benefit to albumin binding for r1 but this falls off quickly with increasing field. At high fields MnL1 displays a relatively low r1 and very high r2 in the presence of HSA. In the absence of protein, the field dependence on r1 and r2 is again flat. As predicted by theory, at 9.4T r1 is actually worse for the protein bound form of MnL1 than the unbound form.
The water exchange rate of MnL1 was previously measured (38) and the mean residency time of water at 20 °C is about 5 ns. This short residency time results in only a modest scalar contribution to r2. We also measured relaxivity for the manganese aqua ion. The rotational correlation time of the aqua ion is very short (10's of picoseconds) while the water residency time was determined to be 50 ns at 20 °C (21). As a result of the very short rotational correlation time, the r1 relaxivity for Mn2+ is very low per coordinated water (1.2 mM−1s−1 per bound H2O). However the aqua ion has 6 waters bound so its T1 relaxivity is reasonable. The very short rotational correlation time insures a flat field dependence on r1. The short τR also means that the dipolar contribution to transverse relaxivity is small. However the relatively long residency time results in a large scalar contribution to r2 and an r2/r1 of ~20 compared to r2/r1 < 2 for MnL1.
The molecular imaging probe EP-2104R is an example of a molecule with internal motion contributing to relaxivity. From the field dependence of fibrin-bound EP-2104R, there is clearly a significant slow motion contribution that has a positive effect on r1 at 1.4T. However at 9.4T, the fibrin-bound T1 relaxivity is slightly lower than that of the unbound EP-2104R. r2 is higher for EP-2104R when it is bound to fibrin as we expect. However the impact of internal motion is such that r2 is not as high as for MS-325/HSA because of the effect of the fast internal motion component on relaxation.
Taken together, the experimental results confirm the results of the simulations. The simulations in turn provide guidance for design of new probes.
Impact of high fields on T1 probes
The simulations and measured high field relaxivities in this paper, along with other high field relaxivity data previously (5) reported highlight the challenges faced at high fields. The receptor induced magnetization enhancement (RIME) strategy (2,45,52) that is effective at low fields is counterproductive at very high fields. It leads to low r1 and large r2. Another factor not explored here is the increasing magnetization of paramagnetic complexes. Unlike iron oxide particles which are fully magnetized by 1T, Gd3+ and Mn2+ magnetize linearly with applied field (53). As a result, susceptibility effects of Gd3+ are higher at higher magnetic fields (susceptibility of Mn is 5/9 that of Gd). A consequence of these r2 and R2* effects is that increasing the contrast agent dose to shorten T1 may not be effective because T2 effects may dominate. This may be less of an issue for targeted probes where the concentrations at the target are relatively low. However as the relaxivities of EP-2104R demonstrate, r1 is much lower at 4.7 or 9.4T than at 1.4T. Increasing the dose here to overcome poor high field relaxivity may also prove ineffective, because the molecular target can become saturated.
Strategies to improve high field relaxivity
The utility of T1 contrast agents has been widely established clinically and in preclinical models. T1 contrast agents can continue to provide benefits at high fields. For new probes, some consideration should be made to how the chelate and the dynamics affect relaxivity. From the simulations, the rotational correlation time should be intermediate between a fast tumbling small molecule like GdDTPA and a slow tumbling protein- or polymer-bound agent. The optimum correlation time will depend on the field strength, but for good relaxivity over the 1.5 to 9.4T range, a correlation time in the 0.5 to 1.5 ns range is effective. In order to achieve these intermediate correlation times, two approaches can be used. One is to place the metal complex at the barycenter of the molecule, Figure 9a. This approach has been employed by Port et al, and Fulton et al. (54,55). This minimizes any effects of internal motion as the chelate should tumble with the overall rate of motion of the molecule. Pendant groups can be added to modulate this rate. The other approach is to make the molecule larger by linking multiple chelates together in a fairly rigid fashion, Figure 9b. There are several examples of this approach and increased high field relaxivity has been noted (15,17,56,57).
Figure 9.

Strategies to achieve high relaxivity compounds at high fields. A) the metal ion is placed at the barycenter of the molecule limiting effects of internal motion; correlation time can be increased by adding pendant groups. B) correlation time and metal content increased by making a rigid multimer. Targeting groups should be linked via flexible linkers to decouple the motion at the metal ion from the macromolecule.
Examples of compounds with improved high field relaxivity have often focused on non-targeted molecules. However there is broader utility in expanding high relaxivity into molecular imaging probes. Protein binding and rotational immobilization is obviously to be avoided for high field, high relaxivity agents. The simulations suggest that the best solution is to decouple the slow rotational dynamics of the target from the internal motion of the metal chelator. The internal motion should be in this sweet spot of 0.5 to 1.5 ns. Targeted agents then should have chelates using the rotational dynamics strategy shown in Figure 9. A targeting vector (e.g. peptide) is tethered via a long flexible linker to metal chelators; upon protein binding, the internal motion of the chelators is decoupled from the slow motion of the protein.
Ideally the complex should have two inner-sphere water molecules (q = 2) since the gain in r1 from multiple waters is independent of field. However, increasing q carries two risks: it can result in a less stable complex where the Gd3+ is more easily released; it can open up a site for anion (HCO3−, HPO42−, citrate) coordination whereby the anion displaces the water molecules (58). However stable q = 2 complexes have been reported with higher relaxivity, e.g. the Gd-HOPO series (59,60) or pyridine analogs of DOTA (50,61). Recently Aime and colleagues reported the AAZTA ligand which forms a thermodynamically stable q=2 complex with Gd resulting in an increased relaxivity that is unaffected by the presence of anions like phosphate (62). An albumin binding version of this complex displayed very high relaxivity at low field (84 mM−1s−1 at 0.47T) (63).
Optimal relaxivity
Relaxivity is clearly field dependent and the correlation times that give the highest r1 at 0.5T are not the same as those that provide the highest relaxivity at 3T. We performed simulations using reasonable molecular parameters to identify what one could expect from an “ideal” high field agent. Figure 10 shows the result of a simulation for a gadolinium complex with 2 inner-sphere water molecules with fast water exchange (τm = 50 ns) and a long lived (τm’ = 1 ns) water at 3.5 Å from Gd. These are reasonable values based on current literature. We assumed isotropic rotation and plotted results for 3 correlation times representing optimal relaxivity at 1.5T, 3T, and 9.4T. Rotational correlation times in the range 0.5 – 4.6 ns give excellent high field relaxivity. The short correlation time 0.5 ns which provides the highest r1 at higher fields would still deliver a relaxivity greater than 30 mM−1s−1 at 1.5T which is better than almost all reported compounds. If the rotational correlation time increases are achieved by making a rigid multimer of n chelates, then the relaxivity of the molecule will be n-fold higher.
Figure 10.

Simulated r1 for a Gd3+ complex with two rapidly exchanging (τm = 50 ns) inner-sphere waters and one water with r’GdH = 3.5 Å, τm’ = 1 ns for three different isotropic rotational correlation times τR = 0.5 ns (•••), 1.6 ns (---), or 4.6 ns (—).
Because of the number of parameters involved, even higher relaxivities may be achievable with q>2 or greater 2nd-sphere hydration. However Figure 10 represents a reasonable, practical upper range of r1 that may be achieved through judicious molecular design, yet still resulting in complexes that are stable and inert enough for in vivo use.
Conclusions
Longitudinal relaxivity will decrease with increasing field but very high relaxivities may still be achieved by judicious molecular design. The design tenets stressed for optimizing r1 at low fields (very slow rotational motion; protein binding with the Gd chelate tethered by short, rigid linker; narrow, optimal water exchange rate) do not apply at higher fields. At 1.5T and higher fields, an intermediate correlation time is desired (0.5 – 4 ns) and the water exchange rate is not as critical to achieving a high r1. Although r1 decreases with increasing field, the parameter space for identifying a compound with high relaxivity becomes greater as the field increases, and thus the likelihood of identifying high field, high relaxivity agents is increased. For targeted applications it may be best to use a multimer of metal chelates with an optimal rotational correlation time that is linked to a protein-targeting group via a long flexible linker to decouple the slow motion of the protein from the water(s) bound to the metal ions. Per ion relaxivities of 80, 45, and 18 mM−1s−1 at 1.5, 3, and 9.4T respectively are feasible for Gd3+ and Mn2+ complexes, making the future bright for T1-contrast at high fields.
Experimental
The compounds EP-2104R (64) and MS-325 (gadofosveset) (45) were gifts of Epix Pharmaceuticals. The albumin binding MnEDTA derivative MnL1 (38) and Gd-DTPA-BSA (65) were prepared as previously described. Human serum albumin (Fraction V) was obtained from Sigma, human fibrinogen was from Calbiochem, and manganese chloride hexahydrate was purchased from Aldrich. Fibrinogen was dialyzed against 50 mM Tris buffered saline (TBS) containing 5 mM sodium citrate. Fibrin gels were prepared by mixing fibrinogen (12 μM final concentration), EP-2104R (0 – 40 μM), and calcium chloride (20 mM) and then adding 0.33 NIH units of human thrombin. All solutions were at pH 7.4.
Relaxation rate measurements were made at ambient temperature (21 °C) using a Bruker mq60 Minispec at 1.4T, or horizontal bore imagers at 4.7 and 9.4T (Bruker, Billerica MA). At 1.4T samples (200 μL) were prepared in small glass cylindrical vials and placed inside a 7.5mm NMR tube inside the Minispec. At 4.7 and 9.4T, samples were prepared in 0.5 mL plastic centrifuge tubes. Five of these tubes were placed in a holder and immersed in an aqueous solution of 10 mM MnSO4 and imaged with a volume coil. T1 was measured using an inversion-recovery sequence with 8 to 10 TI values and care was taken to set TR ≥ 5T1. T2 was measured using a multi echo spin echo sequence with typically 25 echoes.
Acknowledgment
PC thanks Siemens Medical Solutions for funding support. Support was also provided by P41-RR14075 and the MIND Institute.
References
- 1.Vaughan T, DelaBarre L, Snyder C, Tian J, Akgun C, Shrivastava D, Liu W, Olson C, Adriany G, Strupp J, Andersen P, Gopinath A, van de Moortele PF, Garwood M, Ugurbil K. 9.4T human MRI: preliminary results. Magn. Reson. Med. 2006;56:1274–1282. doi: 10.1002/mrm.21073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem. Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 3.Caravan P, Lauffer RB. Contrast Agents: Basic Principles. In: Edelman RR, Hesselink JR, Zlatkin MB, Crues JV, editors. Clinical Magnetic Resonance Imaging. 3rd ed. Vol. 1. Saunders; Philadelphia: 2005. pp. 357–375. [Google Scholar]
- 4.Rooney WD, Johnson G, Li X, Cohen ER, Kim SG, Ugurbil K, Springer CS., Jr. Magnetic field and tissue dependencies of human brain longitudinal 1H2O relaxation in vivo. Magn. Reson. Med. 2007;57:308–318. doi: 10.1002/mrm.21122. [DOI] [PubMed] [Google Scholar]
- 5.Rohrer M, Bauer H, Mintorovitch J, Requardt M, Weinmann HJ. Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Invest. Radiol. 2005;40:715–724. doi: 10.1097/01.rli.0000184756.66360.d3. [DOI] [PubMed] [Google Scholar]
- 6.Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem. Soc. Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 7.Mulder WJ, Strijkers GJ, Briley-Saboe KC, Frias JC, Aguinaldo JG, Vucic E, Amirbekian V, Tang C, Chin PT, Nicolay K, Fayad ZA. Molecular imaging of macrophages in atherosclerotic plaques using bimodal PEG-micelles. Magn. Reson. Med. 2007;58:1164–1170. doi: 10.1002/mrm.21315. [DOI] [PubMed] [Google Scholar]
- 8.Sosnovik DE, Schellenberger EA, Nahrendorf M, Novikov MS, Matsui T, Dai G, Reynolds F, Grazette L, Rosenzweig A, Weissleder R, Josephson L. Magnetic resonance imaging of cardiomyocyte apoptosis with a novel magneto-optical nanoparticle. Magn. Reson. Med. 2005;54:718–724. doi: 10.1002/mrm.20617. [DOI] [PubMed] [Google Scholar]
- 9.Schmieder AH, Winter PM, Caruthers SD, Harris TD, Williams TA, Allen JS, Lacy EK, Zhang H, Scott MJ, Hu G, Robertson JD, Wickline SA, Lanza GM. Molecular MR imaging of melanoma angiogenesis with alphanubeta3-targeted paramagnetic nanoparticles. Magn. Reson. Med. 2005;53:621–627. doi: 10.1002/mrm.20391. [DOI] [PubMed] [Google Scholar]
- 10.Thomsen HS, Marckmann P. Extracellular Gd-CA: differences in prevalence of NSF. Eur. J. Radiol. 2008;66:180–183. doi: 10.1016/j.ejrad.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 11.Marckmann P. An epidemic outbreak of nephrogenic systemic fibrosis in a Danish hospital. Eur. J. Radiol. 2008;66:187–190. doi: 10.1016/j.ejrad.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 12.Grobner T, Prischl FC. Gadolinium and nephrogenic systemic fibrosis. Kidney Int. 2007;72:260–264. doi: 10.1038/sj.ki.5002338. [DOI] [PubMed] [Google Scholar]
- 13.Cowper SE. Nephrogenic systemic fibrosis: a review and exploration of the role of gadolinium. Adv. Dermatol. 2007;23:131–154. doi: 10.1016/j.yadr.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 14.de Sousa PL, Livramento JB, Helm L, Merbach AE, Meme W, Doan BT, Beloeil JC, Prata MI, Santos AC, Geraldes CF, Toth E. In vivo MRI assessment of a novel GdIII-based contrast agent designed for high magnetic field applications. Contrast Media Mol. Imaging. 2008;3:78–85. doi: 10.1002/cmmi.233. [DOI] [PubMed] [Google Scholar]
- 15.Livramento JB, Helm L, Sour A, O'Neil C, Merbach AE, Toth E. A benzene-core trinuclear GdIII complex: towards the optimization of relaxivity for MRI contrast agent applications at high magnetic field. Dalton Trans. 2008:1195–1202. doi: 10.1039/b717390c. [DOI] [PubMed] [Google Scholar]
- 16.Livramento JB, Sour A, Borel A, Merbach AE, Toth E. A starburst-shaped heterometallic compound incorporating six densely packed gd(3+) ions. Chem. Eur. J. 2006;12:989–1003. doi: 10.1002/chem.200500969. [DOI] [PubMed] [Google Scholar]
- 17.Livramento JB, Toth E, Sour A, Borel A, Merbach AE, Ruloff R. High relaxivity confined to a small molecular space: a metallostar-based, potential MRI contrast agent. Angew. Chem. Int. Ed. Engl. 2005;44:1480–1484. doi: 10.1002/anie.200461875. [DOI] [PubMed] [Google Scholar]
- 18.Livramento JB, Weidensteiner C, Prata MI, Allegrini PR, Geraldes CF, Helm L, Kneuer R, Merbach AE, Santos AC, Schmidt P, Toth E. First in vivo MRI assessment of a self-assembled metallostar compound endowed with a remarkable high field relaxivity. Contrast Media Mol. Imaging. 2006;1:30–39. doi: 10.1002/cmmi.92. [DOI] [PubMed] [Google Scholar]
- 19.Hanaoka K, Lubag AJ, Castillo-Muzquiz A, Kodadek T, Sherry AD. The detection limit of a Gd(3+)-based T(1) agent is substantially reduced when targeted to a protein microdomain. Magn. Reson. Imaging. 2008;26:608–17. doi: 10.1016/j.mri.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McConnell HM. Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys. 1958;28:430–431. [Google Scholar]
- 21.Swift TJ, Connick RE. NMR-Relaxation Mechanisms of O[sup 17] in Aqueous Solutions of Paramagnetic Cations and the Lifetime of Water Molecules in the First Coordination Sphere. J. Chem. Phys. 1962;37:307–320. [Google Scholar]
- 22.Solomon I. Relaxation Processes in a System of Two Spins. Phys. Rev. 1955;99:559–565. [Google Scholar]
- 23.Bloembergen N, Morgan LO. Proton Relaxation Times in Paramagnetic Solutions. Effects of Electron Spin Relaxation. J. Chem. Phys. 1961;34:842–850. [Google Scholar]
- 24.Gueron M. Nuclear Relaxation in Macromolecules by Paramagnetic Ions: A Novel Mechanism. J. Magn. Reson. 1975;19:58–66. [Google Scholar]
- 25.Atsarkin VA, Demidov VV, Vasneva GA, Odintsov BM, Belford RL, Raduechel B, Clarkson RB. Direct Measurement of Fast Electron Spin-Lattice Relaxation: Method and Application to Nitroxide Radical Solutions and Gd3+ Contrast Agents. J. Phys. Chem. A. 2001;105:9323–9327. [Google Scholar]
- 26.Borel A, Helm L, Merbach AE, Atsarkin VA, Demidov VV, Odintsov BM, Belford RL, Clarkson RB. T1e in Four Gd3+ Chelates: LODEPR Measurements and Models for Electron Spin Relaxation. J. Phys. Chem. A. 2002;106:6229–6231. [Google Scholar]
- 27.Belorizky E, Fries PH. Simple analytical approximation of the longitudinal electronic relaxation rate of Gd(III) complexes in solutions. PhysChemChemPhys. 2004;6:2341–2351. [Google Scholar]
- 28.Freed JH. Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. II. Finite jumps and independent T[sub 1] processes. J. Chem. Phys. 1978;68:4034–4037. [Google Scholar]
- 29.Hwang L-P, Freed JH. Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. J. Chem. Phys. 1975;63:4017–4025. [Google Scholar]
- 30.Borel A, Helm L, Merbach AE. Molecular dynamics simulations of MRI-relevant GdIII chelates: direct access to outer-sphere relaxivity. Chem. Eur. J. 2001;7:600–610. doi: 10.1002/1521-3765(20010202)7:3<600::aid-chem600>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Greenfield MT, Spiller M, McMurry TJ, Lauffer RB, Caravan P. Multilocus binding increases the relaxivity of protein-bound MRI contrast agents. Angew. Chem. Int. Ed. Engl. 2005;44:6766–6769. doi: 10.1002/anie.200502245. [DOI] [PubMed] [Google Scholar]
- 32.Nicolle GM, Toth E, Schmitt-Willich H, Raduchel B, Merbach AE. The impact of rigidity and water exchange on the relaxivity of a dendritic MRI contrast agent. Chem. Eur. J. 2002;8:1040–1048. doi: 10.1002/1521-3765(20020301)8:5<1040::aid-chem1040>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 33.Wiener EC, Brechbiel MW, Brothers H, Magin RL, Gansow OA, Tomalia DA, Lauterbur PC. Dendrimer-based metal chelates: a new class of magnetic resonance imaging contrast agents. Magn. Reson. Med. 1994;31:1–8. doi: 10.1002/mrm.1910310102. [DOI] [PubMed] [Google Scholar]
- 34.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- 35.Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Albumin Binding, Relaxivity, and Water Exchange Kinetics of the Diastereoisomers of MS-325, a Gadolinium(III)-Based Magnetic Resonance Angiography Contrast Agent. Inorg. Chem. 2007;46:6632–6639. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- 36.Dunand FA, Toth E, Hollister R, Merbach AE. Lipari-Szabo approach as a tool for the analysis of macromolecular gadolinium(III)-based MRI contrast agents illustrated by the [Gd(EGTA-BA-(CH2)12)]nn+ polymer. J. Biol. Inorg. Chem. 2001;6:247–255. doi: 10.1007/s007750000193. [DOI] [PubMed] [Google Scholar]
- 37.Laus S, Sour A, Ruloff R, Toth E, Merbach AE. Rotational dynamics account for pH-dependent relaxivities of PAMAM dendrimeric, Gd-based potential MRI contrast agents. Chem. Eur. J. 2005;11:3064–3076. doi: 10.1002/chem.200401326. [DOI] [PubMed] [Google Scholar]
- 38.Troughton JS, Greenfield MT, Greenwood JM, Dumas S, Wiethoff AJ, Wang J, Spiller M, McMurry TJ, Caravan P. Synthesis and evaluation of a high relaxivity manganese(II)-based MRI contrast agent. Inorg. Chem. 2004;43:6313–6323. doi: 10.1021/ic049559g. [DOI] [PubMed] [Google Scholar]
- 39.Caravan P, Astashkin AV, Raitsimring AM. The gadolinium(III)-water hydrogen distance in MRI contrast agents. Inorg. Chem. 2003;42:3972–3974. doi: 10.1021/ic034414f. [DOI] [PubMed] [Google Scholar]
- 40.Astashkin AV, Raitsimring AM, Caravan P. Pulsed ENDOR Study of Water Coordination to Gd3+ Complexes in Orientationally Disordered Systems. J. Phys. Chem. A. 2004;108:1900–2001. [Google Scholar]
- 41.Raitsimring AM, Astashkin AV, Baute D, Goldfarb D, Caravan P. W-Band 17O Pulsed Electron Nuclear Double Resonance Study of Gadolinium Complexes with Water. J. Phys. Chem. A. 2004;108:7318–7323. [Google Scholar]
- 42.Raitsimring AM, Astashkin AV, Poluektov OG, Caravan P. High field pulsed EPR and ENDOR of Gd3+ complexes in glassy solutions. Appl. Magn. Reson. 2005;28:281–295. [Google Scholar]
- 43.Zech S, Sun W-C, Jacques V, Caravan P, Astashkin AV, Raitsimring AM. Probing the Water Coordination of Protein-Targeted MRI Contrast Agents by Pulsed ENDOR Spectroscopy. ChemPhysChem. 2005;6:2570–2577. doi: 10.1002/cphc.200500250. [DOI] [PubMed] [Google Scholar]
- 44.Richards S, Pedersen B, Silverton JV, Hoard JL. Stereochemistry of Ethylenediaminetetraacetato Complexes. I. The Structure of Crystalline Mn3(HY)2•10H2O and the Configuration of the Seven-Coordinate Mn(OH2)Y(−2) Ion. Inorg. Chem. 1964;3:27–33. [Google Scholar]
- 45.Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JW, Amedio JC, Jr., Looby RJ, Supkowski RM, Horrocks WD, Jr., McMurry TJ, Lauffer RB. The interaction of MS-325 with human serum albumin and its effect on proton relaxation rates. J. Am. Chem. Soc. 2002;124:3152–3162. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- 46.Aime S, Botta M, Terreno E, Anelli PL, Uggeri F. Gd(DOTP)5-outer-sphere relaxation enhancement promoted by nitrogen bases. Magn. Reson. Med. 1993;30:583–591. doi: 10.1002/mrm.1910300509. [DOI] [PubMed] [Google Scholar]
- 47.Caravan P, Greenfield MT, Li X, Sherry AD. The Gd(3+) complex of a fatty acid analogue of DOTP binds to multiple albumin sites with variable water relaxivities. Inorg. Chem. 2001;40:6580–6587. doi: 10.1021/ic0102900. [DOI] [PubMed] [Google Scholar]
- 48.Lebduskova P, Hermann P, Helm L, Toth E, Kotek J, Binnemans K, Rudovsky J, Lukes I, Merbach AE. Gadolinium(III) complexes of mono- and diethyl esters of monophosphonic acid analogue of DOTA as potential MRI contrast agents: solution structures and relaxometric studies. Dalton Trans. 2007:493–501. doi: 10.1039/b612876a. [DOI] [PubMed] [Google Scholar]
- 49.Kotek J, Lebduskova P, Hermann P, Vander Elst L, Muller RN, Geraldes CF, Maschmeyer T, Lukes I, Peters JA. Lanthanide(III) complexes of novel mixed carboxylic-phosphorus acid derivatives of diethylenetriamine: a step towards more efficient MRI contrast agents. Chem. Eur. J. 2003;9:5899–5915. doi: 10.1002/chem.200305155. [DOI] [PubMed] [Google Scholar]
- 50.Aime S, Botta M, Frullano L, Geninatti Crich S, Giovenzana G, Pagliarin R, Palmisano G, Sirtori FR, Sisti M. [GdPCP2A(H(2)O)(2)](−): a paramagnetic contrast agent designed for improved applications in magnetic resonance imaging. J. Med. Chem. 2000;43:4017–4024. doi: 10.1021/jm000983a. [DOI] [PubMed] [Google Scholar]
- 51.Toth E, Burai L, Brucher E, Merbach AE. Tuning water-exchange rates on (carboxymethyl(iminobis(ethylenenitriolo)tetraacetate (dtpa)-type gadolinium(III) complexes. Dalton Trans. 1997:1587–1594. [Google Scholar]
- 52.Lauffer RB. Targeted Relaxation Enhancement Agents for MRI. Magn. Reson. Med. 1991;22:339. doi: 10.1002/mrm.1910220237. [DOI] [PubMed] [Google Scholar]
- 53.Boxerman JL, Hamberg LM, Rosen BR, Weisskoff RM. MR contrast due to intravascular magnetic susceptibility perturbations. Magn. Reson. Med. 1995;34:555–566. doi: 10.1002/mrm.1910340412. [DOI] [PubMed] [Google Scholar]
- 54.Fulton DA, Elemento EM, Aime S, Chaabane L, Botta M, Parker D. Glycoconjugates of gadolinium complexes for MRI applications. Chem. Commun. 2006:1064–1066. doi: 10.1039/b517997a. [DOI] [PubMed] [Google Scholar]
- 55.Port M, Corot C, Raynal I, Idee JM, Dencausse A, Lancelot E, Meyer D, Bonnemain B, Lautrou J. Physicochemical and biological evaluation of P792, a rapid-clearance blood-pool agent for magnetic resonance imaging. Invest. Radiol. 2001;36:445–454. doi: 10.1097/00004424-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 56.Jebasingh B, Alexander V. Synthesis and relaxivity studies of a tetranuclear gadolinium(III) complex of DO3A as a contrast-enhancing agent for MRI. Inorg. Chem. 2005;44:9434–9443. doi: 10.1021/ic050743r. [DOI] [PubMed] [Google Scholar]
- 57.Ranganathan RS, Fernandez ME, Kang SI, Nunn AD, Ratsep PC, Pillai KMR, Zhang X, Tweedle MF. New multimeric magnetic resonance imaging agents. Invest. Radiol. 1998;33:779–797. doi: 10.1097/00004424-199811000-00002. [DOI] [PubMed] [Google Scholar]
- 58.Supkowski RM, Horrocks WD., Jr. Displacement of Inner-Sphere Water Molecules from Eu(3+) Analogues of Gd(3+) MRI Contrast Agents by Carbonate and Phosphate Anions: Dissociation Constants from Luminescence Data in the Rapid-Exchange Limit. Inorg. Chem. 1999;38:5616–5619. doi: 10.1021/ic990597n. [DOI] [PubMed] [Google Scholar]
- 59.Jocher CJ, Botta M, Avedano S, Moore EG, Xu J, Aime S, Raymond KN. Optimized relaxivity and stability of [Gd(H(2,2)-1,2-HOPO)(H2O)]- for use as an MRI contrast agent. Inorg. Chem. 2007;46:4796–4798. doi: 10.1021/ic700399p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Werner EJ, Avedano S, Botta M, Hay BP, Moore EG, Aime S, Raymond KN. Highly soluble tris-hydroxypyridonate Gd(III) complexes with increased hydration number, fast water exchange, slow electronic relaxation, and high relaxivity. J. Am. Chem. Soc. 2007;129:1870–1871. doi: 10.1021/ja068026z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aime S, Botta M, Geninatti Crich S, Giovenzana GB, Jommi G, Pagliarin R, Sisti M. Synthesis and NMR Studies of Three Pyridine-Containing Triaza Macrocyclic Triacetate Ligands and Their Complexes with Lanthanide Ions. Inorg. Chem. 1997;36:2992–3000. doi: 10.1021/ic960794b. [DOI] [PubMed] [Google Scholar]
- 62.Aime S, Calabi L, Cavallotti C, Gianolio E, Giovenzana GB, Losi P, Maiocchi A, Palmisano G, Sisti M. [Gd-AAZTA]-: a new structural entry for an improved generation of MRI contrast agents. Inorg. Chem. 2004;43:7588–7590. doi: 10.1021/ic0489692. [DOI] [PubMed] [Google Scholar]
- 63.Gianolio E, Giovenzana GB, Longo D, Longo I, Menegotto I, Aime S. Relaxometric and modelling studies of the binding of a lipophilic Gd-AAZTA complex to fatted and defatted human serum albumin. Chem. Eur. J. 2007;13:5785–5797. doi: 10.1002/chem.200601277. [DOI] [PubMed] [Google Scholar]
- 64.Overoye-Chan K, Koerner S, Looby RJ, Kolodziej AF, Zech SG, Deng Q, Chasse JM, McMurry TJ, Caravan P. EP-2104R: a fibrin-specific gadolinium-based MRI contrast agent for detection of thrombus. J. Am. Chem. Soc. 2008;130:6025–6039. doi: 10.1021/ja800834y. [DOI] [PubMed] [Google Scholar]
- 65.Schmiedl U, Ogan M, Paajanen H, Marotti M, Crooks LE, Brito AC, Brasch RC. Albumin labeled with Gd-DTPA as an intravascular, blood pool-enhancing agent for MR imaging: biodistribution and imaging studies. Radiology. 1987;162:205–210. doi: 10.1148/radiology.162.1.3786763. [DOI] [PubMed] [Google Scholar]