

Abstract
Fibroblasts do not only serve as matrix-producing reparative cells, but exhibit a wide range of functions in inflammatory and immune responses, angiogenesis and neoplasia. The adult mammalian myocardium contains abundant fibroblasts enmeshed within the interstitial and perivascular extracellular matrix. The current review manuscript discusses the dynamic phenotypic and functional alterations of cardiac fibroblasts following myocardial infarction. Extensive necrosis of cardiomyocytes in the infarcted heart triggers an intense inflammatory reaction. In the early stages of infarct healing, fibroblasts become pro-inflammatory cells, activating the inflammasome and producing cytokines, chemokines and proteases. Pro-inflammatory cytokines (such as Interleukin-1) delay myofibroblast transformation, until the wound is cleared from dead cells and matrix debris. Resolution of the inflammatory infiltrate is associated with fibroblast migration, proliferation, matrix protein synthesis and myofibroblast conversion. Growth factors and matricellular proteins play an important role in myofibroblast activation during the proliferative phase of healing. Formation of a mature cross-linked scar is associated with clearance of fibroblasts, as poorly-understood inhibitory signals restrain the fibrotic response. However, in the non-infarcted remodeling myocardium, local fibroblasts may remain activated in response to volume and pressure overload and may promote interstitial fibrosis. Considering their abundance, their crucial role in cardiac inflammation and repair, and their involvement in myocardial dysfunction and arrhythmogenesis, cardiac fibroblasts may be key therapeutic targets in cardiac remodeling.
Keywords: fibroblast, inflammation, myocardial infarction, cytokine, cardiac remodeling, extracellular matrix, Transforming growth factor (TGF)-β
1. Introduction
Fibroblasts are mesenchymal cells, abundantly distributed in connective tissues of most organs. Although traditionally viewed as matrix-producing cells that become activated following injury and participate in scar formation, fibroblasts have a diverse range of functions and exhibit remarkable plasticity, undergoing dynamic phenotypic alterations in response to changes in their microenvironment. Thus, the role of fibroblasts may extend beyond their contribution to scar formation and matrix remodeling. Experimental studies have suggested important fibroblast-mediated actions in regulating inflammation [1], in modulating oncogenic potential [2] and in stimulating angiogenesis [3]. Unfortunately, the lack of reliable tools for fibroblast-specific gene targeting has hampered efforts to understand the role of fibroblasts in tissue homeostasis and in various pathologic conditions.
The adult myocardium contains a large population of quiescent fibroblasts, enmeshed into the interstitial and perivascular matrix [4]. Due to their abundance, their strategic location and their potential for activation, cardiac fibroblasts may serve as sentinel cells that sense myocardial injury and trigger inflammatory and reparative responses. Because the adult mammalian heart has negligible regenerative capacity, cardiac repair following sudden loss of a large number of cardiomyocytes is dependent on the clearance of dead cells and on the formation of a collagen-based scar. Thus, repair of the infarcted heart requires timely activation of an inflammatory cascade to debride the wound from dead cells and matrix fragments, followed by induction of matrix-preserving signals that induce deposition of extracellular matrix. Tight temporal and spatial regulation of inflammatory and fibrogenic pathways is needed to prevent overactive responses that may accentuate injury and promote adverse remodeling and dysfunction. Fibroblasts undergo dynamic phenotypic changes following myocardial infarction and are capable of regulating the inflammatory and reparative cascade. Our review manuscript discusses the origin of fibroblasts in the healing infarct, the molecular signals responsible for fibroblast activation in the healing infarct and their involvement in repair and remodeling of the infarcted heart. Moreover, we identify potential therapeutic targets that may hold promise for treatment of patients with heart failure by interfering with fibroblast function.
2. Fibroblasts in cardiac homeostasis
Early experimental studies using scanning and transmission electron microscopy as well as gradient centrifugation have suggested that fibroblasts may outnumber cardiomyocytes in adult mammalian hearts [5], [6]. However, it is now appreciated that the relative numbers of cardiomyocytes and non-cardiomyocytes in the myocardium are likely dependent on the species studied, on the age, gender and genetic background of the subjects, and on the technique and marker used for fibroblast identification [5], [6], [7], [8]. Recent investigations using fluorescence activated cell sorting (FACS) analysis demonstrated that the adult murine heart consists of 56% myocytes and 27% fibroblasts [7]. Although fibroblasts are the predominant interstitial cells in normal mammalian myocardium, their function in cardiac homeostasis remains poorly understood. During cardiac development, embryonic fibroblasts may promote cardiomyocyte proliferation through interactions involving β1 integrin signaling [9]. In the absence of injury, cardiac fibroblasts remain quiescent, and are presumably shielded from mechanical stress by the stable interstitial extracellular matrix network. As matrix-producing cells, fibroblasts may be responsible for preservation of the normal interstitial matrix. Loss of the transcription factor TCf21 in mice results in failure to develop a cardiac fibroblast population and is associated with decreased myocardial expression of collagens, highlighting the important role of fibroblasts in generating and maintaining the structure of the cardiac interstitium [10]. Moreover, due to their close association with cardiomyocytes, fibroblasts may transduce survival signals, or may regulate the transmission of mechanical and electrical stimuli, thus contributing to normal systolic and diastolic function of the ventricle. Cardiac fibroblasts exhibit abundant expression of connexins in vivo and form highly coupled fibroblast:fibroblast and fibroblast:cardiomyocyte networks [11]. Moreover, as mechanosensitive cells, fibroblasts may function as independent sensors of alterations in the mechanical environment.
3. Repair and remodeling of the infarcted heart: temporal and spatial considerations
Because fibroblasts promptly respond to alterations in their microenvironment, understanding their phenotypic changes in the infarct requires knowledge of the pathology of cardiac repair. Healing of the infarcted heart can be divided in three distinct, but overlapping phases: the inflammatory phase, the proliferative phase and the maturation phase [12]; each phase is associated with distinct fibroblast phenotypes. Massive necrosis of cardiomyocytes in the infarcted heart triggers the inflammatory phase, which is characterized by activation of innate immune signals that induce cytokine and chemokine expression causing marked infiltration of the infarct with neutrophils and mononuclear cells. Infiltrating leukocytes clear the infarct from dead cells and matrix debris [13]. In the healing wound the inflammatory reaction is programmed to resolve: as neutrophils undergo apoptosis, they are phagocytosed by macrophages that secrete potent suppressors of inflammation, including Transforming Growth Factor (TGF)-β, Interleukin (IL)-10 and proresolving lipid mediators (such as the lipoxins, resolvins, protectins and maresins) [14]. Repression of pro-inflammatory signals and induction of matrix-preserving mediators that activate mesenchymal cells mark the transition to the proliferative phase of infarct healing. At this stage the infarct is infiltrated with abundant fibroblasts and vascular cells; activated myofibroblasts secrete matrix proteins and form the scar (Figure 1). Activation of poorly understood STOP signals inhibits the fibrotic and angiogenic response preventing expansion of fibrosis, and leads to the maturation phase of infarct healing, as the cellular elements undergo apoptosis, and a mature scar comprised of cross-linked collagen is formed.
Figure 1.
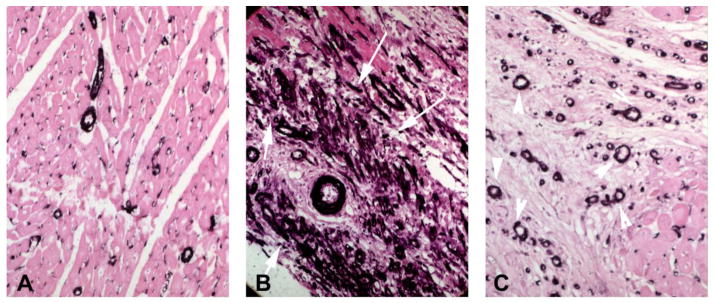
Myofibroblasts in healing myocardial infarction. A–C: Immunohistochemical staining for α-smooth muscle actin (SMA) in control canine myocardium (A) and in infarcted hearts (B – 1h coronary occlusion/7days reperfusion; C – 1h occlusion/28days reperfusion). In control hearts, α-SMA is expressed exclusively by vascular mural cells. After 7 days of reperfusion, abundant spindle-shaped α-SMA+ myofibroblasts are noted in the infarct border zone (arrows). After 28 days of reperfusion, border zone myofibroblasts are markedly reduced and α-SMA immunoreactivity is localized in vascular mural cells (arrowheads).
As the infarct heals, the ventricle undergoes geometric and functional changes, collectively termed “post-infarction ventricular remodeling” [15]. Hypertrophy of the non-infarcted segments, and dilation and increased sphericity of the chamber are the major geometric alterations observed in the remodeling infarcted heart. In human patients, remodeling of the infarcted heart carries important prognostic information and is associated with increased mortality, a high incidence of arrhythmias and heart failure. Infarct size is a major determinant of adverse remodeling (as larger infarcts are generally associated with worse remodeling); however, the severity of post-infarction remodeling is also dependent on ventricular loading conditions and on the qualitative characteristics of the healing wound. For example, prolonged activation of post-infarction inflammation increases protease activity and is associated with enhanced dilative remodeling [16], whereas increased matrix deposition results in a stiffer ventricle and causes diastolic dysfunction [17]. Activation of cardiac fibroblasts in the infarct border zone and in the non-infarcted myocardium may play an important role in the pathogenesis of post-infarction remodeling.
4. Fibroblasts during the inflammatory phase of healing
4.1. Are fibroblasts key inflammatory cells in the infarcted myocardium?
Twenty to thirty minutes of severe ischemia is sufficient to induce irreversible cardiomyocyte injury. Thus, prolonged cessation of blood flow in myocardial infarction causes extensive necrosis of cardiomyocytes in the area at risk. In contrast, interstitial non-cardiomyocytes are much less susceptible to ischemic injury. Because of their resistance to ischemic death, their wide distribution in the cardiac interstitium, their interaction with cardiomyocytes and their potential as sources of pro-inflammatory mediators, fibroblasts are ideally suited as sentinel cells that may sense myocardial injury, triggering an inflammatory reaction (Figure 2). Although the inflammatory potential of cardiac fibroblasts is well-documented in vitro [18], [19], [20], their relative contribution in activation of the post-infarction inflammatory cascade remains unknown. Dissection of the potential role of fibroblasts as inflammatory cells is hampered by two major problems. First, several other cell types, including endothelial cells [21] and resident cardiac mast cells [22] have been proposed as effector cells in triggering the post-infarction inflammatory cascade and are also capable of secreting a wide range of inflammatory mediators. Second, in vivo studies investigating the involvement of fibroblast-specific inflammatory signaling using conditional targeting approaches are limited by the absence of specific and reliable markers for cardiac fibroblasts [23]. The intermediate filament protein vimentin has been used as a fibroblast marker in both normal and injured myocardium; however, other cells of mesenchymal origin (including vascular endothelial and smooth muscle cells) also express vimentin. α-Smooth muscle actin (α-SMA), a marker for transdifferentiated myofibroblasts in the infarcted and remodeling myocardium, is also abundantly expressed in smooth muscle cells. The calcium binding protein S100A4/Fibroblast-specific protein (FSP)-1 has been widely used for fibroblast-specific gene disruption, but lacks specificity and is highly expressed by hematopoietic and vascular cells [23]. The matricellular protein periostin is selectively upregulated in activated fibroblasts infiltrating the infarcted and pressure-overloaded heart and may be a promising marker for labeling activated fibroblasts following cardiac injury [24], but does not identify the abundant fibroblast population in normal hearts [23]. Thus, periostin-Cre mice may be a useful tool to study the role of specific molecular signals in activated fibroblasts infiltrating sites of injury [25].
Figure 2.

During the early stages following myocardial infarction cardiac fibroblasts become inflammatory cells. Infarct fibroblasts activate the inflammasome, a multimolecule complex that cleaves pro-IL-1β leading to secretion of the active cytokine. Reactive oxygen species (ROS), cytokines (such as IL-1β and TNF-α) and matrix fragments induce fibroblast inflammatory activation and promote a matrix-degrading phenotype. IL-1β suppresses α-smooth muscle actin expression delaying myofibroblast conversion.
As a result of these limitations, most of the evidence implicating fibroblasts in the post-infarction inflammatory response is descriptive. In mouse models of reperfused and non-reperfused infarction, fibroblasts exhibit activation of the inflammasome [26], [27] the molecular platform involved in caspase activation and in generation of active IL-1β. In vitro, activation of the inflammasome in cardiac fibroblasts is mediated through reactive oxygen species (ROS) production and potassium efflux. Moreover, cardiac fibroblasts release large amounts of pro-inflammatory cytokines, when stimulated with ATP [28].
4.2. Activators of inflammatory signaling in infarct fibroblasts
Regardless of the relative roles of various cell types in triggering post-infarction inflammation, the pro-inflammatory environment of the early infarct profoundly affects fibroblast phenotype and function. During the first 24–72h following infarction, cardiac fibroblasts in the infarcted heart, acquire a pro-inflammatory phenotype secreting cytokines and chemokines [27] and exhibit matrix-degrading properties. Several pathways may activate inflammatory signaling in infarct fibroblasts. First, ROS may induce pro-inflammatory signals in cardiac fibroblasts, while decreasing extracellular matrix protein synthesis, and enhancing matrix metalloproteinase (MMP) activity [29]. However, experimental in vitro studies do not consistently demonstrate matrix-degrading effects of ROS on cardiac fibroblasts. ROS stimulation enhances collagen synthesis in rat cardiac fibroblasts [30] and may stimulate matrix-preserving pathways by activating TGF-β responses [31]. Second, cytokine stimulation may play a key role in fibroblast activation during the inflammatory phase of cardiac repair. In vitro, IL-1β, Tumor Necrosis Factor (TNF)-α and oncostatin-M promote an inflammatory phenotype in cardiac fibroblasts inducing cytokine and chemokine synthesis [32], [20], [19]. Cytokines also regulate synthesis of extracellular matrix proteins and modulate matrix metabolism by inducing expression of matrix-degrading proteases [33], [34]. IL-1 appears to be a particularly important regulator of fibroblast function in the healing infarct. IL-1β is markedly induced in the infarcted myocardium [35], [36], [37] and mediates inflammatory signaling, while promoting adverse remodeling through protease induction and activation [38], [39]. Recent experiments from our laboratory suggested that IL-1β inhibits conversion of fibroblasts into myofibroblasts in the healing infarct [40]. IL-1β may also inhibit fibroblast proliferation [41], by modulating expression of fibroblast cyclins, cyclin-dependent kinases and their inhibitors [42]. In the early infarct, the anti-fibrotic actions of IL-1 signaling may prevent premature transformation of fibroblasts into matrix-producing cells, until the wound is cleared from dead cells and matrix fragments. Whether other pro-inflammatory signals (such as TNF-α and oncostatin) also regulate infarct fibroblast phenotype in vivo remains unknown. In vitro experiments have demonstrated that, in addition to its pro-inflammatory actions, TNF-α may indirectly favor fibrosis by stimulating upregulation of type 1 angiotensin II (AT1) receptors [43]. These actions highlight the multifaceted effects of some pro-inflammatory cytokines that may promote acute inflammation, while setting the stage for fibrotic responses.
4.3. Resolution of inflammation in cardiac repair. A role for the fibroblasts?
Healing of the infarcted heart is dependent on timely suppression of the inflammatory reaction [13]. Repression of inflammatory gene expression and resolution of inflammation following infarction do not simply reflect cessation of the effects of pro-inflammatory signals, but require activation of endogenous inhibitory pathways that suppress inflammation. Soluble mediators, such as TGF-β and IL-10, and activation of intracellular STOP signals that inhibit innate immune responses (such as Interleukin Receptor Associated Kinase-M) [44] have been implicated in suppression, resolution and containment of the post-infarction inflammatory reaction. Involvement of other mediators (such as pro-resolving lipids) is speculated on the basis of their biological properties [14], but has not been documented by experimental studies. Although all cells participating in cardiac repair are likely involved in suppression and containment of the post-inflammatory reaction, the key cellular effectors for this transition have not been identified. Inhibitory monocyte and lymphocyte subsets and polarized macrophages are ideally suited as negative regulators of the post-infarction inflammatory reaction. Whether fibroblasts become anti-inflammatory cells in the healing infarct has not been established. However, considering their dynamic phenotypic changes during the proliferative phase of healing, their responsiveness to mediators that suppress inflammation, their abundance and strategic location in the infarct border zone, fibroblasts may be important cellular effectors of suppression and containment of post-infarction inflammation.
5. Fibroblasts during the proliferative phase
During the proliferative phase of healing, fibroblasts become the dominant cell type in the infarcted myocardium and undergo dramatic phenotypic changes (Figure 3). Removal of pro-inflammatory signals such as IL-1β and Interferon-γ-inducible Protein (IP)-10, allows unopposed growth factor signaling in cardiac fibroblasts, promoting a matrix-preserving, proliferative myofibroblast phenotype. High proliferative activity, migration through the provisional matrix network of the infarct, expression and incorporation of contractile proteins in the cytoskeleton and synthesis of both structural and matricellular extracellular matrix proteins are the main characteristics of the fibroblasts during the proliferative phase.
Figure 3.

During the proliferative phase of cardiac repair, fibroblasts undergo myofibroblast transdifferentiation expressing contractile proteins and secreting large amounts of matrix proteins. Growth factors, the renin-angiotensin-aldosterone axis, and the mast cell-derived proteases tryptase and chymase are implicated in myofibroblast transformation and proliferation, and stimulate matrix protein synthesis. Specialized matrix proteins contribute to fibroblast activation.
5.1. Border zone myofibroblasts: the main matrix-synthetic cells in the healing infarct
Transition into the proliferative phase of infarct healing is characterized by infiltration of the infarct border zone with activated myofibroblasts, phenotypically modulated fibroblasts that express contractile proteins and exhibit an extensive endoplasmic reticulum, thus secreting large amounts of matrix proteins [45], [46], [47]. Extensive infiltration of the infarct with myofibroblasts is a consistent feature of the reparative response in all experimental models of myocardial infarction. Rat [48], [47], mouse [36], rabbit [49], and canine [50] myocardial infarcts exhibit large numbers of myofibroblasts in the border zone; myofibroblast infiltration is a prominent characteristic in human myocardial scars [51]. Moreover, myofibroblasts have been identified following myocardial injury, even in non-mammalian species that exhibit extensive cardiac regeneration, such as the zebrafish [52]. Although synthesis of α-smooth muscle actin (SMA) is considered the defining characteristic of differentiated myofibroblasts, infarct myofibroblasts also synthesize other contractile proteins (such as non-muscle myosin), but do not express smooth muscle cell-specific proteins, such as smoothelin and the smooth muscle myosin isoforms SM1 and SM2 [50], [53]. In reparative responses, early myofibroblasts may lack α-SMA expression, but have stress fibers containing cytoplasmic actins, and develop mature focal adhesions comprised of β- and γ-actin microfilaments, associated with nonmuscle myosin: these cells are called “proto-myofibroblasts” [54]. Although these cells may represent an intermediate step in the myofibroblast conversion process, α-SMA-negative cells with characteristics of proto-myofibroblasts have not yet been identified in the infarcted myocardium.
5.2. Where do infarct myofibroblasts come from?
Experimental studies provide evidence for several distinct sources of myofibroblasts in the infarcted myocardium. Resident interstitial fibroblasts undergo proliferation and activation [55], [56] in response to the local release of growth factors and represent a large pool of cells capable of myofibroblast transdifferentiation following infarction [57]. Hematopoietic progenitors and endothelial cells have also been proposed as potentially important sources of myofibroblasts in models of infarctive and non-infarctive cardiac fibrosis. In a mouse model of fibrotic interstitial cardiomyopathy induced by brief repetitive ischemia and reperfusion, recruitment of fibroblast progenitors of hematopoietic origin has been documented [58], [59]. In an infarction model, experiments using bone marrow transplantation with enhanced GFP (eGFP)-labeled cells showed infiltration with numerous bone marrow-derived fibroblasts [60]. Experiments using Tie1Cre; R26RstoplacZ mice, in which endothelial cells and their descendants are marked by LacZ, and Fibroblast Specific Protein-1-GFP transgenic mice to label fibroblasts were used to demonstrate that a significant number of cardiac myofibroblasts derive from endothelial cells in models of cardiac fibrosis due to pressure overload or chronic allograft rejection [61]. Unfortunately, because FSP-1 is not a specific fibroblast marker [23], but is abundantly expressed by macrophages and endothelial cells in remodeling hearts, these findings provide no conclusive evidence on the endothelial origin of fibroblasts. Pericytes, smooth muscle cells and epicardial epithelial cells may serve as alternative sources of fibroblasts [62]; however, their relative contribution in models of cardiac injury remains poorly characterized. Although systematic studies examining the source of myofibroblasts in the infarct are lacking, considering their abundance in the cardiac interstitium, resident fibroblasts may be the most important source.
5.3. Molecular signals mediating myofibroblast transdifferentiation in the healing infarct
In the early stages of infarct healing, activation of IL-1 signaling inhibits α-SMA expression by fibroblasts and may delay myofibroblast conversion until the infarct environment is cleared by dead cells and matrix debris. During the proliferative phase, suppression of IL-1 synthesis and activation of pathways that activate α-SMA transcription in cardiac fibroblasts, induce myofibroblast transdifferentiation in the infarct border zone. Several distinct pathways cooperate for the generation of α-SMA-positive differentiated myofibroblasts in the infarct:
TGF-β is induced and activated in the infarcted myocardium and induces α-SMA transcription in fibroblasts by activating Smad-dependent signaling [63] and by mediating translocation of the myocardin-related transcription factor (MRTF)-A into the nucleus [64]. In addition to the established role of canonical TGF-β signaling, activation of non-canonical pathways, such as p38 mitogen-activated protein kinase (MAPK), may also play a role in myofibroblast conversion [65], [66] (Figure 3)
Modulation of the matrix environment plays an important role in myofibroblast conversion. Expression of specialized matrix proteins, such as the ED-A fibronectin variant induces formation of differentiated α-SMA-expressing myofibroblasts [54], [67], [68]. Moreover, deposition of non-fibrillar collagens (such as collagen VI) may also promote myofibroblast transdifferentiation in the infarcted heart [69] (Figure 3).
Expression of proteoglycans by cardiac fibroblasts may be critical for transduction of the growth factor-mediated signals that activate myofibroblast conversion [70] (Figure 3).
Mechanosenstive signaling directly stimulates α-SMA transcription in fibroblasts through activation of Rho/Rho kinase cascades [71]; however, these pathways likely require TGF-β to induce myofibroblast transdifferentiation. Quiescent cardiac fibroblasts in uninjured hearts are protected from mechanical stimuli by a stable matrix network; however, disruption of the structural integrity of the myocardial matrix following infarction may expose interstitial cells to mechanical stress, thus contributing to their conversion into myofibroblasts [54] (Figure 3).
5.4. Fibroblast proliferation in the infarcted myocardium
Intense proliferative activity has been documented in fibroblasts infiltrating the infarcted heart [56], [55]; however, the molecular signals involved in proliferation of infarct fibroblasts remain poorly understood. Several growth factors released by a variety of cells (including macrophages, mast cells and cardiomyocytes) have been implicated in stimulation of the proliferative potential of fibroblasts. Fibroblast growth factor (FGF)-2 exerts potent proliferative effects on infarct fibroblasts [72]. Angiotensin II, Platelet-Derived Growth Factors (PDGF) and the mast cell-derived proteases tryptase and chymase are potentially important activators of proliferative responses in fibroblasts [73],[74]; however their relative role in myocardial infarction remains unknown (Figure 3). The intracellular signaling pathways implicated in fibroblast proliferation following infarction are also poorly defined and may involve activation of protein kinase C, MAPK [73], and β-catenin signaling [75]. In contrast, activation of the Smad3 cascade appears to transduce an anti-proliferative response [63].
5.5. Migration of fibroblasts into the infarct
Moreover, in the dynamic environment of the healing infarct, directed migration of fibroblasts in areas where dead cardiomyocytes have been cleared is important for optimal repair. During the inflammatory phase of infarct healing chemokines provide key molecular signals for recruitment of inflammatory cells [76]; however, whether specific chemokine/chemokine receptor interactions direct fibroblast motility remains unknown. The CC chemokine Monocyte Chemoattractant Protein (MCP)-1/CCL2 is critically involved in the pathogenesis of cardiac fibrosis [58]; however, it is unclear whether its pro-fibrotic actions are mediated through recruitment of fibroblast progenitors, or predominantly reflect its actions on fibrogenic monocytes. Several other mediators have been implicated as regulators of the fibroblast migratory response in the infarcted heart. Leukotrienes, generated through 5-lipoxygenase (5-LOX)-mediated actions may act, at least in part, by promoting fibroblast migration [77]. Cytokines (such as IL-1 and cardiotrophin-1) [78], [79], and growth factors (such as TGF-β and FGFs) [63], may also induce fibroblast migration into the site of infarction. Movement of fibroblasts in the highly plastic and dynamic environment of the infarct requires adhesive interactions of the fibroblasts with the surrounding matrix that may involve integrin signaling. Fibroblast expression of matrix-degrading proteases and deposition of matricellular proteins (such as the thrombospondins and tenascin-C) into the infarct matrix are essential requirements for acquisition of a migratory phenotype [80], [81].
In addition to pro-migratory pathways, inhibitory signals that attenuate fibroblast migration are also activated in the infarcted myocardium, presumably acting to prevent an overactive, or expanded, fibrotic response. The CXC chemokine CXCL10/Interferon-γ-inducible protein (IP)-10 is upregulated in the infarcted myocardium and inhibits growth factor-induced fibroblast migration preventing excessive fibrotic remodeling of the infarcted heart [82].
5.6. Infarct myofibroblasts as modulators of the extracellular matrix
Extensive evidence demonstrates that myofibroblasts are the main source of extracellular matrix proteins in healing myocardial infarcts [83], [84]. In the growth factor-rich environment of the healing infarct, myofibroblasts secrete large amounts of structural matrix proteins (such as collagens and fibronectin), but also deposit matricellular proteins and modulate matrix metabolism by expressing MMPs and their inhibitors. Although several mediators have been implicated in acquisition of a synthetic phenotype by cardiac fibroblasts, their relative importance in the healing infarct remains poorly understood. Experimental studies using pharmacologic inhibitors and genetic loss-of-function strategies suggest that the angiotensin II pathway increases the matrix synthetic capacity of infarct myofibroblasts through actions transduced via AT1 receptors [85]. Mineralocorticoid receptor signaling also induces expression of matrix proteins in cardiac fibroblasts [86]. The pro-fibrotic actions of angiotensin II may be mediated, at least in part, through activation of growth factors, such as TGF-β, FGF-2 and PDGF[87],[74]. TGF-β potently activates a pro-fibrotic and matrix-preserving program in infarct fibroblasts through Smad-dependent actions [63],[17],[88]. The potential role of TGF-β induced Smad-independent signaling in fibrotic remodeling of the infarcted heart is currently unclear.
5.7. Matricellular proteins as key mediators in modulation of fibroblast function
The composition of the extracellular matrix plays a crucial role in regulating the dynamic functional and phenotypic alterations of the fibroblasts during the proliferative phase of healing. Induction of matricellular proteins, a family of structurally unrelated macromolecules that bind to the matrix and do not serve a structural role, but modulate cellular responses by transducing growth factor and cytokine actions, is a key event in the infarcted myocardium. Several members of the matricellular family, including TSP-1, TSP-2, osteopontin, SPARC, periostin, tenascin-C and CCN2 are important modulators of fibroblast phenotype and play an essential role in modulating growth factor signals. The matricellular interactions that regulate cellular phenotype in the remodeling infarcted heart have been recently reviewed in detail [81].
6. The fate of the myofibroblasts during infarct maturation
Formation of a collagen-based matrix marks the end of the proliferative phase and sets the stage for maturation of the scar, a poorly understood process that is characterized by matrix cross-linking and progressive loss of the cellular elements. Myofibroblast density is markedly reduced in mature infarcts; cellular depletion is accentuated and accelerated in mouse models of infarction, where large areas of myocardium are replaced by thin strips of collagenous tissue [37]. Although apoptosis has been identified as a potential mechanism for removal of fibroblasts from the mature scar, the molecular signals that determine the fate of the cells in the infarcted myocardium remain unknown [49]. Withdrawal of growth factors and clearance of matricellular proteins from the mature scar may play an important role in fibroblast deactivation and apoptosis, by depriving infarct myofibroblasts from key signals that promote their survival and stimulate their synthetic activity [89]. However, inhibition of scar formation may also involve active STOP signals that suppress the fibrotic response, inhibiting growth factor mediated fibroblast activation. For example, negative regulation of the TGF-β signaling cascade involves several distinct mechanisms including phosphatases that terminate Smad signaling, expression of pseudoreceptors and activation of inhibitory Smads. A recent investigation demonstrated a role for the TGF-β pseudoreceptor BAMBI in restraining the TGF-β-driven fibrotic response in the pressure overloaded myocardium [90]; however, the significance of these inhibitory pathways following infarction remains unknown.
Alterations in the composition of the matrix may be crucial in promoting fibroblast quiescence during the maturation phase. Certain specialized matrix components, such as the small leucin-rich proteoglycan biglycan, inhibit infarct myofibroblast transdifferentiation [91]; however, whether such mechanisms participate in conversion of myofibroblasts into quiescent fibroblasts during scar maturation remains unknown. The stable mechanical environment of the mature cross-linked scar may transduce signals that deactivate infarct myofibroblasts.
7. The fibroblasts in the remodeling non-infarcted myocardium. Dynamic effectors in heart failure?
While large numbers of infarct myofibroblasts undergo apoptosis, the remote remodeling non-infarcted myocardium is subjected to pressure and volume overload; both pathophysiologic conditions expected to cause chronic activation of the local fibroblast population. Studies systematically examining the characteristics of fibroblasts in the remodeling non-infarcted myocardium are lacking. However, insights derived from experimental animal models suggest that pressure and volume overload have distinct effects on interstitial fibroblasts. Pressure overload induces early activation of matrix synthetic pathways, associated with fibrosis and diastolic dysfunction, followed by activation of matrix-degrading signals, chamber dilation and decompensated heart failure [92]. In contrast, volume overload is predominantly associated with matrix loss and cardiac dilation [93]. The basis for these distinct effects remains unclear. Moreover, how these co-existing pathophysiologic alterations affect the phenotype of cardiac fibroblasts in the remodeling segments of the infarcted heart remains unknown.
8. Do activated myofibroblasts contribute to arrhythmogenesis following myocardial infarction?
A growing body of evidence suggests that cardiac fibroblasts may be involved in the generation and propagation of arrhythmias [94], [95]. Infiltration of the infarcted myocardium with abundant myofibroblasts may alter cardiac electrophysiology by creating a barrier that blocks propagation of the electrical impulse, delaying conduction and promoting formation of reentry circuits [95]. In addition to these mechanical effects, electrical coupling between fibroblasts and cardiomyocytes may promote slow electrotonic conduction through fibrotic regions [96]. The dynamic phenotypic alterations of the fibroblast population following infarction may profoundly influence arrhythmogenesis through several distinct mechanisms. First, secretion of pro-inflammatory mediators by cardiac fibroblasts may exert paracrine effects on cardiomyocytes modulating conduction. Second, induction of connexin43 and connexin45 in infarct fibroblasts exhibit spatially and temporally distinct patterns [97] and may be responsible for dynamic changes in electrical coupling between fibroblasts and cardiomyocytes in the healing infarct. Third, incorporation of α-SMA in the fibroblast cytoskeleton contributes to their arrhythmogenicity [98]; thus, conversion of fibroblasts into myofibroblasts during the proliferative phase may be associated with generation of arrhythmias. The in vivo role of fibroblast modulation in arrythmogenesis remains poorly understood.
9. Fibroblasts as therapeutic targets in myocardial infarction
Because remodeling of the infarcted heart is dependent on the mechanical properties of the scar, the effects of fibroblasts in the infarcted and remodeling myocardium have important functional implications. As the main cellular effectors in matrix remodeling, and as important modulators of the inflammatory and reparative response, fibroblasts are promising therapeutic targets. The actions of certain pharmacologic approaches with well-documented benefit in patients with myocardial infarction may be mediated, at least in part, through effects on fibroblasts. Treatment with angiotensin-converting enzyme inhibitors and AT1 blockers reduces mortality and has protective effects on the development of heart failure; these effects are associated with attenuated cardiac fibrosis [99]. Aldosterone antagonism in patients with acute anterior myocardial infarction attenuates remodeling, reducing plasma levels of pro-fibrotic markers [100]. However, because these approaches have multiple other effects on other cell types, the extent to which their antifibrotic actions are responsible for the observed clinical benefit is unknown.
Animal model studies have identified promising new approaches that may prevent adverse remodeling following myocardial infarction by targeting fibroblast functions. IL-1 inhibition using anakinra may attenuate dilative post-infarction remodeling by limiting fibroblast-mediated inflammatory and matrix-degrading activity [101]. On the other hand, approaches interfering with fibrogenic growth factor-mediated cascades, such as TGF-β1/Smad3 signaling or FGF-2 may hold promise in prevention of fibrotic cardiac remodeling [102], [17] and in attenuation of diastolic heart failure. However, experimental animal studies suggest caution when implementing strategies that interfere with matrix metabolism following myocardial infarction. Excessive matrix deposition in the infarcted heart increases chamber stiffness causing diastolic dysfunction. In contrast, exaggerated matrix degradation due to overactive inflammatory response and increased protease activation, induces ventricular dilation and is associated with systolic dysfunction. Patients who survive an acute myocardial infarction are pathophysiologically diverse. Age, gender, genetic predispositions, the presence of comorbid conditions such as dyslipidemia, hypertension and metabolic disease, and the use of medications that may interfere with fibrogenic signaling have important effects on fibroblast responses. For example, aging is associated with an impaired reparative reserve and with poor responsiveness of fibroblasts to growth factors [103]. In contrast, in diabetic patients, chronic hyperglycemia may accentuate growth factor signaling leading to increased fibrosis and development of diastolic heart failure. Considering the pathophysiologic heterogeneity of patients with myocardial infarction, use of biomarker-based treatment strategies may lead to development of mechanism-guided therapeutic approaches for treatment of patients with myocardial infarction [104], [105]. Thus, measurement of inflammatory markers (such as MCP-1) may identify patients with prominent, accentuated or prolonged inflammatory responses who may benefit from anti-inflammatory approaches (such as anakinra). On the other hand, individuals with overactive TGF-β responses may be identified through biomarkers that reflect matrix synthesis and may benefit from Smad inhibition strategies. In addition to its effects on cardiac remodeling, modulation of fibroblasts may also hold promise in management of infarct-related arrhythmias. Experimental evidence suggests that stress fibers containing α-SMA contribute to the arrhythmogenic potential of myofibroblasts. Thus, modulation of the myofibroblast cytoskeleton may have anti-arrhythmic effects [98].
10. Conclusions
Due to their abundance, strategic location, phenotypic plasticity and ability to secrete a wide range of inflammatory mediators, reparative growth factors, proteases, structural extracellular matrix proteins and matricellular macromolecules, cardiac fibroblasts are ideally suited as key effector cells in healing infarcts. Unfortunately, our understanding of their in vivo role is hampered by the absence of reliable and specific fibroblast markers and by challenges in developing fibroblast-specific gene targeting approaches. To enhance our knowledge on the cell biological effects of fibroblasts in the injured and remodeling heart, significant advances are needed in several important areas. First, identification of new and specific markers of cardiac fibroblasts and cell biological characterization of infarct fibroblast subpopulations with distinct functional properties are needed to understand the biology of the fibrotic response. Second, in vivo dissection of the specific signaling pathways involved in fibroblast activation following infarction will enhance our understanding of their role in cardiac remodeling identifying possible therapeutic targets. Third, systematic study of the fate of myofibroblasts in the healing infarct is needed to understand the endogenous STOP signals that may inhibit their activation restraining the fibrotic response. Finally, understanding the relation between phenotypic modulation of fibroblasts and cardiac function is crucial in order to gain pathophysiologic insights and to design specific strategies for attenuation of post-infarction remodeling.
Highlights.
The adult mammalian heart contains abundant cardiac fibroblasts.
Following myocardial infarction, cardiac fibroblasts undergo dynamic phenotypic changes.
During the proliferative phase fibroblasts undergo myofibroblast conversion.
As the scar matures, myofibroblasts become quiescent and may undergo apoptosis.
Infarct myofibroblasts are implicated in cardiac dysfunction and arrhythmogenesis.
Acknowledgments
Dr Frangogiannis’ laboratory is supported by NIH grants R01 HL76246 and R01 HL85440 and the Wilf Family Cardiovascular Research Institute.
Footnotes
DISCLOSURES: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith RS, Smith TJ, Blieden TM, Phipps RP. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am J Pathol. 1997;151:317–22. [PMC free article] [PubMed] [Google Scholar]
- 2.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 3.Ito TK, Ishii G, Chiba H, Ochiai A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene. 2007;26:7194–203. doi: 10.1038/sj.onc.1210535. [DOI] [PubMed] [Google Scholar]
- 4.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–76. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- 6.Zak R. Development and proliferative capacity of cardiac muscle cells. Circ Res. 1974;35(suppl II):17–26. [PubMed] [Google Scholar]
- 7.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883–91. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 8.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 9.Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, et al. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev Cell. 2009;16:233–44. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–49. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res. 2004;94:828–35. doi: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]
- 12.Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8:1907–39. doi: 10.1089/ars.2006.8.1907. [DOI] [PubMed] [Google Scholar]
- 13.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–73. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. 2010;177:1576–91. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–72. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 16.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–87. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, et al. Essential Role of Smad3 in Infarct Healing and in the Pathogenesis of Cardiac Remodeling. Circulation. 2007;116:2127–38. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 18.Heim A, Zeuke S, Weiss S, Ruschewski W, Grumbach IM. Transient induction of cytokine production in human myocardial fibroblasts by coxsackievirus B3. Circ Res. 2000;86:753–9. doi: 10.1161/01.res.86.7.753. [DOI] [PubMed] [Google Scholar]
- 19.Zymek P, Nah DY, Bujak M, Ren G, Koerting A, Leucker T, et al. Interleukin-10 is not a critical regulator of infarct healing and left ventricular remodeling. Cardiovasc Res. 2007;74:313–22. doi: 10.1016/j.cardiores.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lafontant PJ, Burns AR, Donnachie E, Haudek SB, Smith CW, Entman ML. Oncostatin M differentially regulates CXC chemokines in mouse cardiac fibroblasts. Am J Physiol Cell Physiol. 2006;291:C18–26. doi: 10.1152/ajpcell.00322.2005. [DOI] [PubMed] [Google Scholar]
- 21.Frangogiannis NG, Youker KA, Rossen RD, Gwechenberger M, Lindsey MH, Mendoza LH, et al. Cytokines and the microcirculation in ischemia and reperfusion. J Mol Cell Cardiol. 1998;30:2567–76. doi: 10.1006/jmcc.1998.0829. [DOI] [PubMed] [Google Scholar]
- 22.Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 23.Kong P, Christia P, Saxena A, Su Y, Frangogiannis NG. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am J Physiol Heart Circ Physiol. 2013;305:H1363–72. doi: 10.1152/ajpheart.00395.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–21. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;120:254–65. doi: 10.1172/JCI40295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 27.Sandanger O, Ranheim T, Vinge LE, Bliksoen M, Alfsnes K, Finsen AV, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99:164–74. doi: 10.1093/cvr/cvt091. [DOI] [PubMed] [Google Scholar]
- 28.Lu D, Soleymani S, Madakshire R, Insel PA. ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. Faseb J. 2010;26:2580–91. doi: 10.1096/fj.12-204677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 30.Lijnen P, Papparella I, Petrov V, Semplicini A, Fagard R. Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J Hypertens. 2006;24:757–66. doi: 10.1097/01.hjh.0000217860.04994.54. [DOI] [PubMed] [Google Scholar]
- 31.Barcellos-Hoff MH, Dix TA. Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol. 1996;10:1077–83. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 32.Turner NA, Das A, Warburton P, O’Regan DJ, Ball SG, Porter KE. Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am J Physiol Heart Circ Physiol. 2009;297:H1117–27. doi: 10.1152/ajpheart.00372.2009. [DOI] [PubMed] [Google Scholar]
- 33.Siwik DA, Colucci WS. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev. 2004;9:43–51. doi: 10.1023/B:HREV.0000011393.40674.13. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Schwimmbeck PL, Tschope C, Leschka S, Husmann L, Rutschow S, et al. Collagen degradation in a murine myocarditis model: relevance of matrix metalloproteinase in association with inflammatory induction. Cardiovasc Res. 2002;56:235–47. doi: 10.1016/s0008-6363(02)00546-1. [DOI] [PubMed] [Google Scholar]
- 35.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–28. [PMC free article] [PubMed] [Google Scholar]
- 36.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–77. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christia P, Bujak M, Gonzalez-Quesada C, Chen W, Dobaczewski M, Reddy A, et al. Systematic characterization of myocardial inflammation, repair, and remodeling in a mouse model of reperfused myocardial infarction. J Histochem Cytochem. 2013;61:555–70. doi: 10.1369/0022155413493912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bujak M, Frangogiannis NG. The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp (Warsz) 2009;57:165–76. doi: 10.1007/s00005-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, et al. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J Immunol. 2013;191:4838–48. doi: 10.4049/jimmunol.1300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS. Interleukin-1 beta induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest. 1995;95:2555–64. doi: 10.1172/JCI117956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koudssi F, Lopez JE, Villegas S, Long CS. Cardiac fibroblasts arrest at the G1/S restriction point in response to interleukin (IL)-1beta. Evidence for IL-1beta-induced hypophosphorylation of the retinoblastoma protein. J Biol Chem. 1998;273:25796–803. doi: 10.1074/jbc.273.40.25796. [DOI] [PubMed] [Google Scholar]
- 43.Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91:1119–26. doi: 10.1161/01.res.0000047090.08299.d5. [DOI] [PubMed] [Google Scholar]
- 44.Chen W, Saxena A, Li N, Sun J, Gupta A, Lee DW, et al. Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts. Arterioscler Thromb Vasc Biol. 2012;32:2598–608. doi: 10.1161/ATVBAHA.112.300310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–37. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 46.Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43:146–55. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 47.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–38. [PMC free article] [PubMed] [Google Scholar]
- 48.Vracko R, Thorning D. Contractile cells in rat myocardial scar tissue. Lab Invest. 1991;65:214–27. [PubMed] [Google Scholar]
- 49.Takemura G, Ohno M, Hayakawa Y, Misao J, Kanoh M, Ohno A, et al. Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ Res. 1998;82:1130–8. doi: 10.1161/01.res.82.11.1130. [DOI] [PubMed] [Google Scholar]
- 50.Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb) Cardiovasc Res. 2000;48:89–100. doi: 10.1016/s0008-6363(00)00158-9. [DOI] [PubMed] [Google Scholar]
- 51.Willems IE, Havenith MG, De Mey JG, Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–75. [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez-Rosa JM, Peralta M, Mercader N. Pan-epicardial lineage tracing reveals that epicardium derived cells give rise to myofibroblasts and perivascular cells during zebrafish heart regeneration. Dev Biol. 2012;370:173–86. doi: 10.1016/j.ydbio.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 53.Dobaczewski M, Akrivakis S, Nasser K, Michael LH, Entman ML, Frangogiannis NG. Vascular mural cells in healing canine myocardial infarcts. J Histochem Cytochem. 2004;52:1019–29. doi: 10.1369/jhc.3A6210.2004. [DOI] [PubMed] [Google Scholar]
- 54.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–16. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frangogiannis NG, Perrard JL, Mendoza LH, Burns AR, Lindsey ML, Ballantyne CM, et al. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation. 1998;98:687–98. doi: 10.1161/01.cir.98.7.687. [DOI] [PubMed] [Google Scholar]
- 56.Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. 2003;163:2433–40. doi: 10.1016/S0002-9440(10)63598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yano T, Miura T, Ikeda Y, Matsuda E, Saito K, Miki T, et al. Intracardiac fibroblasts, but not bone marrow derived cells, are the origin of myofibroblasts in myocardial infarct repair. Cardiovasc Pathol. 2005;14:241–6. doi: 10.1016/j.carpath.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 58.Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–92. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 59.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A. 2006;103:18284–9. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mollmann H, Nef HM, Kostin S, von Kalle C, Pilz I, Weber M, et al. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc Res. 2006;71:661–71. doi: 10.1016/j.cardiores.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 61.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–61. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 62.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–28. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sousa AM, Liu T, Guevara O, Stevens J, Fanburg BL, Gaestel M, et al. Smooth muscle alpha-actin expression and myofibroblast differentiation by TGFbeta are dependent upon MK2. J Cell Biochem. 2007;100:1581–92. doi: 10.1002/jcb.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hashimoto S, Gon Y, Takeshita I, Matsumoto K, Maruoka S, Horie T. Transforming growth Factor-beta1 induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c-Jun-NH2-terminal kinase-dependent pathway. Am J Respir Crit Care Med. 2001;163:152–7. doi: 10.1164/ajrccm.163.1.2005069. [DOI] [PubMed] [Google Scholar]
- 67.Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, et al. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res. 2011;108:582–92. doi: 10.1161/CIRCRESAHA.110.224428. [DOI] [PubMed] [Google Scholar]
- 68.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873–81. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Naugle JE, Olson ER, Zhang X, Mase SE, Pilati CF, Maron MB, et al. Type VI collagen induces cardiac myofibroblast differentiation: implications for postinfarction remodeling. Am J Physiol Heart Circ Physiol. 2006;290:H323–30. doi: 10.1152/ajpheart.00321.2005. [DOI] [PubMed] [Google Scholar]
- 70.Matsui Y, Ikesue M, Danzaki K, Morimoto J, Sato M, Tanaka S, et al. Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ Res. 2011;108:1328–39. doi: 10.1161/CIRCRESAHA.110.235689. [DOI] [PubMed] [Google Scholar]
- 71.Zhao XH, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci. 2007;120:1801–9. doi: 10.1242/jcs.001586. [DOI] [PubMed] [Google Scholar]
- 72.Virag JA, Rolle ML, Reece J, Hardouin S, Feigl EO, Murry CE. Fibroblast growth factor-2 regulates myocardial infarct repair: effects on cell proliferation, scar contraction, and ventricular function. Am J Pathol. 2007;171:1431–40. doi: 10.2353/ajpath.2007.070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Booz GW, Baker KM. Molecular signalling mechanisms controlling growth and function of cardiac fibroblasts. Cardiovasc Res. 1995;30:537–43. [PubMed] [Google Scholar]
- 74.Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, et al. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. 2006;48:2315–23. doi: 10.1016/j.jacc.2006.07.060. [DOI] [PubMed] [Google Scholar]
- 75.Hahn JY, Cho HJ, Bae JW, Yuk HS, Kim KI, Park KW, et al. Beta-catenin overexpression reduces myocardial infarct size through differential effects on cardiomyocytes and cardiac fibroblasts. J Biol Chem. 2006;281:30979–89. doi: 10.1074/jbc.M603916200. [DOI] [PubMed] [Google Scholar]
- 76.Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007;97:738–47. [PubMed] [Google Scholar]
- 77.Blomer N, Pachel C, Hofmann U, Nordbeck P, Bauer W, Mathes D, et al. 5-Lipoxygenase facilitates healing after myocardial infarction. Basic Res Cardiol. 2013;108:367. doi: 10.1007/s00395-013-0367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mitchell MD, Laird RE, Brown RD, Long CS. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am J Physiol Heart Circ Physiol. 2007;292:H1139–47. doi: 10.1152/ajpheart.00881.2005. [DOI] [PubMed] [Google Scholar]
- 79.Freed DH, Chilton L, Li Y, Dangerfield AL, Raizman JE, Rattan SG, et al. Role of myosin light chain kinase in cardiotrophin-1-induced cardiac myofibroblast cell migration. Am J Physiol Heart Circ Physiol. 2011;301:H514–22. doi: 10.1152/ajpheart.01041.2010. [DOI] [PubMed] [Google Scholar]
- 80.Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest. 2001;107:785–90. doi: 10.1172/JCI12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–88. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, et al. Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res. 2009;105:973–83. doi: 10.1161/CIRCRESAHA.109.199471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cleutjens JP, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–92. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 84.Squires CE, Escobar GP, Payne JF, Leonardi RA, Goshorn DK, Sheats NJ, et al. Altered fibroblast function following myocardial infarction. J Mol Cell Cardiol. 2005;39:699–707. doi: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 85.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46:250–6. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- 86.Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol. 1994;26:279–92. doi: 10.1006/jmcc.1994.1036. [DOI] [PubMed] [Google Scholar]
- 87.Detillieux KA, Sheikh F, Kardami E, Cattini PA. Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc Res. 2003;57:8–19. doi: 10.1016/s0008-6363(02)00708-3. [DOI] [PubMed] [Google Scholar]
- 88.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–6. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pallero MA, Elzie CA, Chen J, Mosher DF, Murphy-Ullrich JE. Thrombospondin 1 binding to calreticulin-LRP1 signals resistance to anoikis. Faseb J. 2008;22:3968–79. doi: 10.1096/fj.07-104802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Villar AV, Garcia R, Llano M, Cobo M, Merino D, Lantero A, et al. BAMBI (BMP and activin membrane-bound inhibitor) protects the murine heart from pressure-overload biomechanical stress by restraining TGF-beta signaling. Biochim Biophys Acta. 2013;1832:323–35. doi: 10.1016/j.bbadis.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 91.Melchior-Becker A, Dai G, Ding Z, Schafer L, Schrader J, Young MF, et al. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J Biol Chem. 2011;286:17365–75. doi: 10.1074/jbc.M110.192682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol. 2009;131:471–81. doi: 10.1007/s00418-008-0541-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zheng J, Chen Y, Pat B, Dell’italia LA, Tillson M, Dillon AR, et al. Microarray identifies extensive downregulation of noncollagen extracellular matrix and profibrotic growth factor genes in chronic isolated mitral regurgitation in the dog. Circulation. 2009;119:2086–95. doi: 10.1161/CIRCULATIONAHA.108.826230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rohr S. Arrhythmogenic implications of fibroblast-myocyte interactions. Circ Arrhythm Electrophysiol. 2012;5:442–52. doi: 10.1161/CIRCEP.110.957647. [DOI] [PubMed] [Google Scholar]
- 95.Vasquez C, Morley GE. The origin and arrhythmogenic potential of fibroblasts in cardiac disease. J Cardiovasc Transl Res. 2012;5:760–7. doi: 10.1007/s12265-012-9408-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ Res. 2010;107:1011–20. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Camelliti P, Devlin GP, Matthews KG, Kohl P, Green CR. Spatially and temporally distinct expression of fibroblast connexins after sheep ventricular infarction. Cardiovasc Res. 2004;62:415–25. doi: 10.1016/j.cardiores.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 98.Rosker C, Salvarani N, Schmutz S, Grand T, Rohr S. Abolishing myofibroblast arrhythmogeneicity by pharmacological ablation of alpha-smooth muscle actin containing stress fibers. Circ Res. 2011;109:1120–31. doi: 10.1161/CIRCRESAHA.111.244798. [DOI] [PubMed] [Google Scholar]
- 99.Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388–93. doi: 10.1161/01.cir.102.12.1388. [DOI] [PubMed] [Google Scholar]
- 100.Hayashi M, Tsutamoto T, Wada A, Tsutsui T, Ishii C, Ohno K, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents post-infarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation. 2003;107:2559–65. doi: 10.1161/01.CIR.0000068340.96506.0F. [DOI] [PubMed] [Google Scholar]
- 101.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010;105:1371–77. e1. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 102.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–92. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Frangogiannis NG. Biomarkers: hopes and challenges in the path from discovery to clinical practice. Transl Res. 2012;159:197–204. doi: 10.1016/j.trsl.2012.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Christia P, Frangogiannis NG. Targeting inflammatory pathways in myocardial infarction. Eur J Clin Invest. 2013;43:986–95. doi: 10.1111/eci.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]