

Abstract
In the healthy heart, cardiac myocytes form an electrical syncytium, embedded in a supportive fibroblast-rich extracellular matrix designed to optimize electromechanical coupling for maximal contractile efficiency of the heart pump. In the injured heart, however, fibroblasts are activated and differentiate into myofibroblasts that proliferate and generate fibrosis as a component of the wound-healing response. This review discusses how fibroblasts and fibrosis, while essential for maintaining the structural integrity of the heart wall after injury, have undesirable electrophysiological effects by disrupting the normal electrical connectivity of cardiac tissue to increase the vulnerability to arrhythmias. We emphasize the dual contribution of fibrosis in altering source-sink relationships to create a vulnerable substrate while simultaneously facilitating the emergence of triggers such as afterdepolarization-induced premature ventricular complexes– both factors combining synergistically to promote initiation of reentry. We also discuss the potential role of fibroblasts and myofibroblasts in directly altering myocyte electrophysiology in a pro-arrhythmic fashion. Insight into these processes may open up novel therapeutic strategies for preventing and treating arrhythmias in the setting of heart disease as well as avoiding potential arrhythmogenic consequences of cell-based cardiac regeneration therapy. This article is part of a Special Issue entitled “Myocyte-Fibroblast Signaling in Myocardium.”
1. Introduction1
Cardiovascular disease is the leading cause of mortality in industrialized countries, and arrhythmias causing sudden cardiac death constitute a major component. Fortunately, advances in health care have given the injured heart a greater chance to survive injury and heal its wounds. However, a cornerstone of the wound-healing process is scar formation, mediated by activated fibroblasts (myofibroblasts) secreting collagen and producing myocardial fibrosis. Although fibrosis plays a critical role in enhancing mechanical stability to prevent cardiac wall rupture during injury, it also has the undesirable consequence of disrupting the electrical coupling between adjacent strands of myocytes.
In this review, our goal is to highlight how the wound-healing process enhances the risk of potentially lethal cardiac arrhythmias. Our overriding theme is that lethal arrhythmias typically arise from the convergence of two factors: a trigger, such as a premature ventricular complex (PVC), encountering a vulnerable tissue substrate. This trigger-substrate combination promotes the initiation of anatomic or functional reentry that can degenerate to ventricular fibrillation when blood pressure falls, and myocardial ischemia ensues. It has been well-appreciated that fibrosis plays a key role in creating a vulnerable tissue substrate by interposing collagen bundles between strands of myocytes. What is less widely appreciated, but just as important, is the role that fibrosis, and potentially fibroblasts themselves, play in promoting triggers, the other half of this lethal combination. These trigger-promoting effects are mediated through passive effects of fibrosis on the local source-sink relationships that allow triggers to emerge and propagate into normal tissue as PVCs. In addition, emerging but still controversial evidence indicates that activated fibroblasts can exert direct pro-arrhythmic effects on myocytes as a result of myofibroblast-myocyte gap junction coupling [1–3] and/or paracrine factors secreted by myofibroblasts [4–6]. Insight into these mechanisms may lead to new therapeutic approaches to prevent cardiac arrhythmias. Moreover, with the growing focus on cardiac regenerative medicine–in which the therapeutic goal is to induce transplanted stem/progenitor cells or injected biomaterial scaffolds to structurally and functionally integrate with surviving resident myocytes–it is imperative to better understand how endogenous wound-healing mechanisms influence the engraftment process so that the arrhythmogenic effects of myofibroblast proliferation and fibrosis can be minimized.
2. From fibroblasts to myofibroblasts: remodeling the heart in distress
In the normal healthy heart, fibroblasts play a major role in the routine maintenance of myocardial structure. They are the predominant cell type in the heart, exceeding myocytes in number, although not in volume [7]. Primarily responsible for providing myocytes with a 3D mechanical scaffold to integrate the contractile activity of myocytes into the coordinated pumping action of the cardiac chambers, fibroblasts are sentinel cells that tightly coordinate the synthesis and degradation of collagen and other components of the extracellular matrix [8]. Normally quiescent, cardiac fibroblasts are activated by myocardial injury, triggering their differentiation into myofibroblasts to facilitate the wound-healing process, including scar formation and contraction. However, fibroblast heterogeneity and pleiomorphic responses to environmental stress, coupled with the lack of specific lineage markers, present a challenge in analyzing the scope of fibroblast and myofibroblast actions in intact cardiac muscle. Particularly controversial is the extent to which cell culture conditions accurately recapitulate in vivo effects. Indeed, whether fibroblasts and myofibroblasts should be discriminated as separate entities rather than a continuum has been questioned [9, 10]. Nevertheless, it is generally agreed that at either end of the spectrum, fibroblasts and myofibroblasts comprise distinct cell phenotypes and serve different functions at different stages of the heart evolution from birth through disease, injury, and aging. Therefore, the term ‘fibroblasts’ has been used loosely and conveniently at times to refer to both the ‘fibroblasts’ in the normal heart and the ’myofibroblasts’ in the injured heart.
In the diseased, injured, or senescent heart with limited myocyte regenerative capability, myofibroblasts may arise either de novo or from resident quiescent fibroblasts. The former de novo sources may include resident progenitor stem cells, bone-marrow-derived cells, or transformed epithelial and endothelial cells via epithelial and endothelial-mesenchymal transitions. The latter arises from the proliferation of activated resident fibroblasts following a phenotype switch, similar but not identical to the phenotype switch of fibroblasts to myofibroblasts observed in cell culture, such that myofibroblasts gain hybrid characteristics of both smooth muscle cells and fibroblasts [11, 12]. Compared to quiescent fibroblasts, myofibroblasts are much larger [13], proliferate more actively, and deposit collagen at higher rates. They secrete cytokines to recruit other fibroblasts and inflammatory cells, upregulate connexins, readily form both homocellular and heterocellular gap junctions, and express stretch receptors as well as several smooth muscle contractile proteins (such as smooth muscle α-actin and tropomyosin among others). As a result, myofibroblasts can migrate, contract, and respond to a variety of stimuli including mechanical stretch, hypoxia and electrophysiological signals in addition to chemical signals [7]. The increased mobility and collagen-synthesizing function of myofibroblasts is critical for wound closure and maintenance of structural integrity of healing scars in the injured heart [12]. However, the resulting structural remodeling, particularly fibrosis, has important adverse electrophysiological consequences, as described below.
3. Fibrosis Creates a Vulnerable Substrate for Reentry
3.1. Patterns of fibrosis and risk of arrhythmias
Fibrosis is categorized into distinct patterns (or textures): compact, patchy, interstitial, and diffuse (Fig. 1) [14]. These different patterns do not have equivalent arrhythmogenic profiles because they differentially affect the two key features that play a critical role in making cardiac tissue vulnerable to functional and anatomic reentry: slow conduction and susceptibility to unidirectional conduction block.
Fig. 1.
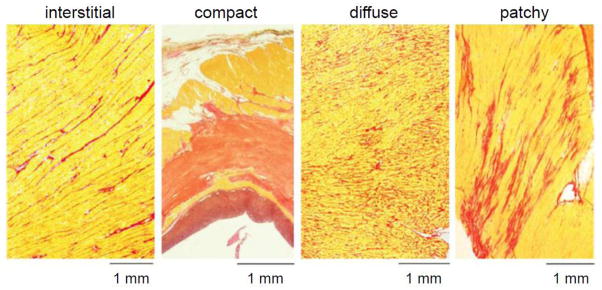
Cardiac fibrosis patterns. Red = collagen; yellow = myocardium. The most arrhythmogenic patterns are interstitial and patchy, which result in interconnected strands of myocytes separated by collagen bundles. Modified from de Jong et al [14] with permission.
Compact fibrosis, defined as large dense areas of collagen that are devoid of cardiac myocytes, e.g. following a myocardial infarction, has the least arrhythmogenic potential because large macroscopic scars, by themselves, neither promote slow conduction nor enhance susceptibility to unidirectional conduction block. Nevertheless, once other conditions initiate reentry, large scars can provide an inexcitable obstacle that anchors a reentry circuit. What makes ischemic heart disease arrhythmogenic is not the compact fibrosis of macroscopic scars per se, but the fact that scars are surrounded by a border zone, in which the other patterns of intermediate fibrosis are present to mixed degrees.
Areas of patchy fibrosis and severe interstitial fibrosis, where myocyte bundles are separated over extended distances by collagenous septa, have the greatest arrhythmogenic potential for initiating reentry, whether associated with the border zone of a compact infarct scar or in the setting of nonischemic heart disease. In these regions, strands of surviving myocytes become tenuously interconnected, predisposing the tissue to slow, “discontinuous”, and “zigzag” conduction [15, 16] (Fig. 2), as well as to unidirectional conduction block due to source-sink mismatches between the strands and adjacent normally coupled tissue (see below). Interconnected strands can also form channels which provide the substrate for anatomic reentry circuits, manifested clinically as monomorphic ventricular tachycardia (VT), because the slowly conducting impulse in the channel exits to the normal myocardium from a consistent site (or sites) during tachycardia.
Fig. 2.

Slow conduction in the border zone of an infarct manifests as fractionated electrograms. Intracellular microelectrode recordings show action potentials from sites A–D in strands of myocytes (solid gray) surrounded by collagen bundles (speckled gray). A simultaneously recorded bipolar electrogram (white circles) is shown below each local action potential recording. Note that the timing of the action potential upstroke at sites A–D each coincides with a spike in the electrogram. Reproduced from Gardner et al [15] with permission.
Diffuse fibrosis, in which short collagen septa are interspersed among myocardial fibers, also has increased arrhythmogenic potential by selectively reducing side-to-side gap junction connections between myocytes, which slows wave propagation transversely and increases anisotropy, predisposing to wave break and anisotropic (functional) reentry [17, 18]. The electrical decoupling can also promote spatially discordant APD alternans and dispersion of refractoriness [19]. When reentry is initiated under these conditions, the reentry circuit is often not anchored to a well-defined anatomical pathway, resulting in polymorphic VT instead of monomorphic VT.
Consistent with these arrhythmogenic properties, the origin of tachycardia in patients with ischemic heart disease who undergo mapping and ablation of recurrent VT is most commonly located in the border zone region of infarcts. Here patchy, interstitial, and diffuse fibrosis patterns coexist to create channels of slowly-conducting myocardium (often indicated by low voltage and fractionated local electrograms during catheter mapping) predisposed to unidirectional conduction block (Fig. 2) [16].
This scenario is supported by recent advances in magnetic resonance imaging using delayed gadolinium enhancement to better define infarct anatomy in patients with ischemic heart disease undergoing VT ablation [20]. Such studies have shown the “gray” regions with intermediate density due to a mixture of viable myocardium (normally dark due to lack of gadolinium uptake) and fibrotic tissue (normally bright due to robust gadolinium uptake), predict arrhythmogenic sites. These findings also highlight the concept that intermediate degrees of fibrosis are more arrhythmogenic than densely fibrotic regions. The reason is that slow conduction eventually transitions to non-conduction (incapable of supporting a reentry circuit) when fibrosis becomes too dense. Both experimental data and computer simulations suggest that <20% and >80% fibrosis are relatively benign, whereas arrhythmogenic potential is maximal at 30–50% fibrosis (by area) [21, 22], as discussed in more detail later.
3.2 Fibrosis and slow conduction
By creating sparsely interconnected strands of myocytes which are electrically isolated from each other by collagen bundles (Fig. 1), fibrosis can force electrical propagation to take a zigzag pattern through the tissue, conducting circuitously from one strand to the other [15, 16] (Fig. 2). The circuitous conduction pathway effectively slows propagation velocity at the macroscopic (millimeter) level, although not necessarily at the microscopic (submillimeter) level. However, the effects of fibrosis are further compounded by both gap junction remodeling and electrical remodeling of ion channels affecting the action potential properties of border zone myocytes.
By reducing the electrical coupling between myocytes within the strands, gap junction remodeling at the microscopic level synergizes with the macroscopic effects of fibrosis to further slow conduction. In hypertrophied [23], ischemic [24], or failing [25] heart, the predominant cardiac connexin, Cx43, is downregulated and Cx43 gap junctions at the intercalated discs at the ends of myocytes are lost or partially redistributed along the sides of myocytes [26]. This lateralization of gap junctions further reduces the longitudinal diffusion coefficient and hence conduction velocity along the major axis of fiber direction (parallel to the myocyte strand). In addition, when gap junction conductance becomes too low, propagation can no longer be mediated by the Na current, which inactivates before sufficient charge can flow into adjacent unexcited myocytes to bring them to their activation threshold. In this case, the more slowly inactivating L-type Ca current mediates propagation, but with a dramatically slower conduction velocity, often referred to as “discontinuous” conduction because of the significant delay between the action potential upstrokes of neighboring myocytes [27]. In the clinical setting, however, discontinuous conduction may be less important in slowing conduction than the circuitous pathway that impulses are forced to take, given the observation that Ca channel blockers have limited effectiveness clinically in preventing reentrant ventricular arrhythmias.
Further compounding the situation, electrical remodeling in border zone myocytes can also directly promote slow conduction, especially at short diastolic intervals due to Na channel remodeling [28], and facilitate unidirectional conduction block by increasing dispersion of refractoriness via ion channel remodeling [28, 29].
In summary, by isolating and electrically insulating bundles of myocytes from each other in diseased hearts, fibrosis is a major factor promoting slow conduction, which is further exacerbated by gap junction and electrical remodeling.
3.3. Fibrosis and unidirectional conduction block: the source-sink mismatch concept
In addition to slow conduction, unidirectional conduction block is the second key factor required to initiate reentry. The classic cause of unidirectional conduction block is dispersion of refractoriness, in which one region of a tissue has longer refractory period than another. In the classic scenario, a PVC with coupling interval that falls in between the long and short refractory periods blocks in the region with the long refractory period and propagates through the region with the short refractory period (Fig. 3). If slow conduction creates a sufficient time delay, the impulse can subsequently reenter the previously blocked (long refractory period) region after that region has regained excitability, thus initiating reentry. In diseased hearts, dispersion of refractoriness is primarily related to electrical remodeling rather than fibrosis per se, but also to a lesser extent, to the paracrine contribution of fibroblasts (see later) and electrical decoupling [19].
Fig. 3.

Classic mechanism by which a PVC initiates reentry in the fibrotic border zone of an infarct due to slow conduction and dispersion of refractoriness. Upper panel: A PVC occurring 250 ms after the previous beat arrives too early to propagate through the upper myocyte strand with a long effective refractory period (ERP) of 275 ms but propagates successfully (red arrows) through the lower strand with a shorter ERP of 225 ms (entry site). The impulse propagates slowly (slow CV), eventually reaching the upper strand from the opposite direction. Lower panel: If the total conduction time is >275 ms, the interface of the upper strand with normal tissue (exit) site has recovered excitability and the impulse can propagate through the region of prior conduction block, initiating reentry. Dispersion of refractoriness is caused by electrical remodeling. The slow propagation is due to zig-zag conduction through the myocyte strands as well as gap junction remodeling.
Less well-appreciated, however, is the fact that fibrosis can directly facilitate unidirectional conduction block by a completely different mechanism unrelated to dispersion of refractoriness. This mechanism depends on the concept of a source-sink mismatch, originally applied to nerve and cardiac conduction in the context of the “liminal length,” defined as the minimal length of a cable required to be excited to elicit a propagating action potential [30, 31]. As illustrated in Fig. 4, when patchy or interstitial fibrosis creates strands of myocytes which remain coupled to normal (nonfibrotic) tissue, the situation is analogous to a 1D cable of cells entering a 3D syncytium of cells. For propagation to succeed through such interfaces, the excited depolarized cells (the current source) must generate enough current flow through gap junctions to depolarize the adjacent repolarized cells (the current sink) to their excitation threshold. When a 1D cable enters a 3D syncytium, the current source from the depolarized cells in the cable is suddenly diluted into a vastly greater number of repolarized cells in the 3D tissue such that, instead of the depolarizing charge being transferred at a 1:1 ratio from depolarized to repolarized cells, the ratio decreases dramatically, thereby creating a source-sink mismatch. If the resulting dilution of the charge transfer is insufficient to bring the repolarized cells to their activation threshold, propagation will fail [32]. In contrast, for propagation from a 3D syncytium into a 1D cable, the source-to-sink ratio is reversed, ensuring successful conduction with a large safety factor. These geometrical source-sink asymmetries in current load set up the conditions for unidirectional conduction block as follows. Suppose that a PVC arises from within a narrow myocyte strand and propagates bidirectionally towards both ends (Fig. 4, upper panel). If one end of the narrow strand makes an abrupt connection to the normal tissue, then propagation may fail due to the source-sink mismatch. If, however, the other end of the strand gradually widens such that the amount of depolarizing source current gradually increases before that strand connects to normal 3D tissue, the source-sink relationship may favor successful propagation through the interface. Thus, the PVC propagates successfully through wide strand interface but blocks at the narrow strand interface due to the source-sink mismatch. With a sufficient time delay afforded by slow conduction, the impulse exiting from the wide strand into normal 3D tissue has a very favorable source-sink relationship to successfully propagate through the previously blocked site in the narrow strand, initiating reentry (Fig. 4, lower panel).
Fig. 4.

Source-sink effects and the initiation of reentry in the border zone of an infarct. Upper panel: A PVC originating from within the lower myocyte strand propagates in both directions (red arrows) but blocks at the interface with normal tissue due to the unfavorable source-sink mismatch (i.e. the small source of the strand faces a large sink at the interface with well-coupled 3D tissue). Meanwhile, the impulse propagates slowly in the other direction (through the scar), eventually reaching the upper strand which widens progressively before the interface with normal tissue. The gradual widening ensures that current density in the strand progressively increases (thickening arrows), creating a more favorable source-sink relationship at the interface, ensuring successfully propagation into the normal tissue. Lower panel: Due to the slow conduction, the lower strand has recovered excitability, allowing the impulse from normal tissue to enter the strand and propagate since the source-sink relationship in the opposite direction is highly favorable. Reentry is thus initiated without requiring any dispersion of refractoriness between the myocyte strands and/or the normal tissue. Note that a PVC arising from the normal tissue (e.g. as during programmed electrical stimulation) will encounter favorable source-sink relationships at the entrance to both strands such that the propagating impulses will collide in the strand and extinguish each other, failing to initiate reentry. Note also that during reentry, the entry site is narrow, whereas the exit site is broad, making the entry site, if identifiable, the more favorable for catheter ablation.
In summary, source-sink mismatches caused by fibrosis and dispersion of refractoriness caused by electrical remodeling both independently contribute to the likelihood of unidirectional conduction block and the initiation of reentry in diseased hearts. Coupled with slow conduction, these effects make fibrosed cardiac tissue highly vulnerable to initiation of reentry by an appropriately timed PVC.
4. Fibrosis promotes triggers
Reentry occurs when an appropriately timed trigger, such as a PVC, encounters a vulnerable tissue substrate. In the last section, we discussed how fibrosis plays a major role in increasing the vulnerability of cardiac tissue. Here we turn to its role in promoting triggers, in which source-sink effects again have very important ramifications [33–35]. Just as impulse propagation depends on the excited depolarized cells (the current source) generating enough current flow through gap junctions to depolarize the adjacent repolarized cells (the current sink) to their threshold of excitation, the same is true for the case of impulse formation. For example, consider diastolic depolarization or a delayed afterdepolarization (DAD) due to a spontaneous sarcoplasmic reticulum Ca release wave in an isolated myocyte. Although the inward current generated by that single myocyte may be fully capable of depolarizing the myocyte to its action potential threshold when that myocyte is not coupled to other myocytes, this situation is very different when that myocyte is coupled via gap junctions to quiescent neighboring myocytes. Since the average myocyte is coupled to 11 neighboring myocytes in normal 3D ventricular tissue [36, 37], any voltage difference causes current to flow into the neighboring cells (the sink), attenuating the depolarization by more than an order of magnitude compared to the isolated uncoupled myocyte. Only when a large collection of neighboring myocytes all decide to generate the inward current synchronously can the depolarization reach sufficient amplitude to reach the threshold for an action potential and generate a PVC.
How many cells are required to trigger a PVC? In normal well-coupled 3D ventricular tissue, computer simulations indicate that the number required is approximately 700,000 [38, 39], in agreement with experimental measurements for creating biological pacemakers [40]. However, the required number of myocytes decreases dramatically in 2D and 1D tissue because the current sink is limited to diffusion into an area or a line of neighboring myocytes rather than a 3D volume. In 2D tissue, the number of contiguous myocytes that must all develop diastolic depolarization synchronously, or a DAD synchronously, or an early afterdepolarization (EAD) synchronously, in order to overcome the source-sink mismatch and generate a PVC decreases to about 7,000–8,000 [39]. In a 1D cable, the number decreases further to about 70–80. Thus, the source-sink mismatch represents a very powerful endogenous mechanism by which normal well-coupled myocardium protects itself from ectopic beats when small groups of myocytes misbehave. This anti-arrhythmic protection provided by the source-sink mismatch has been directly documented experimentally in the case of DADs, in which spontaneous Ca waves in single rat ventricular myocytes in intact rat ventricular tissue failed to cause any detectable change in membrane voltage [41]. Only when the majority of myocytes in the mapped field developed Ca waves synchronously (following rapid pacing) was a measurable DAD observed.
How does fibrosis affect this protective mechanism? By creating interconnected strands of myocytes, patchy and interstitial fibrosis effectively converts a well-coupled 3D syncytium into a tangle of quasi-1D cables. Among any one of these cables, only 70–80 neighboring myocytes need to misbehave to trigger a PVC within the cable [39], which, depending on local source-sink relationships, may then propagate into normal tissue. So, instead of a nonfibrotic 3D syncytium requiring 700,000 cells to generate a PVC, the same volume of fibrotic tissue requires only 70–80 myocytes among any one of a number of quasi-1D cables! For this very same reason, Purkinje fibers, as quasi-1D cables, are also a common source of PVCs compared to ventricular myocardium, although cellular electrophysiological differences such as a reduced inward rectifier K current density also play a role.
In summary, by creating strands of myocytes which resemble a tangle of quasi-1D cables rather than a well-coupled 3D syncytium, patchy and interstitial fibrosis markedly reduces the source-sink mismatch that normally prevents a small numbers of errant myocytes from generating a PVC capable of propagating into surrounding tissue. The effect of fibrosis in promoting slow conduction and unidirectional conduction block, when combined with this effect of fibrosis in reducing the source-sink mismatch, creates the perfect storm for initiation of reentry by the mechanism illustrated in Fig. 4. Alternatively, the effect of fibrosis in slowing conduction, when combined with electrical remodeling to produce dispersion of refractoriness among myocyte strands, can also initiate reentry by the mechanism illustrated in Fig. 3.
5. Direct pro-arrhythmic effects of fibroblasts on myocyte electrophysiology
5.1. Myofibroblast-myocyte gap junction coupling
Cardiac fibroblasts are known to express connexins (primarily Cx43 and Cx45), which are upregulated when fibroblasts are activated in situ and differentiate into myofibroblasts [7, 9, 10]. When quiescent cardiac fibroblasts are cultured in vitro, they activate spontaneously and differentiate into ‘myofibroblasts’ although the extent to which they accurately recapitulate the properties of in situ myofibroblasts in native cardiac tissue remains controversial. Nevertheless, when in situ ‘myofibroblasts’ co-cultured with isolated neonatal cardiac myocytes, they readily form functional heterocellular gap junctions with myocytes as well as homocellular gap junctions with each other. In an elegant series of experiments, Miragoli et al. [1] demonstrated that heterocellular gap junctions directly influence myocyte electrophysiology due to the depolarizing effect of ‘myofibroblasts’ (which typically sit at a relatively depolarized resting potential of −50 mV or less) on the more negative myocyte resting membrane potential. They showed that when the percentage of ‘myofibroblasts’ in the co-culture exceeded 15%, the resulting depolarization of myocyte resting potential was sufficient to induce spontaneous diastolic depolarization and automaticity. In addition, when myocytes were cultured into quasi-1D strands interrupted by defects containing ‘myofibroblasts’, gap junction coupling allowed slow propagation across defects up to 0.3 mm [42].
Despite the unequivocal evidence from co-cultures, however, the presence and extent of functional heterocellular gap junction formation between fibroblasts/myofibroblasts and myocytes in situ in intact cardiac tissue remains controversial. Homocellular gap junctions between cardiac myocytes are abundant and readily identifiable immunohistochemically in intact cardiac tissue whereas heterocellular gap junctions between myocytes and fibroblasts, even when detectable, appear to be relatively rare [43, 44]. Heterocellular gap junction coupling probably does not play an important role in normal cardiac tissue, in which most fibroblasts are quiescent. Whether heterocellular gap junction coupling becomes important in injured hearts in which fibroblasts differentiate into myofibroblasts, particularly in areas of active fibrosis such as the border zone of infarcts, also remains to be convincingly demonstrated [44]. It should also be noted that electrical coupling between fibroblasts and myocytes may be mediated by mechanisms other than gap junctions. Fibroblasts have been shown to project filopodia (nanotubes) that can connect them directly to myocytes via an aqueous–and therefore putatively electrically conductive–channel [45].
Given that significant electrical coupling between myofibroblasts and myocytes in injured hearts remains a possibility, it is relevant to review the potential electrophysiological consequences. Extrapolating from co-culture experiments [1] and computer simulations [46, 47], if the myofibroblast-myocyte ratio in the border zone of an infarct becomes substantial enough (>15%) to significantly depolarize the myocyte resting membrane potential. Such membrane depolarization may slow conduction velocity and induce post-repolarization refractoriness by inactivating Na channels and increasing the electrical load. Sufficient depolarization can also induce automaticity due to spontaneous phase 4 diastolic depolarization causing increased ventricular ectopies [1], especially in quasi-1D myocyte strands in which the number of myocytes required to induce a propagating PVC is much lower than in normal 3D tissue due to the source-sink effects described above. Similarly, by depolarizing the myocyte resting membrane potential and bringing the myocyte closer to the activation threshold, myofibroblast-myocyte coupling in these regions will facilitate spontaneous sarcoplasmic reticulum SR Ca release to generate DADs of sufficient amplitude to elicit triggered action potentials. Finally, EADs may also be promoted by myofibroblast-myocyte coupling (Fig. 5), based on a study using the dynamic clamp technique to simulate the effects of coupling a virtual fibroblast to a real ventricular myocyte with reduced repolarization reserve [3]. Under all of these conditions, the depolarized resting potential of the virtual fibroblast was the most important factor inducing spontaneous diastolic depolarization, reducing the threshold for DAD-mediated triggered activity, and promoting EAD formation. Since fibroblast resting membrane potential changes in response to stretch, hypoxia, and various neurohumeral signals, such factors might dynamically modulate their arrhythmogenic potential.
Fig. 5.
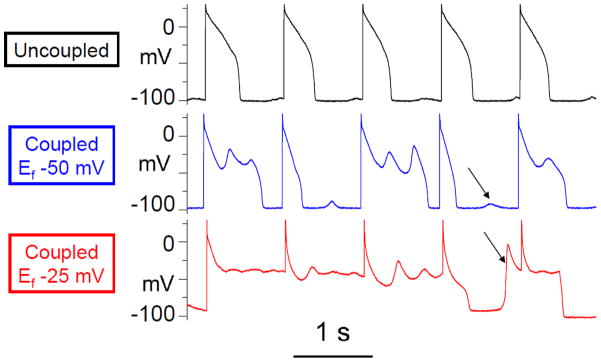
The effect of myofibroblast-myocyte coupling on EAD and DAD formation. A paced rabbit ventricular myocyte exposed to hypokalemia exhibited a normal action potential (upper trace) with very small DADs until coupled to a virtual fibroblast (capacitance 6.3 pF, gap junction conductance 3.0 nS, uncoupled resting membrane potential Ef −50 mV), which caused EADs to appear and DADs (arrows) to grow larger in amplitude (middle trace). Effects were more even more prominent when the virtual fibroblast resting membrane potential Ef was lowered to −25 mV (bottom trace), which resulted in a DAD-triggered premature action potential (arrow) before the pacing spike. Reprinted from Nguyen et al [3] with permission.
Therefore, myofibroblast-myocyte coupling, if it occurs under pathophysiological conditions, could be a significant additional cellular factor synergistically promoting the emergence of triggers due to diastolic depolarization, DADs, or EADs, especially when combined with the structural source-sink effects of fibrosis at the tissue level. Specifically, myofibroblast-myocyte coupling induces myocyte membrane depolarization in areas of patchy and interstitial fibrosis. Such membrane depolarization actively enhances the formation of triggers in the same regions where the source-sink milieu is particularly favorable for the emergence of these triggers and also where slow conduction coexists with increased susceptibility to unidirectional conduction block. The combined effect is a perfect storm for the initiation of reentrant and non-reentrant arrhythmias.
5.2. Paracrine effects of myofibroblasts
In addition to electrical coupling to myocytes, fibroblasts and myofibroblasts also secrete paracrine factors that can modulate myocyte electrophysiology. Pedrotty et al. [4] cultured freshly isolated cardiac fibroblasts and collected the fibroblast-conditioned media (FCM). When cardiac monolayers were exposed to FCM (in the absence of fibroblasts), the action potential lengthened and conduction velocity slowed significantly. Vasquez et al. [5] compared FCM from cardiac fibroblasts isolated from either normal or infarcted hearts. Unlike the normal heart FCM, the infarcted heart FCM depressed conduction velocity and shortened action potential duration of neonatal rat myocyte monolayers. Kaur et al. [6] found that when adult rat myocytes were treated with FCM, the Na current was upregulated, the transient outward current downregulated, the action potential prolonged, and EADs emerged–effects attributed to the cytokine TGF-β1. These findings indicate the paracrine factors (cytokines) secreted by fibroblasts and myofibroblasts can have direct electrophysiological effects that may contribute to conduction slowing and dispersion of refractoriness.
6. Summary and Clinical Implications
Fig. 6 summarizes the major concepts discussed in this review. Cardiac fibroblasts play a key physiological role in maintaining the extracellular scaffold in which the cardiac myocytes must function. In response to acute injury, fibroblasts are activated and differentiate into myofibroblasts to promote wound healing and scar formation, which is essential for maintaining the structural integrity of injured heart walls. Interfering with this process during acute injury such as myocardial infarction is generally counterproductive due to the increased risk of cardiac rupture. However, when the acute phase is over, or when fibrosis develops slowly due to a chronic process, the consequences of the structural remodeling driven by fibroblasts can be highly arrhythmogenic. By interrupting the normal source-sink relationships within a well-coupled 3D syncytium, fibrosis not only creates a vulnerable tissue substrate with slow conduction and unidirectional conduction block but also promotes the emergence of triggers that, on encountering this vulnerable substrate, have a high probability of initiating reentry. In synergy with the pro-arrhythmic effects of fibrosis are those of other aspects of cardiac remodeling–such as electrical, gap junction, excitation-contraction coupling, metabolic, vascular, and neural remodeling–all triggered by cardiac injury. In addition to generating fibrosis, myofibroblasts also can directly affect myocyte electrophysiology in a pro-arrhythmic fashion by forming heterocellular gap junctions with myocytes and/or releasing paracrine factors, although the relevance to native cardiac tissue remains to be established.
Fig. 6.
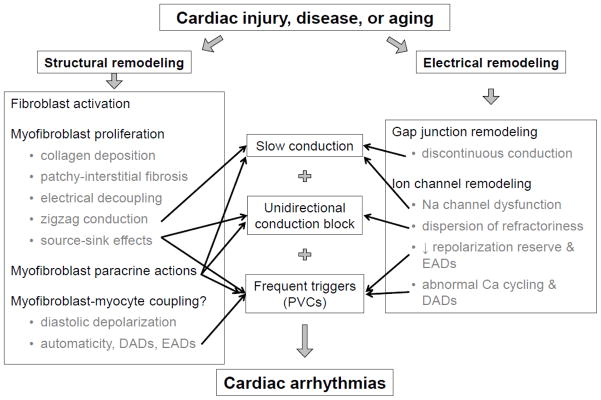
Summary of the role of fibroblasts in cardiac arrhythmogenesis.
It is important to emphasize that the pattern of fibrosis plays a key role. Dense scars are not themselves particularly arrhythmogenic since they are not capable of active electrical propagation. However, dense scars are surrounded by a border zone with intermediate degrees of fibrosis (patchy, interstitial, or diffuse) in which strands of myocytes interdigitate with collagen bundles, creating a tangled web of quasi-1D cables. It is these areas of intermediate fibrosis that cause slow zig-zag anisotropic conduction and alter the local source-sink relationships to promote both triggers and unidirectional conduction block. In non-ischemic cardiomyopathy, macroscopic dense scars with well-defined border zones are less common, but intermediate fibrosis creating arrhythmogenic myocyte strands is also a consequence of chronic tissue injury. In this setting, the lack of macroscopic circuits to anchor reentry tends to favor polymorphic VT over monomorphic VT.
The mechanisms of reentry initiation in fibrotic border-zone tissue illustrated in Figs. 3 and 4 provide insight into the clinical observations using programmed electrical stimulation to induce VT. For the classic dispersion of refractoriness mechanism shown in Fig. 3, a PVC arising from surrounding myocardium initiates reentry by propagating successfully into one myocyte strand but blocking in the other. Clinically, this VT is likely to be inducible by programmed electrical stimulation. However, typically less than half of the sustained monomorphic VTs documented to occur clinically are inducible by programmed electrical stimulation in the electrophysiology laboratory [48]. It is tempting to speculate that in the non-inducible cases, reentry initiation requires a PVC originating from within the myocyte strand, rather than the surrounding tissue, as in Fig. 4. In this case, a PVC induced by programmed electrical stimulation in the surrounding tissue will encounter favorable source-sink relationships and propagate successfully into the strand from both directions (since no unfavorable dispersion of refractoriness is present), colliding and extinguishing in the middle. Rapid pacing may still be effective at inducing reentry, on the other hand, by promoting the emergence of post-pacing DAD-induced PVCs in myocyte strands within the reentry circuit (as favored by the quasi-1D nature of these strands). The notion that the PVC initiating VT often arises from within the reentry circuit is supported by Saeed and colleagues’ [49] analysis of spontaneous episodes of VT recorded by implantable cardioverter-defibrillators. These authors observed that a significant percentage of VT episodes were initiated by a PVC with identical morphology to that of the subsequent VT, suggesting that the PVC arose from within the reentry circuit and exited to the surrounding myocardium using the same exit site as during tachycardia.
The mechanism of unidirectional block for a PVC originating from within the reentry circuit could be due to either asymmetrical local source-sink relationships at the exit sites, as illustrated in Fig. 4, or to dispersion of refractoriness, assuming that the strand has a shorter refractory period than the surrounding tissue at the exit sites. In this case, if a PVC originating in the strand has different conduction times to reach the exit sites, the earlier-arriving impulse may find the surrounding tissue refractory and block, whereas the late-arriving impulse may find the surrounding tissue now excitable and conduct. It remains a matter of pure speculation as to whether the cause of unidirectional conduction block initiating reentry in this setting is more commonly due to dispersion of refractoriness or asymmetrical source-sink mismatches at exit sites (Fig. 4). The geometry of interfaces between strands and surrounding myocardium underlying source-sink mismatches in the border zone is highly variable, but electrical remodeling in the border zone also promotes dispersion of refractoriness, so both factors are likely to contribute.
Although we have focused on ventricular arrhythmias, the same principles apply to atrial arrhythmias. Atrial fibrillation becomes more frequent and less reversible as the atria undergo structural remodeling due to progressive fibrosis associated with hemodynamic stress or injury [50]. It is tempting to speculate that local source-sink relationships play a key role in allowing triggers to emerge from the pulmonary veins and other cardiac vascular-myocardium interfaces, since these structures are characterized by finger-like projections of myocardial tissue (i.e. quasi-1D cables) interdigitating with vascular tissue over centimeter-scale distances [51].
Current imaging techniques, such as gadolinium-enhanced MRI, are now able to detect macroscopic scars, and to some extent intermediate fibrosis (gray zones) in the surrounding border zone. A recent proof-of-concept study combining imaging with computer simulations to identify catheter ablation sites showed encouraging results [20]. However, the ability to detect fibrosis on the submillimeter spatial scale most relevant to arrhythmogenesis has yet to be perfected [52]. In our view, further refinement of imaging technologies to quantify the extent and patterns of fibrosis, especially at the submillimeter scale, has great promise for improving arrhythmia risk assessment in both ischemic and nonischemic heart disease. We anticipate that improved fibrosis definition by cardiac imaging may someday outperform the ejection fraction as the major criterion for device therapy with an implantable cardioverter-defibrillator. Drugs or biological interventions which prevent progression of fibrosis, without compromising essential wound-healing functions, are also a particularly attractive therapeutic avenue to reduce arrhythmia risk in heart disease, particularly since currently recommended drugs for treatment of heart failure which have been shown to reduce sudden death risk, such as angiotensin-converting enzyme inhibitors [53] and aldosterone antagonists [54], are known to suppress fibrosis.
Highlights.
Fibrosis is essential for wound repair but can promote cardiac arrhythmias
Fibrosis creates a tissue substrate favoring initiation of reentry
Fibrosis alters source-sink relationship to promote premature ventricular complexes
Myofibroblasts can alter myocyte electrophysiology in a pro-arrhythmic manner
Together all three factors create a perfect storm for cardiac arrhythmias
Acknowledgments
Funding sources
This work was supported by the National Institutes of Health grant UCLA CTSI UL1TR000124, American Heart Association NCRP Scientist Development Grant, and the Lauren B. Leichtman and Arthur E. Levine Cardiovascular Discovery Fund Investigator Award (to TPN), and National Institutes of Health grant P01 HL078931, and the Laubisch and Kawata endowments (to JNW).
Footnotes
Abbreviations: CV, conduction velocity; Cx, connexin; DAD, delayed afterdepolarization; EAD, early afterdepolarization; ERP, effective refractory period; PVC, premature ventricular complex; VT, ventricular tachycardia
Disclosures
Thao P. Nguyen, None; Zhilin Qu, None; James N. Weiss, None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miragoli M, Salvarani N, Rohr S. Myofibroblasts induce ectopic activity in cardiac tissue. Circ Res. 2007;101:755–8. doi: 10.1161/CIRCRESAHA.107.160549. [DOI] [PubMed] [Google Scholar]
- 2.Zlochiver S, Munoz V, Vikstrom KL, Taffet SM, Berenfeld O, Jalife J. Electrotonic myofibroblast-to-myocyte coupling increases propensity to reentrant arrhythmias in two-dimensional cardiac monolayers. Biophys J. 2008;95:4469–80. doi: 10.1529/biophysj.108.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen TP, Xie Y, Garfinkel A, Qu Z, Weiss JN. Arrhythmogenic consequences of myofibroblast-myocyte coupling. Cardiovasc Res. 2012;93:242–51. doi: 10.1093/cvr/cvr292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pedrotty DM, Klinger RY, Kirkton RD, Bursac N. Cardiac fibroblast paracrine factors alter impulse conduction and ion channel expression of neonatal rat cardiomyocytes. Cardiovasc Res. 2009;83:688–97. doi: 10.1093/cvr/cvp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ Res. 2010;107:1011–20. doi: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaur K, Zarzoso M, Ponce-Balbuena D, Guerrero-Serna G, Hou L, Musa H, et al. TGF-beta1, Released by Myofibroblasts, Differentially Regulates Transcription and Function of Sodium and Potassium Channels in Adult Rat Ventricular Myocytes. PLoS One. 2013;8:e55391. doi: 10.1371/journal.pone.0055391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–26. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 8.Smith RS, Smith TJ, Blieden TM, Phipps RP. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am J Pathol. 1997;151:317–22. [PMC free article] [PubMed] [Google Scholar]
- 9.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 10.van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–7. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 11.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–13. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 12.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009;105:1164–76. doi: 10.1161/CIRCRESAHA.109.209809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chilton L, Ohya S, Freed D, George E, Drobic V, Shibukawa Y, et al. K+ currents regulate the resting membrane potential, proliferation, and contractile responses in ventricular fibroblasts and myofibroblasts. Am J Physiol Heart Circ Physiol. 2005;288:H2931–9. doi: 10.1152/ajpheart.01220.2004. [DOI] [PubMed] [Google Scholar]
- 14.de Jong S, van Veen TA, van Rijen HV, de Bakker JM. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011;57:630–8. doi: 10.1097/FJC.0b013e318207a35f. [DOI] [PubMed] [Google Scholar]
- 15.Gardner PI, Ursell PC, Fenoglio JJ, Jr, Wit AL. Electrophysiologic and anatomic basis for fractionated electrograms recorded from healed myocardial infarcts. Circulation. 1985;72:596–611. doi: 10.1161/01.cir.72.3.596. [DOI] [PubMed] [Google Scholar]
- 16.de Bakker JM, van Capelle FJ, Janse MJ, Tasseron S, Vermeulen JT, de Jonge N, et al. Slow conduction in the infarcted human heart. ‘Zigzag’ course of activation. Circulation. 1993;88:915–26. doi: 10.1161/01.cir.88.3.915. [DOI] [PubMed] [Google Scholar]
- 17.Spach MS, Dolber PC, Heidlage JF. Influence of the passive anisotropic properties on directional differences in propagation following modification of the sodium conductance in human atrial muscle. A model of reentry based on anisotropic discontinuous propagation. Circ Res. 1988;62:811–32. doi: 10.1161/01.res.62.4.811. [DOI] [PubMed] [Google Scholar]
- 18.Ten Tusscher KH, Panfilov AV. Influence of diffuse fibrosis on wave propagation in human ventricular tissue. Europace. 2007;9(Suppl 6):vi38–45. doi: 10.1093/europace/eum206. [DOI] [PubMed] [Google Scholar]
- 19.Qu Z, Karagueuzian HS, Garfinkel A, Weiss JN. Effects of Na+ channel and cell coupling abnormalities on vulnerability to reentry: a simulation study. Am J Physiol Heart Circ Physiol. 2004;286:H1310–21. doi: 10.1152/ajpheart.00561.2003. [DOI] [PubMed] [Google Scholar]
- 20.Ashikaga H, Arevalo H, Vadakkumpadan F, Blake RC, 3rd, Bayer JD, Nazarian S, et al. Feasibility of image-based simulation to estimate ablation target in human ventricular arrhythmia. Heart Rhythm. 2013;10:1109–16. doi: 10.1016/j.hrthm.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDowell KS, Arevalo HJ, Maleckar MM, Trayanova NA. Susceptibility to arrhythmia in the infarcted heart depends on myofibroblast density. Biophys J. 2011;101:1307–15. doi: 10.1016/j.bpj.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qu J, Volpicelli FM, Garcia LI, Sandeep N, Zhang J, Marquez-Rosado L, et al. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res. 2009;104:365–71. doi: 10.1161/CIRCRESAHA.108.184044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–62. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 25.Akar FG, Nass RD, Hahn S, Cingolani E, Shah M, Hesketh GG, et al. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H1223–30. doi: 10.1152/ajpheart.00079.2007. [DOI] [PubMed] [Google Scholar]
- 26.Duffy HS. The molecular mechanisms of gap junction remodeling. Heart Rhythm. 2012;9:1331–4. doi: 10.1016/j.hrthm.2011.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar R, Joyner RW. Calcium currents of ventricular cell pairs during action potential conduction. Am J Physiol. 1995;268:H2476–86. doi: 10.1152/ajpheart.1995.268.6.H2476. [DOI] [PubMed] [Google Scholar]
- 28.Pu J, Boyden PA. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res. 1997;81:110–9. doi: 10.1161/01.res.81.1.110. [DOI] [PubMed] [Google Scholar]
- 29.Dun W, Boyden PA. Diverse phenotypes of outward currents in cells that have survived in the 5-day-infarcted heart. Am J Physiol Heart Circ Physiol. 2005;289:H667–73. doi: 10.1152/ajpheart.00180.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rushton WAH. Initiation of the propagated disturbance. Proc R Soc B. 1937;124:210–43. [Google Scholar]
- 31.Fozzard HA, Schoenberg M. Strength-duration curves in cardiac Purkinje fibres: effects of liminal length and charge distribution. J Physiol. 1972;226:593–618. doi: 10.1113/jphysiol.1972.sp009999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohr S, Kucera JP, Fast VG, Kleber AG. Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science. 1997;275:841–4. doi: 10.1126/science.275.5301.841. [DOI] [PubMed] [Google Scholar]
- 33.Pollard AE, Cascio WE, Fast VG, Knisley SB. Modulation of triggered activity by uncoupling in the ischemic border. A model study with phase 1b-like conditions. Cardiovasc Res. 2002;56:381–92. doi: 10.1016/s0008-6363(02)00598-9. [DOI] [PubMed] [Google Scholar]
- 34.Huelsing DJ, Spitzer KW, Cordeiro JM, Pollard AE. Modulation of repolarization in rabbit Purkinje and ventricular myocytes coupled by a variable resistance. Am J Physiol. 1999;276:H572–81. doi: 10.1152/ajpheart.1999.276.2.H572. [DOI] [PubMed] [Google Scholar]
- 35.Saiz J, Ferrero JM, Jr, Monserrat M, Ferrero JM, Thakor NV. Influence of electrical coupling on early afterdepolarizations in ventricular myocytes. IEEE Trans Biomed Eng. 1999;46:138–47. doi: 10.1109/10.740876. [DOI] [PubMed] [Google Scholar]
- 36.Hoyt RH, Cohen ML, Saffitz JE. Distribution and three-dimensional structure of intercellular junctions in canine myocardium. Circ Res. 1989;64:563–74. doi: 10.1161/01.res.64.3.563. [DOI] [PubMed] [Google Scholar]
- 37.Peters NS, Wit AL. Myocardial architecture and ventricular arrhythmogenesis. Circulation. 1998;97:1746–54. doi: 10.1161/01.cir.97.17.1746. [DOI] [PubMed] [Google Scholar]
- 38.Tveito A, Lines GT. A condition for setting off ectopic waves in computational models of excitable cells. Math Biosci. 2008;213:141–50. doi: 10.1016/j.mbs.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–15. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plotnikov AN, Shlapakova I, Szabolcs MJ, Danilo P, Jr, Lorell BH, Potapova IA, et al. Xenografted adult human mesenchymal stem cells provide a platform for sustained biological pacemaker function in canine heart. Circulation. 2007;116:706–13. doi: 10.1161/CIRCULATIONAHA.107.703231. [DOI] [PubMed] [Google Scholar]
- 41.Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res. 2008;103:509–18. doi: 10.1161/CIRCRESAHA.108.176677. [DOI] [PubMed] [Google Scholar]
- 42.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res. 2003;93:421–8. doi: 10.1161/01.RES.0000089258.40661.0C. [DOI] [PubMed] [Google Scholar]
- 43.Kohl P, Camelliti P. Fibroblast-myocyte connections in the heart. Heart Rhythm. 2012;9:461–4. doi: 10.1016/j.hrthm.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Baum JR, Long B, Cabo C, Duffy HS. Myofibroblasts cause heterogeneous Cx43 reduction and are unlikely to be coupled to myocytes in the healing canine infarct. Am J Physiol Heart Circ Physiol. 2012;302:H790–800. doi: 10.1152/ajpheart.00498.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He K, Shi X, Zhang X, Dang S, Ma X, Liu F, et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc Res. 2011;92:39–47. doi: 10.1093/cvr/cvr189. [DOI] [PubMed] [Google Scholar]
- 46.Jacquemet V, Henriquez CS. Loading effect of fibroblast-myocyte coupling on resting potential, impulse propagation, and repolarization: insights from a microstructure model. Am J Physiol Heart Circ Physiol. 2008;294:H2040–52. doi: 10.1152/ajpheart.01298.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie Y, Garfinkel A, Camelliti P, Kohl P, Weiss JN, Qu Z. Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: A computational study. Heart Rhythm. 2009;6:1641–9. doi: 10.1016/j.hrthm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Issa ZFMJ, Zipes DP. A Companion to Braunwald’s Heart Disease. Elsevier Inc; 2009. Clinical Arrhythmology and Electrophysiology. [Google Scholar]
- 49.Saeed M, Link MS, Mahapatra S, Mouded M, Tzeng D, Jung V, et al. Analysis of intracardiac electrograms showing monomorphic ventricular tachycardia in patients with implantable cardioverter-defibrillators. Am J Cardiol. 2000;85:580–7. doi: 10.1016/s0002-9149(99)00815-2. [DOI] [PubMed] [Google Scholar]
- 50.Burstein B, Nattel S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–9. doi: 10.1016/j.jacc.2007.09.064. [DOI] [PubMed] [Google Scholar]
- 51.Ho SY, Sanchez-Quintana D, Cabrera JA, Anderson RH. Anatomy of the left atrium: implications for radiofrequency ablation of atrial fibrillation. J Cardiovasc Electrophysiol. 1999;10:1525–33. doi: 10.1111/j.1540-8167.1999.tb00211.x. [DOI] [PubMed] [Google Scholar]
- 52.Mewton N, Liu CY, Croisille P, Bluemke D, Lima JA. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011;57:891–903. doi: 10.1016/j.jacc.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohn JN, Johnson G, Ziesche S, Cobb F, Francis G, Tristani F, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med. 1991;325:303–10. doi: 10.1056/NEJM199108013250502. [DOI] [PubMed] [Google Scholar]
- 54.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–17. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]