

Abstract
Background
Bile acid synthesis is regulated by nuclear receptors including farnesoid X receptor (FXR) and small heterodimer partner (SHP), and by fibroblast growth factor15/19 (FGF15/19). Because bile acid synthesis involves amino acid conjugation, we hypothesized that hepatic cysteine sulfinic acid decarboxylase (CSAD) (a key enzyme in taurine synthesis) is regulated by bile acids.
Aims
To investigate CSAD regulation by bile acids and CSAD regulatory mechanisms.
Methods
Mice were fed a control diet or a diet supplemented with either 0.5% cholate or 2% cholestyramine. To gain mechanistic insight into CSAD regulation, we utilized GW4064 (FXR agonist), FGF19, or T-0901317 (LXR agonist) and Shp−/− mice. Tissue mRNA expression was determined by qRT-PCR. Amino acids were measured by HPLC.
Results
Mice supplemented with dietary cholate exhibited reduced hepatic CSAD mRNA expression while those receiving cholestyramine exhibited increased hepatic CSAD mRNA expression. Activation of FXR suppressed CSAD mRNA expression whereas hepatic CSAD mRNA expression was increased in Shp−/− mice. Hepatic hypotaurine concentration (the product of CSAD) was higher in Shp−/− mice with a corresponding increase in serum (but not hepatic) taurine-conjugated bile acids. FGF19 administration suppressed hepatic CYP7A1 mRNA but did not change CSAD mRNA expression. LXR activation induced CYP7A1 mRNA yet failed to induce CSAD mRNA expression.
Conclusion
CSAD mRNA expression is physiologically regulated by bile acids in a feedback fashion via mechanisms involving SHP and FXR but not FGF15/19 or LXR. These novel findings implicate bile acids as regulators of CSAD mRNA via mechanisms shared in part with CYP7A1.
Keywords: Nuclear hormone receptors, taurine synthesis, bile acid metabolism, bile acid conjugation
Introduction
Hepatic bile acid synthesis involves coordinated hydroxylation of the nucleus and oxidation of the cholesterol side chain followed by amino acid conjugation, involving taurine or glycine (1). The initial step in the major pathway of bile acid synthesis is the enzymatic addition of a hydroxyl group to carbon 7 on the B-ring of cholesterol by cholesterol 7-alpha-hydroxylase (CYP7A1). In all, as many as 16 enzymes catalyze 17 steps in bile acid synthesis, with mutations in at least nine enzymes identified as a cause of human disease (1). CYP7A1 gene expression is tightly controlled in a feedback fashion by nuclear receptors farnesoid X receptor (FXR, NR1H4) (2–5) and small heterodimer partner (SHP, NR0B2) (6–8), and by fibroblast growth factor-15/19 (9). This feedback regulation functions to maintain hepatic cholesterol homeostasis, maintain the enterohepatic bile salt pool and also to protect the liver from bile acid toxicity. Mice lacking FXR or SHP are more sensitive to bile acid induced liver injury, manifested by elevated serum aminotransferases (2, 7, 10, 11) and death. Hepatotoxicity in FXR-null mice has been associated with alterations in the ratio of taurine conjugated and unconjugated bile acids in bile (10).
Several studies suggest that the pathways controlling bile acid synthesis play a wide role in regulating hepatic metabolism. For example, FGF15/19 is an insulin-independent regulator of post-prandial hepatic protein and glycogen synthesis (12, 13). In addition, FXR has been shown to have broad effects on triglyceride and cholesterol metabolism as well as hepatic fibrogenesis (14) whereas SHP has been implicated in lipid and glucose metabolism (15). In-vitro and in-vivo studies using the synthetic FXR agonist GW4064 also demonstrate that bile acid-amino acid conjugation is an FXR-regulated process (16) suggesting that coordinated amino acid bile acid conjugation is required for metabolic homeostasis. Nevertheless, the mechanisms controlling hepatic taurine synthesis and availability are poorly understood.
Because bile acid signaling pathways regulate bile acid synthesis and also its conjugation to taurine, we hypothesized that hepatic synthesis of taurine is also tightly regulated. Here we examined transcriptional regulation of cysteine sulfinic acid decarboxylase (CSAD) the enzyme responsible for generation of taurine from cysteinesulfinic acid in liver. We reasoned that hepatic CSAD mRNA expression is controlled by bile acids in a feedback fashion and that CSAD gene expression utilizes regulatory mechanisms shared with cholesterol 7-alpha-hydroxylase.
Experimental Procedures
Animals and Treatments
C57Bl/6J male mice (WT), age 8–12 weeks, from Jackson Labs (Bar Harbor, ME, USA) were housed on a standard 12 hour light cycle in a specific pathogen-free facility at Washington University in St. Louis and were maintained on a cereal-based diet (PicoLab Rodent Diet 20, St. Louis, MO) containing 4.5% total lipid (w/w) and 141 ppm cholesterol (w/w) with free access to food and water, unless otherwise noted. Shp−/− male mice age 10–12 week old mice were generated as described (8), fed a control diet (Harlan Teklad 2020X, Houston, TX) and tissue (along with WT control tissue) was provided for analysis in St. Louis. At the time of sacrifice, tissues were flash frozen in liquid nitrogen and stored at −80°C for later analysis. All animal protocols were approved by the Washington University Animal Studies Committee and conformed to the criteria outlined in the National Institutes of Health “Guide for the Care and Use of Laboratory Animals”.
Bile acid and cholestyramine feeding studies
Experimental diets consisted of control diet supplemented (where indicated) with powdered 0.25% cholate (CA) (Sigma, St. Louis, MO, USA), 0.5% (w/w) CA, or with 2% cholestyramine (Sigma, St. Louis, MO, USA). Mice were fed the assigned diet for 5 days. On the morning of sacrifice, mice were fasted for 4 hours.
FXR agonist treatment
12 week old WT male mice were gavaged with 100 mg/kg body weight of the selective synthetic FXR agonist GW4064 (Tocris Bioscience, Minneapolis, MN, USA; 2473) with corn oil or corn oil alone (vehicle) daily for 5 days. On the morning of sacrifice, mice were fasted for 2 hr, then gavaged with GW4064 or vehicle. Two hours later the mice were sacrificed (2, 17).
LXR agonist treatment
8–10 week old WT male mice were gavaged daily with either 25 mg/kg body weight of the selective synthetic LXR agonist (T-0901317 dissolved in dimethyl sulfoxide and Chremophor) in 5% mannitol/water to a final concentration of 2.5mg/ml (Cayman Chemical Company, Ann Arbor, MI) or vehicle alone (dimethyl sulfoxide and Chremophor in 5% mannitol/water) for 7 days. On the morning of sacrifice mice were gavaged with T-0901317. Four hours later the mice were sacrificed.
FGF19 treatment
13 week old WT male mice were treated by intraperitoneal injection with 1 mg/kg mouse body weight of FGF19 (PA-0273; Bioclone Inc, San Diego, CA, USA) in PBS or vehicle (PBS) and then sacrificed six hours later (18).
mRNA measurements
RNA extraction from liver and other organs was performed using Trizol (Ambion, Grand Island, NY, USA, 15596026). Extracted RNA was treated with RNase-free DNase (Ambion, Grand Island, NY, USA, AM1906) and reverse-transcribed using random hexamers (Applied Biosystems, Carisbad, CA, USA. GAPDH primer sequences were: 5′-TGTGTCCGTCGTGGATCTGA-3′ 5′-CCTGCTTCACCACCTTCTTGA-3′. CSAD primer sequences were as follows: 5′-GGGACTTGGCACCGACAGT-3′ 5′-GGGATCATCCTCCCTCTCTCA-3′. Additional primer sequences are available upon request. Real-time quantitative PCR (qRT-PCR) was performed using SYBR Green PCR Master Mix (Applied Biosystems, Carisbad, CA, USA) on a Step One Plus Sequence Detection System (Applied Biosystems, Carisbad, CA, USA). Relative mRNA fold changes were determined using standard deltaCt calculations. All real-time quantitative PCR data were generated using RNA isolated from tissues of individual animals.
Western blotting
Mouse liver was homogenized and 30 μg of total protein was electrophoresed by denaturing 10% (w/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Separated proteins were transferred to a polyvinlylidene difluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA, USA), which was probed in standard fashion with GAPDH antibody (1:1000) (Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA) or CYP7A1 antibody (1:200) a generous gift from Dr. Simon Hui (San Diego State University).
Serum Lipid Analysis
Biochemical analysis of serum triglyceride and cholesterol concentrations were performed using kits obtained from Wako Chemical USA (Richmond, VA).
Taurine and Hypotaurine Measurement
Serum was acidified with sulfosalicylic acid (SSA) to a final concentration of 5% (w/v). 0.1 g liver tissue from WT or SHP knockout mice was homogenized in 400μl of 6% (w/v) SSA. Homogenate was centrifuged at 16,000 g × 20mins to obtain the acid supernatant. Supernatants were diluted in 200mM borate buffer, pH 10.4, derivatized with o-phthaldialdehyde (OPA) and analyzed by HPLC as previously described (19, 20).
Liver and Serum Bile Acid Measurements
Liver and serum was collected from age matched male 12 week old WT (C57BL/6) or SHP knockout mice. Serum and liver bile acid composition was measured by HPLC as previously described (21, 22).
Data and statistical analysis
Statistical significance was determined with an unpaired, two-tailed Student’s t-test. Data are expressed as the mean ± standard error (SE). Differences in mRNA and protein levels are stated as fold change compared to control group, set at 1.0, unless otherwise noted.
Results
CSAD mRNA distribution
CSAD mRNA expression was surveyed in multiple tissues from C57BL/6 mice, the findings demonstrating highest levels in liver, kidney, white adipose tissue and lung (Fig. 1).
Figure 1.

CSAD mRNA Tissue Distribution
*n=4–5/tissue, p<0.05 for all tissues vs. liver
Bile acid feeding suppresses whereas cholestyramine treatment upregulates hepatic CSAD but not CDO mRNA expression
C57BL/6 mice were fed a chow diet supplemented with 0.25% or 0.5% CA or with 2% cholestyramine. As a positive control for bile acid dependent gene regulation, we measured changes in hepatic CYP7A1 mRNA. CYP7A1 mRNA expression was suppressed in the 0.25% and 0.5% cholate-fed mice (0.08±0.03, p=0.007, 0.09±0.04, p=0.007 respectively) compared to chow-fed control mice (Fig. 2 A). In addition, cholate feeding also led to a dose-dependent suppression of hepatic CSAD mRNA expression in the 0.25% cholate-fed mice (0.23±0.04, p=0.003), and 0.5% cholate-fed mice (0.13±0.02, p=0.001) compared to chow-fed controls (Fig. 2 B). By contrast, bile acid depletion mediated by 2% cholestyramine feeding resulted in significantly higher expression of both CYP7A1 and CSAD mRNAs (4.35±0.65, p=0.001, 2.23±0.28, p=0.006 respectively) compared with control mice (Fig. 2 A, B). Western blotting confirmed that differences in CYP7A1 mRNA level were associated with altered protein levels (Fig. 2 E). As a positive control, we observed a robust increase in hepatic SHP mRNA expression in both 0.25% and 0.5% cholic acid-fed mice (2.26±0.24, p=0.002, 2.23±0.27, p=0.004 respectively) whereas 2% cholestyramine feeding led to reduced hepatic SHP mRNA expression (0.44±0.08, p=0.007) (Fig. 2 D). We observed that 0.25% cholate supplementation led to a 27% decrease in serum triglyceride (TG) levels (p±0.04) (Fig. 2 F), but overall we observed no significant differences in serum triglyceride or cholesterol (TC) between the control and dietary supplemented feeding groups. These findings suggest that alterations in serum lipids are unlikely to be the mechanism for the observed alterations in bile acid synthetic pathways.
Figure 2.
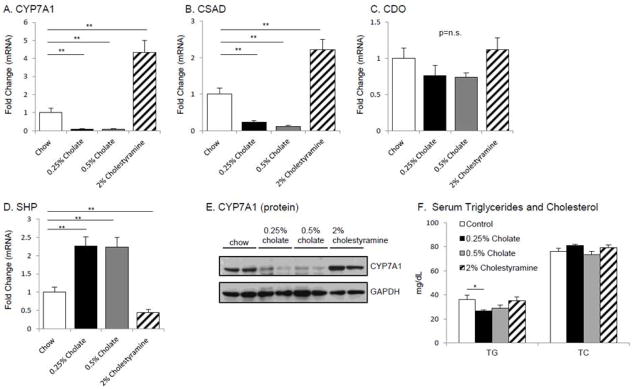
CSAD mRNA regulation by dietary bile acids
We also examined the abundance of mRNA encoding hepatic cysteine dioxygenase (CDO), an enyzyme upstream of CSAD in taurine synthesis. We observed no difference in CDO mRNA expression in 0.25% or 0.5% cholate-fed, or 2% cholestyramine-fed mice (0.76±0.14, p=0.27, 0.74±0.06, p=0.13, and 1.12±0.16, p=0.59 respectively) (Fig. 2 C). These findings suggest that changes in taurine synthesis in the setting of altered bile acid metabolism are being exerted at the level of CSAD expression.
FXR agonist treatment suppresses hepatic CSAD mRNA level
GW4064, a synthetic FXR agonist, is known to suppress hepatic CYP7A1 and CYP8B1 mRNA via FXR and SHP (23, 24). We next examined the role of FXR signaling in taurine synthesis, by treating C57BL/6 mice with either vehicle or GW4064. FXR agonist administration resulted in suppression of CYP7A1 and CYP8B1 mRNA (0.06±0.03, p=0.10 and 0.07±0.03, p=0.03 respectively) compared to control mice (Fig. 3 A). Similarly, hepatic CSAD mRNA abundance was potently suppressed by GW4064 treatment (0.25±0.01, p=0.01) compared to vehicle-treated controls. By contrast, no significant decrease was observed in hepatic CDO mRNA abundance (0.82±0.13, p=0.4833) following GW4064 treatment (Fig. 3 A).
Figure 3.

FXR Regulation of Hepatic and Renal mRNA Levels
We also examined the expression of mRNAs encoding two other genes involved in bile acid conjugation. Bile acid-CoA:amino-acid N-acyltransferase (BAT) mRNA abundance was reduced in GW4064 treated mice (0.55±0.10, p=0.031), while bile acid-CoA synthetase (BACS) mRNA abundance was not significantly changed with GW4064 treatment (0.86±0.14, p=0.61) (Fig. 3 B). In addition, we found no difference in CSAD mRNA abundance in kidney between control and GW4064 treated mice (1.05±0.12) (Fig. 3 C).
Shp−/− mice manifest higher hepatic CSAD mRNA abundance
Previous data have implicated the nuclear receptor SHP in the regulation of CYP7A1 and CYP8B1 expression (6–8). We found CYP7A1 and CYP8B1 mRNA expression was increased in Shp−/− mice (5.90±0.86, p=0.0002, 2.23±0.20, p=0.0003, respectively, Fig. 4 A). We also found that hepatic CSAD mRNA expression was increased in Shp−/− mice (8.49±0.25, p<0.0001, Fig. 4 A), with no difference in hepatic CDO (Fig. 4 A), BAT or BACS mRNA levels (0.96±0.01, p=0.33 and 0.91±0.08, p=0.45, respectively) (Fig. 4 B). In addition, renal CSAD mRNA abundance was not altered (0.95±0.11, p=0.76) in Shp−/− mice (Fig. 4 C).
Figure 4.
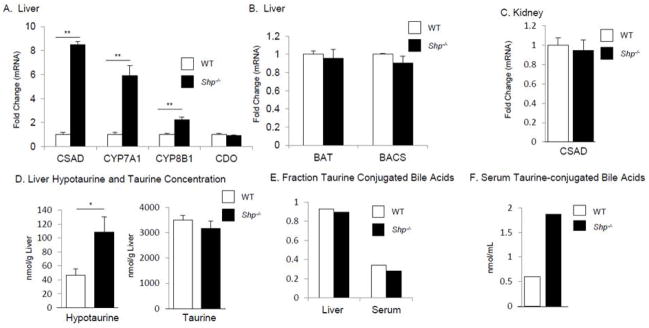
SHP regulation of Hepatic and Renal mRNA Levels and Taurine Metabolism
To explore the biochemical significance of the elevated levels of CSAD mRNA we measured hypotaurine concentrations, the immediate product of CSAD activity. We observed a 2.3 fold elevation in hepatic hypotaurine concentration in Shp−/− mice compared to WT controls (WT 46.4 nmol/g vs. Shp−/− 108.5 nmol/g, p=0.034) (Fig. 4 D). However we did not observe changes in either hepatic (Fig. 4 D) or serum (data not shown) taurine content in Shp−/− mice.
The bile acid pool composition of Shp−/− mice has been previously investigated and includes primarily an increase in cholate (7, 22) as well as a shift in the α-muricholate vs. β-muricholate fraction (22). Measurement of taurine conjugates in liver and serum revealed no difference in the fraction of bile acids that were taurine conjugated (Fig. 4 E). However, we observed increased concentrations of tauro-conjugated bile acids in serum (Fig. 4 F) but not in liver (data not shown).
FGF19 administration differentially regulates CYP7A1 and CSAD mRNA abundance
FGF15/19 is produced by ileal enterocytes and acts in the liver via fibroblast growth factor 4 receptor (FGF4R)/beta klotho to regulate expression of the CYP7A1 gene (25, 26) Hepatic CYP7A1 and CYP8B1 mRNA levels were suppressed in FGF19-treated mice (0.06±0.03, p=0.0001, 0.50±0.13, p=0.005) compared to vehicle-injected control mice (Fig. 5 A). By contrast, hepatic CSAD mRNA abundance was not altered by FGF19 treatment (1.03±0.35, p=0.94). In addition, CDO mRNA abundance in liver and CSAD mRNA abundance in kidney were no different in FGF19 -treated mice (1.14±0.12, p=0.34, 0.98±0.08, p=0.25, respectively) (Fig. 5 A, B).
Figure 5.

FGF19 and LXR Regulation of Hepatic and Renal mRNA Levels
LXR activation differentially regulates CYP7A1 and CSAD mRNA abundance
Excess cholesterol and/or oxysterols in the liver act via the nuclear receptor LXRα to increase CYP7A1 mRNA transcription, resulting in accelerated catabolism of cholesterol to bile acids (27, 28). C57BL/6 mice were gavaged with T-0901317 (a synthetic LXR agonist) for 7 days. We observed that hepatic CYP7A1 mRNA expression was increased in mice treated with T-0901317 (3.4±0.75, p=0.015), but we observed no differences in CSAD mRNA abundance (Fig. 5 C). These findings suggest that LXR mediated activation of bile acid synthesis and CSAD mRNA expression are not coupled.
Discussion
The central findings of this study show that CSAD, a key enzyme in hepatic taurine synthesis, is expressed abundantly in mouse liver and is physiologically regulated by bile acids in both a feedback and tissue-specific fashion. Our novel findings suggest that bile acid regulation of CSAD involves the nuclear hormone receptors SHP and FXR but not FGF19 or LXR. These findings extend our understanding of the integrated regulation of bile acid metabolism beyond the well-established mechanisms of CYP7A1 regulation by bile acids, SHP, FXR, FGF19 and LXR (1). The findings permit us to conclude that bile acid regulation of CSAD gene expression occurs via mechanisms and pathways that are shared, at least in part, with those that regulate the expression of CYP7A1. These new findings suggest a working model for CSAD mRNA regulation in liver (Fig. 6), elements of which are discussed in more detail below.
Figure 6.
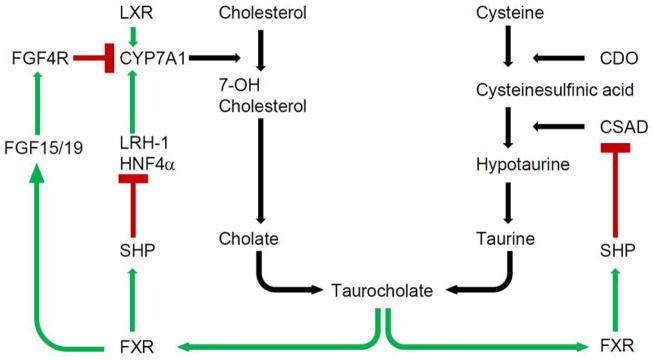
Integrated regulation of hepatic taurocholate metabolism and cysteine sulfinic acid decarboxylase (CSAD) mRNA expression by farnesoid X receptor (FXR) and small heterodimer partner (SHP) (green = promotion or stimulation, red = inhibition). CDO, cysteine dioxygenase; CYP7A1, cholesterol 7-α-hydroxylase; CYP8B1, sterol 12-α-hydroxylase; FGF, fibroblast growth factor; LXR, liver X receptor.
Taurine is the product of cysteine metabolism. Cystine is oxidized to cystine sulfinic acid (CSA) by cysteine dioxygenase (CDO) (29, 30). CSA is then decarboxylated by cysteine sulfinic acid decarboxylase (CSAD) to form hypotaurine, which is oxidized to taurine (31) (Fig. 6). CSAD was first identified in the liver (32, 33) and regulates the partitioning of cysteine sulfinic acid to taurine synthesis (30, 31). Since then, it has been demonstrated that CSAD is expressed not only in liver, but also in kidney (29, 34), brain (35) and male reproductive organs (36, 37). Here we observed that CSAD mRNA abundance was highest in liver and kidney, and that CSAD mRNA was also detected in white adipose tissue, lung, gallbladder, and testis in C57BL/6 mice. To date, limited data is available regarding the factors controlling hepatic CSAD mRNA transcription. Dietary supplementation with sulfur-containing amino acids decreases both CSAD mRNA and enzyme activity (38). Earlier dietary studies using rats suggested that hepatic CSAD enzyme activity, hepatic taurine content, and urinary taurine content was regulated by a variety of dietary components including bile acids, cholesterol, and soluble and insoluble fibers (39). We hypothesized that, because of the importance of taurine in bile acid metabolism, CSAD would be tightly regulated by bile acids at the transcriptional level and share canonical bile acid synthesis regulatory mechanisms with some of the enzymes under bile acid transcriptional control (for example CYP7A1). Our findings reveal potent transcriptional regulation of hepatic but not renal CSAD by enterohepatic bile acids and implicate the nuclear receptors SHP and FXR in this regulatory process (Fig. 6).
Bile acid feedback inhibition of CYP7A1 has been studied for decades. Nevertheless, previous studies have primarily focused on regulation of cholesterol conversion to cholate and other bile acids. By contrast, few studies have investigated the role of bile acid feedback inhibition of taurine synthesis, the “other half” of taurocholate. Our findings demonstrate potent bile acid-mediated suppression of hepatic CSAD mRNA levels and induction with cholestyramine-induced enterohepatic bile acid depletion (Fig. 2 B). Higher CSAD mRNA abundance with cholestyramine feeding suggests that, under physiologic conditions, enterohepatic bile acids exert tonic suppression of CSAD mRNA levels and that CSAD mRNA is not simply suppressed by bile acids at supraphysiologic concentrations. Though we did not directly measure the impact of cholesterol feeding on hepatic CSAD mRNA levels, the lack of response to LXR agonist treatment (T-0901317) (Fig. 5 C) suggests that alterations in cholesterol flux likely would not regulate CSAD mRNA via LXR at the transcriptional level.
FXR and SHP play a canonical role in regulating cholate synthesis (1). Physiologic activation of FXR by enterohepatic bile acids induces expression of SHP, which in turn binds to the orphan nuclear receptors LHR-1 and HNF4α, and potentially other promoter-bound elements, inhibiting transcription of CYP7A1 (1). Previous studies have demonstrated that CYP7A1 gene expression is decreased by FXR agonist treatment (24) and is higher in both Fxr−/− and Shp−/− mice (2, 7, 8, 24) in which the feedback loop has been genetically disrupted. In the current study, we utilize both pharmacologic and genetic approaches to establish that SHP and FXR are also key components of bile-acid mediated suppression of CSAD mRNA level. A role for FXR was established by the finding that GW4064, a FXR agonist, potently suppresses hepatic CSAD mRNA levels (Fig. 3 A). A role for SHP in this feedback loop was established by the finding that CSAD abundance is dramatically increased in Shp−/− mice (Fig. 4 A). Taken together, these results demonstrate that hepatic CSAD mRNA abundance is regulated through genetic mechanisms shared with CYP7A1 (Fig. 6).
Though FXR and SHP are central to CYP7A1 gene expression, the existence of a SHP-independent pathway has been demonstrated by using Shp−/− mice (7, 8). This is now understood to include the FGF15/19 pathway and signaling via. FGFR4/ beta klotho with activation of c-Jun N-terminal kinase (JNK) (9, 25) and transcriptional repression of CYP7A1. Inagaki et al. showed that the FGF15/FGFR4 signaling cooperates with SHP to repress CYP7A1 in liver although FGF19 reduced CYP7A1 mRNA levels without increasing SHP mRNA levels (9) indicating that the FXR/FGF19 pathway is also SHP-independent. The current findings indicate that hepatic CSAD mRNA level is not regulated by FGF19 administration despite potent suppression of CYP7A1 level (Fig. 5 A). In addition, activation of LXR did not result in altered hepatic CSAD mRNA abundance. This divergence in response implies that while CYP7A1 and CSAD mRNA respond similarly to dietary bile acid supplementation, and are both regulated by FXR and SHP, there are important differences in some of the mediators involved. The data further suggest that pharmacologic manipulation of hepatic metabolism via FXR and LXR may have variable effects on genes that are physiologically important for bile acid metabolism.
We also investigated whether pharmacologic interventions that modulated hepatic CSAD mRNA abundance also mediated changes in renal CSAD mRNA abundance. We found that FXR agonist treatment did not alter CSAD abundance in kidney. Furthermore, renal CSAD abundance was not altered in Shp−/− mice. These data suggest that there are liver-specific regulatory components controlling hepatic CSAD response to bile acids, and that these are possibly missing or differentially regulated in kidney. It will be interesting to examine candidate liver-specific components for CSAD transcriptional regulation since both FXR and SHP (though at low levels) are expressed in kidney (40, 41). The possibility of extrahepatic CSAD regulation by FXR agonists should be kept in mind as human clinical trials are underway using pharmacologic FXR agonists.
Bile acid-CoA:amino-acid N-acyltransferase (BAT), and bile acid-CoA synthetase (BACS), two enzymes involved in bile acid conjugation to taurine and glycine, have been demonstrated to undergo FXR-dependent regulation via a promoter inverted repeat (IR-1) (16) In the present study, we observed that pharmacologic FXR agonist activation reduced hepatic BAT but not BACS mRNA expression. In addition, BAT and BACS mRNA abundance were similar in WT and Shp−/− mice. Taken together, these data suggest that BAT and BACS are not potently regulated by nuclear receptors SHP and FXR and neither gene appears to be regulated in tandem with CSAD.
We were surprised to find that, despite >8-fold elevation CSAD in Shp−/− mice, hepatic hypotaurine was only 2–3 fold elevated, and hepatic taurine content did not differ from WT controls. Furthermore, we could not detect a genotype-dependent difference in serum taurine or hypotaurine levels. One explanation could be that the excess taurine generated in the liver is partially utilized to conjugate the excess bile acids produced in Shp−/− mice (7, 8). Because murine bile acids are primarily taurine conjugated (42), we would not expect a substantial alteration in the ratio of taurine: glycine-conjugation of bile acids in Shp−/− mice. In other words, while the fraction of tauro-conjugated bile acids did not differ between WT and Shp−/− mice, the overall concentration of serum tauroconjugated bile acids was elevated in Shp−/− mice because of the increased total bile acid recirculating pool in these mice (Fig. 4 F). It is also possible that additional homeostatic mechanism such as CSAD substrate availability and altered urinary excretion act to modulate any changes in taurine concentrations that could occur in Shp−/− mice. These data suggest the need for further studies of amino acid homeostasis under conditions where bile acid metabolism is perturbed. Furthermore, since taurine is important in non-hepatic tissues (such as in the brain where it serves as an osmolyte) (43), future studies could examine whether the genetic mechanisms controlling CSAD in liver contribute to CSAD control in brain and other tissues.
We recognize several limitations of these data. First, we were not able to measure CSAD protein level because we could not procure an appropriate antibody for western blotting analysis. Second, we did not measure CSAD activity, which we presumed to mirror CSAD mRNA expression (38). It has been reported that brain CSAD activity is activated by phosphorylation and inhibited by dephosphorylation (44). It is intriguing to consider that hepatic CSAD may be additionally controlled at the protein level by phosphorylation status. This could potentially provide a non-transcriptional mechanism for FGF19 to control CSAD activity and taurine availability.
Though the clinical significance of these data remain to be determined, it is worth noting that FXR agonists are currently under development for treatment of non-alcoholic fatty liver disease (NAFLD) (45). The current observation that FXR agonists alters a key enzyme in taurine metabolism suggests that lipid independent consequences of FXR activation be carefully considered in this area of drug development.
In summary, we have demonstrated that hepatic CSAD mRNA abundance is controlled by bile acids in a feedback fashion via mechanisms that include FXR and SHP, but not FGF15/19 or LXR (Fig. 6). We speculate that the coordinate regulation of cholate and taurine availability via shared mechanisms may provide a defense against unconjugated bile acid-induced hepatotoxicity. Nevertheless, the data also expand the range of targets and metabolic consequences for pharmacologic FXR agonists now in clinical development.
Acknowledgments
FINANCIAL SUPPORT
This work was supported in part through awards to TAK (AGA-FFTA); Grants HL-38180; DK-56260, DDRCC DK-52574, to NOD and DK-056649 to MHS.
The authors would like to thank Dr. David W. Russell for his insightful comments that contributed to this project.
ABBREVIATIONS
- FXR
Farnesoid X Receptor
- SHP
Small heterodimer partner
- FGF15/19
Fibroblast growth factor 15/19
- CSAD
Cysteine sulfinic acid decarboxylase
- LXR
Liver X Receptor
- CYP7A1
Cholesterol 7-alpha-hydroxylase
- CYP8B1
Sterol 12-alpha-hydroxylase
- CA
Cholic acid
- SSA
sulfosalicylic acid
- CDO
Cysteine dioxygenase
- BAT
bile acid CoA: amino acid N-acyltransferase
- BACS
bile acid-CoA synthetase
- FGFR4
Fibroblast growth factor receptor 4
- T-compound
T-0901317
Footnotes
| Experimentation | TAK, YM, HM, YX, LLH, SA, SK, MK |
| Preparation of manuscript | TAK, YM, HM, MHS, SA, NOD |
| Provision of Shp−/− tissue samples | SA, DDM |
References
- 1.Russell DW. Fifty years of advances in bile acid synthesis and metabolism. J Lipid Res. 2009;50 (Suppl):S120–125. doi: 10.1194/jlr.R800026-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 3.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 4.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 6.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 7.Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, Shan B, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell. 2002;2:713–720. doi: 10.1016/s1534-5807(02)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, Chua SS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 9.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Miyata M, Tozawa A, Otsuka H, Nakamura T, Nagata K, Gonzalez FJ, Yamazoe Y. Role of farnesoid X receptor in the enhancement of canalicular bile acid output and excretion of unconjugated bile acids: a mechanism for protection against cholic acid-induced liver toxicity. J Pharmacol Exp Ther. 2005;312:759–766. doi: 10.1124/jpet.104.076158. [DOI] [PubMed] [Google Scholar]
- 11.Park YJ, Qatanani M, Chua SS, LaRey JL, Johnson SA, Watanabe M, Moore DD, et al. Loss of orphan receptor small heterodimer partner sensitizes mice to liver injury from obstructive cholestasis. Hepatology. 2008;47:1578–1586. doi: 10.1002/hep.22196. [DOI] [PubMed] [Google Scholar]
- 12.Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, Xu HE, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331:1621–1624. doi: 10.1126/science.1198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kir S, Kliewer SA, Mangelsdorf DJ. Roles of FGF19 in Liver Metabolism. Cold Spring Harb Symp Quant Biol. 2011 doi: 10.1101/sqb.2011.76.010710. [DOI] [PubMed] [Google Scholar]
- 14.Fuchs M. Non-alcoholic Fatty liver disease: the bile Acid-activated farnesoid × receptor as an emerging treatment target. J Lipids. 2012;2012:934396. doi: 10.1155/2012/934396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta. 2011;1812:893–908. doi: 10.1016/j.bbadis.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pircher PC, Kitto JL, Petrowski ML, Tangirala RK, Bischoff ED, Schulman IG, Westin SK. Farnesoid X receptor regulates bile acid-amino acid conjugation. J Biol Chem. 2003;278:27703–27711. doi: 10.1074/jbc.M302128200. [DOI] [PubMed] [Google Scholar]
- 17.Jung D, Inagaki T, Gerard RD, Dawson PA, Kliewer SA, Mangelsdorf DJ, Moschetta A. FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J Lipid Res. 2007;48:2693–2700. doi: 10.1194/jlr.M700351-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, Suino-Powell K, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab. 13:729–738. doi: 10.1016/j.cmet.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ueki I, Roman HB, Valli A, Fieselmann K, Lam J, Peters R, Hirschberger LL, et al. Knockout of the murine cysteine dioxygenase gene results in severe impairment in ability to synthesize taurine and an increased catabolism of cysteine to hydrogen sulfide. Am J Physiol Endocrinol Metab. 2011;301:E668–684. doi: 10.1152/ajpendo.00151.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominy JE, Jr, Hirschberger LL, Coloso RM, Stipanuk MH. Regulation of cysteine dioxygenase degradation is mediated by intracellular cysteine levels and the ubiquitin-26 S proteasome system in the living rat. Biochem J. 2006;394:267–273. doi: 10.1042/BJ20051510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakakura H, Suzuki M, Kimura N, Takeda H, Nagata S, Maeda M. Simultaneous determination of bile acids in rat bile and serum by high-performance liquid chromatography. J Chromatogr. 1993;621:123–131. doi: 10.1016/0378-4347(93)80087-k. [DOI] [PubMed] [Google Scholar]
- 22.Anakk S, Watanabe M, Ochsner SA, McKenna NJ, Finegold MJ, Moore DD. Combined deletion of Fxr and Shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J Clin Invest. 2011;121:86–95. doi: 10.1172/JCI42846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem. 2000;43:2971–2974. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 25.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, Deng CX, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275:15482–15489. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 27.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 28.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 29.Stipanuk MH, Londono M, Lee JI, Hu M, Yu AF. Enzymes and metabolites of cysteine metabolism in nonhepatic tissues of rats show little response to changes in dietary protein or sulfur amino acid levels. J Nutr. 2002;132:3369–3378. doi: 10.1093/jn/132.11.3369. [DOI] [PubMed] [Google Scholar]
- 30.Tappaz ML. Taurine biosynthetic enzymes and taurine transporter: molecular identification and regulations. Neurochem Res. 2004;29:83–96. doi: 10.1023/b:nere.0000010436.44223.f8. [DOI] [PubMed] [Google Scholar]
- 31.Huxtable RJ. Physiological actions of taurine. Physiol Rev. 1992;72:101–163. doi: 10.1152/physrev.1992.72.1.101. [DOI] [PubMed] [Google Scholar]
- 32.Hope DB. Pyridoxal phosphate as the coenzyme of the mammalian decarboxylase for L-cysteine sulphinic and L-cysteic acids. Biochem J. 1955;59:497–500. doi: 10.1042/bj0590497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobsen JG, Thomas LL, Smith LH., Jr Properties and Distribution of Mammalian L-Cysteine Sulfinate Carboxy-Lyases. Biochim Biophys Acta. 1964;85:103–116. doi: 10.1016/0926-6569(64)90171-3. [DOI] [PubMed] [Google Scholar]
- 34.Reymond I, Bitoun M, Levillain O, Tappaz M. Regional expression and histological localization of cysteine sulfinate decarboxylase mRNA in the rat kidney. J Histochem Cytochem. 2000;48:1461–1468. doi: 10.1177/002215540004801103. [DOI] [PubMed] [Google Scholar]
- 35.Tappaz M, Bitoun M, Reymond I, Sergeant A. Characterization of the cDNA coding for rat brain cysteine sulfinate decarboxylase: brain and liver enzymes are identical proteins encoded by two distinct mRNAs. J Neurochem. 1999;73:903–912. doi: 10.1046/j.1471-4159.1999.0730903.x. [DOI] [PubMed] [Google Scholar]
- 36.Li JH, Ling YQ, Fan JJ, Zhang XP, Cui S. Expression of cysteine sulfinate decarboxylase (CSD) in male reproductive organs of mice. Histochem Cell Biol. 2006;125:607–613. doi: 10.1007/s00418-005-0095-8. [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Wu G, Feng Y, Sun C, Lin S, Hu J. CSD mRNA expression in rat testis and the effect of taurine on testosterone secretion. Amino Acids. 2010;39:155–160. doi: 10.1007/s00726-009-0388-7. [DOI] [PubMed] [Google Scholar]
- 38.Bella DL, Hahn C, Stipanuk MH. Effects of nonsulfur and sulfur amino acids on the regulation of hepatic enzymes of cysteine metabolism. Am J Physiol. 1999;277:E144–153. doi: 10.1152/ajpendo.1999.277.1.E144. [DOI] [PubMed] [Google Scholar]
- 39.Ide T. Dietary regulation of hepatic enzymes in taurine biosynthesis in rats. Nutritional Biochemistry. 1998;9:99–105. [Google Scholar]
- 40.Johansson L, Thomsen JS, Damdimopoulos AE, Spyrou G, Gustafsson JA, Treuter E. The orphan nuclear receptor SHP inhibits agonist-dependent transcriptional activity of estrogen receptors ERalpha and ERbeta. J Biol Chem. 1999;274:345–353. doi: 10.1074/jbc.274.1.345. [DOI] [PubMed] [Google Scholar]
- 41.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 42.Alnouti Y, Csanaky IL, Klaassen CD. Quantitative-profiling of bile acids and their conjugates in mouse liver, bile, plasma, and urine using LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;873:209–217. doi: 10.1016/j.jchromb.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butterworth RF. Taurine in hepatic encephalopathy. Adv Exp Med Biol. 1996;403:601–606. doi: 10.1007/978-1-4899-0182-8_66. [DOI] [PubMed] [Google Scholar]
- 44.Tang XW, Hsu CC, Schloss JV, Faiman MD, Wu E, Yang CY, Wu JY. Protein phosphorylation and taurine biosynthesis in vivo and in vitro. J Neurosci. 1997;17:6947–6951. doi: 10.1523/JNEUROSCI.17-18-06947.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adorini L, Pruzanski M, Shapiro D. Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov Today. 2012;17:988–997. doi: 10.1016/j.drudis.2012.05.012. [DOI] [PubMed] [Google Scholar]