

Abstract
Kainate receptors containing the GluK1 subunit have an impact on excitatory and inhibitory neurotransmission in brain regions, such as the amygdala and hippocampus, which are relevant to seizures and epilepsy. Here we used 2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid (ATPA), a potent and selective agonist of kainate receptors that include the GluK1 subunit, in conjunction with mice deficient in GluK1 and GluK2 kainate receptor subunits to assess the role of GluK1 kainate receptors in provoking seizures and in kindling epileptogenesis. We found that systemic ATPA, acting specifically via GluK1 kainate receptors, causes locomotor arrest and forelimb extension (a unique behavioral characteristic of GluK1 activation) and induces myoclonic behavioral seizures and electrographic seizure discharges in the BLA and hippocampus. In contrast, the proconvulsant activity of systemic AMPA, kainate, and pentylenetetrazol is not mediated by GluK1 kainate receptors, and deletion of these receptors does not elevate the threshold for seizures in the 6 Hz model. ATPA also specifically activates epileptiform discharges in BLA slices in vitro via GluK1 kainate receptors. Olfactory bulb kindling developed similarly in wild-type, GluK1, and GluK2 knock-out mice, demonstrating that GluK1 kainate receptors are not required for epileptogenesis or seizure expression in this model. We conclude that selective activation of kainate receptors containing the GluK1 subunit can trigger seizures, but these receptors are not necessary for seizure generation in models commonly used to identify therapeutic agents for the treatment of epilepsy.
Keywords: ATPA, BLA, epilepsy, kainate receptor, kindling, seizure
Introduction
Epileptic seizures occur when the excitability in certain brain circuits exceeds the restraints imposed by inhibitory mechanisms (Nadler, 2012). Ionotropic glutamate receptors of the AMPA and NMDA are the primary mediators of excitation in the CNS (Traynelis et al., 2010) and are critical to seizure generation and spread (Rogawski, 2011). Less is known about role of kainate receptors (KARs). KARs occur predominantly as homomeric or heteromeric combinations of GluK1 and GluK2 or as heteromers of these subunits with GluK4 or GluK5 subunits (Pinheiro and Mulle, 2006). There is evidence that GluK2 KARs, which are widely distributed in the CNS, represent a major target through which the convulsant kainate induces seizures and status epilepticus (Mulle et al., 1998). GluK1 KARs have a more restricted distribution, and their contribution to seizures is uncertain. Studies in various brain regions have demonstrated that GluK1 KARs contribute to postsynaptic excitation of principal neurons (Vignes et al., 1997; Li and Rogawski, 1998; Wu et al., 2005; Koga et al., 2012) and interneurons (Cossart et al., 1998; Wondolowski and Frerking, 2009) and also act presynaptically to modulate GABA (Christensen et al., 2004) and glutamate (Aroniadou-Anderjaska et al., 2012) release. Given that some of these actions would enhance excitation whereas others might suppress it, there is no unified notion of the function of GluK1 KARs in seizures. Some authors have concluded that seizure protection can be achieved through inhibition of GluK1 KARs (Smolders et al., 2002), whereas others have proposed that activation of these receptors is a promising antiepileptic strategy (Khalilov et al., 2002).
The aim of the present study was to elucidate the role of GluK1-containing KARs in seizure generation and epileptogenesis. We used mice in which GluK1 and GluK2 are deleted by gene targeting in conjunction with seizure and epilepsy models. A critical tool in these studies is (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid (ATPA), which is a potent agonist of KARs that include the GluK1 subunit (Clarke et al., 1997; Hoo et al., 1999). ATPA is an analog of AMPA, but in radioligand binding studies it is >2000-fold selective for GluK1 KARs than AMPA receptors (AMPARs) and has no binding affinity for homomeric GluK2 KARs (Clarke et al., 1997; Bleakman et al., 2002), although it weakly interacts with GluK2/GluK5 heteromers (Paternain et al., 2000; Alt et al., 2004). Therefore, ATPA has high selectivity, unlike kainate, which not only nonselectively activates KARs with high potency but can also activate and desensitize AMPARs, and AMPA, which predominantly acts as an AMPAR agonist but can also serve as a partial agonist of some KARs (Kerchner et al., 2001; Park et al., 2010; Traynelis et al., 2010). To assess the role of these receptors in the triggering of seizures, ATPA and other glutamate receptor agonists were administered systemically and ATPA was superfused onto slices of the amygdala, a brain region prone to seizures that densely expresses GluK1 KARs (Bettler et al., 1990; Li et al., 2001). In addition, to determine whether KARs are required for epileptogenesis, we examined olfactory bulb kindling in the knock-out mice.
Materials and Methods
Animals.
Adult mice (8–12 weeks of age) and Sprague Dawley rats were provided free access to food and water in an animal facility with controlled temperature (22°C-24°C), humidity (40%–50%), and lighting (artificial 12 h light/dark cycle). Mice were housed in groups or singly after surgery. Rats were housed in groups of two. The animal facilities are fully accredited by the American Association for Accreditation of Laboratory Animal Care, and the experiments were performed under a protocol approved by the National Institute of Neurological Disorders and Stroke Animal Care and Use Committee, in full compliance with the Guide for Care and Use of Laboratory Animals of the National Research Council.
KAR knock-out mice (GluK1−/− and GluK2−/−) originated from a mixed 129/Sv and C57BL/6 background (Fisahn et al., 2004). They were backcrossed for at least 12 generations to 129Sv/Ev mice to provide an isogenic 129Sv/Ev strain. Mating pairs were genotyped before each backcross. After six generations of mating to 129Sv/Ev, >99% of the genetic background is 129Sv/Ev. Experiments were conducted with male knock-out mice from homozygote crossings and wild-type mice of the same parent generation also from homozygote crossings.
Mouse locomotor activity.
Testing was performed during the light phase of the circadian cycle, between 09:30 and 15:30 h. Locomotor activity was monitored for a period of 30 min in a Plexiglas box equipped with infrared beams aimed at photoelectric detectors placed 2.6 cm apart along the perimeter (Accuscan Instruments). The sensors are capable of sensing movement at a height of up to 2.0 cm off the floor. Immediately after test agent administration, animals are placed in the center of the arena and activity levels are recorded using Versamax software. The total distance traveled was assessed by activity counts in 10 min epochs. These values were used as the basis for the determination of area under the curve. The test chambers were cleaned with 70% ethanol solution after each individual session to prevent subsequent mice from being influenced by deposited odors.
Tail vein infusion of glutamate agonists and pentylenetetrazol (PTZ) in mice.
Testing was conducted during the light cycle after at least 30 min of acclimation to the experimental room. Each mouse was placed in a Rotating Tail Injector (Braintree Scientific). Lidocaine was applied on the tail before catheterization to decrease pain during the procedure, and the lateral tail vein was catheterized with a 0.5-inch-long 30-gauge needle attached to a 45-cm-long polyethylene tube (PE-10). Correct needle placement was verified by the appearance of blood in the tubing. The needle was gently secured to the tail using plastic tape. The tubing was attached to a 12 ml plastic syringe containing the glutamate agonist solutions. The syringe was mounted on an infusion pump (Harvard Apparatus). After catheterization, unrestrained mice were observed for behavioral changes and a subgroup of mice (slow infusion) was additionally connected to the EEG machine for simultaneous video and EEG recordings during the infusion. For glutamate agonists, the slow infusion rate was 0.031 ml/min (with EEG) and the fast infusion rate was 0.125 ml/min (without EEG). The concentration for all glutamate receptor agonists was 25 mm. PTZ solution (10 mg/ml) was infused at a rate of 0.05 ml/min. The time of onset of the following behavioral signs was noted during glutamate receptor agonist infusion: (1) locomotor inactivity (freezing), (2) tonic forelimb extension, (3) myoclonus (single whole-body twitch), (4) clonus (repeated jerking movements of all four limbs lasting at least 5 s with loss of the righting reflex), and (5) tonus (tonic hindlimb extension). The behavioral seizure signs noted during PTZ infusion were as follows: (1) first twitch (rapid upward flick of rigid tail), (2) clonus, and (3) tonus. The threshold doses (TD) of the convulsant substances (in mmol/kg) for each endpoint were calculated as follows: TD = (time of onset [s] × infusion rate [ml/min] × drug concentration [mmol/ml] × 1000)/(60 [s] × weight of mouse [g]).
Video-EEG recording.
Mice were prepared for EEG recording by implantation of a 3 channel EEG electrode assembly (MS333/1; Plastic One). The animals were anesthetized with intraperitoneal ketamine (100 mg/kg) and xylazine (10 mg/kg) and mounted in a stereotaxic frame. One recording electrode tip was implanted in the right BLA (from bregma: anteroposterior −1.4 mm, mediolateral −3.25 mm, dorsoventral −5.0 mm) and the other in the right dorsal hippocampus (from bregma: anteroposterior −1.4 mm, mediolateral −1.25 mm, dorsoventral −1.6 mm). The reference electrode was placed above the cerebellum in the midline directly behind lambda. The electrode assembly was anchored with dental acrylic cement (Geristore resin, gift from DenMat Holdings) to two small stainless steel screws placed in the skull. A period of 7 d was allowed for recovery.
The onset of behavioral seizure signs (as noted above) and first EEG seizure activity was determined during glutamate receptor agonist infusions with a clinical video-EEG monitoring system (Stellate Systems) adapted for the use in rodents (Respitech Medical). After acclimation to the recording cage for 30 min, a baseline recording was acquired for 5 min followed by tail vein infusions of glutamate receptor agonists; video and EEG were acquired continuously during the infusion. Power analysis was conducted offline with Stellate Reviewer software using artifact-free 20 s epochs at 1.5, 3, 4.5, 6, and 7 min.
6 Hz mouse seizure threshold determination.
The threshold for induction of seizures in the 6 Hz model was determined as described by Kaminski et al. (2004). In brief, corneal stimulation (0.2 ms-duration monopolar rectangular pulses at 6 Hz for 3 s) was delivered by a constant-current device (ECT Unit 5780; Ugo Basile) at various current intensities in the range of 10–20 mA. Topical anesthetic (0.5% tetracaine hydrochloride ophthalmic solution) was applied to the corneas 15 min before stimulation. Saline (0.9%) was used to wet the electrodes immediately before testing to ensure good electrical contact. Each animal was stimulated once. During the stimulation, mice were manually restrained and released into the observation cage (27.5 × 20 × 15 cm) immediately after the current application. The seizures were often preceded by a brief period (∼2–3 s) of intense locomotor agitation (wild running and jumping). The animals then exhibited a “stunned” posture associated with rearing (bipedal standing), forelimb clonus, and twitching. At the end of the seizure, animals resumed their normal exploratory behavior. Seizures lasting <10 s were not scored.
Brain slice physiology.
Mice or rats were anesthetized with CO2, decapitated, and the brain rapidly removed and transferred to an ice-cold cutting solution composed of the following (in mm): 202 sucrose, 26 NaHCO3, 3 KCl, 1.25 Na2PO4, 2 CaCl2, 1 MgCl2, and 10 glucose. The cutting solution was maintained at pH 7.4 by bubbling with 95% O2/5% CO2. Coronal amygdala slices (500 μm thick) were prepared using a vibratome tissue slicer. The slices were equilibrated for 1 h at 37°C in aCSF of the same composition as the cutting solution, except that 130 mm NaCl replaced the sucrose. Single slices were placed into a submerged-type recording chamber superfused continuously (3 ml/min) with oxygenated aCSF at 34°C containing various concentrations of ATPA ranging from 1 to 30 μm. ATPA was dissolved in DMSO so that the final concentration in the aCSF was 0.06%. This concentration of DMSO did not affect the cellular behavior of the slices. Glass micropipette electrodes (tip resistance 2–5 mΩ) were pulled from thin-wall borosilicate glass (A-M Systems) with a Sutter PC-80 micropipette puller (Sutter Instrument). The electrodes were filled with aCSF, positioned in the BLA region of the slice with a micromanipulator, and the extracellular electrical activity was monitored with an extracellular preamplifier (Dagan) and Clampex 9.2 software (Axon). After a 10 min equilibration period, the electrode signal was monitored for epileptiform activity. If no epileptiform activity occurred within 3 min, the electrode was repositioned successively at different sites in the BLA with monitoring at each site for 3 min. Slices not exhibiting epileptiform activity at the end of 30 min were scored as negative. In some experiments, LY293558 was added to the superfusion solution to block GluK1 KARs.
Olfactory bulb kindling.
Mice were anesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg). A twisted bipolar stainless steel wire electrode (MS303/1; Plastics One) was stereotaxically implanted in the right olfactory bulb (1 mm anterior to the confluence of sinuses, 1.0 mm lateral to bregma, and 1 mm ventral to the dura mater) and anchored with dental acrylic cement to two small stainless steel screws placed in the skull. A period of 7 d was allowed for recovery.
The afterdischarge threshold was determined by delivering a 2 s duration, 60 Hz train of bipolar, square current pulses at an intensity of 25 μA. The stimulus was repeated at 5 min intervals with increasing amplitude until an afterdischarge of at least 5 s was obtained. The afterdischarge was recorded with a Grass CP511 AC EEG preamplifier (Astro-Med) and stored in digital form using Clampex software (Molecular Devices). Afterdischarge duration was the total duration of amygdala electroencephalographic spike activity (amplitude > 2 × baseline) occurring in a rhythmic pattern at a frequency >1 Hz. Stimulation on subsequent days used a stimulation intensity 125% of the threshold value. The behavioral seizure activity after each stimulation was rated according to the criteria of Racine (1972) as modified for the mouse: Stage 0, no response or behavioral arrest; Stage 1, chewing or head nodding; Stage 2, chewing and head nodding; Stage 3, forelimb clonus; Stage 4, bilateral forelimb clonus and rearing; Stage 5, falling. Stimulation was continued twice daily each morning between 9:00 and 10:30 h and afternoon between 15:30 and 17:00 h for 5 d per week. The day of afterdischarge threshold determination was considered day 1 of kindling. Daily kindling stimulation was continued until Stage 5 behavioral seizures were elicited on 5 consecutive stimulation days. For the purposes of determination of the postkindling afterdischarge threshold, the procedure for prekindling afterdischarge threshold determination was repeated 5 times (twice per day) and averaged or stopped after three determinations if the threshold was stable.
Drugs.
The following convulsant substances were used: (1) ATPA (Tocris Bioscience); (2) kainic acid [kainate; (2S,3S,4R)-carboxy-4-(1-methylethenyl)-3-pyrrolidineacetic acid; Sigma]; (3) AMPA (Tocris Bioscience); and (4) PTZ (Sigma). For in vivo administration, ATPA was dissolved in a 5% solution of hydroxypropyl-β-cyclodextrin (CDT) in PBS. Kainate, AMPA, and PTZ were dissolved in PBS. LY293558 {(3S,4aR,6R,8aR)-6-[2-(1(2)H-tetrazole-5-yl)ethyl]decahydroisoquinoline-3-carboxylic acid)} was a gift from Lilly Research Laboratory.
Statistical analysis.
Group data are expressed as the mean ± SEM. The two-tailed Student's t test was used to determine the statistical significance of differences in the means. The level of significance was p < 0.05. ANOVA, including repeated-measures ANOVA and Tukey's honest significant difference (HSD) test, was performed where appropriate, including in the analysis of EEG power and seizure stage and afterdischarge duration in kindling development. The area under the curve was determined from the EEG power provided by the Stellate Reviewer software and locomotor activity data (total distance in 10 min epochs) provided by the Versamax software using the trapezoidal rule.
CS50 values (stimulation current causing seizures in 50% of animals) and 95% confidence intervals in the 6 Hz model were determined by a logistic fit to the quantal percentage response data for each group constrained between 0% and 100% protection.
Results
Effect of ATPA on locomotor activity in wild-type, GluK1−/−, and GluK2−/− mice
To characterize the gross behavioral actions of systemically administered ATPA and to determine whether systemic ATPA activates GluK1 receptors, we assessed the effects of vehicle or ATPA injection on locomotor activity in wild-type, GluK1−/−, and GluK2−/− mice. After administration of vehicle, wild-type and GluK1−/− mice exhibited a similar travel distance during a 30 min observation period (Fig. 1). GluK2−/− mice exhibited modestly increased travel distance compared with wild-type mice, but the difference did not reach statistical significance. ATPA injected subcutaneously at a dose of 10 mg/kg (0.44 mmol/kg) caused marked impairment of locomotion in wild-type animals but did not induce behavioral seizures. A comparison of the pattern of locomotion after vehicle and ATPA is illustrated in Figure 1B. Wild-type animals receiving ATPA failed to explore the arena and remained confined to a corner. In contrast, ATPA injection did not affect locomotion in GluK1−/− mice, although it did inhibit locomotion in GluK2−/− mice (Fig. 1A,B). The fractional inhibition of locomotion caused by ATPA in wild-type and GluK2−/− mice was similar (respectively, 86% and 83%), and the mean total distance values in the two groups were not statistically different. These results indicate that the action of systemic ATPA on locomotion at the dose used is specifically mediated by GluK1 KARs and confirm the utility of systemically administered ATPA as a tool to activate these receptors.
Figure 1.
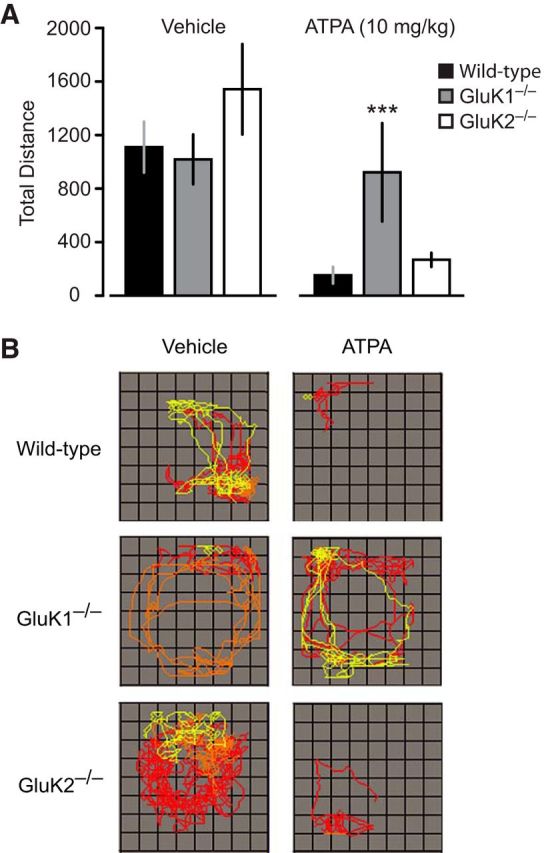
Effects of ATPA on spontaneous locomotor activity in wild-type, GluK1−/−, and GluK2−/− mice. Animals were subcutaneously injected with vehicle or ATPA (10 mg/kg). Locomotion was monitored for 30 min immediately after the injection. A, Bars represent mean ± SEM of total distance traveled (area under the curve) in groups of 8–12 animals. ***p < 0.001 compared with corresponding ATPA-treated wild-type group. There is no significant difference between total distance traveled between wild-type and GluK2−/− mice treated with ATPA; there are no significant differences between the distance traveled between wild-type and GluK1−/− or GluK2−/− mice treated with vehicle. B, Tracings of the infrared beam tracking the typical movements of mice within the arena. Each panel is from a representative wild-type, GluK1−/−, or GluK2−/− mouse treated with either vehicle or ATPA. Each of the 10 min epochs in the 30 min sessions is represented by a different color.
Responses to intravenous ATPA in wild-type, GluK1−/−, and GluK2−/− mice
Having shown that systemically administered ATPA can selectively activate GluK1 KARs, we next sought to determine whether ATPA can elicit behavioral and electrographic seizure activity. ATPA was administered by continuous intravenous infusion, which allows the total exposure to be progressively escalated. The cumulative threshold doses required to evoke individual behavioral seizure signs and the first EEG seizure was determined as described in Materials and Methods. In wild-type animals, intravenous ATPA (25 mm at 0.031 ml/min) caused locomotor inactivity as was observed with subcutaneous ATPA in the experiment of Figure 1 followed by a characteristic progression of seizure signs from bilateral forelimb extension (sometimes associated with a backward movement) to myoclonus (brief twitches) (Fig. 2). At substantially greater dose levels, EEG seizure activity occurred. GluK1−/− mice exhibited a marked elevation in the thresholds for inactivity and myoclonus while forelimb extension was absent, indicating that GluK1 KARs activation can trigger these behaviors. Inactivity and myoclonus did occur in GluK1−/− mice, albeit at greater doses than in wild-type mice, demonstrating that these behaviors can be elicited by an action of ATPA on other brain mechanisms. However, forelimb extension failed to occur in any GluK1−/− animals, even at the highest cumulative dose (0.4 mmol/kg) administered, suggesting that this behavior is specifically triggered by an action of ATPA on GluK1 KARs and cannot be evoked by the action of ATPA on other targets. There was no difference between wild-type and GluK1−/− animals in the threshold for the first EEG seizure, indicating that the induction of electrographic seizures in the hippocampus by ATPA does not occur via an action on GluK1 KARs.
Figure 2.

Thresholds for behavioral signs and first EEG seizure during slow intravenous infusion of ATPA. ATPA (25 mm) was infused at a rate of 0.031 ml/min through the lateral tail vein of wild-type, GluK1−/−, and GluK2−/− mice. Bars indicate mean ± SEM threshold for induction of behavioral arrest (inactivity), forelimb extension (often accompanied by backward movement), myoclonus, and the first EEG seizure recorded in the hippocampus or BLA. Each bar represents 8–10 mice. Forelimb extension did not occur in any of the 10 GluK1−/− mice tested and in only 1 of 8 GluK2−/− mice; the bar represents the value for this animal. **p < 0.01 with respect to wild-type. ***p < 0.001 with respect to wild-type. †No threshold reached at maximum dose.
Analysis of the depth EEG revealed changes consistent with the behavioral effects of intravenous ATPA in wild-type, GluK1−/−, and GluK2−/− animals. Before the onset of behavioral seizure activity or either interictal or ictal epileptiform discharges in the amygdale, there was a marked reduction in EEG power as illustrated in Figure 3. In wild-type and GluK2−/− mice, the power was reduced by approximately one-half at 1.5 min corresponding to a cumulative dose of 0.058 mmol/kg. In contrast, a similar reduction in power required a 6 min infusion in GluK1−/− mice corresponding to a cumulative dose of 0.23 mmol/kg. The reduction in EEG power is interpreted as desynchronization, likely associated with a higher excitable state (Steriade et al., 1990). The occurrence of sharp waves during the EEG suppression supports this interpretation. A distinct EEG pattern could not be discerned for the sudden forelimb extension seen in wild-type mice receiving low doses of ATPA. At greater cumulative intravenous doses of ATPA (threshold 0.07 ± 0.01 mmol/kg in wild-type animals), interictal and ictal epileptiform discharges were recorded in the BLA and hippocampus as illustrated in Figure 4. In the BLA in wild-type mice, the depth EEG signal after ATPA treatment was dominated by sharp waves; whereas in the hippocampus, seizure-like discharges were evident. In those cases where the EEG was not obscured by movement artifacts, sharp waves were occasionally associated with myoclonus. Seizure-like discharges occurred in the BLA after they were initiated in the hippocampus.
Figure 3.

Time-dependent effect of intravenous ATPA (25 mm infused at 0.031 ml/min) on EEG power in the amygdala before the onset of seizure activity in wild-type, GluK1−/−, and GluK2−/− mice. Power spectra were computed from artifact free 20 s epochs of the continuously recorded EEG. Data points indicate the mean ± SEM of the integrated power within the 8–50 Hz frequency band normalized to the preinfusion power. Each point represents values from 4 to 6 animals. EEG power was not quantified in GluK2−/− mice after 4.5 min because EEG seizure discharges occurred after this time point in these animals.
Figure 4.

Sample EEG traces recorded from the right BLA and the right hippocampus in a wild-type mouse during slow infusion of ATPA (25 mm at 0.031 ml/min via lateral tail vein). Traces labeled 1, 2, and 3 are EEG segments that occurred in consecutive order during the infusion; traces 2 and 3 are continuous. BLA trace 1 illustrates the early initiation of sharp wave activity not obtained in the hippocampal recording. A silent period (traces 2) is followed by the initiation of seizures activity in the hippocampus and then with a delay in the BLA (traces 3). Recordings are with respect to a reference electrode placed above the cerebellum.
For comparison, we examined the response of GluK2−/− mice in a similar slow ATPA infusion paradigm. As shown in Figure 2, these animals generally behaved as did wild-type animals, except that there was a significantly reduced threshold to first seizure compared with wild-type or GluK1−/− mice (Fig. 3).
When ATPA was infused intravenously at a more rapid rate (25 mm at 0.125 ml/min) in animals that were not implanted for EEG monitoring, clonic and tonic seizures occurred in rapid progression obscuring the initial myoclonic seizure (Table 1). Also, because of the rapid progression, forelimb extension and behavioral arrest occurred together in wild-type and GluK2−/− mice. Forelimb extension was not observed in GluK1−/− mice, confirming that it is a specific behavior resulting from activation of GluK1−/− KARs. The threshold for forelimb extension was modestly elevated in GluK2−/− mice, possibly because of the absence of ATPA-sensitive GluK1/GluK2 and GluK2/GluK5 heteromers leaving more rapidly desensitizing homomers (Cui and Mayer, 1999; Paternain et al., 2000). There was no difference between wild-type and GluK1−/− animals in the cumulative dose required to elicit clonus and tonus, confirming that unlike myoclonus frank seizures induced by systemic ATPA are not dependent upon GluK1 KARs.
Table 1.
Threshold intravenous doses of glutamate agonists for forelimb extension, behavioral arrest, and clonic and tonic seizure activity with high rate infusiona
| Test substance | Genotype | Forelimb extension (mmol/kg) | Behavioral arrest (mmol/kg) | Clonus (mmol/kg) | Tonus (mmol/kg) |
|---|---|---|---|---|---|
| ATPA | Wild-type | 0.262 ± 0.010 | — | 0.544 ± 0.030 | 0.687 ± 0.027 |
| GluK1−/− | Not present | — | 0.583 ± 0.031 | 0.613 ± 0.032 | |
| GluK2−/− | 0.410 ± 0.025* | — | 0.559 ± 0.027 | 0.613 ± 0.004 | |
| Kainate | Wild-type | — | 0.311 ± 0.009 | 0.397 ± 0.014 | 0.422 ± 0.030 |
| GluK1−/− | — | 0.234 ± 0.015* | 0.281 ± 0.015* | 0.351 ± 0.030 | |
| GluK2−/− | — | 0.411 ± 0.027* | 0.485 ± 0.019* | 0.551 ± 0.028** | |
| AMPA | Wild-type | — | — | 0.354 ± 0.056 | 0.385 ± 0.056 |
| GluK1−/− | — | — | 0.359 ± 0.042 | 0.391 ± 0.042 | |
| GluK2−/− | — | — | 0.317 ± 0.015 | 0.459 ± 0.052 |
aGlutamate receptor agonists at a concentration of 25 mm were infused intravenously at a rate of 0.125 ml/min. Cumulative doses to elicit behavioral signs (threshold) were determined from the time after onset of the infusion, as described in Materials and Methods. Only ATPA caused forelimb extension, which in wild-type and GluK2−/− animals, occurred coincidentally with behavioral arrest. Each group consisted of 7–12 mice. Values are mean ± SEM of the threshold for each of the behavioral signs. Mean values in the GluK1−/− and GluK2−/− groups were not significantly different from the corresponding values in the wild-type groups, except as indicated. —, Not applicable.
*p < 0.01, compared with wild-type group (one-way ANOVA and Tukey's HSD test).
**p < 0.05, compared with wild-type group (one-way ANOVA and Tukey's HSD test).
Induction of seizure signs by intravenous kainate and AMPA
In contrast to ATPA, low-dose intravenous infusion of kainate and AMPA in wild-type mice did not elicit forelimb extension. Rather, wild-type animals exhibited behavioral arrest (kainate but not AMPA) and a sequence of seizure signs consisting of myoclonus, generalized clonus, and tonic seizures with low (Fig. 5) or high (Table 1) rate infusion. In the case of kainate, electrographic seizure activity was observed either beginning in the hippocampus or simultaneously in the hippocampus and BLA before the onset of behavioral seizure signs, whereas with AMPA EEG seizures occurred after initial seizure signs (myoclonus) was observed (Figs. 5 and 6). GluK1−/− animals exhibited a slightly increased threshold to first electrographic seizure and reduced thresholds to the onset of clonus and tonus in response to low-dose intravenous kainate (Fig. 5). Similarly, the thresholds for behavioral arrest and clonic seizures were reduced with high-dose kainate in GluK1−/− mice (Table 1). In contrast, in GluK2−/− mice, there was a significant increase in the threshold for the first EEG seizure with slow infusion of kainate (Fig. 5) and for behavioral arrest and clonic and tonic seizures with fast infusion (Table 1), confirming the results reported previously (Mulle et al., 1998). The threshold for AMPA to evoke clonus and tonus in the slow intravenous infusion experiment in GluK1−/− mice was also reduced (Fig. 6), but this was not obtained with rapid seizure progression with rapid AMPA infusion (Table 1). There were no threshold differences in GluK2−/− mice with either slow (Fig. 6) or fast (Table 1) infusion of AMPA.
Figure 5.

Thresholds for first EEG seizure and behavioral signs during slow intravenous infusion of kainate. Kainate (25 mm) was infused at a rate of 0.03 ml/min through the lateral tail vein of wild-type, GluK1−/−, and GluK2−/− mice. Bars indicate mean ± SEM threshold for induction of the first EEG seizure, myoclonus, clonus, and tonus. Each bar represents 8–10 mice. *p < 0.05 with respect to wild-type group. ***p < 0.001 with respect to wild-type group.
Figure 6.

Thresholds for behavioral signs and first EEG seizure during slow intravenous infusion of AMPA. AMPA (25 mm) was infused at a rate of 0.031 ml/min through the lateral tail vein of wild-type, GluK1−/−, and GluK2−/− mice. Bars indicate mean ± SEM threshold for induction of the first EEG seizure, myoclonus, clonus, and tonus. Each bar represents 6–10 mice. **p < 0.01 with respect to wild-type.
Responses to PTZ in wild-type, GluK1−/−, and GluK2−/− mice
Timed intravenous PTZ infusion is a commonly used model to assess seizure susceptibility (Mandhane et al., 2007). We used the model to determine whether deletion of GluK1 or GluK2 alters the overall seizure threshold. As shown in Table 2, the mean threshold values in GluK1−/− and GluK2−/− mice were not statistically different from the values in wild-type animals, except that the threshold for tonic seizures in GluK2−/− mice was markedly elevated; the significance of this isolated difference is uncertain given that the threshold for other seizure signs in GluK2−/− mice was similar to that in wild-type animals. Nevertheless, GluK2−/− mice may have reduced seizure propensity, at least for tonic seizures.
Table 2.
Threshold intravenous doses of PTZ for first twitch and clonic and tonic seizure activitya
| Genotype | First twitch (mmol/kg) | Clonus (mmol/kg) | Tonus (mmol/kg) |
|---|---|---|---|
| Wild-type | 0.361 ± 0.017 | 0.460 ± 0.022 | 0.471 ± 0.022 |
| GluK1−/− | 0.409 ± 0.031 | 0.459 ± 0.028 | 0.526 ± 0.047 |
| GluK2−/− | 0.314 ± 0.019 | 0.442 ± 0.028 | 0.835 ± 0.043* |
aPTZ at a concentration of 10 mg/ml (72.4 mm) was infused intravenously at a rate of 0.05 ml/min. Cumulative doses to elicit first twitch and clonic and tonic seizures were determined from the time after onset of the infusion, as described in Materials and Methods. Each group consisted of 8 mice. Values are mean ± SEM of the threshold for each of the behavioral signs. Mean values in the GluK1−/− and GluK2−/− groups were not significantly different from the corresponding values in the wild-type groups, except as indicated.
*p < 0.001, compared with wild-type group (one-way ANOVA and Tukey's HSD test).
6 Hz model
The 6 Hz corneal electroshock stimulation induces limbic-like behavioral seizures associated with intense activation of the amygdala as well as the hippocampus (Barton et al., 2001). The model, which avoids the use of chemoconvulsants, is widely used to assess antiseizure agents. Figure 7 shows the results of seizure threshold determinations in wild-type, GluK1−/−, and GluK2−/− mice. The estimated threshold values in wild-type and GluK1−/− mice were not statistically different, whereas the threshold in GluK2−/− mice was elevated, indicating a reduced seizure propensity.
Figure 7.

Incidence of seizures in response to corneal stimulation of various current intensities in the 6 Hz threshold test. Data points indicate fraction of wild-type, GluK1−/−, and GluK2−/− mice exhibiting seizures at the stimulation current level. Curves are logistic fits to the data. Each point represents 6–8 animals; animals were tested only once. Inset, CS50 values in each group with 95% confidence intervals. The CS50 value for the GluK1−/− group is not significantly different from the wild-type group, whereas the CS50 value for the GluK2−/− group is significantly greater than that of the wild-type group (*p = 0.029).
ATPA induction of epileptiform discharges in the BLA slices
In the absence of ATPA in the superfusion solution, extracellular recordings from the BLA in rat brain slices failed to exhibit demonstrable activity. However, when ATPA was included in the superfusion solution, spontaneous synchronized epileptiform discharges were elicited (Fig. 8). The discharges consisted of ∼15–30 s-duration rhythmic, seizure-like runs of predominantly negative-going decrementing spikes (each ∼100 ms in duration; peak-to-peak <100 μV) and isolated negative/positive-going spike-and-waves (100–700 ms in duration; peak-to-peak <200 μV). The threshold concentration was 2.5 μm, and robust responses were obtained at 10 μm. Inclusion of AMPA in the perfusion solution failed to evoke epileptiform discharges similar to those seen with ATPA (data not shown). As illustrated in Figure 9, addition of LY293558, a selective GluK1 KAR antagonist (Traynelis et al., 2010), inhibited ATPA-induced epileptiform activity at 100 nm and completely blocked discharges at 500 nm; discharges resumed upon washout of the antagonist. LY293558 is known to antagonize AMPARs as well as GluK1 KARs, but the affinity for AMPARs is in the micromolar concentration range, whereas the equilibrium dissociation constant (KD) for GluK1 KARs is 220 nm. Thus, the block by LY293558 at concentrations of 100–500 nm suggests that GluK1 KARs mediates the epileptiform activity.
Figure 8.

Extracellular recording from the BLA in a rat brain slice during superfusion with 10 μm ATPA beginning at the point marked with the arrow. The upper two traces are continuous; the two regions boxed are shown expanded below.
Figure 9.
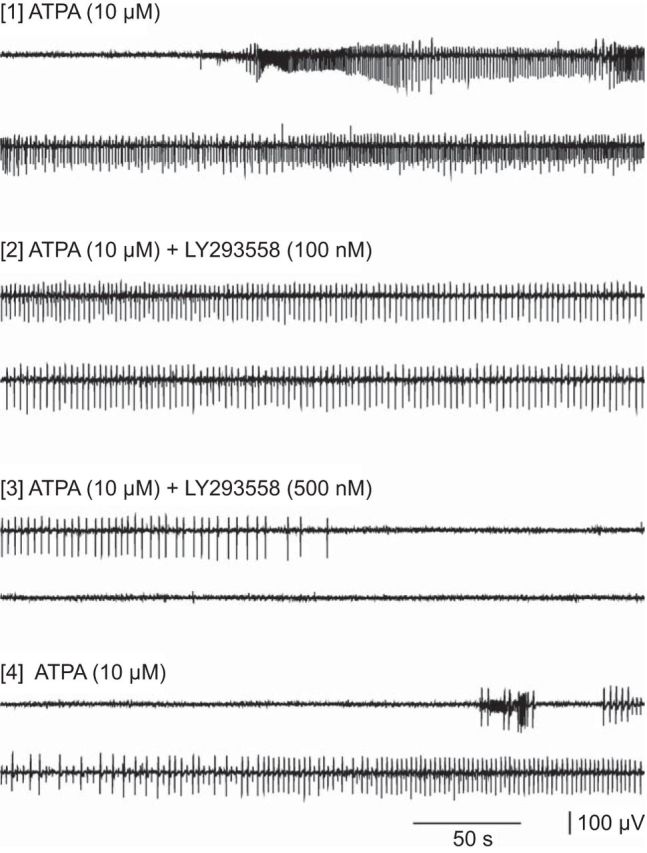
Extracellular recording from the BLA in a rat brain slice during superfusion with 10 μm ATPA. Selected segments are shown after inclusion of 100 and 500 nm LY293558 in the perfusion solution and after washout of the antagonist.
To confirm this conclusion, we used brain slices from wild-type, GluK1−/−, and GluK2−/− mice. As shown in Figure 10B, exposure of wild-type mouse brain slices to increasing concentrations of ATPA was associated with increasing likelihood of recording epileptiform discharges. No slices exhibited discharges when superfused with 1 μm ATPA, whereas 89% and 93% of slices exhibited discharges with 10 or 15 μm ATPA, respectively. The estimated effective concentration at which discharges are evoked in 50% of wild-type slices (EC50) is 7.5 μm (95% confidence limit: 7.4–7.6 μm). The discharges in mouse brain slices were similar to those in rat slices and usually consisted of brief rhythmic, single negative-going deflections with a maximum peak-to-peak amplitude of <200 μV (Fig. 10A). In addition, longer duration (100–1000 ms) biphasic events with peak-to-peak amplitude of 100–400 μV occurred in some slices. Epileptiform events were rarely recorded in slices from GluK1−/− mice. In slices superfused with 7.5 and 10 μm ATPA, discharges were detected in only 1 of 7 and 1 of 10 slices, respectively. At the other concentrations tested, no epileptiform activity was detected. In contrast, ATPA caused a concentration-dependent increase in the frequency of discharges in slices from GluK2−/− mice similar to that in slices from wild-type mice. These observations confirm that ATPA evokes epileptiform discharges via activation of GluK1 KARs.
Figure 10.

Epileptiform discharges induced by ATPA in extracellular recordings in the BLA of brain slices from wild-type, GluK1−/−, and GluK1−/− mice. A, Discharges in a slice from a wild-type mouse after perfusion with 7.5 μm ATPA (top). ATPA fails to evoke such discharges in a slice from a GluK1−/− mouse (bottom). B, Percentage of slices from wild-type and GluK1−/− mice exhibiting epileptiform discharges after superfusion with various concentrations of ATPA. Each point represents 6–10 slices; 50 male 4- to 6-week-old mice were used.
Olfactory bulb kindling in wild-type, GluK1−/− and GluK2−/− mice
To examine the role of the KARs in epileptogenesis, we used olfactory bulb kindling, which has been found to correspond more closely than other kindling models in sensitivity to epilepsy treatments (Fujiwara et., 2010). Twice-daily olfactory bulb stimulation resulted in a fluctuating but a generally progressive rise in mean seizure stage until all animals exhibited Stage 5 seizures by at least the 20th stimulation (Fig. 11A). Despite a brief initial delay in the wild-type group, there was no difference in the rate of kindling development between the wild-type and the GluK1−/− or GluK2−/− groups as assessed by mean seizure stage values (repeated-measures ANOVA, p = 0.09). The mean number of stimulations needed to achieve Stages 2–5 and the fully kindled state (5 consecutive Stage 5 seizures) were similar in the wild-type, GluK1−/−, and GluK2−/− groups (Fig. 11B). There was also a fluctuating but generally progressive rise in mean normalized afterdischarge duration (Fig. 11C). Repeated-measures ANOVA failed to detect any significant difference among the groups in the mean normalized afterdischarge duration (p = 0.38). Finally, as shown in Figure 11D, mean prekindling and postkindling afterdischarge threshold values did not differ significantly in the GluK1−/− and GluK2−/− groups from the mean values in the wild-type group (prekindling: GluK1−/−, p = 0.21; GluK2−/−, p = 0.36; postkindling: GluK1−/−, p = 0.17; GluK2−/−, p = 0.20) and the percentage change values were similar in the wild-type, GluK1−/−, and GluK2−/− groups. The results demonstrate that olfactory bulb kindling development, a form of epileptogenesis, is unaffected by deletion of either GluK1 or GluK2 and that seizure propensity in fully kindled animals that lack these subunits is similar to that of wild-type animals.
Figure 11.

Comparison of olfactory bulb kindling development in wild-type, GluK1−/−, and GluK2−/− mice. A, Mean ± SEM behavioral seizure stage is plotted with respect to stimulation number. B, Mean ± SEM number of stimulations required to achieve the stage indicated. Fully kindled stage is defined as the occurrence of 5 consecutive Stage 5 seizures. C, Mean ± SEM electrographic seizure duration normalized to the duration of the first afterdischarge. D, Mean ± SEM of the prekindling and postkindling afterdischarge threshold and the percentage change in threshold. Afterdischarge threshold is the stimulation intensity (μA) required to evoke an electrographic seizure of at least 5 s duration. Each group consists of 14 mice.
Discussion
A key problem in epilepsy research is the identity of the glutamate receptors at which the widely used convulsant toxin kainate acts to induce seizures (Ben-Ari, 2012). Mulle et al. (1998) found that GluK2−/− mice fail to exhibit seizures in response to low doses of kainate. The present demonstration of an elevated seizure threshold for kainate in GluK2−/− mice (Table 1) confirms this observation and supports the conclusion that low doses of kainate trigger seizures via an action on GluK2 KARs. However, modestly higher doses of kainate do induce seizures in GluK2−/− mice, indicating that other receptors are also involved in the convulsant activity of the toxin. Moreover, in the slow kainate infusion experiment of Figure 5, whereas the threshold for the first EEG seizure was elevated in GluK2−/− mice, the thresholds for other seizure signs did not differ from those in wild-type mice confirming that kainate can effectively trigger seizure behaviors in the absence of GluK2 KARs. Our results indicate that these additional mechanisms are more effectively recruited with slow infusion than with fast infusion. The thresholds for kainate-induced behavioral seizure signs were not elevated in GluK1−/− mice, demonstrating that the GluK1 subunit is not required for kainate-induced seizures. Indeed, the seizure threshold was generally reduced in GluK1−/− mice, which is consistent with the conclusion that, in addition to its proconvulsant actions, kainate can exert anticonvulsant effects because of the activation of GluK1 KARs on interneurons, leading to inhibition of principal cells (Cossart et al., 1998; Khalilov et al., 2002; Braga et al., 2003). Although kainate is modestly (approximately fivefold) less potent and also less efficacious as an agonist of AMPARs than KARs, kainate produces strongly desensitizing responses at KARs and nondesensitizing or very slowly desensitizing responses at AMPARs (Traynelis et al., 2010). Thus, AMPARs activated by kainate carry substantial charge, and it is reasonable to speculate that in GluK2−/− mice kainate-induced seizures occur through effects on AMPARs. The convulsant activity of kainate has not previously been shown to occur independently of GluK1 KARs. However, this conclusion is not surprising given the more restricted expression and lower density of GluK1 KARs in brain regions relevant to seizures than GluK2 KARs (Bahn et al., 1994; Li et al., 2001).
Using the selective agonist ATPA, we have demonstrated that activation of GluK1 KARs can induce certain specific behavioral responses and myoclonic seizures. The initial response to activation of GluK1 KARs in vivo is behavioral arrest. The depth EEG during this state shows desynchronization and sharp waves, indicating local hyperexcitability. The threshold for these events is elevated in GluK1−/− mice, but not in GluK2−/− mice, indicating that they are induced specifically by activation of GluK1 KARs. Modestly greater activation leads to a characteristic forelimb extension, which can be considered a behavioral marker of GluK1 KARs activation as it does not occur in GluK1−/− mice. Generalized myoclonus, a brief seizure discharge, occurs with even greater activation of GluK1 KARs. Although greater ATPA doses do induce sustained electrographic seizures and clonic and tonic behavioral seizures, the responses appear to reflect nonspecific excitatory effects, such as effects on AMPARs, as the thresholds for these more intense seizure events are unaffected by deletion of GluK1. Similarly, we found that deletion of the GluK1 subunit does not produce a general reduction in seizure susceptibility in the PTZ and 6 Hz models. These models are often used for assessing the antiseizure activity of potential antiepileptic drugs (Wilcox et al., 2013). By analogy with the more traditional use of the models in drug screening, the models permit an assessment of the potential of GluK1 KARs as a molecular target in epilepsy therapy. Apart from inhibiting the action of the agonist ATPA, deletion of GluK1 KARs is not reflected in altered seizure susceptibility in these models, implying that pharmacological inhibition of GluK1 KARs is not likely to be a promising antiseizure strategy. An important caveat in the use of knock-out mice is the possibility that changes in receptor expression or localization in the transgenic animals could lead to erroneous conclusions. However, it has been demonstrated that the functional activity and GluK1 selectivity of ATPA are unchanged in GluK2−/− mice, and postsynaptic and presynaptic effects of ATPA are eliminated in GluK1−/− animals, indicating that ATPA remains a useful tool in the knock-out animals (Kerchner et al., 2002; Christensen et al., 2004).
The observation in the present study that deletion of GluK1 KARs fails to alter seizure threshold in diverse in vivo seizure models is at odds with the conclusion of Smolders et al. (2002) that GluK1 antagonists could be potentially useful in epilepsy therapy. These authors showed that two decahydroisoquinoline antagonists of GluK1 KARs inhibit seizure activity in various in vitro and in vivo models, including the 6 Hz model in mice. Although the available information indicates that the antagonists used in this study are relatively selective for GluK1 KARs, they were administered at high doses, and possible effects on other anticonvulsant targets were not excluded. Knock-out mice would be useful in assessing whether the effects are specifically related to blockade of GluK1 KARs. In contrast to the conclusion of Smolders et al. (2002), it has been proposed that activation of GluK1 KARs can protect against seizure generation by activating inhibitory mechanisms (Khalilov et al., 2002). We observed that the thresholds for intravenous kainate to induce behavioral arrest and clonic and tonic seizures (Fig. 5; Table 1) are reduced in GluK1−/− mice. Because kainate is a high potency agonist at both GluK1 and GluK2 KARs (Alt et al., 2004), deletion of GluK1 would allow kainate to act unopposed on GluK2 KARs to evoke seizures. The reduced kainate threshold therefore supports the concept that GluK1 KARs may mediate seizure protection under certain circumstances. Reduced thresholds for intravenous AMPA to induce clonic and tonic seizures were also observed with slow infusion (Fig. 6) but not with rapid infusion (Table 1). The reduced threshold could be the result of enhanced seizure propensity in GluK1−/− mice evoked by an action on AMPARs or, because AMPA is a partial agonist at GluK2 heteromers such as GluK2/GluK5, by an action on GluK2 KARs (Traynelis et al., 2010). Overall, our results indicate that activation of GluK1 KARs can trigger seizures (Rogawski et al., 2003; Aroniadou-Anderjaska et al., 2012), but deletion of these receptors may remove an endogenous mechanism for seizure suppression.
Within the BLA, GluK1 KARs are present on the soma and dendrites of principal neurons and GABAergic interneurons, as well as on the presynaptic terminals of interneurons (Li and Rogawski, 1998; Braga et al., 2003; Gryder and Rogawski, 2003). The complex effects of GluK1 KAR activation on seizure susceptibility in the BLA is the result of the interplay of receptors at these various sites. Activation of principal neuron somatodendritic GluK1 KARs would enhance amygdalar excitability by depolarizing these neurons, leading to glutamate release onto their postsynaptic targets. Recently, it has been observed that activation of GluK1 KARs can also facilitate glutamate release in the BLA via a presynaptic action (Aroniadou-Anderjaska et al., 2012). Activation of somatodendritic GluK1 KARs on interneneurons should suppress amygdalar excitability because of interneuronal depolarization and enhanced GABA release (Wu et al., 2007). This action as noted could, in part, be the basis for antiseizure effects mediated by GluK1 KARs. Additionally, low-level activation of presynaptic GluK1 KARs localized to the terminals of GABAergic neurons facilitates GABA release and could also contribute to seizure suppression (Aroniadou-Anderjaska et al., 2008). However, more intense activation inhibits GABA release, which would be expected to facilitate seizures. In view of the marked reduction in EEG seizure threshold in GluK2−/− mice compared with wild-type and GluK1−/− animals (Fig. 2), it is interesting to speculate that GluK1/GluK2 heteromers could be of particular importance in mediating inhibition because such heteromers are absent in GluK2−/− animals.
Although GluK1 KARs are not necessary for seizure generation in diverse models, they could be required for epileptogenesis. We previously presented pharmacological evidence that GluK1 KARs are not required for the development of amygdala kindling in the mouse (Rogawski et al., 2001). These conclusions are supported by the present results with olfactory bulb kindling in GluK1−/− mice, in which kindling occurred similarly in wild-type and knock-out animals. Our results also indicate that GluK2 KARs are not required for kindling development. In addition, it is apparent that the GluK1 and GluK2 subunits do not contribute to the expression of kindled seizures. The extent to which this observation can be generalized to other epileptogenesis models remains to be determined. There is evidence for enhanced functional activity of KARs at aberrant mossy fiber–granule cell synapses in a poststatus epilepticus model of temporal lobe epilepsy (Epsztein et al., 2005; Artinian et al., 2011), suggesting that there could be differences among epileptogenesis models.
In conclusion, the present results demonstrate that selective activation of GluK1 KARs can elicit seizure activity in vivo and epileptiform discharges in the BLA in vitro but fail to confirm blockade of GluK1 KARs as a likely epilepsy treatment strategy. We recently found that epileptogenesis after kainate-induced status epilepticus is associated with a major loss of interneurons in the amygdala and impaired modulation of GABAergic transmission by GluK1 KARs (Fritsch et al., 2009), diminishing the opportunity for these receptors to serve as a treatment target. Although pharmacological inhibition of GluK1 KARs is unlikely to ameliorate seizures, the present results do not eliminate the possibility that the receptors could play a role in the pathophysiology of certain forms of epilepsy.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NS072094, NS079292) and by the NINDS Intramural Research Program. B.F. was a fellow of the NINDS Intramural Research Program. We thank Wayne D. Yonekawa and Megan Lyle Desai for assistance with experiments, and David Ide and Danny Trang for technical support.
The authors declare no competing financial interests.
References
- Alt A, Weiss B, Ogden AM, Knauss JL, Oler J, Ho K, Large TH, Bleakman D. Pharmacological characterization of glutamatergic agonists and antagonists at recombinant human homomeric and heteromeric kainate receptors in vitro. Neuropharmacology. 2004;46:793–806. doi: 10.1016/j.neuropharm.2003.11.026. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Fritsch B, Qashu F, Braga MF. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 2008;78:102–116. doi: 10.1016/j.eplepsyres.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Pidoplichko VI, Figueiredo TH, Almeida-Suhett CP, Prager EM, Braga MF. Presynaptic facilitation of glutamate release in the basolateral amygdala: a mechanism for the anxiogenic and seizurogenic function of GluK1 receptors. Neuroscience. 2012;221:157–169. doi: 10.1016/j.neuroscience.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artinian J, Peret A, Marti G, Epsztein J, Crépel V. Synaptic kainate receptors in interplay with INaP shift the sparse firing of dentate granule cells to a sustained rhythmic mode in temporal lobe epilepsy. J Neurosci. 2011;31:10811–10818. doi: 10.1523/JNEUROSCI.0388-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahn S, Volk B, Wisden W. Kainate receptor gene expression in the developing rat brain. J Neurosci. 1994;14:5525–5547. doi: 10.1523/JNEUROSCI.14-09-05525.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ME, Klein BD, Wolf HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–227. doi: 10.1016/S0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Kainate and temporal lobe epilepsies: 3 decades of progress. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's basic mechanisms of the epilepsies [Internet] Ed 4. Bethesda, MD: National Center for Biotechnology Information; 2012. http://www.ncbi.nih.gov/books/NBK98166/ [PubMed] [Google Scholar]
- Bettler B, Boulter J, Hermans-Borgmeyer I, O'Shea-Greenfield A, Deneris ES, Moll C, Borgmeyer U, Hollmann M, Heinemann S. Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron. 1990;5:583–595. doi: 10.1016/0896-6273(90)90213-Y. [DOI] [PubMed] [Google Scholar]
- Bleakman D, Gates MR, Ogden AM, Mackowiak M. Kainate receptor agonists, antagonists and allosteric modulators. Curr Pharm Des. 2002;8:873–885. doi: 10.2174/1381612024607108. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Xie J, Li H. Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci. 2003;23:442–452. doi: 10.1523/JNEUROSCI.23-02-00442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JK, Paternain AV, Selak S, Ahring PK, Lerma J. A mosaic of functional kainate receptors in hippocampal interneurons. J Neurosci. 2004;24:8986–8993. doi: 10.1523/JNEUROSCI.2156-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- Cossart R, Esclapez M, Hirsch JC, Bernard C, Ben-Ari Y. GluR5 kainate receptor activation in interneurons increases tonic inhibition of pyramidal cells. Nat Neurosci. 1998;1:470–478. doi: 10.1038/2185. [DOI] [PubMed] [Google Scholar]
- Cui C, Mayer ML. Heteromeric kainate receptors formed by the coassembly of GluR5, GluR6, and GluR7. J Neurosci. 1999;19:8281–8291. doi: 10.1523/JNEUROSCI.19-19-08281.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztein J, Represa A, Jorquera I, Ben-Ari Y, Crépel V. Recurrent mossy fibers establish aberrant kainate receptor-operated synapses on granule cells from epileptic rats. J Neurosci. 2005;25:8229–8239. doi: 10.1523/JNEUROSCI.1469-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisahn A, Contractor A, Traub RD, Buhl EH, Heinemann SF, McBain CJ. Distinct roles for the kainate receptor subunits GluR5 and GluR6 in kainate-induced hippocampal gamma oscillations. J Neurosci. 2004;24:9658–9668. doi: 10.1523/JNEUROSCI.2973-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch B, Qashu F, Figueiredo TH, Aroniadou-Anderjaska V, Rogawski MA, Braga MF. Pathological alterations in GABAergic interneurons and reduced tonic inhibition in the basolateral amygdala during epileptogenesis. Neuroscience. 2009;163:415–429. doi: 10.1016/j.neuroscience.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara A, Watanabe Y, Takechi K, Ishikawa T, Kaida Y, Akagi M, Kamei C. The usefulness of olfactory bulb kindling as a model for evaluation of antiepileptics. Epilepsia. 2010;51:445–453. doi: 10.1111/j.1528-1167.2009.02378.x. [DOI] [PubMed] [Google Scholar]
- Gryder DS, Rogawski MA. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci. 2003;23:7069–7074. doi: 10.1523/JNEUROSCI.23-18-07069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoo K, Legutko B, Rizkalla G, Deverill M, Hawes CR, Ellis GJ, Stensbol TB, Krogsgaard-Larsen P, Skolnick P, Bleakman D. [3H]ATPA: a high affinity ligand for GluR5 kainate receptors. Neuropharmacology. 1999;38:1811–1817. doi: 10.1016/S0028-3908(99)00133-1. [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Livingood MR, Rogawski MA. Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice. Epilepsia. 2004;45:864–867. doi: 10.1111/j.0013-9580.2004.04504.x. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Wilding TJ, Li P, Zhuo M, Huettner JE. Presynaptic kainate receptors regulate spinal sensory transmission. J Neurosci. 2001;21:59–66. doi: 10.1523/JNEUROSCI.21-01-00059.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Wilding TJ, Huettner JE, Zhuo M. Kainate receptor subunits underlying presynaptic regulation of transmitter release in the dorsal horn. J Neurosci. 2002;22:8010–8017. doi: 10.1523/JNEUROSCI.22-18-08010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalilov I, Hirsch J, Cossart R, Ben-Ari Y. Paradoxical anti-epileptic effects of a GluR5 agonist of kainate receptors. J Neurophysiol. 2002;88:523–527. doi: 10.1152/jn.2002.88.1.523. [DOI] [PubMed] [Google Scholar]
- Koga K, Sim SE, Chen T, Wu LJ, Kaang BK, Zhuo M. Kainate receptor-mediated synaptic transmissions in the adult rodent insular cortex. J Neurophysiol. 2012;108:1988–1998. doi: 10.1152/jn.00453.2012. [DOI] [PubMed] [Google Scholar]
- Li H, Rogawski MA. GluR5 kainate receptor mediated synaptic transmission in rat basolateral amygdala in vitro. Neuropharmacology. 1998;37:1279–1286. doi: 10.1016/S0028-3908(98)00109-9. [DOI] [PubMed] [Google Scholar]
- Li H, Chen A, Xing G, Wei ML, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci. 2001;4:612–620. doi: 10.1038/88432. [DOI] [PubMed] [Google Scholar]
- Mandhane SN, Aavula K, Rajamannar T. Timed pentylenetetrazol infusion test: a comparative analysis with s.c.PTZ and MES models of anticonvulsant screening in mice. Seizure. 2007;16:636–644. doi: 10.1016/j.seizure.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Pérez-Otaño I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–605. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- Nadler JV. Plasticity of glutamate synaptic mechanisms. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's basic mechanisms of the epilepsies [Internet] Ed 4. Bethesda, MD: National Center for Biotechnology Information; 2012. http://www.ncbi.nih.gov/books/NBK98204/ [PubMed] [Google Scholar]
- Park SA, Yin H, Bhattarai JP, Park SJ, Lee JC, Kim CJ, Han SK. Postnatal change of GluR5 kainate receptor expression in the substantia gelatinosa neuron of the trigeminal subnucleus caudalis in mice. Brain Res. 2010;1346:52–61. doi: 10.1016/j.brainres.2010.05.065. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Herrera MT, Nieto MA, Lerma J. GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J Neurosci. 2000;20:196–205. doi: 10.1523/JNEUROSCI.20-01-00196.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res. 2006;326:457–482. doi: 10.1007/s00441-006-0265-6. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. Revisiting AMPA receptors as an antiepileptic drug target. Epilepsy Curr. 2011;11:56–63. doi: 10.5698/1535-7511-11.2.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA, Kurzman PS, Yamaguchi SI, Li H. Role of AMPA and GluR5 kainate receptors in the development and expression of amygdala kindling in the mouse. Neuropharmacology. 2001;40:28–35. doi: 10.1016/S0028-3908(00)00112-X. [DOI] [PubMed] [Google Scholar]
- Rogawski M, Gryder D, Castaneda D, Yonekawa W, Banks MK, Li H. GluR5 kainate receptors, seizures, and the amygdala. Ann N Y Acad Sci. 2003;985:150–162. doi: 10.1111/j.1749-6632.2003.tb07079.x. [DOI] [PubMed] [Google Scholar]
- Smolders I, Bortolotto ZA, Clarke VR, Warre R, Khan GM, O'Neill MJ, Ornstein PL, Bleakman D, Ogden A, Weiss B, Stables JP, Ho KH, Ebinger G, Collingridge GL, Lodge D, Michotte Y. Antagonists of GLUK5-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat Neurosci. 2002;5:796–804. doi: 10.1038/nn880. [DOI] [PubMed] [Google Scholar]
- Steriade M, Gloor P, Llinás RR, Lopes de Silva FH, Mesulam MM. Report of IFCN Committee on Basic Mechanisms: basic mechanisms of cerebral rhythmic activities. Electroencephalogr Clin Neurophysiol. 1990;76:481–508. doi: 10.1016/0013-4694(90)90001-Z. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignes M, Bleakman D, Lodge D, Collingridge GL. The synaptic activation of the GluR5 subtype of kainate receptor in area CA3 of the rat hippocampus. Neuropharmacology. 1997;36:1477–1481. doi: 10.1016/S0028-3908(97)00158-5. [DOI] [PubMed] [Google Scholar]
- Wilcox KS, Dixon-Salazar T, Sills GJ, Ben-Menachem E, White HS, Porter RJ, Dichter MA, Moshé SL, Noebels JL, Privitera MD, Rogawski MA. Issues related to development of new antiseizure treatments. Epilepsia. 2013;54(Suppl 4):24–34. doi: 10.1111/epi.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondolowski J, Frerking M. Subunit-dependent postsynaptic expression of kainate receptors on hippocampal interneurons in area CA1. J Neurosci. 2009;29:563–574. doi: 10.1523/JNEUROSCI.4788-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, Zhao MG, Toyoda H, Ko SW, Zhuo M. Kainate receptor-mediated synaptic transmission in the adult anterior cingulate cortex. J Neurophysiol. 2005;94:1805–1813. doi: 10.1152/jn.00091.2005. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Ko SW, Toyoda H, Zhao MG, Xu H, Vadakkan KI, Ren M, Knifed E, Shum F, Quan J, Zhang XH, Zhuo M. Increased anxiety-like behavior and enhanced synaptic efficacy in the amygdala of GluR5 knock-out mice. PLoS One. 2007;2:e167. doi: 10.1371/journal.pone.0000167. [DOI] [PMC free article] [PubMed] [Google Scholar]