

Abstract
Brain-derived neurotrophic factor (BDNF) regulates synaptic activity and behavioral flexibility, and reduction of BDNF strongly predicts psychiatric disorders and cognitive dysfunction. Restoration of BDNF-dependent activity could alleviate these impairments, but BDNF has limited clinical utility due to its pharmacokinetics. Here we demonstrate that activation of a primary BDNF target, the tropomyosin-related kinase B (TrkB) receptor, enhances the amplitude and prolongs the decay kinetics of N-methyl-d-aspartate receptor (NMDAR) currents in male rat infralimbic prefrontal pyramidal neurons. Moreover, these effects were prevented and reversed by blockade of NMDARs containing the GluN2B subunit. Our results show that this signaling cascade bidirectionally regulates extinction of a cocaine-induced conditioned place preference (CPP), a task that requires behavioral flexibility. Blockade of infralimbic TrkB receptors or GluN2B-containing NMDARs disrupted consolidation of extinction of the CPP. In contrast, extinction was strengthened by potentiation of TrkB receptor activity with infralimbic infusions of BDNF or systemic injections of 7,8 dihydroxyflavone (7,8DHF), the newly synthesized TrkB receptor agonist. The 7,8DHF-induced enhancement of extinction was prevented by infralimbic infusions of a GluN2B-specific receptor antagonist, demonstrating that TrkB receptor activation enhances extinction of cocaine-CPP via GluN2B-containing NMDARs. Together, infralimbic TrkB receptor activation strengthens GluN2B-containing NMDAR currents to support extinction learning. TrkB receptor agonists would therefore be useful as pharmacological adjuncts for extinction-based therapies for treatment of psychiatric disorders associated with reduced BDNF activity.
Keywords: brain-derived neurotrophic factor, extinction learning, medial prefrontal cortex, NR2B-containing NMDA receptor, patch-clamp electrophysiology, TrkB receptor
Introduction
Brain-derived neurotrophic factor (BDNF) is critical for synaptic plasticity (Lohof et al., 1993; Korte et al., 1995) and behavioral flexibility (Sakata et al., 2013). Abnormal BDNF expression is prevalent among psychiatric disorders that cause a lack of behavioral flexibility, such as addiction and stress disorders (Dwivedi et al., 2003; Schumacher et al., 2005; D'Sa et al., 2011; Chen et al., 2012). Although restoration of BDNF could alleviate these disorders (Govindarajan et al., 2006; Berglind et al., 2007), BDNF has low clinical utility due to its pharmacokinetics (Zuccato and Cattaneo, 2009). Thus, identification of downstream BDNF targets that regulate behavioral flexibility could lead to the development of efficacious therapeutics. Recent work has aimed to create small lipid-soluble agonists for a major BDNF target, the tropomyosin-related kinase B (TrkB) receptor (Jang et al., 2010). However, whether TrkB receptor activation facilitates or is necessary for behavioral flexibility is unknown.
Drug addiction is characterized by a lack of behavioral flexibility, wherein extinction of drug seeking is difficult (Conklin and Tiffany, 2002). Although relatively little is known regarding the neural mechanisms underlying extinction of drug seeking, studies on fear extinction demonstrate that extinction training induces the formation of a new memory that is consolidated in the infralimbic medial prefrontal cortex (IL-mPFC; Quirk et al., 2000; Quirk and Mueller, 2008). Moreover, fear extinction is enhanced by IL-mPFC BDNF (Peters et al., 2010) and requires both N-methyl-d-aspartate receptor (NMDAR) activation (Burgos-Robles et al., 2007; Sotres-Bayon et al., 2009) and synaptic plasticity (Pattwell et al., 2011; Sepulveda-Orengo et al., 2013). However, whether BDNF, TrkB receptor, or NMDAR activation are necessary for extinction of drug seeking is unknown. Despite a dearth of knowledge regarding the necessity of BDNF for extinction of drug seeking, BDNF has been shown to bidirectionally regulate drug-seeking behaviors. For example, increasing BDNF expression within the nucleus accumbens or ventral tegmental area increases cocaine seeking (Lu et al., 2004; Graham et al., 2007; Bahi et al., 2008), whereas BDNF infusions into the dorsal prelimbic mPFC (PL-mPFC) reduces cocaine seeking (Berglind et al., 2007, 2009). However, how BDNF interacts with TrkB and NMDAR activation to support extinction of drug seeking has not been studied.
We assessed the effects of TrkB receptor activation on synaptic NMDAR signaling in IL-mPFC and determined whether this interaction supports extinction learning. Using whole-cell patch-clamp electrophysiology, we identified the effects of IL-mPFC TrkB receptor activation on NMDAR-dependent EPSCs and determined the particular NMDAR subunit that is modified by TrkB receptor activation. Behaviorally, we tested whether this interaction within IL-mPFC enhances or is necessary for extinction. Using a conditioned place preference (CPP) procedure, rats were conditioned to associate one chamber with cocaine and another with saline. Following conditioning, daily cocaine-free extinction tests were administered resulting in reduced time spent within the previously cocaine-paired chamber across extinction tests. Results reveal that IL-mPFC TrkB receptor-dependent enhancement of GluN2B-containing NMDAR channel conductance is necessary for extinction of cocaine-CPP.
Materials and Methods
Patch-clamp electrophysiology.
Patch-clamp recordings were performed as previously described (Otis et al., 2013). Male rats aged 3–6 months were anesthetized with pentobarbital, and brains were quickly removed and transferred into ice-cold (0–2°C) oxygenated (95%O2/5%CO2) artificial CSF (aCSF) composed of the following (in mm): 124 NaCl, 2.8 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 20 dextrose. Coronal slices (300 μm) containing the mPFC were taken using a vibratome (Leica, VT1200) and recovered in warm aCSF (32°C) for 30 min, followed by incubation in aCSF at room temperature for 0.5–6 h. Slices were transferred into a recording chamber and continuously perfused with warm aCSF (2 ml/min; 32°C). IL-mPFC layer II/III or V neurons were visualized with differential interference contrast using a 60× water-immersion lens mounted on an upright Eclipse FN1 microscope (Nikon Instruments). Pyramidal neurons were identified based on the presence of an apical dendrite, and GABAergic interneurons were identified based on the lack of an apical dendrite. This morphology was confirmed by staining biocytin-filled neurons with a green florescent antibody (streptavidin, 1:250; Invitrogen) as previously described (Otis et al., 2013). Whole-cell recordings were obtained using borosilicate glass pipettes with low resistance tips (2–4 ΜΩ) containing internal solution composed of the following (in mm): 110 K-gluconate, 20 KCl, 10 HEPES, 2 MgCl2, 2 ATP, 0.3 GTP, 10 phosphocreatine, 0.2% biocytin, pH 7.3, and 280 mOsm. Recordings were made in voltage-clamp mode using the MultiClamp 700B amplifier connected to a Digidata 1440A digitizer (Molecular Devices). The liquid–liquid junction potential of 13 mV was compensated for throughout all recordings.
Monosynaptic NMDAR-dependent EPSCs were recorded from IL-mPFC pyramidal neurons. All neurons were voltage-clamped at −40 mV in the presence of the GABA receptor antagonist picrotoxin (100 μm) and AMPA receptor antagonist DNQX (10 μm). Presynaptic stimulation was applied (50–500 μA; 0.067 Hz) using a bipolar microelectrode (FHC) placed ∼250 μm proximal to the cell soma.
To determine the relative amount of NMDAR currents in pyramidal and GABAergic interneurons, a series of presynaptic pulses were applied (50–350 μA, 50 μA steps) in brain slices taken from naive rats. Next, to determine the specificity of our recordings to NMDAR currents, the nonselective NMDAR antagonist dl-2-amino-5-phosphonopentanoic acid (APV; 25 μm) or GluN2B-selective NMDAR antagonist ifenprodil (3 μm) were applied following a minimum of 5 min of stable recording. To determine the effect of TrkB receptor activation on NMDAR-dependent EPSCs, baseline recordings were performed in the presence or absence of ifenprodil for 10–15 min before application of the TrkB receptor agonist 7,8 dihydroxyflavone (7,8DHF; 10 μm) or vehicle (0.02% DMSO in aCSF). Recordings continued for a minimum of 45 min following 7,8DHF application. We also determined the effect of ifenprodil on NMDAR-dependent EPSCs in neurons previously treated with 7,8DHF or vehicle for a minimum of 45 min. We next examined the effect of TrkB receptor activation on NMDAR-dependent EPSCs in brain slices from rats with a history of cocaine experience. Rats were killed 1–3 d following cocaine-CPP conditioning, and electrophysiological recordings of IL-mPFC pyramidal neurons were obtained. The TrkB receptor agonist 7,8DHF was then applied for 45 min in the presence or absence of ifenprodil, as described above.
Electrophysiological data were analyzed using Clampfit (Molecular Devices). EPSC amplitudes were calculated by comparing four averaged EPSCs 35 ms poststimulus (2 ms period) compared with baseline (2 ms period). EPSC amplitudes were compared across time and between groups using a two-way ANOVA. Post hoc tests were then used to compare EPSC amplitudes before and after drug treatment. Decay kinetics of NMDAR-dependent EPSCs were calculated as time from peak to one-half peak of 20 averaged EPSCs. ANOVA was also used to analyze the decay half times across time and between groups. Following a time by group interaction, planned comparisons were used to compare specific groups based on hypotheses made from the EPSC amplitude data.
CPP.
Male rats aged 3–6 months were housed in clear plastic cages and maintained on a 14/10 h light/dark cycle (lights on at 7:00 A.M.), during which they had access to both water and standard laboratory rat chow ad libitum. Rats were weighed and handled daily for 3 d before surgery and before the start of experiments. All experimental protocols were performed in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Milwaukee.
Surgeries were performed as previously described (Otis et al., 2013). Rats were anesthetized with a mixture of ketamine/xylazine (90 mg/kg, 10.5 mg/kg, i.p.) and implanted with a double-barrel guide cannula aimed bilaterally at IL-mPFC (anteroposterior, +2.8; mediolateral ±0.6, and dorsoventral, −4.4 mm relative to bregma). Rats recovered for a minimum of 7 d before behavioral testing. Following all experiments, cannula placements were revealed using a cresyl violet stain.
Place conditioning and testing were performed as described in detail (Otis and Mueller, 2011). Following surgery, rats were exposed to two distinguishable conditioning chambers that were separated by a center chamber for 15 min. Following this baseline test, an unbiased conditioning procedure was used in which rats were assigned to receive cocaine or saline in one of the two conditioning chambers in a pseudorandom and counterbalanced fashion over 8 d. Following conditioning rats were given daily CPP extinction trials, during which free access to all chambers was allowed. CPP extinction trials were 15 min in length to prolong extinction across trials when the hypothesis was drug-induced facilitation of extinction. Under these conditions, extinction continued until drug-treated rats expressed extinction compared with the initial CPP test. In contrast, extinction trials were 30 min in length when the hypothesis was drug-induced impairment of extinction. Under these conditions, extinction continued until vehicle-treated rats expressed extinction compared with the initial CPP test.
Cocaine HCl (National Institute on Drug Abuse) was dissolved in 0.9% saline at 10 mg/ml and administered (i.p.) at a dose of 10 mg/kg. 7,8DHF (Tokyo Chemical Industry) was dissolved in a vehicle of 33% dimethylsulfoxide in PBS at 5 mg/ml (i.p.) and administered systemically at 5 mg/kg (Andero et al., 2011). BDNF (R&D Systems) was mixed in a vehicle of PBS and infused at a dose of 0.75 μg/0.3 μl/side (Peters et al., 2010). Ifenprodil (Sigma-Aldrich) was dissolved in a vehicle of distilled water and infused into IL-mPFC at 1.2 μg/0.3 μl/side. ANA-12 (Sigma-Aldrich) was dissolved in a vehicle of 50% dimethylsulfoxide in PBS and infused into IL-mPFC at 3 μg/0.3 μl/side.
To determine whether BDNF enhances extinction of cocaine-CPP, we infused BDNF or vehicle into IL-mPFC 1 h before an initial CPP extinction trial. Next, we assessed whether GluN2B-containing NMDARs are necessary for extinction by infusing the selective GluN2B receptor antagonist ifenprodil or vehicle into IL-mPFC immediately after daily CPP extinction trials. To identify whether TrkB receptors are necessary for extinction, we administered the TrkB receptor antagonist ANA-12 or vehicle directly into IL-mPFC 15 min before daily CPP extinction trials. We then examined whether direct TrkB receptor activation enhances extinction by systemically administering the selective TrkB receptor agonist 7,8DHF or vehicle 1 h before a CPP extinction trial. Moreover, we determined whether this effect required GluN2B activation by infusing ifenprodil or vehicle into IL-mPFC immediately after the extinction trial.
Drug-seeking behavior was analyzed using a three-way ANOVA to compare time within each chamber across trials and between groups (Otis et al., 2014). In experiments with more than three CPP trials (see Fig. 3), the first two extinction trials and last two extinction trials were combined and directly compared to limit variance across trials. Tukey's post hoc tests were used to compare time within the previously cocaine-paired chamber versus the saline-paired chamber during each CPP trial for each group.
Figure 3.

TrkB receptor activation enhances IL-mPFC GluN2B-containing NMDAR EPSCs in brain slices taken from cocaine-conditioned rats. A, Example traces of IL-mPFC NMDAR-dependent EPSCs before (gray) and after (black) 7,8DHF (n = 3) or 7,8DHF+ifenprodil (n = 4) treatment. B, C, 7,8DHF enhanced NMDAR currents and this effect was prevented by ifenprodil. The orange line represents EPSC data from 7,8DHF-treated neurons in brain slices taken from naive rats. ***p < 0.001 compared with 7,8DHF+ifenprodil-treated neurons at 40 min. Scale bars: vertical, 50 pA; horizontal, 50 ms. Error bars indicate SEM. Ifen, Ifenprodil.
Results
We first characterized NMDAR currents within the adult rat IL-mPFC. Using patch-clamp electrophysiology, we isolated synaptic NMDAR currents from IL-mPFC layer II/III and V pyramidal neurons and GABAergic interneurons (Fig. 1A). The electrophysiological characteristics of these neurons allowed for confirmation that these were distinct groups of neurons. Specifically, the membrane resistance of pyramidal neurons (107 ± 4 MΩ) was greatly reduced compared with GABAergic interneurons (274 ± 35 MΩ; t(18) = 8.17, p < 0.001), consistent with previous observations within mPFC (Otis et al., 2013). We next examined the relative level of NMDAR channel conductance between pyramidal neurons and GABAergic interneurons. Presynaptic stimulation provoked larger NMDAR-dependent currents in pyramidal neurons compared with GABAergic interneurons (Fig. 1B,C). ANOVA revealed a group by stimulation intensity interaction, and Tukey's post hoc tests confirmed that the amplitude of NMDAR-dependent EPSCs was greater for pyramidal neurons compared with GABAergic interneurons (at stimulation intensities >100 μA; ps < 0.05). Thus, NMDAR-dependent EPSCs in the adult rat IL-mPFC are highly elevated in pyramidal neurons compared with GABAergic interneurons. Subsequent experiments were therefore conducted in IL-mPFC pyramidal neurons exclusively.
Figure 1.

Characterization of NMDAR EPSCs in IL-mPFC. A, Photomicrographs of a biocytin-filled pyramidal neuron and GABAergic interneuron. Scale bar, 100 μm. B, Example traces of NMDAR EPSCs evoked by 50, 150, and 350 μA stimulation of IL-mPFC neurons. C, NMDAR EPSCs were larger in pyramidal neurons as compared with GABAergic interneurons. D, Example traces of IL-mPFC NMDAR-dependent EPSCs before (gray) and after (black) pyramidal neurons were treated with ifenprodil (n = 7) or APV (n = 3). E, Ifenprodil significantly reduced evoked EPSCs in pyramidal neurons, and APV abolished evoked EPSCs. F, Baseline and final 5 min of recording were individually averaged; **p < 0.01 compared with baseline, ∧p < 0.01 compared with ifenprodil-treated neurons. Scale bars: vertical, 50 pA; horizontal, 50 ms. Error bars indicate SEM.
We next characterized NMDAR-dependent EPSCs in IL-mPFC pyramidal neurons. These currents were abolished by the NMDAR antagonist APV, and were reduced to 63% by the GluN2B-selective NMDAR antagonist ifenprodil (Fig. 1D–F). ANOVA revealed a time by treatment interaction (F(24,192) = 13.69, p < 0.001) and post hoc tests confirmed that neurons had significantly reduced EPSC amplitudes after APV or ifenprodil application as compared with baseline (p < 0.003). Moreover, neurons treated with APV had significantly reduced EPSC amplitudes as compared with neurons treated with ifenprodil after (p = 0.007) but not before (p = 0.90) drug application. Thus, we isolated NMDAR-dependent EPSCs from the adult rat IL-mPFC pyramidal neurons, and those currents are composed of GluN2B-containing NMDAR EPSCs (37%) and GluN2B-lacking NMDAR EPSCs (63%).
Infralimbic TrkB receptor activation enhances GluN2B-NMDAR channel conductance
To test whether TrkB receptor activation modulates NMDAR currents in naive rats, IL-mPFC neurons were treated with vehicle, the TrkB receptor agonist 7,8DHF, or 7,8DHF+ifenprodil. NMDAR currents were enhanced by 7,8DHF and this effect was prevented by ifenprodil (Fig. 2A–C). ANOVA revealed a time by treatment interaction (F(108,972) = 3.90, p < 0.001), and post hoc tests confirmed that neurons treated with 7,8DHF had significantly larger EPSC amplitudes as compared with vehicle-treated or 7,8DHF+ifenprodil-treated neurons after 7,8DHF treatment (p < 0.02), but not before (p > 0.89). Thus, TrkB receptor activation enhances IL-mPFC NMDAR currents, and this effect requires the GluN2B subunit.
Figure 2.
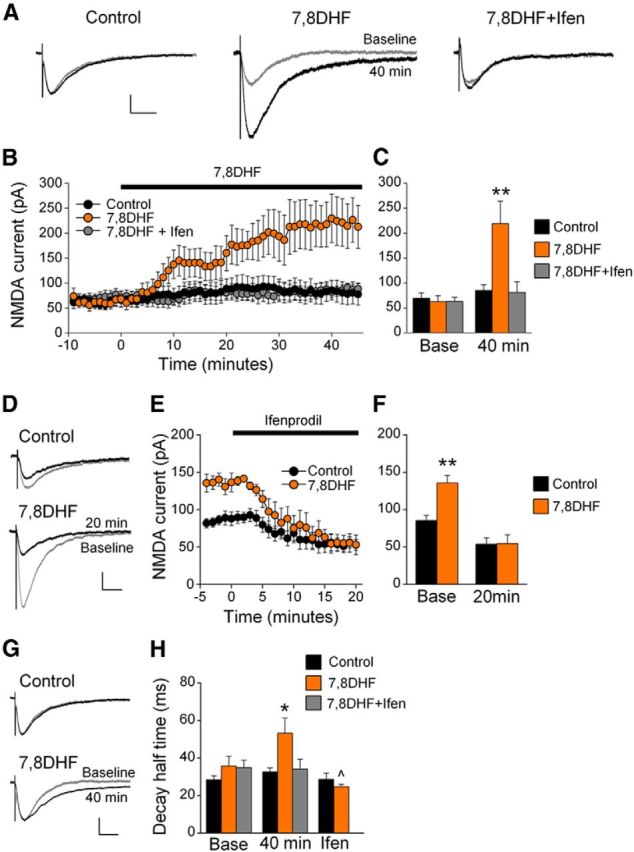
TrkB receptor activation enhances IL-mPFC GluN2B-containing NMDAR EPSCs in brain slices taken from naive rats. A, Example traces of IL-mPFC NMDAR-dependent EPSCs before (gray) and after (black) vehicle (n = 8), 7,8DHF (n = 7), or 7,8DHF+ifenprodil (n = 6) treatment. B, C, 7,8DHF enhanced NMDAR currents, and this effect was prevented by ifenprodil. D, Example traces from neurons that were previously treated with vehicle (n = 7) or 7,8DHF (n = 3) before (gray) and after (black) ifenprodil application. E, F, Ifenprodil reversed the 7,8DHF-induced potentiation of NMDAR-dependent EPSCs. G, Example traces before (gray) and after (black) 7,8DHF treatment. H, 7,8DHF (orange bars; n = 10) prolonged the decay half-time compared with control (black bars; n = 8), and this effect was both prevented by ifenprodil (gray bars; n = 10) and reversed by ifenprodil; **p < 0.01 compared with control at the same time point, ∧p < 0.01 compared with 7,8DHF-treated neurons at 40 min. Scale bars: vertical, 50 pA; horizontal, 50 ms. Error bars indicate SEM. Ifen, Ifenprodil.
We next determined whether ifenprodil would reverse the 7,8DHF-induced potentiation of NMDAR currents. Following vehicle or 7,8DHF application, ifenprodil was also applied. Ifenprodil reversed the 7,8DHF-induced enhancement of NMDAR-dependent currents (Fig. 2D–F). ANOVA revealed a time by treatment interaction (F(24,192) = 6.83; p < 0.001), and post hoc tests confirmed that neurons treated with 7,8DHF had significantly larger EPSC amplitudes compared with neurons treated with vehicle before (p < 0.01) but not after ifenprodil application (p = 0.97). Thus, TrkB receptor activation enhanced the amplitude of GluN2B-containing NMDAR currents.
Slow decay kinetics is a hallmark of GluN2B-dependent EPSCs (Vicini et al., 1998). Considering that TrkB receptor activation enhances GluN2B-dependent EPSCs in brain slices from both naive and cocaine-conditioned rats (Fig. 3), we pooled the data from these rats for analysis of EPSC decay kinetics. We found that 7,8DHF increased the decay half time of NMDAR-dependent EPSCs, and this effect was both prevented and reversed by ifenprodil (Fig. 2G,H). Comparing EPSCs between baseline and 40 min following treatment, ANOVA revealed a time by treatment interaction (F(2,24) = 4.979; p = 0.02). The 7,8DHF-treated neurons had prolonged EPSC decay half times compared with vehicle and 7,8DHF+ifenprodil-treated neurons at 40 min (p < 0.05) but not at baseline (p > 0.24). Furthermore, this potentiation was reversed by subsequent ifenprodil application. Comparing EPSC decay half times before (at 40 min) and after application of ifenprodil, ANOVA revealed a time by treatment interaction (F(1,9) = 8.65; p < 0.02), and EPSCs from 7,8DHF-treated neurons had prolonged decay half times compared with control neurons before (p = 0.04) but not after (p = 0.42) ifenprodil application. Thus, TrkB receptor activation prolonged the decay of NMDAR-dependent EPSCs, and this effect is due to potentiation of GluN2B-containing NMDARs.
We further examined whether TrkB receptor activation enhances GluN2B-containing NMDAR currents in cocaine-conditioned rats. Following CPP conditioning, rats were killed and IL-mPFC neurons were treated with 7,8DHF or 7,8DHF+ifenprodil. Similar to EPSCs from naive rats, NMDAR-dependent EPSCs were enhanced by 7,8DHF and this effect was prevented by ifenprodil (Fig. 3). ANOVA revealed a time by treatment interaction (F(54,270) = 26.48, p < 0.001), and post hoc tests confirmed that neurons treated with 7,8DHF had significantly larger EPSC amplitudes as compared with 7,8DHF+ifenprodil-treated neurons after 7,8DHF treatment (p < 0.001), but not before (p = 0.96). Moreover, the effects of 7,8DHF and 7,8DHF+ifenprodil treatment were similar between naive and cocaine-conditioned rats (7,8DHF: F(54,432) = 0.28, p = 1.00; 7,8DHF+ifenprodil: F(54,132) = 1.26, p = 0.11). Thus, TrkB receptor activation enhances IL-mPFC NMDAR currents in both naive and cocaine-conditioned rats.
BDNF signaling facilitates extinction of cocaine-CPP
Although IL-mPFC TrkB receptor signaling enhances evoked GluN2B-containing NMDAR currents, whether this interaction is important for extinction learning is unknown. BDNF endogenously activates TrkB receptors, and we therefore first determined the effect of IL-mPFC BDNF on extinction of cocaine-CPP. Following conditioning in a cocaine-induced CPP paradigm, rats were given shortened CPP extinction trials (15 min) to limit extinction across trials, as we hypothesized that BDNF would facilitate extinction. Rats were given a bilateral IL-mPFC microinfusion (Fig. 4A) of vehicle or BDNF (0.75 μg/side) before the first extinction trial. BDNF facilitated extinction of the cocaine-CPP (Fig. 4B). ANOVA revealed a chamber by trial by group interaction (F(2,30) = 3.30; p = 0.05), and subsequent two-way ANOVAs indicated a chamber by trial interaction for BDNF-treated rats (F(2,14) = 7.33, p < 0.01), but not vehicle-treated rats (F(2,16) = 0.51, p = 0.61). Post hoc analyses confirmed that both BDNF-treated and vehicle-treated rats expressed a CPP during the first trial (p < 0.001) and second trial (p < 0.01). However, only vehicle-treated rats expressed a CPP during subsequent trials (i.e., 3, 4, and 6; p < 0.03), whereas BDNF-treated rats did not (p > 0.09). Thus, IL-mPFC BDNF facilitated extinction of the cocaine-CPP.
Figure 4.

IL-mPFC BDNF and GluN2B-containing NMDARs mediate consolidation of extinction of a cocaine-CPP. A, Coronal drawings (bregma, +3.72 mm) showing injector tip placements for vehicle (n = 8) and BDNF (n = 8) IL-mPFC infusions. B, IL-mPFC infusions (arrows) of BDNF before the first CPP extinction trial enhanced extinction of the CPP. C, Coronal drawings (bregma, +3.72 mm) showing injector tip placements for vehicle (n = 11) and ifenprodil (n = 12) IL-mPFC infusions. D, IL-mPFC infusions (arrows) of ifenprodil immediately after each CPP extinction trial prevented extinction of the CPP; ***p < 0.001, **p < 0.01, *p < 0.05. Error bars indicate SEM.
GluN2B activation in infralimbic cortex is necessary for consolidation of extinction of cocaine-CPP
We found that TrkB receptor activation enhances GluN2B-dependent currents in IL-mPFC, but whether GluN2B activity mediates extinction of cocaine-CPP is unknown. Thus, we determined the necessity of IL-mPFC GluN2B-containing NMDAR activation for the consolidation of extinction of cocaine-CPP. Following conditioning, rats were given lengthened extinction trials (30 min) to promote learning, as we hypothesized that GluN2B blockade would prevent extinction. IL-mPFC infusions (Fig. 4C) of vehicle or ifenprodil (1.2 μg/side) were administered immediately after the first four extinction trials. IL-mPFC ifenprodil prevented consolidation of extinction (Fig. 4D). ANOVA revealed a chamber by trial by group interaction (F(2,42) = 3.40; p = 0.04), and subsequent two-way ANOVAs indicated a chamber by trial interaction for both vehicle-treated rats (F(2,20) = 6.73; p < 0.01) and ifenprodil-treated rats (F(2,22) = 4.69; p = 0.02). Post hoc analyses confirmed that vehicle-treated and ifenprodil-treated rats expressed a CPP during the first trial (p < 0.001) and second trial (p < 0.01). However, only ifenprodil-treated rats expressed a CPP during subsequent trials (i.e., trials 3–5; p < 0.01), whereas vehicle-treated rats did not (p > 0.58). Thus, IL-mPFC GluN2B-containing NMDAR blockade prevented consolidation of extinction of the CPP.
TrkB receptor activation in infralimbic cortex is necessary for extinction of cocaine-CPP
Although TrkB receptor activation enhances GluN2B-containing NMDAR currents, and GluN2B-containing NMDARs are critical for extinction of the CPP, whether TrkB receptors are required for extinction is unknown. Thus, we next determined the necessity of TrkB receptor activation for extinction of a cocaine-induced CPP. IL-mPFC microinfusions (Fig. 5A) of vehicle or the TrkB receptor antagonist ANA-12 (3 μg/side) were administered before the first 30 min extinction trial. ANA-12 treatment prevented extinction of the CPP (Fig. 5B). ANOVA revealed a chamber by trial by group interaction (F(2,36) = 3.89; p = 0.03), and subsequent two-way ANOVAs indicated a chamber by trial interaction for vehicle-treated rats (F(2,22) = 8.58, p < 0.01), but not ANA-12-treated rats (F(2,14) = 0.15, p=0.86). Post hoc analyses confirmed that both vehicle-treated and ANA-12-treated rats expressed a CPP during the first trial (p < 0.001). However, only ANA-12-treated rats expressed a CPP during the second trial (p < 0.001), whereas vehicle-treated rats did not (p = 0.99). Thus, IL-mPFC TrkB receptor blockade prevented consolidation of extinction of the CPP.
Figure 5.

Extinction of a cocaine-CPP requires and is enhanced by TrkB receptor activation of GluN2B-containing NMDA receptors. A, Coronal drawings (bregma, +3.72 mm) showing injector tip placements for IL-mPFC infusions. B, IL-mPFC infusions (arrows) of ANA-12 (n = 8), but not vehicle (n = 11), before the first CPP trial prevented extinction of the CPP. C, Systemic injections (black arrows) of 7,8DHF (n = 21) before the first CPP trial enhanced extinction compared with vehicle (n = 9), an effect blocked by IL-mPFC infusions (gray arrows) of ifenprodil (n = 10) immediately after the first CPP trial; ***p < 0.001, **p < 0.01. Error bars indicate SEM.
TrkB signaling facilitates extinction via GluN2B activation
We next identified whether TrkB receptor activation would facilitate extinction, and whether this effect is GluN2B-dependent. Following conditioning, rats were systemically treated with vehicle or 7,8DHF (5 mg/kg) before the first 15 min extinction trial. Rats were also given IL-mPFC microinfusions of vehicle or ifenprodil (1.2 μg/side) immediately after extinction. Rats treated with 7,8DHF/vehicle expressed extinction of the CPP by the second CPP trial, whereas rats treated with vehicle/vehicle or 7,8DHF/ifenprodil did not (Fig. 5C). ANOVA revealed a chamber by group interaction (F(4,74) = 3.69; p < 0.01), and post hoc analysis confirmed that all groups expressed a CPP during the first CPP trial (p < 0.01). However, only rats treated with vehicle/vehicle and 7,8DHF/ifenprodil expressed a CPP during the second trial (p < 0.01), whereas rats treated with 7,8DHF/vehicle did not (p = 0.94). Thus, TrkB receptor activation facilitated extinction of the CPP, and this effect was prevented by GluN2B blockade in IL-mPFC.
Discussion
We demonstrate that TrkB receptor activation enhances GluN2B-containing NMDAR channel conductance at adult IL-mPFC pyramidal neuron synapses. The TrkB receptor agonist 7,8DHF increased the amplitude and prolonged the decay kinetics of NMDAR-dependent EPSCs. These effects were blocked and reversed by application of the GluN2B-selective antagonist ifenprodil. We also reveal that the interaction between BDNF/TrkB receptor signaling and GluN2B-containing NMDARs regulates extinction of cocaine-CPP. IL-mPFC microinfusions of BDNF and systemic injections of the TrkB receptor agonist 7,8DHF enhanced extinction of cocaine-CPP, whereas IL-mPFC microinfusions of the TrkB receptor antagonist ANA-12 prevented extinction. Moreover, IL-mPFC microinfusions of the GluN2B-selective antagonist ifenprodil prevented consolidation of extinction and blocked the effects of 7,8DHF on extinction. Thus, IL-mPFC TrkB receptor activation enhances GluN2B-containing NMDAR channel conductance to promote extinction of cocaine-CPP.
Our findings support the hypothesis that IL-mPFC-projecting ventral hippocampal (vHipp) neurons release BDNF to support extinction. vHipp or IL-mPFC infusions of BDNF induce fear extinction (Peters et al., 2010; Rosas-Vidal et al., 2014), and the effect of vHipp BDNF infusions is prevented by sequestration of IL-mPFC BDNF (Peters et al., 2010). Moreover, vHipp CA1 neurons synthesize BDNF following fear extinction (Rosas-Vidal et al., 2014), and vHipp synaptic inputs to mPFC are strengthened following extinction (Farinelli et al., 2006; Hugues and Garcia, 2007). Finally, vHipp infusions of BDNF increase the firing rate of IL-mPFC neurons (Rosas-Vidal et al., 2014), suggesting that the excitability of IL-mPFC neurons and/or excitatory synaptic inputs from vHipp to IL-mPFC are strengthened by BDNF. Together, vHipp neurons secrete BDNF within IL-mPFC to promote the plasticity required for extinction. We now advance these findings by demonstrating that IL-mPFC BDNF/TrkB receptor activation facilitates extinction via potentiation of GluN2B-containing NMDARs. Enhancement of GluN2B-containing NMDARs likely occurs via TrkB-dependent phosphorylation of GluN2B. BDNF promotes phosphorylation of the tyrosine-1472 residue on the GluN2B C-terminal domain (Xu et al., 2006). Moreover, this phosphorylation requires Fyn (Xu et al., 2006), a tyrosine kinase which coimmunoprecipitates with TrkB receptors (Mizuno et al., 2003). Together, our data indicate that TrkB receptor activation promotes extinction by enhancing Fyn-mediated phosphorylation of GluN2B.
BDNF/TrkB enhancement of GluN2B currents may regulate extinction by enhancing synaptic plasticity. BDNF and GluN2B-containing NMDARs contribute to long-term synaptic potentiation within the adult medial prefrontal cortex (Zhao et al., 2005; Pattwell et al., 2012). Moreover, synaptic plasticity and learning require NMDAR-dependent calcium influx and ensuing phosphorylation of calcium-dependent calmodulin protein kinase II (CaMKII; Morris et al., 1986; Silva et al., 1992a,b; Barria et al., 1997; Shen and Meyer, 1999). Following phosphorylation, CaMKII binds with particularly high affinity to the GluN2B C-terminal domain at postsynaptic densities (Leonard et al., 1999; Bayer et al., 2001). Synaptic CaMKII induces AMPA receptor insertion and phosphorylation (Barria et al., 1997; Hayashi et al., 2000), which is critical for synaptic plasticity and learning (Barria et al., 1997; Lee et al., 2003). Furthermore, the binding of CaMKII and GluN2B is required for phosphorylation of AMPA receptors (Zhou et al., 2007), synaptic plasticity (Barria and Malinow, 2005; Zhou et al., 2007; Sanhueza et al., 2011), and memory consolidation (Zhou et al., 2007; Halt et al., 2012). BDNF also increases AMPA receptor phosphorylation (Narisawa-Saito et al., 1999; Wu et al., 2004) and glutamatergic currents in cultured hippocampal neurons (Levine et al., 1998; Crozier et al., 1999), and these effects are prevented by NMDAR blockade (Levine et al., 1998; Crozier et al., 1999; Wu et al., 2004). Together, BDNF/TrkB enhancement of infralimbic NMDAR currents is likely to induce CaMKII phosphorylation, CaMKII/GluN2B binding, and AMPA receptor modifications. In turn, these AMPA receptor modifications are likely to be critical for the consolidation of extinction. In support of this, recent data reveal that successful extinction learning is associated with AMPAR insertion at IL-mPFC synapses (Pattwell et al., 2011; Sepulveda-Orengo et al., 2013).
Enhancement of BDNF/TrkB receptor signaling enhanced extinction of cocaine-CPP. Interestingly, IL-mPFC infusions of BDNF facilitated extinction by the third day of testing, whereas systemic injections of the TrkB receptor agonist 7,8DHF facilitated extinction by the second day of testing. Why the effects of BDNF were not observed until the third day is unclear. However, one hypothesis is that BDNF was taken up by IL-mPFC neurons following microinfusions, a phenomenon that occurs in ventral hippocampus (Peters et al., 2010). BDNF could then be released upon subsequent extinction tests, resulting in a gradual enhancement of extinction. Although this is the first demonstration within a drug model that BDNF/TrkB receptor signaling facilitates extinction, BDNF has also been demonstrated to modify drug seeking within the cocaine self-administration paradigm. Interestingly, BDNF/TrkB signaling in reward-related brain regions (e.g., the nucleus accumbens and ventral tegmental area) promotes drug seeking (Lu et al., 2004; Graham et al., 2007; Bahi et al., 2008). In contrast, BDNF/TrkB signaling in the dorsal medial prefrontal cortex can reduce drug seeking (Berglind et al., 2007, 2009). Thus, although the effects of BDNF/TrkB signaling on drug seeking varies depending on the brain region, our results demonstrate that enhancement of BDNF or TrkB signaling strengthens extinction of a cocaine-CPP.
Our data reveal that BDNF/TrkB receptor activation potentiates GluN2B-containing NMDARs for extinction of cocaine-CPP. This interaction likely regulates extinction across paradigms. Prefrontal BDNF and GluN2B levels are decreased in rats with fear extinction deficits (Gourley et al., 2009). Moreover, fear extinction is enhanced by IL-mPFC infusion of BDNF (Peters et al., 2010) and systemic administration of 7,8DHF enhances fear extinction in mice with a history of stress (Andero et al., 2011). In contrast, disruption of NMDAR activity prevents extinction of aversively motivated behavior (Burgos-Robles et al., 2007; Sotres-Bayon et al., 2009; Wang et al., 2012), and more specifically GluN2B-containing NMDAR blockade prevents the consolidation of fear extinction (Sotres-Bayon et al., 2009). Together, BDNF, TrkB receptors, and GluN2B-containing NMDARs regulate extinction of both aversively motivated and appetitive behaviors.
Research regarding a single nucleotide polymorphism of the BDNF gene (val66→met) suggests that disruption of BDNF signaling impairs NMDAR signaling and extinction. The BDNFmet polymorphism affects 20–30% of the Caucasian population (Shimizu et al., 2004) and is associated with generalized anxiety, depression, and cognitive dysfunction (Egan et al., 2003; Jiang et al., 2005; Schumacher et al., 2005; Hwang et al., 2006). Moreover, BDNFmet decreases evoked BDNF secretion (Egan et al., 2003; Chen et al., 2006), such that synaptic plasticity is impaired (Ninan et al., 2010; Pattwell et al., 2012) and NMDAR-dependent EPSCs are reduced (Pattwell et al., 2012). Accordingly, both mice and humans with BDNFmet have fear extinction impairments (Soliman et al., 2010; Pattwell et al., 2012). BDNF restoration would likely reduce symptoms of the BDNFmet polymorphism, such as the attenuation of NMDAR channel conductance (Pattwell et al., 2012). However, BDNF has a low therapeutic index because of poor blood–brain barrier permeability, limited diffusion, short half-life, and nonspecific receptor binding (Zuccato and Cattaneo, 2009). Thus, our research supports the use of 7,8DHF for the restoration of BDNF/TrkB and GluN2B-containing NMDAR signaling in patients with BDNFmet-related disorders.
Pharmacological enhancement of BDNF/TrkB/GluN2B signaling is also clinically relevant for treatment of disorders associated with cognitive dysfunction and behavioral inflexibility. For example, anxiety-related disorders and drug addiction have been treated with extinction-based exposure therapy in attempt to overcome the inappropriate expression of fear or drug-seeking behaviors, respectively. However, such programs would be more successful using pharmacological adjuncts that enhance extinction learning (Conklin and Tiffany, 2002). Our results support the use of 7,8DHF as a pharmacological adjunct to exposure therapy. Such treatment would enhance BDNF/TrkB/GluN2B signaling, promoting the synaptic and behavioral plasticity required for extinction learning.
Footnotes
This work was supported by DA027870 and a grant from the University of Wisconsin-Milwaukee Research Growth Initiative to D.M. We thank Brittany Mattson and Julia Wong for technical assistance.
The authors declare no competing financial interests.
References
- Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry. 2011;168:163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi A, Boyer F, Chandrasekar V, Dreyer JL. Role of accumbens BDNF and TrkB in cocaine-induced psychomotor sensitization, conditioned-place preference, and reinstatement in rats. Psychopharmacology. 2008;199:169–182. doi: 10.1007/s00213-008-1164-1. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, McGinty JF. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- Berglind WJ, Whitfield TW, Jr, LaLumiere RT, Kalivas PW, McGinty JF. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29:3715–3719. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- Chen PH, Huang MC, Lai YC, Chen PY, Liu HC. Serum brain-derived neurotrophic factor levels were reduced during methamphetamine early withdrawal. Addict Biol. 2012 doi: 10.1111/j.1369-1600.2012.00444.x. in press. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin CA, Tiffany ST. Applying extinction research and theory to cue-exposure addiction treatments. Addiction. 2002;97:155–167. doi: 10.1046/j.1360-0443.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- Crozier RA, Black IB, Plummer MR. Blockade of NR2B-containing NMDA receptors prevents BDNF enhancement of glutamatergic transmission in hippocampal neurons. Learn Mem. 1999;6:257–266. doi: 10.1101/lm.6.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Sa C, Fox HC, Hong AK, Dileone RJ, Sinha R. Increased serum brain-derived neurotrophic factor is predictive of cocaine relapse outcomes: a prospective study. Biol Psychiatry. 2011;70:706–711. doi: 10.1016/j.biopsych.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60:804–815. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/S0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Farinelli M, Deschaux O, Hugues S, Thevenet A, Garcia R. Hippocampal train stimulation modulates recall of fear extinction independently of prefrontal cortex synaptic plasticity and lesions. Learn Mem. 2006;13:329–334. doi: 10.1101/lm.204806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourley SL, Kedves AT, Olausson P, Taylor JR. A history of corticosterone exposure regulates fear extinction and cortical NR2B, GluR2/3, and BDNF. Neuropsychopharmacology. 2009;34:707–716. doi: 10.1038/npp.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan A, Rao BS, Nair D, Trinh M, Mawjee N, Tonegawa S, Chattarji S. Transgenic brain-derived neurotrophic factor expression causes both anxiogenic and antidepressant effects. Proc Natl Acad Sci U S A. 2006;103:13208–13213. doi: 10.1073/pnas.0605180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 2012;31:1203–1216. doi: 10.1038/emboj.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hugues S, Garcia R. Reorganization of learning-associated prefrontal synaptic plasticity between the recall of recent and remote fear extinction memory. Learn Mem. 2007;14:520–524. doi: 10.1101/lm.625407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JP, Tsai SJ, Hong CJ, Yang CH, Lirng JF, Yang YM. The Val66Met polymorphism of the brain-derived neurotrophic-factor gene is associated with geriatric depression. Neurobiol Aging. 2006;27:1834–1837. doi: 10.1016/j.neurobiolaging.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Xu K, Hoberman J, Tian F, Marko AJ, Waheed JF, Harris CR, Marini AM, Enoch MA, Lipsky RH. BDNF variation and mood disorders: a novel functional promoter polymorphism and Val66Met are associated with anxiety but have opposing effects. Neuropsychopharmacology. 2005;30:1353–1361. doi: 10.1038/sj.npp.1300703. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/S0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, He J, Nakajima A, Nabeshima T. Involvement of BDNF TrkB in spatial memory formation. Learn Mem. 2003;10:108–115. doi: 10.1101/lm.56003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Silva AJ, Yamaguchi T, Hayashi T, Yamamoto T, Nawa H. Growth factor-mediated Fyn signaling regulates alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor expression in rodent neocortical neurons. Proc Natl Acad Sci U S A. 1999;96:2461–2466. doi: 10.1073/pnas.96.5.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninan I, Bath KG, Dagar K, Perez-Castro R, Plummer MR, Lee FS, Chao MV. The BDNF Val66Met polymorphism impairs NMDA receptor-dependent synaptic plasticity in the hippocampus. J Neurosci. 2010;30:8866–8870. doi: 10.1523/JNEUROSCI.1405-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Mueller D. Inhibition of beta-adrenergic receptors induces a persistent deficit in retrieval of a cocaine-associated memory providing protection against reinstatement. Neuropsychopharmacology. 2011;36:1912–1920. doi: 10.1038/npp.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Dashew KB, Mueller D. Neurobiological dissociation of retrieval and reconsolidation of cocaine-associated memory. J Neurosci. 2013;33:1271–1281a. doi: 10.1523/JNEUROSCI.3463-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Fitzgerald MK, Mueller D. Inhibition of hippocampal beta-adrenergic receptors impairs retrieval but not reconsolidation of cocaine-associated memory and prevents subsequent reinstatement. Neuropsychopharmacology. 2014;39:303–310. doi: 10.1038/npp.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattwell SS, Bath KG, Casey BJ, Ninan I, Lee FS. Selective early-acquired fear memories undergo temporary suppression during adolescence. Proc Natl Acad Sci U S A. 2011;108:1182–1187. doi: 10.1073/pnas.1012975108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I. The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci. 2012;32:2410–2421. doi: 10.1523/JNEUROSCI.5205-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Vidal LE, Do-Monte FH, Sotres-Bayon F, Quirk GJ. Hippocampal–prefrontal BDNF and memory for fear extinction. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.64. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata K, Martinowich K, Woo NH, Schloesser RJ, Jimenez DV, Ji Y, Shen L, Lu B. Role of activity-dependent BDNF expression in hippocampal-prefrontal cortical regulation of behavioral perseverance. Proc Natl Acad Sci U S A. 2013;110:15103–15108. doi: 10.1073/pnas.1222872110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, Otmakhov N, Hell JW, Lisman J. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011;31:9170–9178. doi: 10.1523/JNEUROSCI.1250-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher J, Jamra RA, Becker T, Ohlraun S, Klopp N, Binder EB, Schulze TG, Deschner M, Schmäl C, Höfels S, Zobel A, Illig T, Propping P, Holsboer F, Rietschel M, Nöthen MM, Cichon S. Evidence for a relationship between genetic variants at the brain-derived neurotrophic factor (BDNF) locus and major depression. Biol Psychiatry. 2005;58:307–314. doi: 10.1016/j.biopsych.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Sepulveda-Orengo MT, Lopez AV, Soler-Cedeño O, Porter JT. Fear extinction induces mGluR5-mediated synaptic and intrinsic plasticity in infralimbic neurons. J Neurosci. 2013;33:7184–7193. doi: 10.1523/JNEUROSCI.5198-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shimizu E, Hashimoto K, Iyo M. Ethnic difference of the BDNF 196G/A (val66met) polymorphism frequencies: the possibility to explain ethnic mental traits. Am J Med Genet B Neuropsychiatr Genet. 2004;126B:122–123. doi: 10.1002/ajmg.b.20118. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992a;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992b;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, Jing D, Tottenham N, Amso D, Somerville LH, Voss HU, Glover G, Ballon DJ, Liston C, Teslovich T, Van Kempen T, Lee FS, Casey BJ. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Diaz-Mataix L, Bush DE, LeDoux JE. Dissociable roles for the ventromedial prefrontal cortex and amygdala in fear extinction: NR2B contribution. Cereb Cortex. 2009;19:474–482. doi: 10.1093/cercor/bhn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Wang WS, Kang S, Liu WT, Li M, Liu Y, Yu C, Chen J, Chi ZQ, He L, Liu JG. Extinction of aversive memories associated with morphine withdrawal requires ERK-mediated epigenetic regulation of brain-derived neurotrophic factor transcription in the rat ventromedial prefrontal cortex. J Neurosci. 2012;32:13763–13775. doi: 10.1523/JNEUROSCI.1991-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Len GW, McAuliffe G, Ma C, Tai JP, Xu F, Black IB. Brain-derived neurotrophic factor acutely enhances tyrosine phosphorylation of the AMPA receptor subunit GluR1 via NMDA receptor-dependent mechanisms. Brain Res Mol Brain Res. 2004;130:178–186. doi: 10.1016/j.molbrainres.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Xu F, Plummer MR, Len GW, Nakazawa T, Yamamoto T, Black IB, Wu K. Brain-derived neurotrophic factor rapidly increases NMDA receptor channel activity through Fyn-mediated phosphorylation. Brain Res. 2006;1121:22–34. doi: 10.1016/j.brainres.2006.08.129. [DOI] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Takahashi E, Li W, Halt A, Wiltgen B, Ehninger D, Li GD, Hell JW, Kennedy MB, Silva AJ. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci. 2007;27:13843–13853. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]