

Abstract
Trk neurotrophin receptor ubiquitination in response to ligand activation regulates signaling, trafficking, and degradation of the receptors. However, the in vivo consequences of Trk ubiquitination remain to be addressed. We have developed a mouse model with a mutation in the TrkA neurotrophin receptor (P782S) that results in reduced ubiquitination due to a lack of binding to the E3 ubiquitin ligase, Nedd4-2. In vivo analyses of TrkAP782S indicate that defective ubiquitination of the TrkA mutant results in an altered trafficking and degradation of the receptor that affects the survival of sensory neurons. The dorsal root ganglia from the TrkAP782S knock-in mice display an increased number of neurons expressing CGRP and substance P. Moreover, the mutant mice show enhanced sensitivity to thermal and inflammatory pain. Our results indicate that the ubiquitination of the TrkA neurotrophin receptor plays a critical role in NGF-mediated functions, such as neuronal survival and sensitivity to pain.
Keywords: neurotrophins, pain, TrkA neurotrophin receptor, ubiquitination
Introduction
Neurotrophins play a pivotal role in the proper functioning of the nervous system by modulating survival, proliferation, differentiation, axonal growth, synaptic plasticity, and sensitivity to pain. The actions of neurotrophins depend on two different transmembrane receptors: Trk neurotrophin receptors and the p75 neurotrophin receptor. Nerve growth factor (NGF) binds specifically to TrkA (Chao, 2003; Huang and Reichardt, 2003), the first member of the Trk neurotrophin receptor family identified (Martin-Zanca et al., 1986), which is mainly expressed in different ganglia from the peripheral nervous system (PNS), such as the dorsal root ganglia (DRG), the superior cervical ganglia (SCG), and the trigeminal ganglia (Martin-Zanca et al., 1990; Tessarollo et al., 1993). trkA- and ngf-deficient mice (KO) display a significant loss of neurons from these ganglia (Crowley et al., 1994; Smeyne et al., 1994).
In addition to regulating sensory neuron survival during development, NGF- and TrkA-mediated signaling also plays a major role in pain processing after primary nociceptors have lost their absolute dependency on NGF in the postnatal period. Several observations support this view: (1) trkA and ngf KO mice exhibit several sensory defects (Crowley et al., 1994; Smeyne et al., 1994), (2) NGF administration produces behavioral hyperalgesia (Lewin et al., 1993; McMahon, 1996), (3) NGF acts as an inflammatory mediator (Woolf et al., 1994; McMahon et al., 1995), (4) NGF modulates the levels of substance P and calcitonin gene-related peptide (CGRP) in sensory neurons (Otten et al., 1980; Lindsay and Harmar, 1989; Crowley et al., 1994), (5) nearly all human cases of sensory and autonomic neuropathy type IV are caused by mutations in the TrkA gene (Indo et al., 1996; Indo, 2001), and (6) promising results have been obtained with recent clinical trials targeting NGF for pain relief (Lane et al., 2010). All these findings highlight the relevance of NGF-TrkA in pain modulation, but the molecular mechanisms are not fully understood.
It has been shown that TrkA ubiquitination controls the turnover of TrkA (Geetha et al., 2005; Makkerh et al., 2005; Arévalo et al., 2006). TrkA, but not TrkB or TrkC, binds to and is ubiquitinated by Nedd4-2 (Arévalo et al., 2006). Moreover, depletion of the endogenous levels of Nedd4-2 in DRG neurons affects TrkA trafficking, degradation and NGF-mediated signaling as well as the in vitro survival of sensory neurons (Yu et al., 2011). However, the in vivo consequences of TrkA ubiquitination on NGF-mediated neuronal survival and nociceptive functions have not been addressed.
Here we report that ubiquitination of TrkA plays an influential role in NGF-mediated neuronal survival and nociceptive functions in vivo. We observed that TrkA ubiquitination was impaired in a knock-in (KI) mouse model expressing a mutant TrkA protein (TrkAP782S) in which the binding site for Nedd4-2 was mutated. Consequently, in these mice TrkAP782S afforded increased tyrosine kinase activity, but was inefficiently degraded. This KI mouse exhibited an increased number of sensory neurons, enhanced levels of substance P and CGRP and augmented skin innervation. Thus, this mouse model pointed to an enhanced sensitivity to pain mediated by thermal and inflammatory stimuli. Our data provide strong support for an in vivo physiological role for Nedd4-2-mediated TrkA ubiquitination.
Materials and Methods
Materials
NGF was obtained from Alomone Labs. The following antibodies were used: anti-203, RTA, and p75 (9992); [generous gifts from Dionisio Martín-Zanca (IBFG, Salamanca, Spain), Louis Reichardt (UCSF, San Francisco, CA), and Moses Chao (NYU, New York, NY), respectively]; Nedd4-2 was previously described (Arévalo et al., 2006); Trk (C-14), P4D1 (ubiquitin), and PY99 (Santa Cruz Biotechnology); Rab7, anti-FLAG, actin, and βIII-Tubulin (Sigma-Aldrich); phosphoTrk (Y490), (Cell Signaling Technology); EEA1 (Beckton Dickinson); c-fos (Oncogene Research Products); and PGP9.5 (Thermo Scientific).
DNA transfections, preparation of cell lysates, and immunoblotting
Plasmid DNA was transiently transfected into HEK293 cells using the calcium phosphate method. Cells were lysed in a lysis buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 1 mm PMSF, 1 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mm vanadate, 10 mm NaF, and 20 mm β-glycerophosphate) for 40 min at 4°C with gentle shaking and centrifuged at 12,000 rpm for 15 min to eliminate the debris (Arévalo et al., 2004). Lysates were resuspended in 5× SDS-buffer and boiled for 7 min to denature the proteins. Proteins were resolved by SDS-PAGE, and Western blots were performed with antibodies against different proteins.
Quantitative RT-PCR analysis
Total RNA was isolated from wild-type (WT) TrkA or KI E13.5 mouse DRGs using TRIzol reagent (Invitrogen). cDNA was reverse-transcribed using Superscript II reverse transcriptase and random primers. cDNA sample concentrations were determined by measuring absorbance at 260 nm using a NanoDrop 2000C spectrophotometer (Thermo Scientific). Quantification of the PCR products (trkA and β-actin, as a reference gene) was accomplished with a standard curve using SYBR-Green. The SYBR-Green was included in a 2× Master Mix from Applied Biosystems (SYBR Green dye, dNTPs, Passive, ROX, AmpliTaq Gold DNA polymerase). The final volume of each reaction was 20 μl: 10 μl of Master Mix, 0.8 μl (16 pmols) of each oligonucleotide, 7.4 μl of distilled water and 1 μl of cDNA at a concentration of 100 ng/μl. A standard curve was constructed for each experiment by serial dilutions of cDNA: 0.01, 0.001, 0.0001, and 0.00001 ng/μl. The amplification reaction was performed in an ABI Prism 7300 detection system (Applied Biosystems), with the following conditions: 10 min at 95°C followed by 36 cycles of 15 s at 95°C, and 1 min at 53°C (trkA and β-actin). The primers for trkA were: 5′-TCTCCTTCTCGCCAGTGGAC-3′ and 5′-ACAGCCACAGAGACCCCAAA-3′; for β-actin: 5′-AGCCATGTACGTAGCCATCC-3 and 5′-ACCCTCATAGATGGGCACAG-3. The amplicons for trkA and actin were 79 bp and 104 bp long, respectively. Three PCRs were performed for each sample per plate, and each experiment was repeated three times. Data are represented as means ± SEM and were analyzed using the two-tailed Student's t test (n = 3).
DRG neuron culture
DRGs were dissected from embryonic day (E)13.5 WT TrkA and KI mouse embryos following the previously described protocol (Yu et al., 2011). Briefly, dissected DRGs were incubated and dissociated with 0.25% trypsin in L-15 media for 45 min at 37°C. Trypsin was eliminated after centrifugation and cells were resuspended in plating medium (MEM, 10% FBS, 0.4% glucose, 2 mm glutamine, 100 U/ml pen/strep). A single-cell suspension was obtained by pipetting up and down 20 times sequentially with a 1000 and 200 μl tip. The same number of cells were plated using plating medium and NGF (50 ng/ml) overnight on plastic plates coated with Growth Factor Reduced Matrigel (BD Biosciences) as substrate. On the following day the medium was changed to neurobasal medium (B-27, 0.4% glucose, 2 mm glutamine), NGF, and 5-fluorodeoxyuridine (2.44 μg/ml), and uridine (2.44 μg/ml). Proliferating cells disappeared after 3–4 d and >95% of cells at 8 d in vitro (DIV) were neurons.
Immunofluorescence and quantification
The DRG neurons used to perform immunofluorescence analysis were cultured on coverslips coated with 1 mg/ml poly-d-lysine and growth factor reduced matrigel. NGF was withdrawn in the presence of 20 μm Z-VAD-FMK overnight and on the following day 50 ng/ml NGF was added to induce TrkA activation and internalization. Cells were then fixed with 4% paraformaldehyde (PFA) in PBS for 5 min, quenched with 50 mm NH4Cl for 10 min, blocked, and permeabilized with PBS containing 5% normal goat serum, 0.1% Tween 20, and 0.1% Triton X-100 for 30 min, and incubated with primary antibodies overnight at 4°C. The following day, cells were washed three times with PBS, incubated with the corresponding fluorescent secondary antibody at room temperature for 40 min, and washed with PBS three times. Images were collected with a Leica confocal microscope in RGB color and with identical format and sizes (1024 × 1024 pixels). To avoid bleed-through between channels, each channel was acquired separately in the colocalization experiments. For colocalization analyses, images of the cells were processed with the functions of ImageJ (NIH). Processing consisted of saving the original color images as 8-bit black and white images and applying similar threshold settings for all pictures analyzed to remove the background. Background pixels were set to zero. Thus, the image pairs for pTrkA and EEA1 or Rab7 show specific signals for each marker. Quantification of the processed images was performed with a custom-written program using custom-made MATLAB scripts. Image pairs were multiplied in MATLAB so that the multiplication image would contain only signal pixels that were present in both images. The amount of colocalization was then expressed as a percentage of the number of pixels with values different from zero in the multiplied image relative to the number of pixels in the corresponding image for pTrkA.
Surface and degradation assays
Surface labeling and degradation assays were performed using cultured mouse DRG neurons, as previously described (Yu et al., 2011). Briefly, to detect the amount of surface proteins in response to NGF, WT and KI cultured DRG neurons were used. Cells were stimulated with or without NGF (50 ng/ml) for 60 min to induce the internalization of surface proteins. Subsequently, cells were washed sequentially using room temperature PBS and cold PBS, chilled on ice, and incubated in 0.5 μg/ml Sulfo-NHS-SS-biotin (Pierce) dissolved in biotinylation buffer (PBS, 1 mm CaCl2, 0.5 mm MgCl2) for 20 min at 4°C to label the membrane proteins. Free biotin was quenched with 0.1 m glycine for 15 min at 4°C. Cells were then washed twice with cold PBS and lysed as indicated above. Biotinylated proteins were isolated from the total cell lysate by immobilization on Neutravidin beads (Pierce) for at least 3 h at 4°C. The beads were washed three times with lysis buffer and 20 μl of 2× SDS sample buffer was added before boiling for 7 min. Proteins were subjected to SDS-PAGE and immunoblotted with the corresponding antibodies. Degradation assays of surface proteins were performed using cultured WT and KI DRG neurons. Cells were NGF-starved and on the following day were washed with PBS, chilled on ice, and biotinylated using 0.5 μg/ml Sulfo-NHS-SS-biotin for 20 min at 4°C. Neurons were sequentially washed with cold and room-temperature PBS and incubated in prewarmed medium at 37°C with or without NGF (50 ng/ml) for different times (15 and 60 min) to allow the biotinylated receptors to become internalized and degraded. Subsequently, cells were washed and lysed using lysis buffer and the biotinylated proteins were precipitated with Neutravidin beads, washed, and subjected to SDS-PAGE, and immunoblotted using different antibodies. With this method, it was possible to detect surface-labeled proteins that had not been degraded at different time points, regardless of whether the proteins had been internalized or whether they had returned to the plasma membrane.
In situ hybridization
Brains from postnatal day (P)0 WT and KI mice were fixed in 4% PFA for 48 h and incubated with 30% sucrose in PBS. Tissue sections (12 μm) were obtained, postfixed with 4% PFA for 10 min, treated with PBS containing 0.1% DEPC for 5 min, and then with PBS containing 20 μg/ml proteinase K for 6 min, followed by acetylation for 10 min. Sections were prehybridized for 30 min, and then hybridized with the corresponding probe (0.25–1 μg/ml) overnight at 55°C. On the next day, sections were rinsed with 5× SSC at 65°C, washed with 2× SSC/50% formamide for 30 min at 65°C, treated with 20 μg/ml RNase for 30 min at 37°C, washed with 2× SSC and 0.1 × SSC, and then subjected to anti-DIG-AP-conjugated antibody (Roche) staining and a BCIP/NBT (Roche) color reaction.
DRG neuron counting
Tissues from P0 WT and KI mice were fixed in PBS containing 4% PFA, cryoprotected overnight in PBS containing 30% sucrose, and frozen. Cyrostat sections (12 μm) from DRGs were prepared and stained with a solution containing 0.5% cresyl violet, and cells with visible nucleoli were counted as neurons independently by four investigators blind to the genotype.
Immunohistochemistry
WT and KI mice of 3–5 month of age were perfused with PBS containing 4% PFA and spinal cord and skin from hindpaws were dissected and postfixed in 4% PFA O/N. The tissue was cryoprotected overnight in PBS containing 30% sucrose. It was then sectioned (20 μm), and skin and spinal cord were stained with c-fos and PGP9.5 antibodies, respectively. Images were taken with an Olympus AX70 equipped with an Olympus DP70 camera. The number of c-fos-positive cells from formalin-injected mice in 17 and 19 sections, respectively, corresponding to three WT and three KI mice were quantified. Using NIH ImageJ software, the total area of the PGP9.5-positive fibers from 41 and 44 sections corresponding respectively to four WT and four KI mice was quantified.
Behavioral assays
All behavioral tests were performed with an experimenter blind to the genotype. For all behavioral experiments, mice (3–5 month males, 25–35 g) were placed in individual cages for 30 min and habituated to the test procedure for at least 3 d before experiments were performed. The mice used were backcrossed for at least six generations to C57BL/6.
Heat-test response.
To assess acute heat sensitivity, we performed a semiautomated tail flick test (Ugo Basile). The experiment was performed using a radiant heat source focused at 3 cm from the tail tip, and the time from the initiation of the radiant heat until tail withdrawal was measured. Three separate determinations with 10 min interval between each determination were performed per animal and the numbers were averaged. A maximum cutoff of 15 s was used to prevent tissue damage.
Cold test.
For the cold plate, we followed the protocol previously described (Guo et al., 2011b) with slight modifications. Briefly, a clear Plexiglas cylinder was placed on an ice/water bath equilibrated to 0°C and mice were placed inside the cylinder. The latency to the onset of brisk and persistent paw withdrawal and/or licking of the paw was measured.
Paw pressure test.
Mechanical nociceptive thresholds were determined by the Randall–Selitto test on the left hindpaw as described previously (Arsequell et al., 2009), with slight modifications. Pain thresholds were measured using an analgesymeter (Ugo-Basile). The apparatus was used to apply a force of between 0 and 745 g, increasing from zero at a rate of 48 g/s. The nociceptive threshold was taken as the point at which the mouse vocalized or struggled vigorously. Mice that did not attempt to remove their paws or vocalize before the 745 g point was reached (when pressure exerted is 420 g/mm2) were recorded as having reached the cutoff pressure.
Inflammatory pain.
Twenty microliters of 5% formalin was injected subcutaneously into the plantar surface of the right hindpaw. The mouse was recorded in a cage for the following 60 min to monitor licking and biting of the injected paw. Phases I (acute phase) and II (inflammatory phase) were considered from 0 to 15 min and from 20 to 55 min, respectively.
All animals were housed and bred in the SPF Animal Facility of the University of Salamanca. Proper measures were taken to reduce the pain or discomfort to the experimental animals. Animal care and its procedures were done in accordance with protocols approved by the Bioethics Committee of the University of Salamanca and following European Community guidelines.
Results
Impaired TrkAP782S ubiquitination in vivo
The ubiquitination of Trk tyrosine kinase receptors depends on the activation and autophosphorylation of the receptor in response to neurotrophins. Use of the K252a kinase inhibitor or kinase-dead mutant Trk neurotrophin receptors effectively abolishes ubiquitination (Geetha et al., 2005; Makkerh et al., 2005; Arévalo et al., 2006). To address whether TrkA ubiquitination levels depend on the degree of receptor activation, we stimulated PC12-615 cells (Hempstead et al., 1991) with different amounts of NGF. TrkA ubiquitination levels were directly correlated with the level of receptor activation because PC12-615 cells stimulated with increasing concentrations of NGF resulted in a dose-dependent effect on TrkA ubiquitination (Fig. 1A). Accordingly, TrkA ubiquitination can be said to require the activation of the receptor and the ubiquitination levels correlate with the receptor activation level.
Figure 1.

Impaired TrkAP782S ubiquitination in cultured DRG neurons. A, The ubiquitination of TrkA is directly dependent on its activation levels. PC12-615 cells were stimulated with different amounts of NGF for 10 min and TrkA was immunoprecipitated. Western blot was performed to detect ubiquitination, tyrosine phosphorylation, and protein levels. A representative experiment is shown. Note the correlation between TrkA activation and ubiquitination. B, Nedd4-2 ubiquitinates and binds to WT TrkA but not to TrkAP782S. Lysates from HEK293 cells transfected with WT TrkA or TrkAP782S receptors and FLAG-Nedd4-2 were immunoprecipitated using Trk antibodies. Western blots were performed to assess TrkA ubiquitination, Nedd4-2 coimmunoprecipitation and TrkA. The expression levels of Nedd4-2 and tubulin as a control loading are shown. C, TrkAP782S is less ubiquitinated than WT TrkA. Equal amounts of lysates from cultured WT and KI DRG neurons treated with NGF (50 ng/ml) were immunoprecipitated with TrkA antibodies. Western blot analyses were performed to assess the levels of ubiquitination, expression, and the phosphorylation of TrkA proteins. A representative experiment is shown. WT, Wild-type TrkA; KI, TrkAP782S. D, Quantification of TrkA ubiquitination was performed using ImageJ (NIH). The intensity of the bands for the ubiquitination and phosphorylation of TrkA neurotrophin receptors were quantified and normalized to the amount in WT samples (100%). Results are means ± SEM; p values were calculated using a two-tailed Student's t test (n = 4). Note the decrease in the ubiquitination of TrkAP782S despite its increased phosphorylation.
To address the in vivo effects of TrkA ubiquitination, a KI mouse was generated which expresses a mutant TrkA protein with a single change in proline782 to serine (TrkAP782S; Yu et al., 2011). Proline782 is part of the PPVY785 motif present in TrkA, but not TrkB or TrkC, that is responsible for the binding of Nedd4-2 to TrkA and the ubiquitination of this latter (Fig. 1B; Arévalo et al., 2006). We first addressed whether the ubiquitination of TrkAP782S expressed in KI DRG neurons was altered. NGF-dependent sensory neurons from WT and KI mouse embryos were cultured. Upon stimulation with NGF (50 ng/ml) for 10 min, we observed a decrease in the ubiquitination of TrkAP782S compared with WT TrkA (Fig. 1C). Quantification of four independent experiments revealed a significant reduction in the ubiquitination of TrkAP782S compared with WT TrkA in sensory neurons (68.6 vs 100%; Fig. 1D). To rule out the possibility that the reduced TrkAP782S ubiquitination observed in neurons might have been a consequence of impaired activation of the mutant receptor in response to NGF, we tested its activation. We observed that TrkAP782S activation was increased in comparison with WT TrkA (138 vs 100%; (Fig. 1C,D). Together with previous data, this suggests that the reduced ubiquitination of TrkAP782S was not due to the impairment of TrkAP782S activation, but to the lack of Nedd4-2 binding and ubiquitination.
Because the TrkAP782S protein levels appeared to be reduced in the immunoprecipitation experiments (Fig. 1C), we assessed TrkAP782S levels in the DRGs isolated from KI mice. We analyzed TrkAP782S expression using lysates from cultured DRG neurons and observed a decrease in the protein levels of TrkAP782S compared with WT TrkA (Fig. 2A). The quantification of several independent experiments using two different antibodies, anti-203 and RTA, which recognize different epitopes of TrkA, indicated a 32 and 23% reduction, respectively (Fig. 2B). The differences in the results may be explained in terms of the antibodies used: the RTA antibody is directed to the extracellular domain of TrkA and the 203 antibody was generated against a peptide that includes the PPVY motif, which may display altered affinity for the TrkAP782S receptor. In any case, KI neurons expressed a lower level of TrkAP782S protein. To determine whether the differences in protein levels were a consequence of different levels of WT and KI trkA mRNA, we obtained total RNA from DRGs and performed quantitative real time PCR. To our surprise, there was a 40% reduction in the trkA mRNA levels obtained from the KI DRGs (Fig. 2C), whereas there were no differences in the actin mRNA levels (Fig. 2D). The differences observed in protein levels between TrkA and TrkAP782S were less prominent than at the mRNA level (23 vs 40% reduction; Fig. 2B,C). Normalization of TrkA protein levels to the corresponding mRNA levels indicated that there was an increased amount of TrkAP782S versus WT TrkA (Fig. 2D). Therefore, KI mice have reduced TrkAP782S protein levels, possibly due to a reduced amount of mRNA synthesis or stability (see Discussion), which is somehow compensated at the post-translational level.
Figure 2.
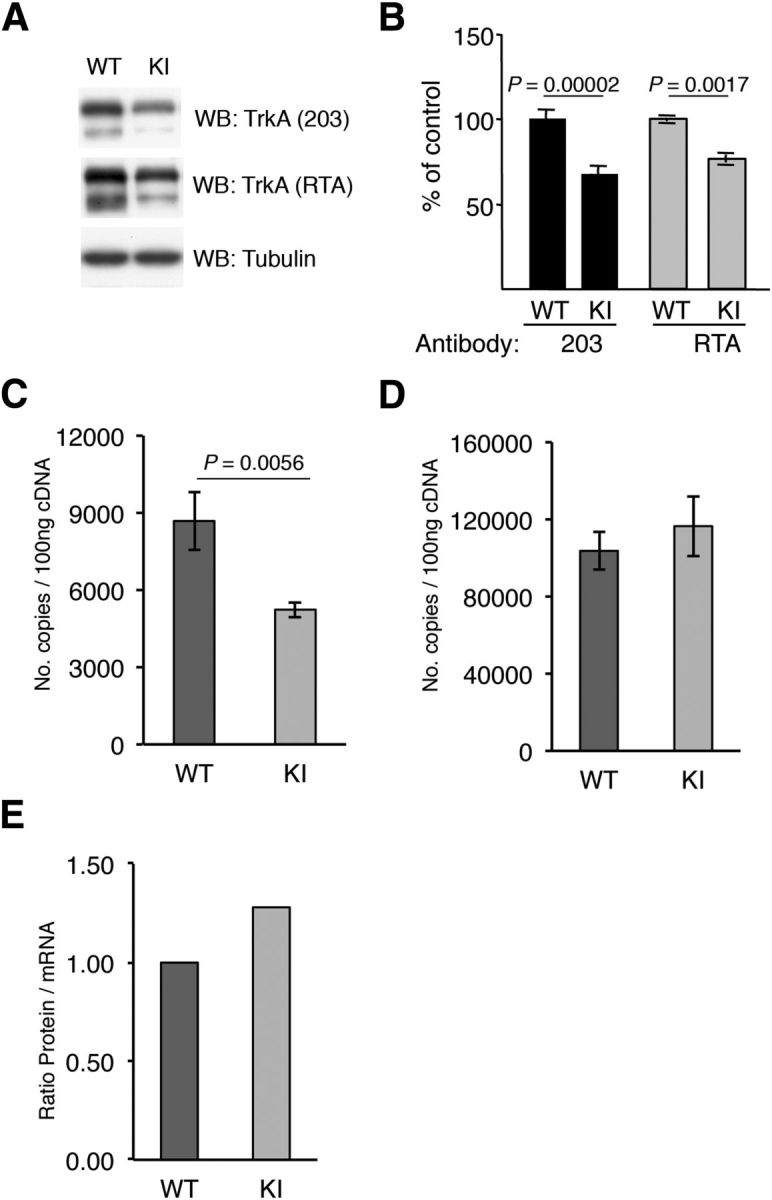
Reduced amounts of TrkAP782S protein due to reduced mRNA levels. A, Reduced amount of TrkAP782S protein in KI mice. Lysates from cultured DRG neurons were subjected to Western blot analysis with the indicated antibodies. B, Quantification of TrkA protein levels in WT and KI DRG neurons. Western blots were scanned and quantified using ImageJ software. Data are presented as means ± SEM; p values were calculated using a two-tailed Student′s test (n = 12). C, qPCR analysis showing the reduced expression of trkAP782S mRNA. The trkA transcripts for WT and KI were 8683 ± 1123 and 5236 ± 314 per 100 ng of cDNA, respectively. Results are means ± SEM of three independent experiments performed in triplicate. The p value was calculated using a two-tailed Student's t test. D, Actin mRNA is not altered in KI DRG neurons. Actin transcripts for WT and KI were 103,700 ± 9707 and 116,500 ± 15,500 per 100 ng of cDNA, respectively. Results are means ± SEM of three independent experiments performed in triplicate. E, Increased protein/mRNA ratio in KI mice. The ratio of the amount of protein vs mRNA levels was normalized to the ratio of the WT sample. Note the increase in the ratio in the KI samples.
Impaired degradation and enhanced recycling of TrkAP782S
Previously, more TrkA receptor protein was observed along with enhanced TrkA stability in neurons depleted of Nedd4-2 (Yu et al., 2011). We surmised that TrkAP782S protein might be more resistant to degradation than WT TrkA protein in response to NGF. To address this issue, we performed degradation experiments using biotinylation in response to NGF with WT and KI DRG neurons (Fig. 3A). Surface TrkA was labeled with biotin to follow the fate of the receptors from the plasma membrane when stimulated with NGF. After 60 min of NGF treatment, there was a clear decrease in the amount of biotin-labeled WT TrkA as a result of TrkA degradation, but not in the amount of TrkAP782S (Fig. 3B). Quantification of three independent experiments indicated no significant degradation of surface TrkAP782S upon NGF stimulation for 60 min, whereas a 29% reduction was noted in surface WT TrkA (Fig. 3C). No differences in the response to NGF were detected in the degradation rate of p75 obtained from WT or KI neurons (Fig. 3B,C). Therefore, TrkAP782S has a lower degradation rate than WT TrkA in response to NGF.
Figure 3.

Decreased degradation and increased surface expression of TrkAP782S in response to NGF in DRG neurons. A, A receptor degradation assay was performed as depicted in the schematic diagram. Cultured DRG neurons from WT and KI mice were biotinylated to label surface proteins and were treated or not with NGF (50 ng/ml) for 60 min. Cells were processed as previously described (Yu et al., 2011). Subsequently, Western blot analyses with Trk, p75, and tubulin antibodies were performed. B, Representative Western blots are shown. Actin was used as a negative control for biotinylated proteins and tubulin as a loading control for lysates. C, Quantification of biotin-labeled TrkA and p75. Data are normalized to the amount of biotinylated TrkA and p75 in nontreated neurons. Results are presented as means ± SEM; p values were calculated using a two-tailed Student's t test (n = 3). Note that the levels of TrkAP782S at 60 min are similar to 0 min. D, A schematic diagram of the surface expression assay using the biotinylation procedure in response to NGF is shown. Mouse DRG neurons were treated or not with NGF (50 ng/ml) for 60 min, and then biotinylated as previously described (Yu et al., 2011). E, Representative Western blots are shown. Actin and tubulin were used as a negative controls for biotinylated proteins and as a loading control for lysates respectively. F, Quantification of surface TrkA and p75 upon NGF treatment. Data are presented as means ± SEM; p values were calculated using a two-tailed Student's t test (n = 4). Note that the surface levels of TrkAP782S at 60 min are similar to 0 min.
What is the fate of the TrkAP782S protein after NGF stimulation? To address this question, we assessed the amount of TrkA at the cell surface upon NGF treatment in a biotin-labeling experiment, as outlined in Figure 3D and described previously (Yu et al., 2011). We observed a reduction over time in the amount of surface TrkA from WT neurons, whereas the reduction in TrkAP782S surface levels from KI neurons was attenuated in response to NGF stimulation for 60 min (Fig. 3E,F). The levels of p75 in the cell surface in response to NGF were similar in WT and KI neurons (Fig. 3E,F). Previous results indicated that the internalization rate of TrkAP782S in transfected cells in response to NGF was similar to that of WT TrkA (Arévalo et al., 2006). Accordingly, the biotinylation results suggest that TrkAP782S recycling may be enhanced in sensory neurons in response to NGF.
Defective trafficking of TrkAP782S in response to NGF
The alterations in the ubiquitination and degradation of TrkAP782S observed allow us to speculate that the trafficking of this mutant receptor in response to NGF may be affected. To assess the trafficking of activated TrkAP782S, we performed immunofluorescence analysis in WT and KI neurons using phospho-specific TrkA antibodies and different endosomal markers (Rajagopal et al., 2004). Because these sensory neurons were maintained in the presence of NGF, they were first starved to assess the trafficking of TrkA in response to acute NGF treatment. After starvation, neurons were stimulated with NGF for different times and stained. Upon NGF treatment for 10 and 30 min, internalized WT TrkA and TrkAP782S colocalized with the early endosomal marker EEA1 (Fig. 4A). No differences were observed at early time points (10 min), suggesting that both receptors reached this compartment similarly upon activation. However, in early endosomes we found a 42% increase in TrkAP782S compared with WT TrkA after 30 min of NGF treatment (Fig. 4B), suggesting an accumulation of the mutant receptor in this compartment. In addition, we performed colocalization studies of active WT TrkA and TrkAP782S with the late endosomal marker Rab7 (Fig. 4C). Quantification of the data indicated 43 and 49% decreases in the amount of pTrkAP782S compared with WT pTrkA present in the late endosomes after 10 and 30 min of NGF treatment, respectively (Fig. 4D). Together, the colocalization experiments suggest that TrkAP782S exhibits a trafficking defect between early and late endosomal compartments that may be a result of its impaired ubiquitination.
Figure 4.
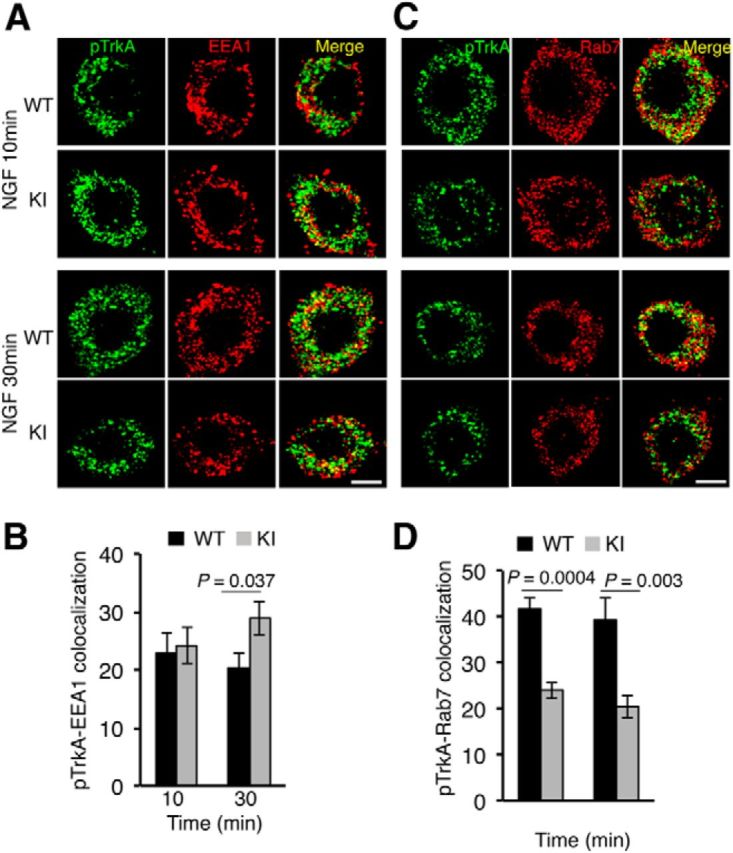
Defective trafficking of active TrkAP782S in NGF-stimulated DRG neurons. A, Colocalization of pTrkA with EEA1 compartments upon NGF treatment in WT and KI DRG neurons. Immunofluorescence was performed as described in Material and Methods. Images were taken with a confocal microscope. Scale bar, 10 μm. B, Quantification of pTrkA colocalization in EEA1 endosomes. Images were processed using ImageJ and the percentage of colocalization was quantified. Data are presented as means ± SEM; p values were calculated using a two-tailed Student's t test (n = 9–10 neurons/time point). C, Colocalization of pTrkA with Rab7 compartments upon NGF treatment in WT and KI DRG neurons was performed as described in A. Scale bar, 10 μm. D, Quantification of pTrkA colocalization in Rab7 endosomes was performed as described in B. Data are presented as means ± SEM; p values were calculated using a two-tailed Student's t test (n = 10 neurons/time point).
Enhanced numbers of DRG neurons in TrkAP782S mice
The dependence of most DRG neurons on NGF-TrkA signaling during development is evident from the substantial cell loss in mice harboring null mutations in the ngf and trkA genes (Crowley et al., 1994; Smeyne et al., 1994). Therefore, we assessed whether the number of L3 DRG neurons in KI mice at P0 was altered. Upon Nissl staining of L3 DRGs from WT and KI mice (Fig. 5A), cell counts revealed a significant increase in the numbers of DRG neurons from KI compared with WT mice. The mean number of neurons was 4578 ± 553 and 3100 ± 382 for KI and WT DRGs, respectively (Fig. 5B). Thus, the expression of TrkAP782S protein, which presents reduced ubiquitination and enhanced receptor activation, confers survival advantages to DRG neurons.
Figure 5.

Increased amount of DRG neurons in TrkAP782S mice. A, Nissl staining of L3 DRGs from P0 WT and KI mice. Representative pictures are shown. Scale bar, 100 μm. B, L3 DRGs from P0 KI mice have more neurons than sibling WT mice. Heterozygous females were mated with heterozygous males to obtain WT and KI mice. Results are presented as means ± SEM; p value was calculated using a two-tailed Student's t test (n = 4). Note the increase number of neurons in DRGs from KI mice.
Increased thermal and inflammatory pain in mice expressing TrkAP782S
To address the physiological consequences of the P782S mutation, we addressed the role of TrkA in pain sensation. There is a considerable body of evidence to support a role for NGF-TrkA in hyperalgesia (Mantyh et al., 2011; Indo, 2012). Thus, we tested whether WT and KI mice have differential sensitivities to pain by assessing standard nociception tests. First, we assessed the susceptibility of KI mice to noxious heat using the tail-flick assay. KI mice showed an increased sensitivity to noxious heat compared with WT mice (Fig. 6A). The mean latency of tail withdrawal was 3.09 ± 0.2 s and 5.2 ± 0.66 s for KI and WT mice, respectively (Fig. 6A). Similar results were obtained when the mice were exposed to cold pain. The mean latency of paw withdrawal or paw licking was 4 ± 0.55 s and 6.33 ± 0.58 s for KI and WT mice, respectively (Fig. 6B). However, the WT and KI mice did not display significant differences in the susceptibility to mechanical stimuli as studied with the Randall–Selitto test (Fig. 6C). Thus, TrkAP782S mice exhibit an enhanced sensitivity to thermal pain but not to mechanical pain.
Figure 6.
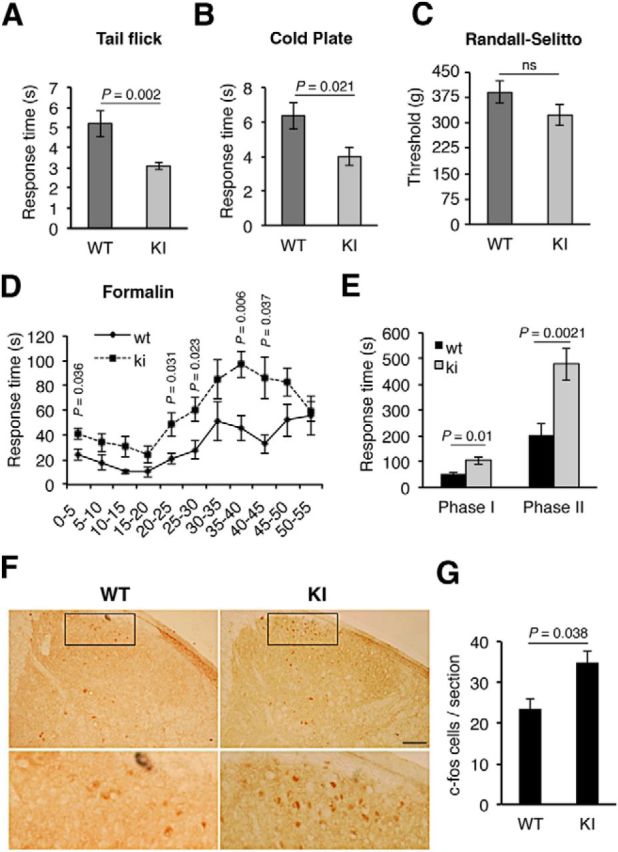
Enhanced sensitivity to thermal and inflammatory pain in TrkAP782S mice. A, TrkAP782S mice are more sensitive to thermal pain as compared with WT mice, assessed by tail-flick test (n = 11 for WT and n = 15 for KI). B, TrkAP782S mice are more sensitive to cold pain compared with WT mice (n = 9 for WT; n = 5 for KI). C, TrkAP782S mice display a similar noxious mechanical pain to WT mice assessed with the Randall–Selitto assay (n = 20 for WT and KI). D, TrkAP782S mice are more sensitive to inflammatory pain induced by formalin injections. Licking and biting times upon formalin injection in the right hindpaw were recorded and quantified in cumulative periods of 5 min (n = 7 for WT; n = 13 for KI). E, Cumulative licking and biting times in Phases I (acute, 0–15 min) and II (inflammatory, 20–55 min). The mean observed response time in Phase I was 51.0 ± 7.65 s and 105.38 ± 14.58 s for the WT and KI mice, whereas the mean observed response time in Phase II was 205.89 ± 43.03 s and 478.77 ± 63.04 s. F, c-fos staining in the spinal cord from WT and KI mice 3 h after formalin injection. TrkAP782S mice showed an increased c-fos staining in lamina I and II after formalin injection compared with WT mice. Note the increased number of c-fos-positive neurons in the section from a KI mouse. Scale bar, 200 μm. G, Quantification of c-fos-positive neurons. c-fos staining in the spinal cord from WT and KI mice after formalin injection (n = 3). Results for all panels are presented as means ± SEM; p values for A–C, G were calculated using a two-tailed Student's t test, and for D, E using the Mann–Whitney test, two-tailed. ns, Nonsignificant. Note the increased sensitivity of KI mice to thermal- and inflammatory-mediated pain.
We next examined whether the WT and KI mice showed differential sensitivities to inflammatory pain induced by formalin injection. KI mice displayed an increased responsiveness compared with WT mice (Fig. 6D). Both WT and KI mice exhibited the typical biphasic response induced by formalin injection (Fig. 6D), although the responses were significantly higher in the KI than in WT mice (105.4 ± 14.6 s vs 51 ± 7.65 s and 478.7 ± 63 s vs 205.9 ± 43 s for Phases I and II, respectively; Fig. 6E). To address neuronal activity within the spinal cord of WT and KI mice in response to inflammatory pain, we tested the levels of c-fos, a well established activity-dependent gene, in spinal cord sections 3 h after formalin injection. The expression of c-fos has been connected directly to increased neuronal activity in response to extracellular stimuli (Greenberg et al., 1985). Injection of formalin in the right hindpaw induced a higher number of c-fos-positive neurons in laminae I and II of the ipsilateral dorsal horn of KI mice compared with WT mice (Fig. 6F). The amounts of c-fos-positive cells were 23 and 35 per section in the WT and KI animals, respectively (Fig. 6G). The expression of c-fos was specific to formalin injection because it was not detected at the contralateral side of the injection (data not shown). Thus, KI mice exhibit an enhanced sensitivity to inflammatory pain that can be monitored by increased neuronal activity in the spinal cord. Together, these data indicate that KI mice exhibit an enhanced response to noxious thermal and inflammatory stimuli but not to mechanical pain.
Increased numbers of substance P- and CGRP-positive neurons and enhanced skin innervation in TrkAP782S mice
To verify that the effects of the mutation of P782S in mice was associated with a pain phenotype, we performed in situ hybridization experiments on DRGs from WT and KI P0.5 pups to detect substance P and CGRP mRNA levels. Both genes are induced in response to NGF signaling (Otten et al., 1980; Lindsay and Harmar, 1989; Crowley et al., 1994; McMahon et al., 1995). We observed a significant increase in the amount of CGRP- and substance P-positive cells in DRGs from KI compared with WT mice (Fig. 7A,B), equivalent to the numbers of neurons observed in both genotypes. To assess whether the increase numbers of DRG neurons had an impact on skin innervation, we performed immunohistochemistry assays using skin from hindpaws. The pan-neuronal marker PGP9.5 (Guo et al., 2011a) was used to detect the nerve terminals that innervate the skin (Fig. 7C). The skin from the KI mice displayed an enhanced area of innervation compared with the WT mice (Fig. 7D). Thus, the expression of TrkAP782S leads to an increase in CGRP- and substance P-positive neurons and to an enhanced innervation of the skin.
Figure 7.

Increased CGRP and substance P (SP) expression and skin innervation in TrkAP782S mice. A, Enhanced expression of CGRP and substance P in KI mice. In situ hybridizations using antisense probes against CGRP and substance P in lumbar DRGs from P0.5 pups. Sense probes for CGRP and substance P did not show any staining. Representative pictures of DRGs from WT and KI mice. Scale bar, 100 μm. B, Quantification of positive neurons for CGRP and substance P in L3-5 DRGs from WT and KI P0.5 pups. The area of each DRG section was quantified with ImageJ software and the amount of positive neurons was counted. The number of sections quantified for CGRP was five for WT and nine for KI, and the number of sections for substance P was five for WT and six for KI. Results are presented as means ± SEM; p value was calculated using a two-tailed Student's t test. C, Skin innervation in TrkAP782S mice is increased. Skin from the hindpaws of WT and KI mice was obtained and stained with the pan-neuronal marker PGP9.5. A representative image of the staining of skin from WT and KI mice is shown (n = 4). Note the increased signal exhibited by the skin from a KI mouse. Scale bar, 50 μm. D, Quantification of PGP9.5 staining in the hindpaw skin from WT and KI mice. The area of PGP9.5 staining was quantified as described in Material and Methods. Results are presented as means ± SEM; p value was calculated using a two-tailed Student's t test (n = 4).
Discussion
In this report, we provide evidence that the TrkAP782S mutant protein results in impaired ubiquitination of TrkA that plays a critical role in the degradation and trafficking of the receptor in response to NGF. To address in vivo the role of TrkA ubiquitination, we used a KI mouse model that expresses the mutant TrkA neurotrophin receptor, TrkAP782S, which is insensitive to Nedd4-2 E3 ubiquitin ligase action (Fig. 1B; Yu et al., 2011). Although TrkAP782S is more active than WT TrkA, it is less ubiquitinated in response to NGF, resulting in impaired trafficking and degradation. KI mice exhibit an enhanced sensitivity to thermal and inflammatory pain, but not to mechanical pain. This is due to an increased amount of DRG neurons expressing CGRP and substance P as well as enhanced skin innervation and neuronal activity present in mutant mice.
The development of a mouse model expressing a mutant TrkA receptor insensitive to Nedd4-2 has allowed us to determine in vivo a role for Nedd4-2-mediated TrkA ubiquitination. Unexpectedly, the levels of TrkAP782S protein in this mouse are reduced compared with WT TrkA because of the reduced trkAP782S mRNA levels. Several different possibilities may be invoked to explain these results: (1) changes in the nucleotides required to mutate proline to serine may affect the stability of the mRNA; (2) insertion of the loxP and FRT sites required to generate the construct to target the trkA gene may have affected the transcriptional activity of the trkA promoter, and therefore, mRNA expression; and (3) a potential negative-feedback loop may reduce trkAP782S mRNA expression due to its high levels of activation, as previously suggested (Deppmann et al., 2008). Nonetheless, TrkAP782S protein is less ubiquitinated than WT TrkA despite its higher activation levels. One consequence of the reduced TrkAP782S ubiquitination is the impairment of receptor degradation (Fig. 3B,C). Remarkably, the DRG from this KI mouse exhibited an increased number of neurons in vivo (Fig. 5A,B). A higher activation of TrkAP782S protein and downstream signaling cascades in response to NGF may account for these in vivo effects. It has previously been reported that the expression of this mutant receptor results in an increased in vitro survival of cultured sensory neurons (Yu et al., 2011). Moreover, these data are supported by previous findings, such as that the overexpression of Nedd4-2 decreases the levels of TrkA (Arévalo et al., 2006) and that the depletion of Nedd4-2 in sensory neurons results in a more stable TrkA (Yu et al., 2011). All these data indicate that ubiquitination is a key modification to TrkA that controls the signal duration regulating the neuronal survival dependent on NGF.
Although the degradation of TrkAP782S was reduced, no differences were observed in the transit of active TrkAP782S to the early endosomal compartment after 10 min of NGF stimulation (Fig. 4A,B), supporting previous data indicating that the internalization of this mutant TrkA was not altered (Arévalo et al., 2006). However, after 30 min of NGF stimulation, an accumulation of TrkAP782S was observed in early endosomes compared with WT TrkA (Fig. 4A,B), which may have been caused by the impaired trafficking of the mutant receptor to the late endosomes upon NGF stimulation (Fig. 4C,D). Early endosomes represents a sorting station to redirect internalized proteins to degradation via late endosomes or to recycling via recycling endosomes (Sorkin and von Zastrow, 2009). The observed accumulation of TrkAP782S in early endosomes may explain the increased amount of biotin-labeled receptor found in the plasma membrane after 60 min of NGF treatment (Fig. 3E,F). Together, all these results suggest an important role for TrkA ubiquitination mediated by Nedd4-2 in the proper trafficking of the receptor in response to NGF.
Nedd4-2 has been related directly to the modulation of several different proteins, including channels (ENaC, voltage-gated sodium channels, chloride ion channels, and voltage-gated potassium channels), transporters (dopamine, glutamate, and amino acid and glucose transporters), kinases (Sgk1, ACK1), adaptor proteins (14-3-3), and receptors (TrkA; Yang and Kumar, 2010). Recently, the influence of Nedd4-2 in Nav1.7 channels has been reported in a conditional Nedd4-2 KO mouse in Nav1.8-positive neurons (Laedermann et al., 2013). Nav1.7, which is expressed in nociceptive neurons, has been implicated in the modulation of human pain perception because mutations in SCN9A, the gene encoding Nav1.7, cause either congenital insensitivity or hypersensitivity to pain (Cox et al., 2006; Fertleman et al., 2006; Reimann et al., 2010). In the study by Laedermann et al. (2013), the authors concluded that Nav1.7 currents in DRG neurons depleted of Nedd4-2 are enhanced, and as a consequence, there was an increase in thermal hypersensitivity and also in the inflammatory response in formalin-injected mice. However, considering that Nedd4-2 also modulates TrkA levels in these nociceptive neurons (Arévalo et al., 2006; Yu et al., 2011), the levels of TrkA in those Nedd4-2-depleted DRG neurons and the potential role of NGF-TrkA in the described phenotypes remains to be addressed.
TrkA and NGF have been directly implicated in pain modulation because the mice knocked-out for both genes show remarkable pain phenotypes (Crowley et al., 1994; Smeyne et al., 1994), and mutations in TrkA and/or NGF genes are responsible for pathological conditions in humans, leading to congenital insensitivity to pain (Indo, 2012). The results obtained with our KI mouse model and another very recently described TrkA mouse mutant (Kiris et al., 2014) further support this role and to our knowledge provide the first evidence of a gain-of-function mutation in the TrkA neurotrophin receptor. TrkAP782S mice are hypersensitive to thermal and inflammatory pain, but not to mechanical pain (Fig. 6). The lack of Nedd4-2 binding and the ubiquitination of TrkAP782S result in a more active and stable receptor that elicits an increased amount of nociceptive neurons and enhanced skin innervation (Figs. 5, 7). Therefore, the ubiquitination of the TrkA neurotrophin receptor is a seminal modification regulating NGF-mediated functions in vivo.
The potential analgesic use of NGF-neutralizing antibodies has been recently assessed in a clinical trial in patients suffering for chronic pain due to osteoarthritis (Lane et al., 2010). Although blocking NGF actions by neutralizing antibodies has been shown to have beneficial effects, such as a potent analgesic effect against knee pain (Lane et al., 2010), the side effects observed preclude the use of this approach to modulate neurotrophin signaling. In view of the results presented here, pointing to TrkA ubiquitination as a major regulator of nociception, the modulation of TrkA ubiquitination may provide a novel target for the treatment of pain. Further research to fully elucidate the molecular machinery governing TrkA ubiquitination may in the future allow us to design drugs to control pain without side effects.
Footnotes
This work was supported by Ministerio de Ciencia e Innovación Grant BFU2008-00162, by Ministerio de Economía y Competitividad (BFU2011-22898), by a Marie Curie International Reintegration Grant within the VII European Community Framework Programme by Conserjería de Educación (SA074A08), and Conserjería de Sanidad de Junta Castilla y León (J.C.A.). J.C.A. was a “Ramón y Cajal” Investigator from the University of Salamanca and NARSAD 2009 Young Investigator Awardee. L.T. was supported by the Intramural Research Program of the NCI, Center for Cancer Research, NIH. We thank Dionisio Martín-Zanca, Louis Reichardt, and Moses Chao for the anti-203 Trk, RTA, and p75 antibodies, respectively; Moses Chao and Francis Lee for critical review of the paper and helpful discussions; Lino Tessarollo for sharing unpublished results; and Synphen Wu and Nicholas Skinner for corrections to the paper.
The authors declare no competing financial interests.
References
- Arévalo JC, Yano H, Teng KK, Chao MV. A unique pathway for sustained neurotrophin signaling through an ankyrin-rich membrane-spanning protein. EMBO J. 2004;23:2358–2368. doi: 10.1038/sj.emboj.7600253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arévalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50:549–559. doi: 10.1016/j.neuron.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Arsequell G, Salvatella M, Valencia G, Fernández-Mayoralas A, Fontanella M, Venturi C, Jiménez-Barbero J, Marrón E, Rodríguez RE. Synthesis, conformation, and biological characterization of a sugar derivative of morphine that is a potent, long-lasting, and nontolerant antinociceptive. J Med Chem. 2009;52:2656–2666. doi: 10.1021/jm8011245. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- Deppmann CD, Mihalas S, Sharma N, Lonze BE, Niebur E, Ginty DD. A model for neuronal competition during development. Science. 2008;320:369–373. doi: 10.1126/science.1152677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Geetha T, Jiang J, Wooten MW. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol Cell. 2005;20:301–312. doi: 10.1016/j.molcel.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Greene LA, Ziff EB. Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J Biol Chem. 1985;260:14101–14110. [PubMed] [Google Scholar]
- Guo G, Kan M, Martinez JA, Zochodne DW. Local insulin and the rapid regrowth of diabetic epidermal axons. Neurobiol Dis. 2011a;43:414–421. doi: 10.1016/j.nbd.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Guo T, Mandai K, Condie BG, Wickramasinghe SR, Capecchi MR, Ginty DD. An evolving NGF-Hoxd1 signaling pathway mediates development of divergent neural circuits in vertebrates. Nat Neurosci. 2011b;14:31–36. doi: 10.1038/nn.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Indo Y. Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): mutations and polymorphisms in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Hum Mutat. 2001;18:462–471. doi: 10.1002/humu.1224. [DOI] [PubMed] [Google Scholar]
- Indo Y. Nerve growth factor and the physiology of pain: lessons from congenital insensitivity to pain with anhidrosis. Clin Genet. 2012;82:341–350. doi: 10.1111/j.1399-0004.2012.01943.x. [DOI] [PubMed] [Google Scholar]
- Indo Y, Tsuruta M, Hayashida Y, Karim MA, Ohta K, Kawano T, Mitsubuchi H, Tonoki H, Awaya Y, Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet. 1996;13:485–488. doi: 10.1038/ng0896-485. [DOI] [PubMed] [Google Scholar]
- Kiris E, Wang T, Yanpallewar S, Dorsey SG, Becker J, Bavari S, Palko ME, Coppola V, Tessarollo L. TrkA in vivo function is negatively regulated by ubiquitination. J Neurosci. 2014;34:4090–4098. doi: 10.1523/JNEUROSCI.4294-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laedermann CJ, Cachemaille M, Kirschmann G, Pertin M, Gosselin RD, Chang I, Albesa M, Towne C, Schneider BL, Kellenberger S, Abriel H, Decosterd I. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J Clin Invest. 2013;123:3002–3013. doi: 10.1172/JCI68996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane NE, Schnitzer TJ, Birbara CA, Mokhtarani M, Shelton DL, Smith MD, Brown MT. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med. 2010;363:1521–1531. doi: 10.1056/NEJMoa0901510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Ritter AM, Mendell LM. Nerve growth factor-induced hyperalgesia in the neonatal and adult rat. J Neurosci. 1993;13:2136–2148. doi: 10.1523/JNEUROSCI.13-05-02136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;337:362–364. doi: 10.1038/337362a0. [DOI] [PubMed] [Google Scholar]
- Makkerh JP, Ceni C, Auld DS, Vaillancourt F, Dorval G, Barker PA. p75 neurotrophin receptor reduces ligand-induced Trk receptor ubiquitination and delays Trk receptor internalization and degradation. EMBO Rep. 2005;6:936–941. doi: 10.1038/sj.embor.7400503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW, Koltzenburg M, Mendell LM, Tive L, Shelton DL. Antagonism of nerve growth factor-TrkA signaling and the relief of pain. Anesthesiology. 2011;115:189–204. doi: 10.1097/ALN.0b013e31821b1ac5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319:743–748. doi: 10.1038/319743a0. [DOI] [PubMed] [Google Scholar]
- Martin-Zanca D, Barbacid M, Parada LF. Expression of the trk proto-oncogene is restricted to the sensory cranial and spinal ganglia of neural crest origin in mouse development. Genes Dev. 1990;4:683–694. doi: 10.1101/gad.4.5.683. [DOI] [PubMed] [Google Scholar]
- McMahon SB. NGF as a mediator of inflammatory pain. Philos Trans R Soc Lond B Biol Sci. 1996;351:431–440. doi: 10.1098/rstb.1996.0039. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nat Med. 1995;1:774–780. doi: 10.1038/nm0895-774. [DOI] [PubMed] [Google Scholar]
- Otten U, Goedert M, Mayer N, Lembeck F. Requirement of nerve growth factor for development of substance P-containing sensory neurones. Nature. 1980;287:158–159. doi: 10.1038/287158a0. [DOI] [PubMed] [Google Scholar]
- Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, Cox JJ, Belfer I, Diatchenko L, Zaykin DV, McHale DP, Drenth JP, Dai F, Wheeler J, Sanders F, Wood L, Wu TX, Karppinen J, Nikolajsen L, Männikko M, Max MB, Kiselycznyk C, Poddar M, Te Morsche RH, Smith S, Gibson D, Kelempisioti A, Maixner W, Gribble FM, Woods CG. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc Natl Acad Sci U S A. 2010;107:5148–5153. doi: 10.1073/pnas.0913181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–249. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Martin-Zanca D, Gilbert DJ, Jenkins NA, Copeland NG, Parada LF. trkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in non-neuronal tissues. Development. 1993;118:463–475. doi: 10.1242/dev.118.2.463. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Yang B, Kumar S. Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010;17:68–77. doi: 10.1038/cdd.2009.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Calvo L, Anta B, López-Benito S, Southon E, Chao MV, Tessarollo L, Arévalo JC. Regulation of trafficking of activated TrkA is critical for NGF-mediated functions. Traffic. 2011;12:521–534. doi: 10.1111/j.1600-0854.2010.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]