

SUMMARY
Steven Smith and co-workers probe how the Flemish mutation in amyloid precursor protein (APP) affects its conformation and cleavage by γ-secretase (Tang et al., 2014). They provide molecular insight into how an extracellular inhibitory element and cholesterol interactions affect the generation of Aβ peptides.
Various neurodegenerative diseases are characterized by protein misfolding aggregation, which may be preceded or accompanied by protease cleavage of the affected proteins. Disease-related mutations can thus affect not only the aggregation process, but also this pivotal cleavage event. In Alzheimer’s Disease (AD), cleavage of membrane-associated APP by β- and γ-secretases generates aggregation-prone peptide fragments, including Aβ40 and Aβ42 (Fig. 1). The study by Smith et al in this issue of Structure (Tang et al., 2014)focuses on key structural and genetic aspects of this crucial step. The so-called Flemish mutation A21G (A692G in full-length APP) leads to AD accompanied by extensive Aβ deposition. Although a priori it may be tempting to attribute this to changed aggregation propensity of the mutant Aβ peptide, previous work indicates that it actually increases the formation of Aβ (e.g. (De Jonghe et al., 1998)). Despite a closer proximity to the α- and β-secretase cleavage sites (Fig. 2), it turns out that this mutation actually increases γ-secretase activity by disrupting an extracellular APP substrate inhibition domain (ASID) (Tian et al., 2010). Alas, the molecular underpinnings of the inhibition of γ-secretase, and the effect of the Flemish mutation, remained unclear.
Figure 1.
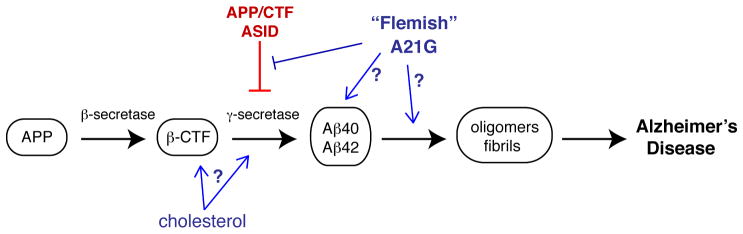
Chain of cleavage and self-assembly events in Alzheimer’s Disease. Full-length APP is cleaved by β- and γ-secretases to give disease-causing Aβ peptides that are prone to self-assembly and aggregation. An extracellular inhibitory ASID domain in APP (and its c-terminal fragment β-CTF) protects against γ-secretase cleavage. AD risk factors like cholesterol and the Flemish mutation increase γ-secretase activity, in part by disrupting the ASID.
Figure 2.

Structural features.(a) Sequence of APP around the TM and inhibitory ASID domains (boxed), showing the Flemish mutation A21G. α-, β-, and γ-secretase cleavage sites are marked with blue arrows. The Aβ-40/42 sequences are shown in bright/dark red. (b) Secondary structure elements for wild type and the A21G mutant. (c) Helical wheel plots of the segment marked in (b), showing the extended helix in the mutant (blue residues), as well as the G33 dimerization site that is active in lipid bilayers. A black arrow indicates A42, which is a key dimerization site in micellar environments. Although the TM domain is shown as a single α-helix, other studies suggest local distortions in both bilayers and micelles.
Steven Smith and colleagues now confirm the inhibitory effect of the extracellular ASID and the fact that the Flemish mutation disrupts the γ-secretase inhibition. Importantly, they go on to probe structural features of membrane-bound APP in both wild-type and mutant proteins to address the structural mechanisms involved. To do so in the context of a lipid bilayer environment, they leverage Fourier- Transform Infrared (FTIR) and magic-angle-spinning (MAS) solid-state nuclear magnetic resonance (ssNMR) spectroscopies, complemented with mutational and isotopic labeling experiments. Thus, they provide insight into the structural features of the extracellular domain, and in particular the inhibitory ASID. They find the latter to adopt a β-sheet structure that is disrupted by mutations, including the A21G Flemish mutation. This secondary structure change is accompanied by an apparent extension of the transmembrane (TM) α-helix, and an enhanced propensity of this helix to dimerize within the membrane.
Dimerization of the APP TM domain has been investigated by different structural methods before, leading to divergent models of the point of interaction between the helices. Conflicting reports suggest dimerization via different GxxxG or GxxxA motifs (see e.g. (Chen et al., 2014; Song et al., 2013a)). Crucial MAS ssNMR measurements by Smith et al. reveal that within their lipid bilayer samples, the dimerization involves an intimate interaction between Gly33 residues, which is in contrast with detergent-based studies that indicate a closer contact involving A42.
Importantly, they also probe the effect of cholesterol, which a risk factor in AD and appears to modulate the ability of γ-secretase to generate Aβ. It is shown that cholesterol enhances the TM domains’ α-helicity, in a way that is similar to (and synergistic with) the A21G mutation. Cholesterol also modulates the propensity for the TM helices to dimerize, reminiscent of earlier work(Barrett et al., 2012; Song et al., 2013a). As an aside, and likely simply coincidence, the (unexpected) β/α transition in context of cholesterol binding reminds us of our recent observations(Hoop et al., 2012)applying similar methods to the presumed cholesterol-binding “CRAC” motif of caveolin-1. It seems like in APP, cholesterol-interacting residues span an analogous β/α transition, which might reflect a “CRAC-like” motif found in this exact part of the protein (Abad et al., 2009; Song et al., 2013b).
The study by Tang et al. helps to clarify a number of ongoing debates regarding the role of APP mutations, TM dimerization, and the effects of cholesterol. These new insights are specifically enabled by their use of structural methods that allow one to employ lipid bilayers featuring various lipid mixtures as well as cholesterol. This combination of FTIR and ssNMR is shown to be powerful and highly complementary, with a key role for isotopic labeling that benefits both methods. The explicit comparison of different lipid bilayers as well as micellar membrane mimics makes it clear that the latter abolish the ASID β-sheet structure, a reinforces previous observations of sensitivity to the nature of the membrane environment (Lu et al., 2011).
It appears that cholesterol and the Flemish mutation both act through similar mechanisms, involving modulation of the ASID secondary structure, helicity of the TM domain, and dimerization. This raises “chicken and egg” type questions regarding the interplay and sequence of these structural effects. Moreover: how do these structural features affect γ-secretase, both in terms of its propensity to do the cleavage (cause increased Aβ levels) and the location of cleavage (changing the Aβ40/42 ratios)? The current work provides a starting point to address these questions, and highlights the importance of the choice of membrane mimics(Zhou and Cross, 2013)when doing so.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Abad C, Martínez-Gil L, Tamborero S, Mingarro I. BBA - Biomembranes. 2009;1788:2132–2141. doi: 10.1016/j.bbamem.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR. Science (New York, NY) 2012;336:1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Gamache E, Rosenman DJ, Xie J, Lopez MM, Li Y-M, Wang C. Nat Commun. 2014;5:Article number: 3037. doi: 10.1038/ncomms4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jonghe C, Zehr C, Yager D, Prada CM, Younkin S, Hendriks L, Van Broeckhoven C, Eckman CB. Neurobiol Dis. 1998;5:281–286. doi: 10.1006/nbdi.1998.0202. [DOI] [PubMed] [Google Scholar]
- Hoop CL, Sivanandam VN, Kodali R, Srnec MN, van der Wel PCA. Biochemistry. 2012;51:90–99. doi: 10.1021/bi201356v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JX, Yau WM, Tycko R. Biophys J. 2011;100:711–719. doi: 10.1016/j.bpj.2010.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Hustedt EJ, Brandon S, Sanders CR. Biochemistry. 2013a;52:5051–5064. doi: 10.1021/bi400735x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Kenworthy AK, Sanders CR. Protein Science. 2013b;23:1–22. doi: 10.1002/pro.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang T-C, Hu Y, Kienlen-Campard P, El Haylani L, Decock M, Van Hees J, Fu Z, Octave J-N, Constantinescu SN, Smith SO. Structure. 2014 doi: 10.1016/j.str.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Bassit B, Chau D, Li YM. Nat Struct Mol Biol. 2010;17:151–158. doi: 10.1038/nsmb.1743. [DOI] [PubMed] [Google Scholar]
- Zhou HX, Cross TA. Annu Rev Biophys. 2013;42:361–392. doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]