

Abstract
Purpose
Both Hsp90 and checkpoint kinase 1 (Chk1) have emerged as novel therapeutic targets. We conducted a phase I study of irinotecan and the Hsp90 inhibitor 17AAG, which can also down-regulate Chk1, in patients with solid tumors.
Experimental Design
During the dose-escalation phase, patients received intravenous irinotecan followed by 17AAG once weekly for 2 weeks in a 21-day cycle. At the maximum tolerated dose (MTD), additional patients were enrolled to undergo pre- and post-17AAG tumor biopsies for pharmacodynamic evaluation. The pharmacokinetics of irinotecan, 17AAG, and their metabolites were characterized. Tumor p53 status as determined by immunohistochemistry was correlated with antitumor activity.
Results
Twenty-seven patients with a variety of solid tumors were enrolled. Four patients developed dose-limiting toxicity (DLT) at dose level 4 (100 mg/m2 irinotecan and 375 mg/m2 17AAG) including nausea, vomiting, diarrhea, and pulmonary embolism. The pharmacokinetics of 17AAG and its metabolite were not significantly affected by coadministration of irinotecan, and vice versa. There was no partial response although tumor shrinkage was observed in 6 patients. Five of 10 patients with p53-mutant tumor had stable disease as the best response compared with 2 of 6 patients with p53-wildtype tumor (P=0.63). Evidence for Hsp90 inhibition by 17AAG, resulting in phospho-Chk1 loss, abrogation of the G2/M cell cycle checkpoint, and cell death could be demonstrated in tumor biopsy samples obtained at the MTD.
Conclusions
The combination of irinotecan and 17AAG can be given to patients with acceptable toxicity. The recommended phase II dose of the combination is 100 mg/m2 irinotecan and 300 mg/m2 17AAG.
Introduction
The molecular chaperone Hsp90 is essential for promoting stability and functional maturation of a number of signaling proteins including Her2, Raf-1, Akt, C-Met, and HiF-1α (1). 17AAG inhibits the amino-terminus ATPase activity of Hsp90, resulting in destabilization of its chaperoned “clients”, thereby allowing simultaneous targeting of multiple oncogenic signaling pathways in tumors. Preclinical studies of 17AAG revealed promising antitumor activities in cell culture and xenograft models (2–4). Phase I studies of 17AAG showed acceptable toxicity, and down-regulation of Hsp90 clients in normal and tumor tissues following treatment (5–9).
Preclinical data also showed that 17AAG sensitizes tumors to chemotherapy including nucleoside analogues (10, 11) and topoisomerase I poisons (12). One possible mechanism by which 17AAG enhances the cytotoxicity of chemotherapy is by depleting Chk1, an Hsp90 client kinase that is critical for the S and G2/M cell cycle checkpoints. We have shown that 17AAG selectively abrogates the G2/M checkpoint induced by a topoisomerase I poison and promotes apoptosis in colon cancer cells that lack p53. We therefore conducted a phase I study of 17AAG and irinotecan to determine the toxicity profile, MTD, pharmacokinetics (PKs), and pharmacodynamics (PDs) of the combination.
Patients and Methods
Patient selection
Patients with histologically confirmed solid tumors refractory to standard treatment, or for which no standard therapy exists, were considered. Other eligibility criteria included age ≥ 18 years; KPS ≥ 60; WBC count ≥ 3000/mm3; absolute neutrophil count (ANC) ≥ 1500/mm3; platelet count ≥ 100,000/mm3; creatinine ≤ 1.5 mg/dl; total bilirubin ≤ 1.5 mg/dl; AST and ALT levels ≤ 3 times the upper limit of normal (ULN) (or ≤ 5 times ULN if there were known liver metastases); and normal serum potassium, magnesium, and calcium (or ionized calcium). Prior treatment with irinotecan but not 17AAG was allowed.
Patients were excluded if they were pregnant or lactating, had new untreated central nervous system metastases, primary brain tumor, HIV infection, severe egg allergy, or were taking potent CYP3A4 inducers or inhibitors. Because of supraventricular arrhythmias and pulmonary toxicity associated with 17AAG, patients at risk for cardiopulmonary toxicities were excluded, including those with a history of prior chest radiation, active ischemic heart disease or cardiac arrhythmias, left bundle branch block, heart failure, prolonged QTc, baseline diffusion-limited carbon monoxide ≤80%, baseline ejection fraction < 50%, and debilitating pulmonary diseases.
Study design and treatment plan
This phase I study consisted of a dose escalation and an expansion phase. During dose escalation, 3 to 6 patients per dose level were treated with escalating doses of irinotecan and 17AAG from 85 mg/m2 and 220 mg/m2, respectively, up to the highest levels of 125 mg/m2 and 450 mg/m2, respectively, or until a DLT was observed in ≥ 2/3 or ≥ 2/6 patients. Irinotecan was given as a 30-minute infusion followed by a 120-minute infusion of 17AAG once weekly for 2 consecutive weeks in a 21-day cycle. To study possible PK interactions between the 2 agents, irinotecan alone was given on day 1 and 17AAG on day 2 during the second cycle (see Supplemental Fig. 1 for study design schema).
Toxicity was graded according to National Cancer Institute Common Toxicity Criteria Version 3.0. A DLT was defined as the occurrence of grade 4 hematological toxicity, febrile neutropenia (ANC <1,000/mm3 and fever ≥ 38.5°C), ≥ grade 3 nonhematological toxicity, and any delay in treatment for > 7 days. Patients received treatment at the same dose in the absence of DLT if, on the day of scheduled treatment, the ANC was ≥ 1500/mm3, and platelet count ≥ 100,000/mm3; otherwise, therapy was held. A minimum of 3 patients were followed for at least 1 complete cycle of therapy before dose was escalated to the next level. If a DLT was observed, the cohort was expanded to 3 additional patients. The MTD was defined as the highest dose level with DLTs seen in ≤ 1/6 patients.
After the MTD had been determined, 12 additional patients were treated with the MTD at the expansion cohort to assure tolerability. A decision rule based on Bayesian analysis dictated that ≤ 5 DLTs out of 15 patients treated at a given dose level would define a dose acceptable for further study (13).
Patients treated at the expansion cohort also underwent tumor biopsies for PD evaluation. On day 1 of cycle 1, patients received irinotecan alone and underwent tumor biopsy 24–48 hours later; on day 8 of cycle 1, patients received both irinotecan and 17AAG, with a second biopsy performed 24–48 hours after (see Supplemental Fig. 1 for study design schema). Since patients received only one combination treatment during cycle 1 at the MTD, DLT evaluation was extended to cycle 2 in this cohort to ensure adequate assessment of tolerability of the MTD.
Because of reported cardiopulmonary toxicities associated with 17AAG, the protocol was amended to include pulmonary function tests and an echocardiogram or MUGA scan at baseline. Laboratory evaluation for potassium, magnesium, and calcium (or ionized calcium) performed within 24 hours of each 17AAG treatment had to be within institutional normal limits. Any electrolytes disturbance necessitated correction to within the institutional normal range prior to initiation of treatment. A post-17AAG infusion EKG was performed during the first week of therapy in addition to that obtained at baseline.
Measurement of tumor indicator lesion(s) was performed within 4 weeks of beginning study treatment and every 2 cycles thereafter. Treatment responses were evaluated using Response Evaluation Criteria in Solid Tumors (RECIST) (14).
Drug supply
Irinotecan (Camptosar®, Pfizer, New York, NY), commercially available, was reconstituted in 5% dextrose solution. 17AAG (NSC 330507, National Cancer Institute, Bethesda, MD) was prepared using the 2% dimethyl sulfoxide (DMSO)/egg phospholipid diluent (NSC 704057, National Cancer Institute, Bethesda, MD) as previously described (15).
Pharmacokinetics
During dose escalation, PK blood samplings were obtained during week 1 (cycle 1) when both drugs were given on the same day. Samplings were performed at specified time-points up to 24 hours after initiation of 17AAG. During week 1 (cycle 2), when irinotecan was given on day 1 and 17AAG on day 2, serial samples were collected at specified time-points up to 24 hours after initiation of 17AAG.
Concentrations of 17AAG and its active metabolite 17-amino-17-demethoxy geldanamycin (17AG) in plasma were quantitated using a validated HPLC assay (7). The time courses of 17AAG and 17AG in plasma were analyzed non--compartmentally. Pharmacokinetic parameters were estimated using the LaGrange function (16), implemented by the LAGRAN computer program LAGRAN (16, 17).
The simultaneous assay of irinotecan, and its metabolites SN-38 and SN-38 glucuronideate (SN-38G), was used with slight modifications (18). Acetonitrile was used to remove proteins in the samples. A 2-compartment model was fit to the data, and standard PK parameters were calculated using WinNonlin software (Version. 5.2, Pharsight Corporation, Mountain View, CA).
For PK interaction analysis, differences between the PK parameters of 17AAG (or irinotecan) and its metabolites obtained with or without irinotecan (or 17AAG) co-administration were compared using a paired signed rank test (paired Wilcoxon test). None of the expansion cohort patients were included in the PK data for irinotecan and its metabolites.
Pharmacodynamics
During cycle 1 in the expansion cohort, patients underwent tumor biopsies 24–48 hours after irinotecan given alone on day 1, and after the combination of irinotecan and 17AAG on day 8. Tumor biopsy at baseline was optional. Up to three 18–20 gauge core needle biopsies were obtained under computer tomography (CT) guidance or using the Tru-Cut technique. Automated IHC stainings were performed on 5-μm formalin-fixed paraffin-embedded sections using established protocols on a Discovery XT processor (Ventana Medical Systems, Tucson, AZ). Antibody binding was detected by the avidin-biotin peroxidase complex method (Vector Laboratories, Burlingame, CA) using diaminobenzidine as a chromogen. Slides were counterstained with hematoxylin. The primary antibodies and their concentrations were: Hsp70 (StressGene; 5 μg/ml), p-Chk1 (Ser 345; Cell Signaling; 2.5 μg/ml), p-H2AX (Ser 139; Upstate; 5 μg/ml), p-histone H3 (Ser 10; Upstate; 5 μg/ml), and cleaved caspase-3 (Cell Signaling; 0.1 μg/ml). P-Chk1 (Ser 345) was used as a surrogate for total Chk1 because as commercially available antibodies for total Chk1 do not perform well with IHC. The expression of p-histone H3 was analyzed by immunofluorescence confocal microscopy using the antibody employed for IHC.
Sample sections were compared in a pair-wise fashion by a pathologist (D. S. K.) who was blinded to the clinical data. For Hsp70, p-Chk1, and p-H2AX, immunoreactivity was evaluated semiquantitatively based on a staining score calculated by multiplying the percentage of positive tumor cells (0% to 100%) by the staining intensity (0, 1+, 2+, 3+, and 4+). For p-histone H3 and cleaved caspase-3, immunostaining was scored based on the number of positively stained tumor cells per high powered field.
Analysis of p53 status
Tumor p53 status was determined on tissue sections from archived materials by IHC using the monoclonal antibody (DO-7; DAKO; 1:500). Greater than 20% nuclear staining for p53 was considered indicative of mutant protein. The association between p53 status and treatment response was assessed using a Fisher’s exact test.
RESULTS
Patient characteristics
Of 31 patients enrolled between May 2005 and March 2007, 27 received treatment. Four patients were ineligible because of prolonged QTc (n=2) and elevated total bilirubin (n=2; Table 1). Ten patients had previously received irinotecan.
Table 1.
Patient Demographics
| Total no. of patients treated | 27 |
| Median age, years (range) | 57 (32–73) |
| Male:Female ratio | 15:12 |
| Median baseline KPS, % (range) | 80 (70–90) |
| Median # of prior chemotherapy treatments (range) | 3 (0–6) |
| Primary disease sites | |
| Colorectal | 6 |
| Pancreatic/ampullary | 3 |
| Breast | 3 |
| Gastric | 2 |
| Adrenocortical | 2 |
| Sarcoma | 2 |
| Gall bladder | 1 |
| Anal | 1 |
| Carcinoid | 1 |
| Germ cell | 1 |
| Head & Neck | 1 |
| High-grade neuroendocrine | 1 |
| Liver | 1 |
| Non-small cell lung | 1 |
| Carcinoma of unknown primary | 1 |
Abbreviations: KPS, Karnofsky performance status.
Sixty-seven cycles of treatment were given (median 2; range 1–12) over 4 dose levels. Twenty-six of 27 patients who commenced treatment were evaluable for toxicity. One patient in the MTD cohort withdrew consent after receiving one treatment and was replaced. Four patients were removed from study because of DLTs during cycle 1, leaving 22 patients assessable for tumor response.
Dose escalation and toxicity
Common toxicities of grade 2 that occurred in cycle 1 and all cycles and were at least possibly related to drug treatment are summarized in Tables 2 and 3, respectively. Patients enrolled in the first 3 cohorts completed cycle 1 without DLT. Four patients experienced DLTs in cohort 4 (irinotecan 100 mg/m2 and 17AAG 375 mg/m2). One patient developed grade 3 nausea, vomiting, and dehydration in part related to the odor from the DMSO used for solubilizing 17AAG. One patient experienced grade 3 diarrhea and dehydration, and another patient had grade 4 diarrhea. A fourth patient developed a symptomatic pulmonary embolism. This patient was managed with anticoagulation and continued study treatment at one lower dose level.
Table 2.
Incidence of Grade 2 or higher Adverse Events (Cycle 1 only)
| Cohort | 1 | 2 | 3 | 4 | MTD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CPT11, mg/m2 | 85 | 100 | 100 | 100 | 100 | ||||||
| 17AAG, mg/m2 | 220 | 220 | 300 | 375 | 300 | ||||||
| No. of patients | 3 | 3 | 3 | 5 | 13 | ||||||
|
|
|||||||||||
| Event | G2 | G3 | G2 | G3 | G2 | G3 | G2 | G3 | G4 | G2 | G3 |
|
|
|||||||||||
| Hematological | |||||||||||
| Leukopenia | — | — | 1 | — | 1 | — | — | 1 | — | 1 | — |
| Neutropenia | — | — | — | — | — | — | 1 | — | — | 1 | — |
| Anemia | 2 | — | 1 | — | 1 | — | 1 | — | — | 2 | — |
| Thrombocytopenia | — | — | — | — | — | — | — | — | — | — | — |
| Lymphopenia | — | — | — | — | — | — | — | 2 | — | — | 2 |
| Non-hematological | |||||||||||
| Diarrhea | 1 | — | — | — | — | — | — | 1 | 1 | — | 1 |
| Fatigue | 1 | — | — | — | — | — | — | — | — | 2 | 1 |
| Nausea | — | — | — | — | — | — | 1 | 2 | — | 1 | — |
| Vomiting | — | — | — | — | — | — | 1 | 1 | — | 1 | — |
| Dehydration | — | — | — | — | — | — | 1 | 2 | — | — | — |
| Abdominal pain | — | — | — | — | — | — | — | — | — | — | 1 |
| Neuropathy | — | — | — | — | — | — | — | — | — | — | — |
| AST/ALT | — | — | — | — | — | — | — | — | — | — | — |
| Total bilirubin | — | — | — | — | — | — | 1 | — | — | — | — |
| Alkaline phosphatase | — | — | — | — | 2 | — | 1 | — | — | — | — |
| Thromboembolism | — | — | — | — | — | — | — | — | 1 | — | — |
Numbers in bold indicate DLTs that occurred during cycle 1 in the dose escalation phase or during cycles 1 and 2 in the MTD expansion cohort. See text for DLT definitions. Abbreviations: G, grade; AST, aspartate aminotransferase; ALT, alanine aminotransferase
Table 3.
Incidence of Grade 2 or higher Adverse Events (All Cycles)
| Cohort | 1 | 2 | 3 | 4 | MTD | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Irinotecan, mg/m2 | 85 | 100 | 100 | 100 | 100 | |||||
| 17AAG, mg/m2 | 220 | 220 | 300 | 375 | 300 | |||||
| No. of patients | 3 | 3 | 3 | 5 | 13 | |||||
|
| ||||||||||
| Event | G2 | G4 | G2 | G2 | G3 | G2 | G3 | G4 | G2 | G3 |
|
|
||||||||||
| Hematological | ||||||||||
| Leukopenia | — | — | 1 | 1 | — | — | 1 | — | 5 | — |
| Neutropenia | — | — | — | 1 | — | 1 | — | — | 1 | 2 |
| Anemia | 3 | — | 1 | 1 | 1 | 1 | — | — | 4 | — |
| Thrombocytopenia | — | — | — | — | — | — | — | — | — | — |
| Lymphopenia | — | — | — | — | — | — | 2 | — | — | 3 |
| Non-hematological | ||||||||||
| Diarrhea | 1 | — | — | — | — | — | 1 | 1 | 2 | 2 |
| Fatigue | 2 | — | — | — | 1 | — | — | — | 2 | 2 |
| Nausea | — | — | — | — | — | 1 | 2 | — | 3 | — |
| Vomiting | — | — | — | — | — | 1 | 1 | — | 2 | — |
| Dehydration | — | — | — | — | — | 1 | 2 | — | 1 | — |
| Abdominal pain | — | — | — | — | — | — | — | — | 2 | 1 |
| Neuropathy | — | — | — | — | — | — | — | — | 1 | — |
| AST/ALT | — | — | — | 1 | — | — | — | — | — | — |
| Total bilirubin | — | — | — | 1 | — | 1 | — | — | — | — |
| Alkaline phosphatase | — | — | — | 1 | 1 | 1 | — | — | — | — |
| Thromboembolism | — | 1 | — | — | — | — | — | 1 | — | — |
Abbreviations: G, grade; AST, aspartate aminotransferase; ALT, alanine aminotransferase.
Cohort 3 (irinotecan 100 mg/m2 and 17AAG 300 mg/m2) was therefore designated as the MTD. We expanded this cohort to enroll 12 additional patients. One patient withdrew consent after developing grade 2 gastrointestinal toxicities before undergoing the second tumor biopsy; this patient was replaced. Four patients experienced DLTs, including one grade 3 diarrhea, one grade 3 abdominal pain, one grade 3 fatigue, and one with dose delay ≥ 7 days because of grade 1 thrombocytopenia. Thus, the incidence of protocol-defined DLT was 4 of 15 patients (27%) treated at the MTD. Using a Bayesian approach, the probability that DLT rate exceeds 33% is calculated to be 0.34, within acceptable limits of toxicity tolerance in Phase I trials. There is 95% probability that the true DLT rate is between 0.11 and 0.52 (13).
Among other significant toxicities, grade 2 hepatotoxicity was noted in one patient during cycle 2. One patient developed dyspnea after 4 cycles of treatment and was found to have a pulmonary embolism. One patient experienced grade 3 fatigue during cycle 3, one patient had grade 2 (but progressive) neuropathy that required discontinuation of therapy after 2 cycles. Post-17AAG infusion EKGs performed during the first week of cycle 1 showed no cardiac dysrhythmias.
Anti-tumor activity
Of the 22 accessible patients, none had a complete or partial response by RECIST. Eleven patients had stable disease as the best response. The median duration of stable disease was 3 months (range, 1.5 – 8 months). One patient with Her2-negative breast cancer had a 29% reduction in target lesions after 2 cycles (Fig, 1). Minor responses in target lesions were seen in 4 other patients: non-small cell lung (−13%), head and neck squamous cell (−10%), colorectal (−9%), and pancreatic adenocarcinoma (−8%). One patient with ampullary adenocarcinoma had improvement of carcinomatosis and a decrease in CA19-9 from 1346 to 174 units/ml. All patients who showed tumor measurement reductions were irinotecan-naïve. One patient with irinotecan-refractory colorectal cancer was treated on the current study for 5 months. Tumor p53 status was available for 16 patients. Two of 6 patients with wild-type p53 had stable disease as compared with 5 of 10 patients with mutant p53 (P=.63; Fisher’s exact test).
PK studies
During dose escalation, PK samplings were performed both in cycle 1 when irinotecan and 17AAG were given on the same day, and in cycle 2, when the two agents were given sequentially on days 1 and 2. Pair-wise comparison of the PK parameters of 17AAG and 17AG obtained between the 2 cycles for each patient showed no significant differences, suggesting that the PK of 17AAG was not affected by irinotecan coadministration (Supplemental Table 1). Likewise, the PK parameters of irinotecan, SN-38, and SN-38G obtained during cycle 1 were not significantly different from those obtained in cycle 2, indicating no effect of 17AAG treatment on the PK of irinotecan (Supplemental Table 2). The mean plasma concentration versus-time curves of 17AAG and 17AG in patients treated at the MTD are shown in Supplemental Fig. 2. The mean area under the curve (AUC), half-life, and clearance for 17AAG are 35.9 ± 15.9 μM·h, 2.0 ± 1.2 h, and 17.1 ± 10.6 L/h/m2, respectively. The mean AUC and half-life of 17AG are 36.7 ± 21.2 μM·h and 2.3 ±1.5 h, respectively. These values agree with those reported in two phase I studies of single-agent 17AAG given at a similar dose in one of their cohorts (6, 19).
PD analyses
During the first cycle of the MTD expansion cohort, 12 additional patients underwent tumor biopsies after treatment with irinotecan only in week 1 and with irinotecan and 17AAG in week 2. All but one paired set of biopsies were obtained within 24 hours after drug treatment. There were no biopsy-related complications. Eight of 12 paired samples contained tumor cells deemed adequate for PD evaluation. Markers were examined for these cellular effects: (1) Hsp90 inhibition as measured by induction of the co-chaperone Hsp70; (2) loss of p-Chk1 as a surrogate for depletion of the Hsp90 client protein, Chk1; (2) abrogation of the G2/M checkpoint as measured by staining of the mitosis-specific marker p-histone H3; (4) p-H2AX for DNA damage response; and (5) cleaved caspase-3 for apoptosis. Hsp70 was up-regulated following treatment with irinotecan and 17AAG in 6 of 8 patients (Fig. 2). Of the 6 patients whose tumors showed an induction of the co-chaperone, 4 also showed a decrease in p-Chk1 (Fig. 2). The expression of p-H2AX was elevated following combination treatment in 3 patients, 2 of whom also had Hsp70 induction and p-Chk1 down-regulation (Fig. 2). An increase in p-histone H3 was demonstrated in 2 patients whereas an increase in cleaved caspase-3 was found in 3 patients (Fig. 2). Fig. 3A illustrates a case of p53-mutant (positive p53 staining) gastric cancer metastasized to the liver in which Hsp90 inhibition was associated with a depletion of its client Chk1, leading to an abrogation of the G2/M checkpoint, as well as an increase in DNA damage and apoptosis. Examination of the nuclear morphology of tumor cells under fluorescence microscopy showed mitotic cells with fragmented chromatin characteristic of cells undergoing mitotic catastrophe in the post-combination sample (Fig. 3B) (20).
Figure 2.
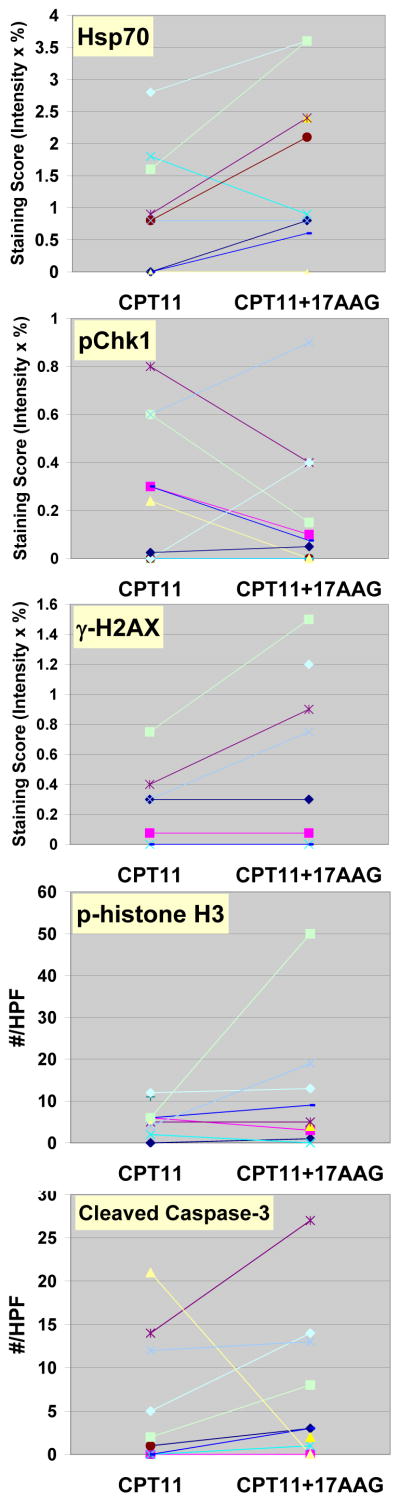
Semiquantitative analysis of changes in pharmacodynamic markers in tumor biopsy samples obtained from patients treated at the MTD after irinotecan (CPT11) alone and after irinotecan and 17AAG (CPT11+17AAG). Different marker studies performed for the same patient are represented by the same symbol. HPF, high power field.
Figure 3.


A, Immunohistochemistry analysis of paired biopsies in a patient with p53-mutant gastric adenocarcinoma, showing in the post-combination (CPT11+17AAG) samples an induction of Hsp70, a suppression of p-Chk1 nuclear staining, an increase in p-H2AX nuclear staining, an increase in p-histone H3 indicative of G2/M checkpoint abrogation, as well as an increase in cleaved caspase-3. Scale bar, 100 μm. B, Confocal immunofluorescence microscopy of tumor samples stained for p-histone H3 (pH3; green) showing tumor cells with fragmented chromatin characteristic of those undergoing mitotic catastrophe after irinotecan and 17AAG treatment (arrows). Chromatin was counterstained using 4′-6-diamidino-2-phenylindole (DAPI; blue). Scale bar, 20 μm.
Of the limited number of patients with paired biopsies who were evaluable for tumor response (n=7), we found no correlation between changes of biomarkers and treatment response (see Discussion).
Discussion
Both Hsp90 and Chk1 have emerged as novel anticancer targets. Of considerable interest to both therapeutic areas is the identification of Chk1 as an Hsp90 client protein.(11) We and others have shown that treatment with 17AAG results in depletion of cellular Chk1 and abrogation of the S and/or G2/M checkpoints induced by chemotherapy, prompting us to embark on this phase I study of 17AAG and irinotecan (11, 12, 21).
In three single-agent phase I studies, the MTD of 17AAG given once weekly or weekly x3 in a 4-week cycle (two studies) was determined to be 450, 308, and 295 mg/m2, respectively (5–7). In the current combination study, the recommended phase II dose is 300 mg/m2 17AAG and 100 mg/m2 irinotecan. Thus, 17AAG can be delivered at essentially its full singe-agent dose. As predicted from the toxicity profile of each individual agent, gastrointestinal toxicities are dose-limiting for the combination. It is possible that some of these adverse effects are from the DMSO required for formulating 17AAG.
No complete or partial responses were noted in this study. However, one patient with breast cancer showed a 29% reduction in index lesions, and 5 other patients demonstrated tumor shrinkage. While it may be difficult to separate the activity of the combination from that of irinotecan alone, as all patients who showed tumor shrinkage were irinotecan-naïve, we were encouraged to see signs of antitumor activity in this heavily pretreated group. Preclinical studies have suggested that tumors with intrinsic checkpoint defects, by virtue of p53 loss, may be more susceptible to undergoing G2/M checkpoint abrogation and apoptosis induced by treatment with 17AAG and irinotecan. In our trial, 2 of 6 patients with wild-type p53 had stable disease compared with 5 of 10 patients with mutant p53. The relationship between tumor p53 status and response to this drug combination therefore deserves further exploration. Given the encouraging response data seen in a recently completed phase II study of 17AAG and trastuzumab in trastuzumab-refractory Her2-positive breast cancer (22) and the 29% tumor shrinkage observed in a patient with Her2-negative disease in the current study, breast cancer would potentially be a tumor type in which the combination of irinotecan and 17AAG be further developed.
Validation of the PD effects of Hsp90 inhibition presents a unique challenge in this combination study, as it requires demonstration of successful suppression of the Hsp90 client Chk1 and abrogation of the irinotecan-induced G2/M checkpoint as a result of Chk1 inhibition. Pharmacodynamic evaluations at the MTD were designed to discern the biological effects of combined 17AAG and irinotecan (week 2) from that of irinotecan alone (week 1). One caveat is that it may not be possible to determine whether biomarker changes seen in the second biopsy are from 17AAG or a delayed effect of irinotecan given in the previous week. However, given the short-lived nature of the cellular processes relevant to G2/M checkpoint abrogation (i.e. mitosis and apoptosis) and the advantage of using the patient as his or her own control, this concern is probably minor. Ideally, a third biopsy obtained at baseline (optional for the study) would allow assessment of the PD effects induced by irinotecan alone. However, we were unable to obtain any baseline biopsies because of logistic reasons.
In preclinical studies, a 24-hour exposure of tumor cells in culture to 200–500 nM 17AAG results in Chk1 depletion and abrogation of the G2/M checkpoint induced by SN-38.(21) At the MTD, the mean combined plasma concentration of 17AAG and its active metabolite 17AG measured at 24 hours was approximately 300 nM, suggesting that biologically relevant drug concentrations can be achieved in some patients. This was supported by PD validation studies. Of the biomarkers examined, Hsp70 induction was mostly consistently seen after 17AAG treatment (6/8 patients). Chk1, the Hsp90 client of interest, was down-regulated in 4 samples. Evidence for abrogation of the G2/M checkpoint, as measured by induction of the M-phase specific marker p-histone H3, was demonstrated 2 patients; both were mutant for p53. To our knowledge, these results provide the first demonstration of the biological effects of an Hsp90 inhibitor causing down-regulation of the client protein, Chk1, and abrogation of the G2/M checkpoint in human tumors.
Of note, the patient with the PD responses shown in Fig. 3 was taken off study for “early disease progression” after experiencing increased fatigue and abdominal pain during cycle 1. An imaging study performed after only one combination treatment of irinotecan and 17AAG revealed tumor progression in the liver. However, retrospective examination of the CT performed for tumor biopsy after irinotecan alone showed already substantial tumor progression when compared with baseline. It is possible that the PD effects observed in this patient preceded subsequent radiological response if this patient was allowed to continue on study.
In summary, the combination of irinotecan and 17AAG can be given with acceptable toxicity. At this juncture, further clinical development of this combination is unclear as non-DMSO formulation of 17AAG and newer generations of Hsp90 as well as more selective Chk1 inhibitors are now available (23, 24). Nonetheless, our PD studies show for the first time that Chk1-mediated signaling can be disrupted by an Hsp90 inhibitor in human tumors, providing the proof-of-mechanism of this therapeutic approach. We envisioned that this strategy of combining cytotoxic agents with checkpoint inhibitor can be further optimized with improved formulation of 17AAG and/or selective Chk1 inhibitors that are currently undergoing clinical testing.
Supplementary Material
Figure 1.

Computed tomography of a patient with Her2-negative p53-mutant metastatic breast cancer before (A) and after 2 cycles (B) of 17AAG and irinotecan given at the MTD, demonstrating regression of chest wall metastases (arrows).
Statement of Clinical Relevance.
Heat shock protein 90 (Hsp90) and checkpoint kinase 1 (Chk1) are two novel targets for cancer therapy. Of interest to both areas of targeted therapeutics is the identification of Chk1 as an Hsp90 client protein. We embarked on this phase I study based on our pre-clinical work showing that, 17AAG, the first Hsp90 inhibitor in clinical trial, could enhance the cytotoxicity of topoisomerase I poison, by depleting Chk1 and abrogating the cyto-protective G2/M cell cycle checkpoint.
We showed in this combination study that 17AAG can be given at its full single-agent dose with acceptable toxicity. Although there was no partial response by RECIST, we observed minor tumor shrinkage in 6 patients treated by the combination. We obtained pre- and post-17AAG tumor biopsies to evaluate the pharmacodynamic effects of combined irinotecan and 17AAG. Evidence for Hsp90 inhibition by 17AAG, resulting in phospho-Chk1 loss, abrogation of the G2/M cell cycle checkpoint, and cell death could be demonstrated in some tumor biopsy samples. To our knowledge, these pharmacodynamic studies showed for the first time that Chk1-mediated signaling can be disrupted by an Hsp90 inhibitor in human tumors, providing the proof-of-mechanism of this therapeutic approach and pave the way for future Hsp90/Chk1 inhibitor studies.
Acknowledgments
We are indebted to members of the Molecular Cytology Core Facility at Memorial Sloan-Kettering Cancer Center for their technical assistance in immunohistochemistry and immunofluorescence analysis. We thank Carol Pearce for her editorial assistance. This work was supported in part by K08CA116612 (A.N.T.), U01CA069856, UO1CA099168 (M.J.E.), and 2P30 CA47904 (M.J.E.) (National Cancer Institute), and a Career Development Award from the American Society of Clinical Oncology (A.N.T.).
References
- 1.Solit DB, Rosen N. Hsp90: a novel target for cancer therapy. Current topics in medicinal chemistry. 2006;6:1205–14. doi: 10.2174/156802606777812068. [DOI] [PubMed] [Google Scholar]
- 2.Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61:4003–9. [PubMed] [Google Scholar]
- 3.Solit DB, Zheng FF, Drobnjak M, et al. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res. 2002;8:986–93. [PubMed] [Google Scholar]
- 4.Munster PN, Marchion DC, Basso AD, Rosen N. Degradation of HER2 by ansamycins induces growth arrest and apoptosis in cells with HER2 overexpression via a HER3, phosphatidylinositol 3′-kinase-AKT-dependent pathway. Cancer Res. 2002;62:3132–7. [PubMed] [Google Scholar]
- 5.Banerji U, O’Donnell A, Scurr M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23:4152–61. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 6.Goetz MP, Toft D, Reid J, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23:1078–87. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 7.Ramanathan RK, Trump DL, Eiseman JL, et al. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clin Cancer Res. 2005;11:3385–91. doi: 10.1158/1078-0432.CCR-04-2322. [DOI] [PubMed] [Google Scholar]
- 8.Solit DB, Ivy SP, Kopil C, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin Cancer Res. 2007;13:1775–82. doi: 10.1158/1078-0432.CCR-06-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grem JL, Morrison G, Guo XD, et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23:1885–93. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 10.Mesa RA, Loegering D, Powell HL, et al. Heat shock protein 90 inhibition sensitizes acute myelogenous leukemia cells to cytarabine. Blood. 2005;106:318–27. doi: 10.1182/blood-2004-09-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J Biol Chem. 2003 doi: 10.1074/jbc.M309054200. [DOI] [PubMed] [Google Scholar]
- 12.Flatten K, Dai NT, Vroman BT, et al. The role of checkpoint kinase 1 in sensitivity to topoisomerase I poisons. J Biol Chem. 2005;280:14349–55. doi: 10.1074/jbc.M411890200. [DOI] [PubMed] [Google Scholar]
- 13.Gonen M. A Bayesian evaluation of enrolling additional patients at the maximum tolerated dose in Phase I trials. Contemp Clin Trials. 2005;26:131–40. doi: 10.1016/j.cct.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 15.Bagatell R, Gore L, Egorin MJ, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-N-allylamino-17-demethoxygeldanamycin in pediatric patients with recurrent or refractory solid tumors: a pediatric oncology experimental therapeutics investigators consortium study. Clin Cancer Res. 2007;13:1783–8. doi: 10.1158/1078-0432.CCR-06-1892. [DOI] [PubMed] [Google Scholar]
- 16.Yeh KC, Kwan KC. A comparison of numerical integrating algorithms by trapezoidal, Lagrange, and spline approximation. Journal of pharmacokinetics and biopharmaceutics. 1978;6:79–98. doi: 10.1007/BF01066064. [DOI] [PubMed] [Google Scholar]
- 17.Rocci ML, Jr, Jusko WJ. LAGRAN program for area and moments in pharmacokinetic analysis. Computer programs in biomedicine. 1983;16:203–16. doi: 10.1016/0010-468x(83)90082-x. [DOI] [PubMed] [Google Scholar]
- 18.Rivory LP, Robert J. Reversed-phase high-performance liquid chromatographic method for the simultaneous quantitation of the carboxylate and lactone forms of the camptothecin derivative irinotecan, CPT-11, and its metabolite SN-38 in plasma. J Chromatogr B Biomed Appl. 1994;661:133–41. doi: 10.1016/0378-4347(94)00340-8. [DOI] [PubMed] [Google Scholar]
- 19.Banerji U, Walton M, Raynaud F, et al. Pharmacokinetic-pharmacodynamic relationships for the heat shock protein 90 molecular chaperone inhibitor 17-allylamino, 17-demethoxygeldanamycin in human ovarian cancer xenograft models. Clin Cancer Res. 2005;11:7023–32. doi: 10.1158/1078-0432.CCR-05-0518. [DOI] [PubMed] [Google Scholar]
- 20.Tse A, Schwartz G. Potentiation of cytotoxicity of topoisomerase I poison by concurrent and sequential treatment with the checkpoint inhibitor 7-hydroxystaurosporine involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res. 2004;64:6635–44. doi: 10.1158/0008-5472.CAN-04-0841. [DOI] [PubMed] [Google Scholar]
- 21.Tse AN, Sheikh T, Ho A, Schwartz GK. Hsp90 inhibition abrogates the G2/M checkpoint in p53-null tumor cells by depleting Chk1 and Wee1. Cancer Res. 2008 doi: 10.1124/mol.108.050807. In Revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Modi S, Sugarman S, Stopeck A, et al. Phase II trial of the Hsp90 inhibitor tanespimycin (Tan) + trastuzumab (T) in patients (pts) with HER2-positive metastatic breast cancer (MBC) Proc Am Soc Clin Oncol. 2008;26 [Google Scholar]
- 23.Solit DB, Chiosis G. Development and application of Hsp90 inhibitors. Drug Discov Today. 2008;13:38–43. doi: 10.1016/j.drudis.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Tse AN, Carvajal R, Schwartz GK. Targeting checkpoint kinase 1 in cancer therapeutics. Clin Cancer Res. 2007;13:1955–60. doi: 10.1158/1078-0432.CCR-06-2793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.