

Abstract
Cellular quiescence is a reversible non-proliferating state. The reactivation of ‘sleep-like’ quiescent cells (e.g. fibroblasts, lymphocytes and stem cells) into proliferation is crucial for tissue repair and regeneration and a key to the growth, development and health of higher multicellular organisms, such as mammals. Quiescence has been a primarily phenotypic description (i.e. non-permanent cell cycle arrest) and poorly studied. However, contrary to the earlier thinking that quiescence is simply a passive and dormant state lacking proliferating activities, recent studies have revealed that cellular quiescence is actively maintained in the cell and that it corresponds to a collection of heterogeneous states. Recent modelling and experimental work have suggested that an Rb-E2F bistable switch plays a pivotal role in controlling the quiescence–proliferation balance and the heterogeneous quiescent states. Other quiescence regulatory activities may crosstalk with and impinge upon the Rb-E2F bistable switch, forming a gene network that controls the cells’ quiescent states and their dynamic transitions to proliferation in response to noisy environmental signals. Elucidating the dynamic control mechanisms underlying quiescence may lead to novel therapeutic strategies that re-establish normal quiescent states, in a variety of hyper- and hypo-proliferative diseases, including cancer and ageing.
Keywords: cellular quiescence, cell cycle, bistable switch, gene network
1. Introduction
There are 1013–1014 cells in our human body. At any given time, the vast majority of these cells are non-dividing and outside of an active cell cycle. Some of these non-dividing cells (e.g. senescent or terminally differentiated cells) are irreversibly arrested; they can no longer re-enter the cell cycle to proliferate under normal physiological conditions. By contrast, a subset of non-dividing cells is ‘reactivatable’ and can enter the proliferative cell cycle in response to physiological growth signals; these cells are called quiescent cells (figure 1). Examples of quiescent cells include many adult stem cells, progenitor cells, fibroblasts, lymphocytes, hepatocytes and some epithelial cells. The exact number of quiescent cells in the body is not well characterized.
Figure. 1.
Cellular quiescence. Quiescence differs from other non-dividing states in that it can be reverted into proliferation. The molecular mechanisms underlying quiescence and its reversibility are unknown. (Online version in colour.)
Unlike many unicellular organisms in which the cell can reside in either a quiescent dormant state or a growth state depending on the environment, in adult multicellular organisms the quiescent cellular state is unique to higher organisms. In lower multicellular organisms, such as Caenorhabditis elegans, all cells in the adult body are ‘post-mitotic’ and no longer able to proliferate. However, in higher multicellular organisms with prolonged lifespans, such as mammals, reversible quiescent cells are fundamental to forming tissues capable of renewal and regeneration, which is critical for the tissue homoeostasis of the adult body. Cellular quiescence also provides protection against stress and toxicities, which is especially important for long-lived cells, such as stem cells [1,2]. On the other hand, the balance between cellular quiescence and proliferation needs to be carefully regulated in tissues of higher organisms; misregulation of the quiescence–proliferation balance can lead to a wide range of hypo- and hyper-proliferative diseases, such as fibrosis, autoimmune diseases, cancer and ageing.
Regarding the two sides of the quiescence–proliferation balance, while cell proliferation and its regulation have been well studied, cellular quiescence is poorly understood. One recent PubMed search with ‘cell proliferation’ returned over 190 000 articles, while searches for ‘cell quiescence’ and ‘cellular quiescence’ returned about 200 hits each. The lack of study of cellular quiescence is at least partially owing to the fact that, for a long time, quiescence had been simply regarded as a passive and dormant cellular state lacking proliferation activities.
Studies in the past few years have revealed that, instead of being a passive state, quiescence is actively maintained in the cell. Quiescent cells are transcriptionally active: in addition to the downregulation of proliferation-related genes, quiescent cells actively express a group of genes that are distinct from those in proliferating cells or in cell cycle arrested cells forced by the overexpression of cyclin-dependent kinase inhibitors (CKIs) p21 or p27 [3–6]. At least some of the actively expressed genes in quiescent cells contribute to the reversibility of quiescence (e.g. by suppressing terminal differentiation and apoptosis) [3]. As a result, quiescent cells, but not forced cell cycle arrested cells, are resistant to the inductions of differentiation and senescence [3,7]. In addition, while many quiescent cell types exhibit reduced metabolic activities, quiescent fibroblasts maintain comparable metabolic rates to their proliferating counterparts [8,9].
Recent studies including ours have further shown that quiescence is not a unique state but rather a collection of heterogeneous states. Quiescence has traditionally been considered a single non-proliferating state (also called ‘G0’ [10–13]) outside of the cell cycle. However, cells induced to quiescence by different signals (e.g. serum starvation, loss of adhesion and cell contact inhibition at high cell density) exhibit overlapping yet distinct gene expression profiles, suggesting that cells may enter different quiescent states depending on the initiating signals [3]. We found that fibroblasts that are serum starved for longer periods of time appear to access ‘deeper’ states of quiescence: the longer serum-starved cells (e.g. those starved for 6 versus 2 days) were less likely to exit quiescence in response to growth signals at non-saturating levels (e.g. 1% serum; Kwon & Yao 2014, unpublished data); furthermore, among those cells that exited quiescence, longer serum-starved cells did so at a slower speed than shorter serum-starved cells (similar to earlier observations [14,15]). Yet, the longer starved cells could still re-enter the cell cycle to proliferate under normal growth conditions (e.g. 10% serum), demonstrating that they reside in a reversible quiescent state, not an irreversibly arrested state, such as senescence. Together, accumulating evidence indicates that quiescence goes beyond a single homogeneous state.
What mechanisms underlie the actively maintained, heterogeneous quiescent states? Answers to this question will help in uncovering a currently underappreciated layer of complexity in growth control and provide novel insights into the cause and treatment of hyper- and hypo-proliferative diseases. High-throughput studies in the past few years have suggested a long list of cellular activities associated with quiescence, particularly in fibroblasts and adult stem cells, such as haematopoietic stem cells (HSCs) [3–6]. Meanwhile, recent computational modelling studies have begun to provide an effective framework to integrate descriptive ‘parts lists’ into a mechanistic understanding of the basic controls of cellular quiescence, which will be reviewed below.
2. Phenotypic quiescence–proliferation transition
2.1. Restriction point
The proliferation of normal, untransformed mammalian cells in culture requires serum, which contains a combination of growth factors. Specifically, cells are sensitive to extracellular serum signals in the G1 phase of the cell cycle prior to a critical time point, namely the ‘restriction point’ or ‘R-point’ [16]. If serum starvation occurs in the G1 phase before the R-point, cells will withdraw from the cell cycle and enter quiescence. After passing the R-point, however, the cell cycle becomes autonomous—even when serum is removed, the cell cycle will continue until it is completed. Therefore, it appears that the R-point is a ‘point of no return’ at which a cell commits to proliferation. In actively growing Swiss 3T3 cells, whose average cycle time is 16 h with an average 7 h G1 phase, the R-point is approximately 3.5 h after cell birth [17].
There are two key characteristics of the R-point: first, it sets a high threshold that separates quiescence from proliferation, serving as a noise filter against uncontrolled and accidental cell growth; second, it provides a low-maintenance mechanism to ensure that, once initiated, the cell cycle will be completed regardless of later fluctuations in the extracellular environment, which is fundamental to maintaining genome integrity.
2.2. G0 versus G1: two non-proliferating phases before DNA replication
There is an R-point in every cell cycle before the start of cell proliferation. However, the first R-point that cells encounter when they exit quiescence is distinct from the other R-points in actively growing cells. Despite a historical debate regarding whether quiescent cells reside in an extended G1 phase at positions where they last see growth signals [18–20], quiescence is typically considered a distinct G0 state outside of the cell cycle. Quiescent G0 cells are similar to the cells in the G1 phase of a proliferative cell cycle (G1 cells): they both reside between M- and S-phases and contain non-replicated 2n DNA content. However, compared with G1 cells, G0 cells experience a significant delay in re-entering S-phase. In Swiss 3T3 cells, such a delay lasts about 8 h [17]. The reasons behind this delay in quiescent cells are not entirely clear, but may be owing to the fact that proteins (e.g. CDC6) required for creating the DNA replication origins are removed from chromatin in G0 (but not G1) cells [21].
2.3. Historical models on the quiescence–proliferation transition
In the 1970s and 1980s, a number of mathematical models were proposed to describe the transition between cellular quiescence and proliferation and the apparent cell-to-cell variations in this process within a mammalian cell population. One class of models, ‘transition probability’ (TP) models [22–25], assume that a cell has two distinct phases: non-replication (NR-phase) and replication (R-phase). Cells leave the NR-phase randomly but with a constant probability; they then enter the R-phase in which replicative activities are deterministic. In TP models, it is the random transition from the NR-phase to the R-phase that is thought to create the variations in growth rates of cells in a population. The NR-phase in original TP models referred primarily to the G1 phase of actively proliferating cells; it was soon extended to the quiescent state (the G0 state) in serum-starved cells (and, correspondingly, an additional random transition from the G0 to G1 phase was proposed) [26].
Another class of models, ‘growth controlled’ (GC) models [27,28] or ‘continuum’ models [18,29], proposed that the different growth rates (i.e. different cell cycle durations) of cells in a population do not result from the probabilistic transition from the NR-phase to the R-phase, but rather from the cell-to-cell variations in biomass and cell metabolism as well as the related time required to complete essential steps in the cell cycle. Integrating TP and GC models, the hybrid ‘sloppy size control’ (SSC) model [30,31] proposed that the quiescence–proliferation transition is a random process with cell-size-dependent probability. Interestingly, all of these different models (TP, GC and SSC) fit well with various types of experimental data; however, they are all descriptive only at the population distribution level. Interest in these models gradually faded but was later reinvigorated by findings in molecular and cell biology, particularly in the genes that regulate the quiescence–proliferation transition.
3. Rb-E2F bistable switch: a mechanistic framework underlying quiescence
3.1. Rb-E2F pathway
Among cellular activities that regulate the quiescence–proliferation transition in mammalian cells, the Rb-E2F pathway plays a pivotal role (figure 2). Rb (retinoblastoma) was the first identified tumour suppressor gene [32]. The Rb protein family also contains p130 and p107; these so-called pocket proteins (pRb, p107 and p130) regulate proliferation in most, if not all, cell types [33]. E2F refers to a family of transcription factors (E2F1–8), among which E2F1, 2, 3a are considered ‘E2F activators’ and E2F3b–8 are ‘E2F repressors’ [34]. E2F proteins regulate a large battery of target genes involved in DNA replication and cell cycle progression [34–38]. In quiescent cells, E2F activators (which we will refer to as ‘E2F’ below for simplicity) are bound to and repressed by Rb family proteins. In addition, E2F repressors form complexes with Rb family members, which recruit chromatin modification factors and actively repress E2F target genes [36,39–43]. With sufficient growth stimulation, the levels of Myc and cyclin D (CycD) increase. Myc activates E2F expression [44]. CycD activates cyclin-dependent kinases (Cdks) 4 and 6 [45–48]. Cdk4,6 activities phosphorylate Rb family proteins and remove their repression of E2F. Subsequently, E2F transactivates CycE, which forms a complex with Cdk2 to further remove Rb repression by phosphorylation [49], establishing a positive-feedback loop. E2F also activates its own transcription, forming another positive-feedback loop. E2F is a master regulator of mammalian cell-cycle entry and has been shown both necessary and sufficient for this process [50,51]. Because of its critical role in cell growth control, the Rb-E2F pathway is frequently mutated in most, if not all, cancers [52].
Figure 2.
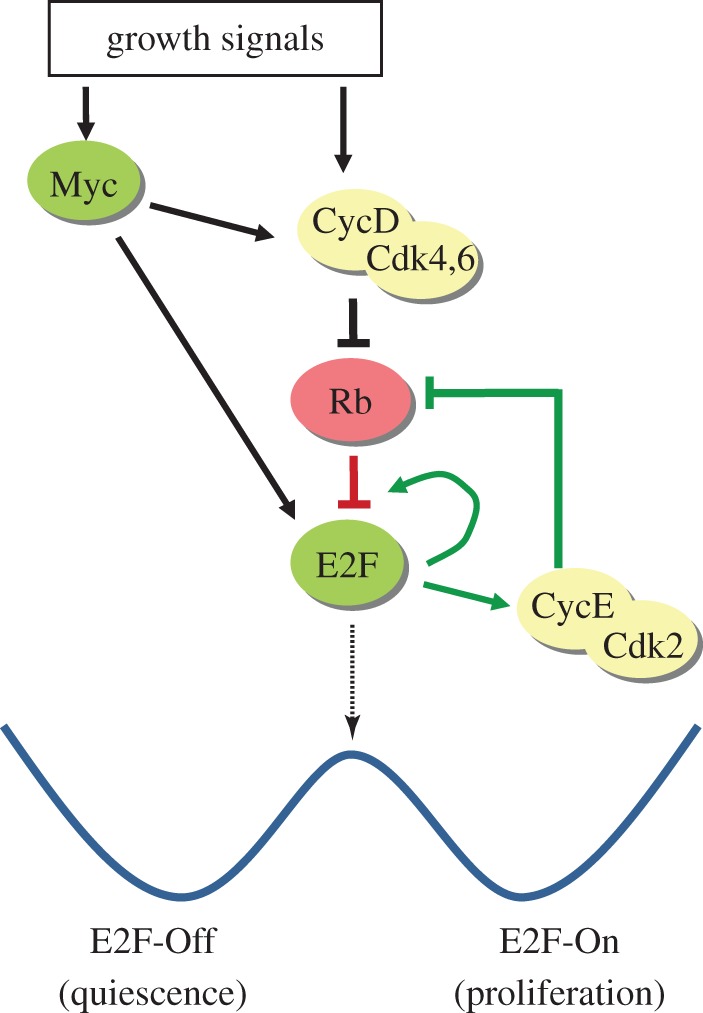
The Rb-E2F bistable switch. A simplified view of the Rb-E2F pathway is shown (at the top). The Rb-E2F pathway functions as a bistable switch, represented by the double-well potential wave (at the bottom). The two wells represent the two stable steady states of the bistable system (E2F-Off and E2F-On), which underlie cellular quiescence and proliferation, respectively. The ‘energy barrier’ that separates the two wells corresponds to the R-point, which is an unstable steady state of the Rb-E2F bistable system. (Online version in colour.)
Quiescent cells typically feature lower levels of Rb-E2F pathway activators (e.g. CycD [53–55], Cdk2 [56]) and high levels of pathway repressors, such as Cdk inhibitors (e.g. p21 [57], p27 [58,59]), Rb family proteins (especially p130 [43,60,61]) and E2F repressors that are either dependent (e.g. E2F4 [43,62]) or independent (e.g. E2F6 [63]) of Rb family proteins to silence E2F target genes. Downregulation of Cdk inhibitor activities (e.g. p27 [59,64], p21 [55,57,65,66]) can lead to quiescence exit and cell cycle re-entry. Similarly, the disruption of all three Rb family proteins [67–69] and the acute loss of the Rb function in quiescent cells lead to cell proliferation [70]. Very recently, it was shown that a low versus increasing Cdk2 activity (as directly controlled by p21) at mitotic exit marks the bifurcation of a cell population into quiescent versus continual proliferating cells [56].
3.2. Rb-E2F bistable switch
The ‘high-threshold, low-maintenance’ transition between quiescence and proliferation at the R-point is reminiscent of a bistable switch, as seen in several decision-making processes [71–74]. Regulation by positive feedback is a hallmark of a bistable switch [75,76]. Despite the fact that positive-feedback loops are under-represented in biological networks [77], they are enriched in the Rb-E2F pathway, suggesting functional non-randomness (e.g. the mutual inhibition between Rb and E2F and the E2F self-activation mentioned above and others, including the mutual inhibition between CycE/Cdk2 and p27 [78]). Several mathematical models of the Rb-E2F-cyclin/Cdk network were proposed and they suggested the theoretical possibility that the mammalian R-point is controlled by a bistable mechanism [79–83].
By coupling single-cell experiments (measuring E2F expression dynamics in individual cells using a destabilized GFP reporter) with a simplified Rb-E2F model, we have recently shown that the Rb-E2F pathway indeed functions as a bistable switch [84]. This Rb-E2F bistable switch converts graded and transient serum growth signals into a binary (Off and On) and hysteretic E2F activity in individual cells (figure 2). Once turned ‘On’ by strong growth simulation, E2F remains On even when the serum simulation is diminished [84]. The all-or-none E2F activation directly underlies the all-or-none transition between quiescence and proliferation. At the single-cell level, the very cells that re-entered the cell cycle in response to serum growth signals were those that switched On E2F [84]. Importantly, the Rb-E2F bistable switch is resettable: if the serum concentration drops below a ‘deactivation threshold’ (corresponding to a serum concentration that is lower than the switch's activation threshold; see fig. 1 in [84]), the final E2F level eventually returns to the monostable Off state after a time delay [84,85]. By creating a wide hysteresis loop, the resettable Rb-E2F switch (in the steady-state domain) drives the irreversible cell cycle progression after the R-point (in the temporal domain) [85].
Using a computational search of all possible topologies derived from a simplified Rb-E2F circuit, a 3-node module (containing coarse-grained nodes EE, RP and MD, which correspond to the E2F activators, the Rb family proteins and the linear signalling cascade from Myc to CycD, respectively; figs 1 and 2 in [85]) has been identified as the minimal module responsible for creating the Rb-E2F bistable switch. This core module contains a mutual inhibition loop between Rb and E2F and a feed-forward motif from Myc to E2F (figure 2). Experimental disruption of this core motif (by a small chemical inhibitor of Cdk2) abolishes the bistability in the Rb-E2F circuit [85].
The Rb-E2F bistable switch model provides a mechanistic explanation for the R-point transition between quiescence and proliferation. Interestingly, the historical TP and GC models can be reconciled into the same Rb-E2F bistable switch model: the simulated stochastic E2F activation based on a stochastic Rb-E2F model with a given set of parameters (e.g. parameters related to protein synthesis, turnover and modification as well as the degree of stochasticity) can be uniquely mapped to coarse-grained parameters defining the TP and GC models, respectively [86]. Therefore, the stochastic Rb-E2F model can be considered as a common mechanistic basis for the seemingly different TP and GC models, each of which holds true in describing particular aspects of the cell cycle transition.
4. Activation threshold of the Rb-E2F switch defines heterogeneous quiescent states
4.1. An analogue-to-digital convertor
Extracellular quiescence and growth signals are ‘analogue’ as they can vary continuously in degrees and durations. By contrast, the final quiescence–proliferation decision of each individual cell is ‘digital’ (all or none) because otherwise incomplete cell proliferation would disrupt genome integrity. A bistable system provides a necessary analogue-to-digital conversion mechanism allowing cells to respond to a complex environment yet still have a discrete cell-fate output. Along these lines, the Rb-E2F bistable switch is the only experimentally verified bistable system in mammalian cell cycle entry; it serves as a ‘core’ control system enabling the necessary analogue-to-digital conversion in the cell. Consistent with this notion, the few genes whose exogenous expression alone is sufficient to drive quiescent cells into proliferation (e.g. Myc, E2F, CycE) [51,87,88] are all part of the Rb-E2F bistable switch (figure 2).
4.2. E2F activation threshold underlies heterogeneous quiescent states
The E2F-Off state of the Rb-E2F bistable switch defines cellular quiescence versus proliferation. Between the different quiescent states induced by different durations of serum starvation (e.g. 2 versus 6 days), there is little difference in the E2F-Off state (i.e. the basal E2F expression level); yet, the serum strength required to turn On the Rb-E2F bistable switch varies (Kwon & Yao 2014, unpublished data). We define the minimum serum concentration required to turn On the Rb-E2F bistable switch in over half of a cell population within a time frame (e.g. 48 h) as the ‘E2F activation threshold’. A deeper quiescent state (e.g. in longer starved cells) features a higher E2F activation threshold (figure 3a), which can also be illustrated from computer simulations based on the Rb-E2F bistable switch model (figure 3b,c). Therefore, an emerging model is that a unique E2F activation threshold (in terms of minimal serum stimulation) defines each quiescent state. This model also predicts that, given the same growth stimulation, cells at a deeper quiescent state will exhibit a slower speed of switching E2F from the Off to On state (figure 3b), consistent with earlier observations that longer serum-starved cells are slower to re-enter the cell cycle [14,15].
Figure 3.

E2F activation threshold defines quiescent state depth. (a) Different quiescent states correspond to the same E2F-Off steady state (indicated by the same horizontal position of the two left wells, relative to the horizontal position of the E2F-On well on the right); however, they correspond to different E2F activation thresholds (indicated by the different depths of the two left E2F-Off wells, from which different serum stimulation strengths are required to turn On the Rb-E2F bistable switch). (b) Stochastic simulations of the Rb-E2F bistable switch model [84]. The parameter kp refers to the phosphorylation rate constants of Rb family proteins by G1 cyclin/Cdk activities. The deeper and shallower quiescent states (determined by the activator/inhibitor balance in the Rb-E2F pathway) are implemented with kp values 17 and 17.3, respectively. A total of 500 stochastic simulations with 1% serum stimulation are shown under each condition. (c) The percentage of E2F-On cells is graphed against time (with 1% serum stimulation) from simulations in (b). For the condition of kp = 17 (the deeper quiescent state), the E2F-On% reaches 50% by 48 h and thus its E2F activation potential is 1%; for the condition of kp = 17.3 (the shallower quiescent state), the E2F activation potential would be less than 1%. (Online version in colour.)
In our Rb-E2F bistable switch model, the E2F activation threshold and the switch's resettability (discussed in the previous section) are dynamic properties intrinsic to the Rb-E2F pathway, determined by the balance between the pathway's activators (e.g. cyclin/Cdks) and inhibitors (e.g. Cdk inhibitors and Rb family proteins). Cells under different environmental conditions or with different cell types are likely to display different profiles of these activators and inhibitors (e.g. in terms of their abundance as well as their synthesis and turnover rates), which will affect the parameter values in the Rb-E2F model that determine the final E2F activation threshold. Future studies are needed to further test the multi-state quiescence model and its underlying control mechanisms (e.g. potentially on the gene regulatory, epigenetic or metabolic level), as well as to characterize the Rb-E2F switch behaviours that are likely to be qualitatively identical but quantitatively different in different settings (e.g. in stem cells versus fibroblasts, in tissues in vivo versus under culture conditions in vitro).
4.3. E2f activation threshold may define the reversibility of quiescence
Under senescence and terminal differentiation, the expression of Cdk inhibitors (e.g. p16 and p21) is greatly increased [88–92], which raises the E2F activation threshold. When the E2F activation threshold increases above physiological serum levels, the Rb-E2F switch becomes irreversible under normal growth conditions. However, such ‘irreversible’ states can be reverted by decreasing the E2F activation threshold, which can be accomplished by reducing the high level of Cdk inhibitors [55] or their activators, such as p53 and oncogenic Ras [93], or by the forced expression of exogenous cyclin/Cdks [94]. Therefore, an E2F activation threshold within the physiological range is likely to distinguish reversible quiescent states from irreversibly arrested non-proliferating states, such as senescence and terminal differentiation.
5. Other quiescence regulators
Besides the Rb-E2F pathway, several other cellular pathways have been reported to regulate cellular quiescence and the quiescence–proliferation transition. Such quiescence regulators include Notch-HES1, p53, and stress and metabolic response pathways, as well as autophagy, microRNA (miRNA) and epigenetic mechanisms. These quiescence regulators crosstalk with the Rb-E2F pathway: some directly impinge on the Rb-E2F pathway and affect its bistable switch dynamics (and thus the final E2F activation threshold); some are regulated by the Rb-E2F bistable switch and serve as its ‘effectors’ to regulate cellular quiescence.
5.1. Notch-HES1 pathway
The Notch signalling pathway is an important regulator in tissue maintenance and regeneration during development [95]. Notch is also involved in the regulation of cellular quiescence through its effector proteins, transcriptional regulators RBPJ and HES1. The deletion of RBPJ leads to spontaneous cell cycle re-entry and eventually the depletion of quiescent muscle stem cells [96,97]. The expression of HES1 is upregulated in quiescent fibroblasts, but not in the same cell type that undergoes cell cycle arrest by ectopic p21 [3]. HES1 has been shown to be required for maintaining the reversible quiescent states by preventing premature senescence and differentiation [7,98]. Interestingly, HES1 is a transcriptional repressor of several key players in the Rb-E2F pathway, including E2F1 [99], p21 [100] and p27 [101].
5.2. P53 pathway
P53 is a master regulator of many cellular responses, most notably DNA damage response and apoptosis. P53 is important for quiescence maintenance of human fibroblasts and HSCs; p53 disruption can lead to quiescence exit in these cells [102,103]. The role of p53 in quiescence maintenance may involve its target genes, Gfi-1 and Necdin [103,104], and such a role is negatively regulated by the transcription factor MEF/ELF4 [105]. Notably, p53 activates p21, a Cdk inhibitor and critical component of the Rb-E2F pathway.
5.3. Stress and metabolic response pathways
As quiescent cells may stay in the non-proliferating state for a prolonged time under suboptimal conditions, they need to cope with accumulated cellular stresses and metabolic changes. Cellular activities such as those of the FOXO [106,107] and ATM-BID [108] pathways protect quiescent cells from the oxidative stress caused by the accumulation of reactive oxygen species (ROS); depletion of these cellular activities leads to quiescence exit in HSCs and an increase in their proliferation and apoptosis. Quiescent cells often exhibit low metabolic activities characterized by a decrease in glucose uptake and glycolysis, reduced translation rates, as well as reduced PI3K and mTOR pathway activities [9]. The PI3K-Akt pathway crosstalks with the Rb-E2F pathway by inhibiting p21 activity through phosphorylation [109,110]. Similarly, the deletion of HIF1α [111] (involved in hypoxia regulation), LKB1 [112–114] (a regulator of AMPK that is an mTOR and FOXO pathway target) and negative regulators of mTOR (such as Fbxw7 [115], PTEN [116,117], PML [118] and TSC [119]) leads to the depletion of HSCs, suggesting the importance of metabolic response pathways in maintaining cell quiescence. Interestingly, E2F was recently found acting as a regulatory switch coordinating proliferation and metabolic pathways by repressing key genes involved in oxidative metabolism [120].
5.4. Autophagy
Quiescent cells are associated with activation of autophagy pathways [121]. Autophagy is a process of ‘self-eating’ that recycles organelles and removes damaged components through a lysosomal degradation pathway. Autophagy provides nutrients for cell survival under suboptimal growth conditions [122–124] and is essential for maintaining HSC quiescence [125]. Autophagy is actively repressed by mTOR [126–128] and is regulated by the Rb-E2F pathway [129,130].
5.5. MicroRNAs
miRNAs have also been implicated in the regulation of quiescence at the post-translational level. The miRNA profiles of quiescent stem cells and fibroblasts are different from their proliferative or differentiated progenies [131–134]. Several miRNAs, such as miR-126 [135] and miR-489 [131], are important for maintaining quiescent stem cells, while let-7, miR-125 and miR-29 regulate key aspects of fibroblast quiescence [134]. Knockout of Dicer, the miRNA processing factor, leads to quiescence exit of muscle stem cells [131]. There is also recent evidence showing that cellular quiescence is regulated by mechanisms that alter the length of the 3’ untranslated region (UTR) of mRNA and thus the potential miRNA susceptibility [1,136–138]. The miRNA pathways crosstalk with the Rb-E2F pathway: for example, miR-221 and miR-222 that regulate the quiescence–proliferation transition are known to target Cdk inhibitors p27 and p57 [139,140]; the let-7 family of miRNAs antagonizes Myc [141]; the miR-17–92 cluster that is induced by Myc negatively regulates E2F1 [142–145].
5.6. Epigenetic regulators
Epigenetic regulations including DNA methylation and histone modifications modify the chromatin states (e.g. accessible or repressed) and the corresponding cellular gene expressions [146–148]. Quiescence may be associated with a unique epigenetic state and chromatin structure that help in maintaining the reversibility of quiescence against terminal differentiation or senescence [98,149]. For example, polycomb group genes EZH1 and EZH2—methyltransferases for histone H3 lysine 27 (H3K27)—are essential for the maintenance of quiescent haematopoietic and hair follicle stem cells [150–152]; specific methylation states of histone H4 lysine 20 (H4K20me2 and H4K20me3) are found to promote chromatin compaction and quiescence entry in primary human fibroblasts [153]. The Rb-E2F pathway also plays an important role in epigenetic modifications [154]. For example, the RB/E2F family proteins interact with various chromatin-modifying complexes that are involved in stable transcription repressions [36,39–42]. The E2F proteins also interact with HCF-1, which recruits the MLL family of histone H3 lysine 4 (H3K4) methyltransferases to induce transcriptional activation [155,156].
6. Conclusion
Cellular quiescence and its reversible transition into proliferation in response to environmental signals are critical to the physiological functions of many important cell types in our body (e.g. stem and progenitor cells). A model is emerging from recent studies that the Rb-E2F bistable switch serves as a core analogue-to-digital converter between environmental signals and quiescence–proliferation decisions. Meanwhile, other quiescence regulatory pathways crosstalk with the Rb-E2F bistable switch: some modulate the activation/inhibition balance of the Rb-E2F switch and thus the final E2F activation threshold, and others are regulated by the Rb-E2F switch and serve as its ‘effectors’ to regulate cellular quiescence. The crosstalk between the Rb-E2F bistable switch and these quiescence regulatory pathways (and possibly between these pathways themselves) forms a gene network. This gene network integrates environmental signals into the final control of cellular quiescence and its heterogeneous, reversible state. Integrating quantitative modelling and detailed experimentation (e.g. single-cell measurements) holds the promise to dissect and reconstruct the quiescence regulatory gene network. Elucidating this quiescence regulatory network is fundamental to understanding the accurate control of cellular quiescence. This new knowledge could profoundly impact therapeutic strategies by suggesting how to re-establish normal quiescent states in a variety of hyper- and hypo-proliferative diseases, such as cancer and ageing.
Acknowledgements
I thank Dr Lingchong You and Dr Hilary Coller and two anonymous reviewers for critical readings and comments on the manuscript. This work was partially supported by the American Cancer Society (G.Y., IRG7400134) and a Faculty Seed Grant from the University of Arizona Foundation (G.Y.).
References
- 1.Cheung TH, Rando TA. 2013. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 14, 329–340. ( 10.1038/nrm3591) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Legesse-Miller A, et al. 2012. Quiescent fibroblasts are protected from proteasome inhibition-mediated toxicity. Mol. Biol. Cell 23, 3566–3581. ( 10.1091/mbc.E12-03-0192) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coller HA, Sang L, Roberts JM. 2006. A new description of cellular quiescence. PLoS Biol. 4, e83 ( 10.1371/journal.pbio.0040083) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu H, Adler AS, Segal E, Chang HY. 2007. A transcriptional program mediating entry into cellular quiescence. PLoS Genet. 3, 996–1008. ( 10.1371/journal.pgen.0030091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yusuf I, Fruman DA. 2003. Regulation of quiescence in lymphocytes. Trends Immunol. 24, 380–386. ( 10.1016/S1471-4906(03)00141-8) [DOI] [PubMed] [Google Scholar]
- 6.Venezia TA, Merchant AA, Ramos CA, Whitehouse NL, Young AS, Shaw CA, Goodell MA. 2004. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2, e301 ( 10.1371/journal.pbio.0020301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sang L, Coller HA, Roberts JM. 2008. Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science 321, 1095–1100. ( 10.1126/science.1155998) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemons JMS, et al. 2010. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 8, e1000514 ( 10.1371/journal.pbio.1000514) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valcourt JR, Lemons JMS, Haley EM, Kojima M, Demuren OO, Coller HA. 2012. Staying alive: metabolic adaptations to quiescence. Cell Cycle 11, 1680–1696. ( 10.4161/cc.19879) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Temin HM. 1971. Stimulation by serum of multiplication of stationary chicken cells. J. Cell. Physiol. 78, 161–170. ( 10.1002/jcp.1040780202) [DOI] [PubMed] [Google Scholar]
- 11.Sander G, Pardee AB. 1972. Transport changes in synchronously growing CHO and L cells. J. Cell. Physiol. 80, 267–271. ( 10.1002/jcp.1040800214) [DOI] [PubMed] [Google Scholar]
- 12.Rovera G, Baserga R. 1973. Effect of nutritional changes on chromatin template activity and non-histone chromosomal protein-synthesis in Wi-38 and 3t6 cells. Exp. Cell Res. 78, 118–126. ( 10.1016/0014-4827(73)90045-1) [DOI] [PubMed] [Google Scholar]
- 13.Baserga R. 1976. Multiplication and division in mammalian cells. New York, NY: M. Dekker. [Google Scholar]
- 14.Augenlicht LH, Baserga R. 1974. Changes in the G0 state of WI-38 fibroblasts at different times after confluence. Exp. Cell Res. 89, 255–262. ( 10.1016/0014-4827(74)90789-7) [DOI] [PubMed] [Google Scholar]
- 15.Soprano KJ. 1994. WI-38 cell long-term quiescence model system: a valuable tool to study molecular events that regulate growth. J. Cell Biochem. 54, 405–414. ( 10.1002/jcb.240540407) [DOI] [PubMed] [Google Scholar]
- 16.Pardee AB. 1974. A restriction point for control of normal animal cell proliferation. Proc. Natl Acad. Sci. USA, 71, 1286–1290. ( 10.1073/pnas.71.4.1286) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zetterberg A, Larsson O. 1985. Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl Acad. Sci. USA 82, 5365–5369. ( 10.1073/pnas.82.16.5365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper S. 1987. On G0 and cell cycle controls. Bioessays 7, 220–223. ( 10.1002/bies.950070507) [DOI] [PubMed] [Google Scholar]
- 19.Cooper S. 2003. Reappraisal of serum starvation, the restriction point, G0, and G1 phase arrest points. Faseb J. 17, 333–340. ( 10.1096/fj.02-0352rev) [DOI] [PubMed] [Google Scholar]
- 20.Coller HA. 2011. Cell biology. The essence of quiescence. Science 334, 1074–1075. ( 10.1126/science.1216242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coller HA. 2007. What's taking so long? S-phase entry from quiescence versus proliferation. Nat. Rev. Mol. Cell Biol. 8, 667–670. ( 10.1038/nrm2223) [DOI] [PubMed] [Google Scholar]
- 22.Burns FJ, Tannock IF. 1970. On existence of a G0-phase in cell cycle. Cell Tissue Kinetics 3, 321. [DOI] [PubMed] [Google Scholar]
- 23.Smith JA, Martin L. 1973. Do cells cycle. Proc. Natl Acad. Sci. USA 70, 1263–1267. ( 10.1073/pnas.70.4.1263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demaertelaer V, Galand P. 1975. Some properties of a G0 model of cell-cycle investigation on possible existence of natural constraints on theoretical model in steady-state conditions. Cell Tissue Kinetics 8, 11–22. [PubMed] [Google Scholar]
- 25.Shields R, Smith JA. 1977. Cells regulate their proliferation through alterations in transition-probability. J. Cell. Physiol. 91, 345–355. ( 10.1002/jcp.1040910304) [DOI] [PubMed] [Google Scholar]
- 26.Brooks RF, Bennett DC, Smith JA. 1980. Mammalian cell cycles need two random transitions. Cell 19, 493–504. ( 10.1016/0092-8674(80)90524-3) [DOI] [PubMed] [Google Scholar]
- 27.Koch AL. 1980. Does the variability of the cell-cycle result from one or many chance events. Nature 286, 80–82. ( 10.1038/286080a0) [DOI] [PubMed] [Google Scholar]
- 28.Castor LN. 1980. A G1 rate model accounts for cell-cycle kinetics attributed to transition-probability. Nature 287, 857–859. ( 10.1038/287857a0) [DOI] [PubMed] [Google Scholar]
- 29.Cooper S. 1982. The continuum model—statistical implications. J. Theor. Biol. 94, 783–800. ( 10.1016/0022-5193(82)90078-9) [DOI] [PubMed] [Google Scholar]
- 30.Tyson JJ, Hannsgen KB. 1985. The distributions of cell size and generation time in a model of the cell cycle incorporating size control and random transitions. J. Theor. Biol. 113, 29–62. ( 10.1016/S0022-5193(85)80074-6) [DOI] [PubMed] [Google Scholar]
- 31.Tyson JJ, Diekmann O. 1986. Sloppy size control of the cell-division cycle. J. Theor. Biol. 118, 405–426. ( 10.1016/S0022-5193(86)80162-X) [DOI] [PubMed] [Google Scholar]
- 32.Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. 1986. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323, 643–646. ( 10.1038/323643a0) [DOI] [PubMed] [Google Scholar]
- 33.Cobrinik D. 2005. Pocket proteins and cell cycle control. Oncogene 24, 2796–2809. ( 10.1038/sj.onc.1208619) [DOI] [PubMed] [Google Scholar]
- 34.Attwooll C, Lazzerini Denchi E, Helin K. 2004. The E2F family: specific functions and overlapping interests. EMBO J. 23, 4709–4716. ( 10.1038/sj.emboj.7600481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sears RC, Nevins JR. 2002. Signaling networks that link cell proliferation and cell fate. J. Biol. Chem. 277, 11 617–11 620. ( 10.1074/jbc.R100063200) [DOI] [PubMed] [Google Scholar]
- 36.Frolov MV, Dyson NJ. 2004. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 117, 2173–2181. ( 10.1242/jcs.01227) [DOI] [PubMed] [Google Scholar]
- 37.DeGregori J, Johnson DG. 2006. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 6, 739–748. [DOI] [PubMed] [Google Scholar]
- 38.Macaluso M, Montanari M, Giordano A. 2006. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene 25, 5263–5267. ( 10.1038/sj.onc.1209680) [DOI] [PubMed] [Google Scholar]
- 39.Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D. 2001. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol. Cell. Biol. 21, 6484–6494. ( 10.1128/MCB.21.19.6484-6494.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nielsen SJ, et al. 2001. Rb targets histone H3 methylation and HP1 to promoters. Nature 412, 561–565. ( 10.1038/35087620) [DOI] [PubMed] [Google Scholar]
- 41.Trojer P, et al. 2007. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 129, 915–928. ( 10.1016/j.cell.2007.03.048) [DOI] [PubMed] [Google Scholar]
- 42.Litovchick L, et al. 2007. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 26, 539–551. ( 10.1016/j.molcel.2007.04.015) [DOI] [PubMed] [Google Scholar]
- 43.Smith EJ, Leone G, DeGregori J, Jakoi L, Nevins JR. 1996. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol. Cell. Biol. 16, 6965–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leung JY, Ehmann GL, Giangrande PH, Nevins JR. 2008. A role for Myc in facilitating transcription activation by E2F1. Oncogene 27, 4172–4179. ( 10.1038/onc.2008.55) [DOI] [PubMed] [Google Scholar]
- 45.Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J. 1996. Cyclin D1 expression is regulated positively by the p42/p44(MAPK) and negatively by the p38/HOG(MAPK) pathway. J. Biol. Chem. 271, 20 608–20 616. ( 10.1074/jbc.271.34.20608) [DOI] [PubMed] [Google Scholar]
- 46.Aktas H, Cai H, Cooper GM. 1997. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol. Cell. Biol. 17, 3850–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerkhoff E, Rapp UR. 1997. Induction of cell proliferation in quiescent NIH 3T3 cells by oncogenic c-Raf-1. Mol. Cell. Biol. 17, 2576–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng M, Sexl V, Sherr CJ, Roussel MF. 1998. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase (MEK1). Proc. Natl Acad. Sci. USA 95, 1091–1096. ( 10.1073/pnas.95.3.1091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harbour JW, Luo RX, Santi AD, Postigo AA, Dean DC. 1999. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98, 859–869. ( 10.1016/S0092-8674(00)81519-6) [DOI] [PubMed] [Google Scholar]
- 50.Wu L, et al. 2001. The E2F1–3 transcription factors are essential for cellular proliferation. Nature 414, 457–462. ( 10.1038/35106593) [DOI] [PubMed] [Google Scholar]
- 51.Johnson DG, Schwarz JK, Cress WD, Nevins JR. 1993. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 365, 349–352. ( 10.1038/365349a0) [DOI] [PubMed] [Google Scholar]
- 52.Nevins JR. 2001. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10, 699–703. ( 10.1093/hmg/10.7.699) [DOI] [PubMed] [Google Scholar]
- 53.Ladha MH, Lee KY, Upton TM, Reed MF, Ewen ME. 1998. Regulation of exit from quiescence by p27 and cyclin D1-CDK4. Mol. Cell. Biol. 18, 6605–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boonstra J. 2003. Progression through the G(1)-phase of the on-going cell cycle. J. Cell. Biochem. 90, 244–252. ( 10.1002/jcb.10617) [DOI] [PubMed] [Google Scholar]
- 55.Pajalunga D, Mazzola A, Salzano AM, Biferi MG, De Luca G, Crescenzi M. 2007. Critical requirement for cell cycle inhibitors in sustaining nonproliferative states. J. Cell Biol. 176, 807–818. ( 10.1083/jcb.200608109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spencer SL, Cappell SD, Tsai F-C, Overton KW, Wang CL, Meyer T. 2013. The proliferation–quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. ( 10.1016/j.cell.2013.08.062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M. 2000. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287, 1804–1808. ( 10.1126/science.287.5459.1804) [DOI] [PubMed] [Google Scholar]
- 58.Sherr CJ, Roberts JM. 1995. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 9, 1149–1163. ( 10.1101/gad.9.10.1149) [DOI] [PubMed] [Google Scholar]
- 59.Coats S, Flanagan WM, Nourse J, Roberts JM. 1996. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 272, 877–880. ( 10.1126/science.272.5263.877) [DOI] [PubMed] [Google Scholar]
- 60.Mayol X, Grana X. 1998. The p130 pocket protein: keeping order at cell cycle exit/re-entrance transitions. Front. Biosci. 3, d11–d24. [DOI] [PubMed] [Google Scholar]
- 61.Stevaux O, Dyson NJ. 2002. A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 14, 684–691. ( 10.1016/S0955-0674(02)00388-5) [DOI] [PubMed] [Google Scholar]
- 62.Vidal A, Koff A. 2000. Cell-cycle inhibitors: three families united by a common cause. Gene 247, 1–15. ( 10.1016/S0378-1119(00)00092-5) [DOI] [PubMed] [Google Scholar]
- 63.Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. 2002. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G(0) cells. Science 296, 1132–1136. ( 10.1126/science.1069861) [DOI] [PubMed] [Google Scholar]
- 64.Rivard N, L'Allemain G, Bartek J, Pouyssegur J. 1996. Abrogation of p27Kip1 by cDNA antisense suppresses quiescence (G0 state) in fibroblasts. J. Biol. Chem. 271, 18 337–18 341. ( 10.1074/jbc.271.31.18337) [DOI] [PubMed] [Google Scholar]
- 65.Nakanishi M, Adami GR, Robetorye RS, Noda A, Venable SF, Dimitrov D, Pereira-Smith OM, Smith JR. 1995. Exit from G0 and entry into the cell cycle of cells expressing p21Sdi1 antisense RNA. Proc. Natl Acad. Sci. USA 92, 4352–4356. ( 10.1073/pnas.92.10.4352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kippin TE, Martens DJ, van der Kooy D. 2005. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 19, 756–767. ( 10.1101/gad.1272305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dannenberg JH, van Rossum A, Schuijff L, te Riele H. 2000. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 14, 3051–3064. ( 10.1101/gad.847700) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, Butte AJ, Passegué E, Sage J. 2008. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell 3, 416–428. ( 10.1016/j.stem.2008.07.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. 2000. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 14, 3037–3050. ( 10.1101/gad.843200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. 2003. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424, 223–228. ( 10.1038/nature01764) [DOI] [PubMed] [Google Scholar]
- 71.Xiong W, Ferrell JE., Jr 2003. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature 426, 460–465. ( 10.1038/nature02089) [DOI] [PubMed] [Google Scholar]
- 72.Sha W, Moore J, Chen K, Lassaletta AD, Yi C-S, Tyson JJ, Sible JC. 2003. Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc. Natl Acad. Sci. USA 100, 975–980. ( 10.1073/pnas.0235349100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ozbudak EM, Thattai M, Lim HN, Shraiman BI, Van Oudenaarden A. 2004. Multistability in the lactose utilization network of Escherichia coli. Nature 427, 737–740. ( 10.1038/nature02298) [DOI] [PubMed] [Google Scholar]
- 74.Paliwal S, Iglesias PA, Campbell K, Hilioti Z, Groisman A, Levchenko A. 2007. MAPK-mediated bimodal gene expression and adaptive gradient sensing in yeast. Nature 446, 46–51. ( 10.1038/nature05561) [DOI] [PubMed] [Google Scholar]
- 75.Tyson JJ, Chen KC, Novak B. 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 15, 221–231. ( 10.1016/S0955-0674(03)00017-6) [DOI] [PubMed] [Google Scholar]
- 76.Ferrell JE., Jr 2002. Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr. Opin. Cell Biol. 14, 140–148. ( 10.1016/S0955-0674(02)00314-9) [DOI] [PubMed] [Google Scholar]
- 77.Ma'ayan A, Cecchi GA, Wagner J, Rao AR, Iyengar R, Stolovitzky G. 2008. Ordered cyclic motifs contribute to dynamic stability in biological and engineered networks. Proc. Natl Acad. Sci. USA 105, 19 235–19 240. ( 10.1073/pnas.0805344105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. 1997. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 11, 1464–1478. ( 10.1101/gad.11.11.1464) [DOI] [PubMed] [Google Scholar]
- 79.Aguda BD, Tang Y. 1999. The kinetic origins of the restriction point in the mammalian cell cycle. Cell Prolif. 32, 321–335. ( 10.1046/j.1365-2184.1999.3250321.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Novak B, Tyson JJ. 2004. A model for restriction point control of the mammalian cell cycle. J. Theor. Biol. 230, 563–579. ( 10.1016/j.jtbi.2004.04.039) [DOI] [PubMed] [Google Scholar]
- 81.Hatzimanikatis V, Lee KH, Bailey JE. 1999. A mathematical description of regulation of the G1-S transition of the mammalian cell cycle. Biotechnol. Bioeng. 65, 631–637. () [DOI] [PubMed] [Google Scholar]
- 82.Qu Z, MacLellan WR, Weiss JN. 2003. Dynamics of the cell cycle: checkpoints, sizers, and timers. Biophys. J. 85, 3600–3611. ( 10.1016/S0006-3495(03)74778-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thron CD. 1997. Bistable biochemical switching and the control of the events of the cell cycle. Oncogene 15, 317–325. ( 10.1038/sj.onc.1201190) [DOI] [PubMed] [Google Scholar]
- 84.Yao G, Lee TJ, Mori S, Nevins JR, You L. 2008. A bistable Rb-E2F switch underlies the restriction point. Nat. Cell Biol. 10, 476–482. ( 10.1038/ncb1711) [DOI] [PubMed] [Google Scholar]
- 85.Yao G, Tan C, West M, Nevins JR, You L. 2011. Origin of bistability underlying mammalian cell cycle entry. Mol. Syst. Biol. 7, 485 ( 10.1038/msb.2011.19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee T, Yao G, Bennett DC, Nevins JR, You L. 2010. Stochastic E2F activation and reconciliation of phenomenological cell-cycle models. PLoS Biol. 8, e1000488 ( 10.1371/journal.pbio.1000488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eilers M, Schirm S, Bishop JM. 1991. The MYC protein activates transcription of the alpha-prothymosin gene. EMBO J. 10, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blomen VA, Boonstra J. 2007. Cell fate determination during G1 phase progression. Cell Mol. Life Sci. 64, 3084–3104. ( 10.1007/s00018-007-7271-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. 1996. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl Acad. Sci. USA 93, 13 742–13 747. ( 10.1073/pnas.93.24.13742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stein GH, Drullinger LF, Soulard A, Dulic V. 1999. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell Biol. 19, 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang W, Martindale JL, Yang X, Chrest FJ, Gorospe M. 2005. Increased stability of the p16 mRNA with replicative senescence. EMBO Rep. 6, 158–164. ( 10.1038/sj.embor.7400346) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pajalunga D, Mazzola A, Puggioni E, Crescenzi M. 2007. Non-proliferation as an active state—conceptual and practical implications. Cell Cycle 6, 1415–1418. ( 10.4161/cc.6.12.4378) [DOI] [PubMed] [Google Scholar]
- 93.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. 2003. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 22, 4212–4222. ( 10.1093/emboj/cdg417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Latella L, Sacco A, Pajalunga D, Tiainen M, Macera D, D'Angelo M, Felici A, Sacchi A, Crescenzi M. 2001. Reconstitution of cyclin D1-associated kinase activity drives terminally differentiated cells into the cell cycle. Mol. Cell Biol. 21, 5631–5643. ( 10.1128/MCB.21.16.5631-5643.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Artavanis-Tsakonas S, Rand MD, Lake RJ. 1999. Notch signaling: cell fate control and signal integration in development. Science 284, 770–776. ( 10.1126/science.284.5415.770) [DOI] [PubMed] [Google Scholar]
- 96.Bjornson CRR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA. 2012. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30, 232–242. ( 10.1002/stem.773) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mourikis P, Sambasivan R, Castel D, Rocheteau P, Bizzarro V, Tajbakhsh S. 2012. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 30, 243–252. ( 10.1002/stem.775) [DOI] [PubMed] [Google Scholar]
- 98.Sang L, Coller HA. 2009. Fear of commitment: Hes1 protects quiescent fibroblasts from irreversible cellular fates. Cell Cycle 8, 2161–2167. ( 10.4161/cc.8.14.9104) [DOI] [PubMed] [Google Scholar]
- 99.Hartman J, Müller P, Foster JS, Wimalasena J, Gustafsson J, Ström A. 2004. HES-1 inhibits 17beta-estradiol and heregulin-beta1-mediated upregulation of E2F-1. Oncogene 23, 8826–8833. ( 10.1038/sj.onc.1208139) [DOI] [PubMed] [Google Scholar]
- 100.Kabos P, Kabosova A, Neuman T. 2002. Blocking HES1 expression initiates GABAergic differentiation and induces the expression of p21(CIP1/WAF1) in human neural stem cells. J. Biol. Chem. 277, 8763–8766. ( 10.1074/jbc.C100758200) [DOI] [PubMed] [Google Scholar]
- 101.Murata K, Hattori M, Hirai N, Shinozuka Y, Hirata H, Kageyama R, Sakai T, Minato N. 2005. Hes1 directly controls cell proliferation through the transcriptional repression of p27(Kip1). Mol. Cell. Biol. 25, 4262–4271. ( 10.1128/MCB.25.10.4262-4271.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Itahana K, et al. 2002. A role for p53 in maintaining and establishing the quiescence growth arrest in human cells. J. Biol. Chem. 277, 18 206–18 214. ( 10.1074/jbc.M201028200) [DOI] [PubMed] [Google Scholar]
- 103.Liu Y, et al. 2009. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4, 37–48. ( 10.1016/j.stem.2008.11.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, Orkin SH. 2004. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431, 1002–1007. ( 10.1038/nature02994) [DOI] [PubMed] [Google Scholar]
- 105.Lacorazza HD, Yamada T, Liu Y, Miyata Y, Sivina M, Nunes J, Nimer SD. 2006. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell 9, 175–187. ( 10.1016/j.ccr.2006.02.017) [DOI] [PubMed] [Google Scholar]
- 106.Kops GJ, et al. 2002. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321. ( 10.1038/nature01036) [DOI] [PubMed] [Google Scholar]
- 107.Tothova Z, et al. 2007. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339. ( 10.1016/j.cell.2007.01.003) [DOI] [PubMed] [Google Scholar]
- 108.Maryanovich M, Oberkovitz G, Niv H, Vorobiyov L, Zaltsman Y, Brenner O, Lapidot T, Jung S, Gross A. 2012. The ATM-BID pathway regulates quiescence and survival of haematopoietic stem cells. Nat. Cell Biol. 14, 535–541. ( 10.1038/ncb2468) [DOI] [PubMed] [Google Scholar]
- 109.Rossig L, Jadidi AS, Urbich C, Badorff C, Zeiher AM, Dimmeler S. 2001. Akt-dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells. Mol. Cell Biol. 21, 5644–5657. ( 10.1128/MCB.21.16.5644-5657.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou BP, Liao Y, Xia W, Spohn B, Lee M-H, Hung M-C. 2001. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 3, 245–252. ( 10.1038/35060032) [DOI] [PubMed] [Google Scholar]
- 111.Takubo K, et al. 2010. Regulation of the HIF-1 alpha level is essential for hematopoietic stem cells. Cell Stem Cell 7, 391–402. ( 10.1016/j.stem.2010.06.020) [DOI] [PubMed] [Google Scholar]
- 112.Gurumurthy S, et al. 2010. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 468, 659–663. ( 10.1038/nature09572) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gan B, et al. 2010. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 468, 701–704. ( 10.1038/nature09595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nakada D, Saunders TL, Morrison SJ. 2010. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 468, 653–658. ( 10.1038/nature09571) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, Zavadil J, Nimer SD, Aifantis I. 2008. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J. Exp. Med. 205, 1395–1408. ( 10.1084/jem.20080277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. 2006. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482. ( 10.1038/nature04703) [DOI] [PubMed] [Google Scholar]
- 117.Zhang J, et al. 2006. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441, 518–522. ( 10.1038/nature04747) [DOI] [PubMed] [Google Scholar]
- 118.Ito K, et al. 2008. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 453, 1072–1078. ( 10.1038/nature07016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen C, Liu Y, Liu R, Ikenoue T, Guan K-L, Liu Y, Zheng P. 2008. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 205, 2397–2408. ( 10.1084/jem.20081297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Blanchet E, et al. 2011. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol. 13, 1146–1152. ( 10.1038/ncb2309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Valentin M, Yang E. 2008. Autophagy is activated, but is not required for the G(0) function of BCL-2 or BCL-x(L). Cell Cycle 7, 2762–2768. ( 10.4161/cc.7.17.6595) [DOI] [PubMed] [Google Scholar]
- 122.Dunn WA., Jr 1994. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 4, 139–143. ( 10.1016/0962-8924(94)90069-8) [DOI] [PubMed] [Google Scholar]
- 123.Klionsky DJ, Emr SD. 2000. Cell biology—autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721. ( 10.1126/science.290.5497.1717) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rabinowitz JD, White E. 2010. Autophagy and metabolism. Science 330, 1344–1348. ( 10.1126/science.1193497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mortensen M, et al. 2011. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 208, 455–467. ( 10.1084/jem.20101145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chang YY, Neufeld TP. 2009. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 20, 2004–2014. ( 10.1091/mbc.E08-12-1250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jung CH, Jun CB, Ro S-H, Kim Y-M, Otto NM, Cao J, Kundu M, Kim D-H. 2009. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. ( 10.1091/mbc.E08-12-1249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kamada Y, Yoshino K-I, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y. 2010. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 30, 1049–1058. ( 10.1128/MCB.01344-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jiang H, et al. 2010. The RB-E2F1 pathway regulates autophagy. Cancer Res 70, 7882–7893. ( 10.1158/0008-5472.CAN-10-1604) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Polager S, Ofir M, Ginsberg D. 2008. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 27, 4860–4864. ( 10.1038/onc.2008.117) [DOI] [PubMed] [Google Scholar]
- 131.Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, Rando TA. 2012. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 482, 524–528. ( 10.1038/nature10834) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Arnold CP, et al. 2011. MicroRNA programs in normal and aberrant stem and progenitor cells. Genome Res. 21, 798–810. ( 10.1101/gr.111385.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang L, Stokes N, Polak L, Fuchs E. 2011. Specific microRNAs are preferentially expressed by skin stem cells to balance self-renewal and early lineage commitment. Cell Stem Cell 8, 294–308. ( 10.1016/j.stem.2011.01.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Suh EJ, et al. 2012. A microRNA network regulates proliferative timing and extracellular matrix synthesis during cellular quiescence in fibroblasts. Genome Biol. 13, R121 ( 10.1186/gb-2012-13-12-r121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lechman ER, et al. 2012. Attenuation of miR-126 activity expands hsc in vivo without exhaustion. Cell Stem Cell 11, 799–811. ( 10.1016/j.stem.2012.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Boutet SC, et al. 2012. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell 10, 327–336. ( 10.1016/j.stem.2012.01.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. 2008. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science 320, 1643–1647. ( 10.1126/science.1155390) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mayr C, Bartel DP. 2009. Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684. ( 10.1016/j.cell.2009.06.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Medina R, Zaidi SK, Liu C-G, Stein JL, vanWijnen AJ, Croce CM, Stein GS. 2008. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 68, 2773–2780. ( 10.1158/0008-5472.CAN-07-6754) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gillies JK, Lorimer IA. 2007. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 6, 2005–2009. ( 10.4161/cc.6.16.4526) [DOI] [PubMed] [Google Scholar]
- 141.Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ. 2007. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 67, 9762–9770. ( 10.1158/0008-5472.CAN-07-2462) [DOI] [PubMed] [Google Scholar]
- 142.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. 2005. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839–843. ( 10.1038/nature03677) [DOI] [PubMed] [Google Scholar]
- 143.Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, Ferbeyre G, Chartrand P. 2007. An E2F/miR-20a autoregulatory feedback loop. J. Biol. Chem. 282, 2135–2143. ( 10.1074/jbc.M608939200) [DOI] [PubMed] [Google Scholar]
- 144.Li Y, Zhang H, Chen Y. 2011. MicroRNA-mediated positive feedback loop and optimized bistable switch in a cancer network involving miR-17–92. PLoS ONE 6, e26302 ( 10.1371/journal.pone.0026302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB. 2008. MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR-17–92, E2F, and Myc. Proc. Natl Acad. Sci. USA 105, 19 678–19 683. ( 10.1073/pnas.0811166106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bird A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. ( 10.1101/gad.947102) [DOI] [PubMed] [Google Scholar]
- 147.Bernstein BE, Meissner A, Lander ES. 2007. The mammalian epigenome. Cell 128, 669–681. ( 10.1016/j.cell.2007.01.033) [DOI] [PubMed] [Google Scholar]
- 148.Turner BM. 2007. Defining an epigenetic code. Nat. Cell Biol. 9, 2–6. ( 10.1038/ncb0107-2) [DOI] [PubMed] [Google Scholar]
- 149.Srivastava S, Mishra RK, Dhawan J. 2010. Regulation of cellular chromatin state: insights from quiescence and differentiation. Organogenesis 6, 37–47. ( 10.4161/org.6.1.11337) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kamminga LM, et al. 2006. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood 107, 2170–2179. ( 10.1182/blood-2005-09-3585) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hidalgo I, Herrera-Merchan A, Ligos JM, Carramolino L, Nuñez J, Martinez F, Dominguez O, Torres M, Gonzalez S. 2012. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell 11, 649–662. ( 10.1016/j.stem.2012.08.001) [DOI] [PubMed] [Google Scholar]
- 152.Ezhkova E, Lien W-H, Stokes N, Pasolli HA, Silva JM, Fuchs E. 2011. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 25, 485–498. ( 10.1101/gad.2019811) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Evertts AG, Manning AL, Wang X, Dyson NJ, Garcia BA, Coller HA. 2013. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 24, 3025–3037. ( 10.1091/mbc.E12-07-0529) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Blais A, Dynlacht BD. 2007. E2F-associated chromatin modifiers and cell cycle control. Curr. Opin. Cell Biol. 19, 658–662. ( 10.1016/j.ceb.2007.10.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Takeda S, et al. 2006. Proteolysis of MLL family proteins is essential for Taspase1-orchestrated cell cycle progression. Genes Dev. 20, 2397–2409. ( 10.1101/gad.1449406) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tyagi S, Chabes AL, Wysocka J, Herr W. 2007. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol. Cell 27, 107–119. ( 10.1016/j.molcel.2007.05.030) [DOI] [PubMed] [Google Scholar]