

Abstract
Cocaine is a widely abused and addictive drug without an FDA-approved medication. Our recently designed and discovered cocaine hydrolase, particularly E12-7 engineered from human butyrylcholinesterase (BChE), has the promise of becoming a valuable cocaine abuse treatment. An ideal anti-cocaine therapeutic enzyme should have not only a high catalytic efficiency against cocaine, but also a sufficiently long biological half-life. However, recombinant human BChE and the known BChE mutants have a much shorter biological half-life compared to the native human BChE. The present study aimed to extend the biological half-life of the cocaine hydrolase without changing its high catalytic activity against cocaine. Our strategy was to design possible amino-acid mutations that can introduce cross-subunit disulfide bond(s) and, thus, change the distribution of the oligomeric forms and extend the biological half-life. Three new BChE mutants (E364–532, E377–516, and E535) were predicted to have a more stable dimer structure with the desirable cross-subunit disulfide bond(s) and, therefore, a different distribution of the oligomeric forms and a prolonged biological half-life. The rational design was followed by experimental tests in vitro and in vivo, confirming that the rationally designed new BChE mutants, i.e. E364–532, E377–516, and E535, indeed had a remarkably different distribution of the oligomeric forms and prolonged biological half-life in rats from ~7 hr to ~13 hr without significantly changing the catalytic activity against (−)-cocaine. This is the first demonstration that rationally designed amino-acid mutations can significantly prolong the biological half-life of a high-activity enzyme without significantly changing the catalytic activity.
Introduction
Cocaine is a widely abused and addictive drug which blocks dopamine reuptake in the central nervous system (CNS).1 Currently, there is no FDA-approved medication specific for cocaine abuse treatment.2, 3 The disastrous medical and social consequences of cocaine abuse have made the development of an anti-cocaine medication a high priority. However, despite decades of efforts, traditional pharmacodynamic approach has failed to yield a truly useful small-molecule drug due to the difficulties inherent in blocking a blocker like cocaine without affecting the normal functions of dopamine transporter. An alternative approach is to interfere with the delivery of cocaine to its receptors or accelerate its metabolism in the body.2, 4–8 It would be an ideal anti-cocaine medication to accelerate cocaine metabolism producing biologically inactive metabolites via a route similar to the primary cocaine-metabolizing pathway, i.e. cocaine hydrolysis catalyzed by human butyrylcholinesterase (BChE) in plasma.9, 10 Unfortunately, the catalytic efficiency of wild-type BChE against naturally occurring, biologically active (−)-cocaine is low (kcat = 4.1 min−1 and KM = 4.5 μM).11 In order to improve the catalytic efficiency, BChE mutants were designed and discovered in our group based on the understanding of the catalytic mechanism of cocaine hydrolysis using an integrated computational-experimental approach.12–16 One of our designed and discovered BChE mutants, i.e. the A199S/F227A/S287G/A328W/Y332G mutant (denoted as enzyme E12-7 here for convenience), has a ~2000-fold improved catalytic efficiency against (−)-cocaine compared to the wild-type BChE and, therefore, is known as the cocaine hydrolase (CocH).16 It has been known that E12-7 can be used to fully protect mice from the acute toxicity of a lethal dose of cocaine (180 mg/kg, LD100).16
In order to effectively suppress cocaine reward for a long period of time after administration of an exogenous CocH, the therapeutic enzyme (CocH) should have not only a high catalytic efficiency against cocaine, but also a sufficiently long circulation time (biological half-life). We note that the long biological half-life might be unnecessary for cocaine overdose treatment. However, for cocaine addiction treatment using a cocaine-metabolizing enzyme, it is desired to have a highly efficient cocaine-metabolizing enzyme circulating in the body for a long time. With a highly efficient cocaine-metabolizing enzyme circulating in the body, whenever a cocaine user takes cocaine again, the enzyme will metabolize cocaine rapidly such that the user will not receive the reward effects of the drug. Native human BChE has a biological half-life of ~24 hours in mice and ~7 to 12 days in humans.17 However, recombinant forms of wild-type human BChE and the known BChE mutants have a much shorter biological half-life compared to the native human BChE.18 The difference between the native and recombinant human BChE proteins in biological half-life is associated with the difference in the distribution of the oligomeric forms and the post-translational modification. Native BChE consists of more than 95% of tetramer, whereas predominant forms of recombinant BChE are monomer and dimer.18, 19 In addition, the native BChE is fully glycosylated with whole nine N-linked oligosaccharides, whereas recombinant BChE is either not fully glycosylated or glycosylated differently.20–23
The present study aimed to extend the biological half-life of E12-7, i.e. the A199S/F227A/S287G/A328W/Y332G mutant of human BChE, without changing its catalytic activity against cocaine. It is very popular for extending the biological half-life of a protein to chemically modify the protein surface with polyethylene glycol (PEG). However, it is well-known that PEG can elicit an immune response in humans.24–26 For this reason, we are reluctant to PEGylate the human protein which is accommodated perfectly in humans. Lockridge et al.18, 27–31 demonstrated that co-expression of a proline-rich peptide with recombinant human BChE can form a complex between the proline-rich peptide and BChE tetramer, and the complex has a longer biological half-life. For the purpose of practical protein drug development, we prefer to extend the biological half-life of E12-7 through an alternative method without co-expression of another peptide, i.e. rationally designed further mutations on its amino acid residues that are not available for intermolecular interactions with any other proteins in the body; such type of aminoacid mutations is not expected to produce immune response.
On the other hand, it is a grand challenge to design amino-acid mutations that can extend the biological half-life of a protein, although rational or computational design has been used successfully to identify thermostable mutants of proteins.32–36 This is because the biological half-life of a protein is determined by many factors, in addition to the thermostability. For a protein to have a desirably long biological half-life, the protein must be thermostable enough at the body temperature (37°C) to have an in vitro half-life at 37°C which is at least not shorter than the desirable biological half-life. So, when a protein has a short biological half-life because the protein is thermally unstable, one may design a thermostable mutant of the protein to extend the biological half-life.33 However, for most proteins, their short biological half-lives are due to factors other than the thermostability. Specifically for the therapeutically interesting protein concerned in the present study, BChE (wild-type or the mutant) is very thermostable at 37°C. But the recombinant form of the thermostable protein is quickly eliminated from the body. So, further improving the protein thermostability is not expected to extend its biological half-life. One must account for other factors affecting the biological half-life.
In the present study, we aimed to design further amino-acid mutations that can favorably change the distribution of the oligomeric forms of E12-7 and, thus, extend the biological half-life. In particular, our design was focused on possible mutations that may introduce disulfide bond(s) between different subunits in the multimeric forms (dimer and tetramer) and, thus, stabilize the multimeric forms and decrease the concentration of the monomer. This rational design strategy was based on the observation that the multimeric forms of BChE have a longer biological half-life compared to the monomer, and that the formation of the multimeric forms of wild-type human BChE relies on an interchain disulfide bond (C571a-C571b) that, once formed, can covalently link two subunits.37 Our rational design was followed by experimental studies in vitro and in vivo, leading to discovery of novel BChE mutants that not only retain the high catalytic activity of E12-7 against cocaine, but also have a significantly prolonged biological half-life, demonstrating that it is possible to rationally design amino-acid mutations on therapeutic proteins with a prolonged biological half-life without changing the biological functions.
Material and Methods
Molecular dynamics simulation
The initial BChE structure modeled in the present study came from our previously reported computational study on the BChE tetramer.38 Based on the tetramer structure, the tetramer consists of two equivalent dimers, and our rational design was focused on the amino-acid residues on the inter-subunit interface within a dimer. For this reason, we only needed to model a BChE dimer, instead of a tetramer, for the present rational design. The general procedure for carrying out the molecular dynamics (MD) simulation on the dimer in this study was similar as that used in our previously reported computational studies.13, 14, 16 All molecular mechanics optimization and MD simulation were carried out by using the AMBER 9 program package.39, 40 The Amber ff03 force field was used to establish the potentials of protein.41 Counter ions (Na+) were used to neutralize the system and, then, the system was immersed in an orthorhombic box of TIP3P water molecules with a minimum solute-wall distance of 10 Å.42 The whole system was carefully equilibrated and fully energy-minimized. After that, the system was gradually heated in the NPT ensemble from 10 K to 300 K over 60 ps. Then, the MD simulation was performed under normal temperature (300 K) for 50 ns. During the MD simulation, Particle Mesh Ewald (PME) method was employed to treat the long-range electrostatic interactions.43 The SHAKE procedure was applied to constrain the bond lengths of all covalent bonds involving hydrogen atoms, with the time step of 2 fs.44 The atomic coordinates were saved every 1 ps for the productive sampling and analysis.
Materials for in vitro activity assays
Cloned Pfu DNA polymerase and DpnI endonuclease were obtained from Stratagene (La Jolla, CA). All oligonucleotides were synthesized by the Eurofins (Huntsville, AL). The QIAprep Spin Plasmid Miniprep Kit was obtained from QIAGEN (Valencia, CA). Chinese hamster ovary (CHO) cells and culture medium were from life technologies (Grand Island, NY). The transfection kit was from Mirus Bio LLC (Madison, WI). Anti-BChE (mouse monoclonal antibody) was purchased from AntibodyShop (Gentofte, Denmark), and goat anti-mouse IgG HRP conjugate was from Zymed Laboratories (South San Francisco, CA). [3H](−)-Cocaine (50 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). QFF ion exchanger was from GE Healthcare.
Site-directed mutagenesis
Site-directed mutagenesis of full-length human BChE cDNA was performed by using the QuikChange method.45 Further mutations required to produce a new BChE mutant cDNA were generated from the cDNA corresponding to the A199S/F227A/S287G/A328W/Y332G mutant of human BChE in a pRc/CMV expression plasmid.46 Using plasmid DNA as template and primers with specific base-pair alterations, mutations were made by polymerase chain reaction with Pfu DNA polymerase for replication fidelity. The PCR product was treated with DpnI endonuclease to digest the parental DNA template. The digested product was transformed into Escherichia coli, amplified, and purified. The DNA sequences of the mutants were confirmed by DNA sequencing.
Protein expression
The A199S/F227A/S287G/A328W/Y332G BChE and newly designed BChE mutants were expressed in CHO-S cells in free-style CHO expression medium. Cells were first grown to a density of ~1.0 × 106 in 1L shake flask and transfected using TransIT-PRO Transfectio Kit. Cells were incubated at 37°C in a CO2 incubator for 6 days. The culture medium was then harvested for the BChE activity assays.
Protein purification
Purification of each enzyme mutant in medium was achieved by using an ion exchange chromatography. In brief, 1L of medium with a BChE mutant was diluted with the same volume of 20 mM Tris-HCl, pH 7.4. Pre-Equilibrated QFF anion exchanger was added to the diluted medium in 1% of its volume and incubated at 4°C with shaking (100 rpm) overnight. More than 95% enzyme activity was found to bind to the resin after the incubation. The suspension was then packed in a column (5 × 50 cm), and the medium was allowed to flow through rapidly with the aid of suction (50–100 ml/min). The QFF resin was repacked again in a washing buffer after all of the medium was excluded. After washing the column with 20 mM Tris-HCl, pH 7, the enzyme was eluted by 20 mM Tris-HCL, pH 7.0, plus 0.3 M NaCl. The eluate was concentrated and changed to phosphate-buffered saline by Millipore centrifugal filter device. Finally, the purified enzyme was stored at 4°C or −80°C. The entire purification process was carried out in cold room at 4°C.
Gel stained for enzyme activity
8% non-denaturing polyacrylamide gel was run at constant current of 8 mA at 4 °C, overnight. The gel was stained for BChE activity with 1 mM butyrylthiocholine iodide as substrate in the Karnovsky and Roots staining procedure.47
Enzyme activity assays
To measure (−)-cocaine and benzoic acid, the product of (−)-cocaine hydrolysis catalyzed by BChE, we used sensitive radiometric assays based on toluene extraction of [3H](−)-cocaine labeled on its benzene ring.17 In brief, to initiate the enzymatic reaction, 100 nCi of [3H](−)cocaine was mixed with the enzyme. The enzymatic reactions proceeded at room temperature (25°C) with varying concentrations of (−)-cocaine. The reactions were stopped by adding 200 μl of 0.1 M HCl, which neutralized the liberated benzoic acid whereas ensuring a positive charge on the residual (−)-cocaine. [3H]Benzoic acid was extracted by 1 ml of toluene and measured by scintillation counting. Finally, the measured (−)-cocaine concentration-dependent radiometric data were analyzed by using the standard Michaelis-Menten kinetics.48
In vivo tests
Male rats (~300 g) were obtained from Harlan Sprague-Dawley Inc. (Indianapolis, IN) and were housed in groups of 3 per cage. All rats were allowed ad libitum access to food and water, and were maintained on a 12-h light-dark cycle with lights on at 8 AM in a room kept at a temperature of 21–22°C. Each rat was used only once. Experiments were performed in the same colony room in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. The experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky.
Purified BChE mutants were injected intravenously (i.v.) to rats (0.15 mg/kg). Following anesthesia by isoflurane, prior to the enzyme injection, a blood sample was taken as the control. For each rat, following the i.v. injection of the enzyme, ~30 to 50 μl blood sample was taken at 2, 15, and 30 min, and 1, 2, 3, 5, 8, 12, 24, 48, and 72 hr from the saphenous veins (punctured at each time point). Plasma isolated by centrifugation (5000 g, 15 min) from the drawn blood sample will be used for analysis using the in vitro enzyme activity assays mentioned above. The time-dependent concentrations of the enzyme in plasma were fitted to a double-exponential equation described by Kronman.49
Results and Discussion
BChE mutant design: Insights from molecular modeling
Our goal of the present study was to design new BChE mutants that have a prolonged biological half-life without a significant change in the high catalytic activity of E12-7 against (−)cocaine. Our new design strategy relies on the MD-simulated dynamic structure of the BChE dimer and the idea that the BChE monomers can be covalently bonded to form covalent dimers by introducing intermolecular disulfide bond(s). So, we have carried out a long MD simulation (50 ns) on the BChE dimer structure in order to obtain a dynamically stable BChE dimer structure. To search for appropriate mutational sites to introduce the cross-subunit disulfide bond(s), a self-developed script was used to scan the key internulcear distances between the Cα atoms of the residues on the dimer interface from the snapshots extracted from MD trajectory. Essentially, each pair of residues from different subunits was evaluated for the simulated Cα–Cα distance. If the simulated Cα–Cα distance was within 7 Å, the pair of the residues would be checked manually for further evaluation of the detailed interactions. The most hopeful pairs of residues may be mutated to cysteines for introducing possible cross-subunit disulfide bond(s).
As seen in Table 1, the detailed analysis of the MD trajectory predicted that mutations F364C/M532C, V377C/A516C, and N535C may introduce the desirable cross-subunit disulfide bond(s). Depicted in Figure 1 are the simulated time-dependent Cα–Cα distances for these pairs of the residues. Depicted in Figure 2(A) is the MD-simulated BChE dimer structure consisting of subunits a and b. Figure 2(B)–(G) shows the details of the MD-simulated BChE dimer structure concerning these important pairs of residues. The interface is mainly composed of 4 α helices (A516a – M532a, E363a – Y373a, A516b – M532b, and E363a – Y373b) and several loop parts. F364 is located on one α helix (E363a – Y373a), M532 is located on the end of one α helix (A516b – M532b) as shown in Figure 2(B) and (C). V377 is located on the loop part of the interface. The location of A516 is similar to that of M532. Both A516 and M532 are located on the same α helix (A516 – M532), with A516 being on the head and M532 being on the end of the α helix.
Table 1.
The Cα-Cα distances between key residues in various dimer/tetramer BChE structures obtained from the modeling/simulation.
| Cα-Cα distance (Å) | F364a-M532bd | F364b-M532a | V377a-A516b | V377b-A516a | N535a-N535b | |
|---|---|---|---|---|---|---|
| BChE structure | ||||||
| BChE model Aa | 9.02 | 9.31 | 5.83 | 6.20 | N/A | |
| BChE model Bb | 9.98 | 10.29 | 8.15 | 8.82 | 9.07 | |
| MD-simulated BChE structure (50 ns MD)c | Maximum | 10.71 | 8.08 | 15.70 | 10.47 | 10.84 |
| Minimum | 5.33 | 4.42 | 4.71 | 5.56 | 4.53 | |
| Average | 7.93 | 5.61 | 7.73 | 6.89 | 6.83 | |
A simple homology model of BChE tetramer obtained from the homology modeling using the X-ray crystal structure of Electrophorus electricus AChE (EeAChE) tetramer (PDB ID: 1C2O)50 as a template. Because the simple homology modeling was based on the sequence alignment only (without any energy minimization or MD simulation on the backbone), the Cα-Cα distances between F364 and M532 in the BChE tetramer are simply the corresponding Cα-Cα distances between L373 and A542 in the EeAChE tetramer, and the Cα-Cα distances between V377 and A516 in the BChE tetramer are simply the corresponding Cα-Cα distances between L386 and A526 in the EeAChE tetramer. The sequence alignment indicates that N535 in BChE corresponds to T545 in EeAChE which is not available missing in the X-ray crystal structure.
BChE tetramer model obtained from the combined use of the X-ray crystal structures of the EeAChE tetramer (PDB ID: 1C2O) and the BChE monomer (PDB ID: 1P0P)9; each subunit of the EeAChE tetramer was superimposed with the BChE monomer38 in order to obtain the BChE tetramer (with the backbone frozen during the energy minimization and MD simulation).
Fully relaxed MD simulation of the BChE dimer structure starting from the BChE model B. The maximum, minimum, and average values of the Cα-Cα distances refer to the stable MD trajectory (10 to 50 ns).
Letters a and b after the residue numbers refer to subunits a and b, respectively.
Figure 1.
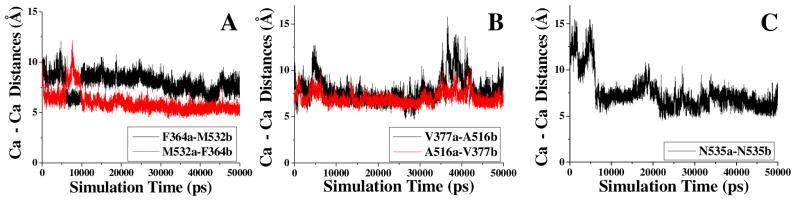
The time-dependent Cα–Cα distances between key residues from different subunits (a and b) in the MD-simulated BChE dimer structure: (A) F364 and M532; (B) V377 and A516; and (C) N535.
Figure 2.

(A) The modeled BChE dimer structure (subunits a and b). (B) Key residues F364 (a/b) and M532 (a/b) on the dimer interface. (C) Cross-subunit disulfide bonds formed on the interface of E364–532 by C364a-C532b and C532a-C364b. (D) Key residues V377 (a/b) and A516 (a/b) in the dimer interface. (E) Cross-subunit disulfide bonds formed on the interface of E377–516 by C377a-C516b and C516a-C377b. (F) Key residues N535 (a/b) on the dimer interface. (G) Cross-subunit disulfide bond formed on the interface of E535 by C535a-C535b. Different subunits are shown in different colors: subunit a in green and subunit b in orange. The active-site residues are shown in pink. Key residues are colored in blue and red using the ball-and-stick model.
According to the locations of these residues, it is possible to introduce a pair of cross-subunit disulfide bonds (C364a-C532b and C532a-C364b) through the F364C/M532C mutations on E12-7. The designed new mutant, i.e. the A199S/F227A/S287G/A328W/Y332G/F364C/M532C mutant (denoted as E364–532 for convenience), may have a pair of cross-subunit disulfide bonds: one between C364 of subunit a (C364a) and C532 of subunit b (C532b), and the other between C532 of subunit a (C532a) and C364 of subunit b (C364b). Similarly, the V377C/A516C mutations may also introduce a pair of cross-subunit disulfide bonds (C377a-C516b and C516a-C377b), and the designed new mutant, i.e. the A199S/F227A/S287G/A328W/Y332G/V377C/A516C mutant, is denoted as E377–516 for convenience. In addition, we noted in the simulated E12-7 structure that N535 was right in the middle of the interface, so that N535a and N535b from the two subunits are close to each other. For this reason, a single mutation on N535 (i.e. the N535C mutation) may introduce a single cross-subunit disulfide bond (C535a-C535b). The designed new mutant, i.e. the A199S/F227A/S287G/A328W/Y332G/N535C mutant, is denoted as E535 for convenience. It is also interesting to note that residues V377, M532, and N535 are either on the loop or next to the loop, implying that the positions of these residues have some flexibility which may help to form the desirable cross-subunit disulfide bonds.
As noted above, our prediction of the possible cross-subunit disulfide bonds was based on the MD-simulated dynamically stable BChE dimer structure. For comparison, we also examined two static models of the BChE tetramer structures. One was a simple model of BChE tetramer (BChE model A in Table 1) obtained from the homology modeling using the X-ray crystal structure of Electrophorus electricus AChE (EeAChE) tetramer (PDB ID: 1C2O)50 as a template. The other one was a BChE tetramer model (BChE model B in Table 1)38 obtained from the combined use of the X-ray crystal structures of the EeAChE tetramer (PDB ID: 1C2O) and the BChE monomer (PDB ID: 1P0P).9 As seen in Table 1, based on BChE model A, we could only predict the possible cross-subunit disulfide bonds that can be introduced by the V377C/A516C mutations. As seen in Table 1, based on BChE model B, we could not predict any of the aforementioned cross-subunit disulfide bonds.
Oligomeric forms of the proteins
Based on the insights from the modeling, we carried out in vitro experimental tests, including site-directed mutagenesis, protein expression and purification, on the E12-7, E364–532, E377–516, and E535 under the same experimental conditions. Depicted in Figure 3 is the nondenaturing gel (8%) stained for the BChE activity. As shown in Figure 3, E12-7 exists in a mixture of the monomer (~70%), dimer (~10%), and tetramer (~20%), whereas E364–532, E377–516, and E535 only exist in the dimer (~80%) and tetramer (~20%) without the monomer at all. The data shown in Figure 3 indicate that the desirable cross-subunit disulfide bonds were likely formed in the designed enzymes E364–532, E377–516, and E535, and that formation of the cross-subunit disulfide bond(s) can help to completely eliminate the monomer and increase the dimer concentration.
Figure 3.
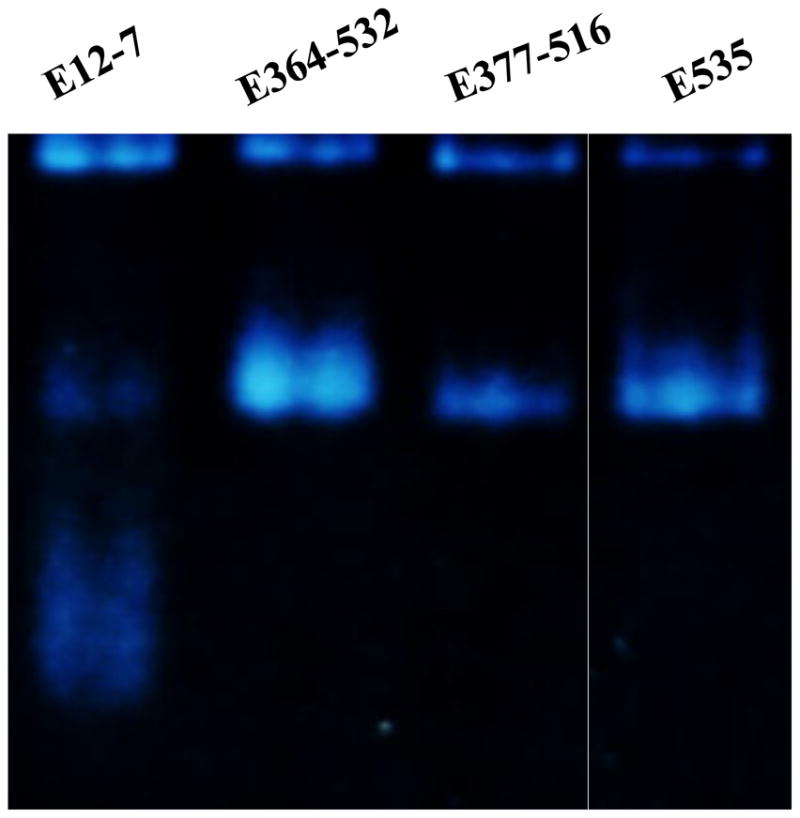
Nondenaturing gel (8%) stained for the BChE activity of (A) E12-7, (B) E364–532, (C) E377–516, and (D) E535. The four lanes came from the same gel where lane B was not adjacent to lane C. The gel was run with the constant current of 8 mA at 4°C overnight. The gel was stained for the BChE activity with butyrylthiocholine iodide as substrate at room temperature for 1 to 4 hr until the protein bands with the enzymatic activity were clearly identified.
Catalytic activity against (−)-cocaine
Knowing the effects of the designed mutations on the distribution of the oligomeric forms of the enzyme, we further carried out the in vitro enzyme activity assays to determine the effects of the same mutations on the catalytic activity of the enzyme against (−)-cocaine. To minimize the possible systematic experimental errors of in vitro kinetic analysis, we expressed the enzymes and performed kinetic studies on E12-7 and the new mutants (E364–532, E377–516 and E535) at the same time under the same experimental conditions, and compared the catalytic activity of the new mutants to that of E12-7 against (−)-cocaine. Depicted in Figure 4 are the kinetic data, and summarized in Table 2 are the determined kinetic parameters. As summarized in Table 2, the catalytic parameters of the designed new BChE mutants are very close to or essentially the same as corresponding catalytic parameters of E12-7 against (−)-cocaine. In terms of the KM values, only E377–516 has a slightly larger KM value (3.8 μM) compared to that (3.1 μM) of E12-7. Concerning the kcat values, E535 and E12-7 have a very similar kcat value (5700 min−1 for E12-7 and 5770 min−1 for E535) with a negligible difference (70 min−1 which might be within the possible experimental errors). The kcat values of E364–532 and E377–516 are slightly lower than that of E12-7. So, we may say that the rationally designed mutations have remarkably changed the distribution of the oligomeric forms of the enzyme without significantly affecting the high catalytic activity of E12-7 against (−)-cocaine.
Figure 4.

Kinetic data for the hydrolysis of (−)-cocaine catalyzed by BChE mutants: (A) E12-7; (B) E364–532; (C) E377–516; and (D) E535. The reaction rates were determined by using a sensitive radiometric assays based on toluene extraction of [3H](−)-cocaine labeled on its benzene ring. All of the enzyme activity tests were performed in triplicate (n=3), and the data are present with the error bars in standard deviation (SD).
Table 2.
Kinetic parameters determined for (−)-cocaine hydrolysis catalyzed by the BChE mutants.
| Enzyme | KM (μM) | kcat (min−1) | kcat/KM (M−1 min−1) |
|---|---|---|---|
| E12-7 | 3.1±0.2 | 5700±107 | 1.8×109 |
| E364–532 | 3.1±0.2 | 4650±70 | 1.5×109 |
| E377–516 | 3.8±0.2 | 4980±64 | 1.3×109 |
| E535 | 3.1±0.2 | 5770±101 | 1.9×109 |
Biological half-life
Based on the encouraging in vitro data discussed above, we further tested E12-7, E364–532, E377–516, and E535 in vivo for their pharmacokinetic in rats. First of all, five rats (n=5) were tested for E12-7 to determine its biological half-life as the standard reference. For comparison, the other three enzymes (E364–532, E377–516, and E535) were also tested for their biological half-lives in rats. Depicted in Figure 5 are the time-dependent average concentrations of the enzymes in plasma after the i.v. injection of the enzymes. It has been well-known that measured time-dependent concentrations of the active enzyme [E] in plasma follows a double exponential equation, i.e. [E] = Aexp(−α1t) + Bexp(−α2t), which accounts for both the enzyme distribution process (the fast phase, associated with α1) and elimination process (the slow phase, associated with α2). The elimination half-life is also known as the biological half-life. Analysis of the time courses depicted in Figure 5 revealed the biological half-lives of the enzymes. The determined biological half-life of E12-7 was 7.3 hr in rats. With the cross-subunit disulfide bond(s), the rationally designed new mutants all had a significantly prolonged biological half-life which was 13.2, 13.1, and 13.3 hr for E364–532, E377–516, and E535, respectively. So, the designed new enzymes (E364–532, E377–516, and E535) indeed had a significantly prolonged biological half-life in rats without significantly changing the high catalytic activity of E12-7 against (−)-cocaine.
Figure 5.

Time-dependent concentrations of the active enzyme in rat plasma (n=5) after the i.v. injection of the enzyme at a dose of 0.15 mg/kg: (A) E12-7, (B) E364–532, (C) E377–516, and (D) E535. The data are present with the error bars (SD).
The in vivo data suggest that the new mutants, particularly E535, could be more valuable than E12-7 in the future further development of a cocaine abuse treatment. It has been known that, for a given therapeutically useful enzyme, the biological half-life in humans is usually much longer than that in rats. As noted above, the biological half-life of the native human BChE is ~24 hours in mice and ~7 to 12 days in humans. Assuming that the biological half-life of a BChE mutant in humans is at least 7-fold longer than that in mice and rats, then we may theoretically predict the biological half-lives of the BChE mutants in humans from their biological half-lives in rats. Thus, E12-7 (with a biological half-life of ~7 hr in rats) is expected to have a biological half-life of at least two days in humans, and the newly designed enzymes (E364–532, E377–516, and E535, all with a biological half-life of ~13 hr in rats) are expected to have a biological half-life of about four days or longer in humans.
Conclusion
Three new mutants of human BChE (E364–532, E377–516, and E535) have been designed to have a more stable dimer structure with the desirable cross-subunit disulfide bond(s) and, therefore, a different distribution of the oligomeric forms and a prolonged biological half-life. In vitro and in vivo experimental studies have demonstrated that the rationally designed new BChE mutants, i.e. E364–532, E377–516, and E535, indeed have a remarkably different distribution of the oligomeric forms (dimer and tetramer) without monomer at all and a significantly prolonged biological half-life in rats. The designed new mutations (F364C/M532C, V377C/A516C, and N535C) have successfully extended the biological half-life of E12-7 in rats from ~7 hr to ~13 hr, without significantly changing the high catalytic activity of E12-7 against (−)-cocaine. The newly designed and discovered BChE mutants, particularly E535, could be more valuable than E12-7 in the future development of a novel enzyme therapy for cocaine abuse. The encouraging outcomes of the present study suggest that the structure-and-mechanism-based design and integrated computational-experimental approach are promising for rational protein design, discovery, and development. As well-known, most recombinant proteins have very short biological half-lives and, thus, it is highly desirable to prolong their biological half-lives, particularly for therapeutic development. The general protein design strategy and approach may also be valuable for engineering other proteins.
Research Highlights.
We design mutants of a cocaine hydrolase with cross-subunit disulfide bond(s).
The designed mutations stabilize the dimer structure without changing the activity.
The designed mutations extend the biological half-life of the cocaine hydrolase.
Acknowledgments
This work was supported by the NIH (grants R01 DA035552, R01 DA032910, R01 DA013930, and R01 DA025100 to Zhan). The authors also acknowledge the Computer Center at the University of Kentucky for supercomputing time on a Dell X-series Cluster with 384 nodes or 4,768 processors. This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.UNODC. World Drug Report 2010. United Nations Publication; 2010. Sales No. E.10.XI.13. [Google Scholar]
- 2.Karila L, Gorelick D, Weinstein A, Noble F, Benyamina A, Coscas S, Blecha L, Lowenstein W, Martinot JL, Reynaud M, Lépine JP. The International Journal of Neuropsychopharmacology. 2008;11:425–438. doi: 10.1017/S1461145707008097. [DOI] [PubMed] [Google Scholar]
- 3.Xi ZX, Gardner LE. Current Drug Abuse Review. 2008;1:303–327. doi: 10.2174/1874473710801030303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landry DW, Zhao K, Yang GX, Glickman M, Georgiadis TM. Science. 1993;259:1899–1901. doi: 10.1126/science.8456315. [DOI] [PubMed] [Google Scholar]
- 5.Kamendulis LM, Brzezinski MR, Pindel EV, Bosron WF, Dean RA. Journal of Pharmacology and Experimental Therapeutics. 1996;279:713–717. [PubMed] [Google Scholar]
- 6.Carrera MRA, Kaufmann GF, Mee JM, Meijler MM, Koob GF, Janda KD. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10416–10421. doi: 10.1073/pnas.0403795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meijler MM, Kaufmann GF, Qi L, Mee JM, Coyle AR, Moss JA, Wirsching P, Matsushita M, Janda KD. Journal of the American Chemical Society. 2005;127:2477–2484. doi: 10.1021/ja043935e. [DOI] [PubMed] [Google Scholar]
- 8.Zhan CG, Deng SX, Skiba JG, Hayes BA, Tschampel SM, Shields GC, Landry DW. Journal of Computational Chemistry. 2005;26:980–986. doi: 10.1002/jcc.20241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. Journal of Biological Chemistry. 2003;278:41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- 10.Zheng F, Zhan CG. Future Medicinal Chemistry. 2012;4:125–128. doi: 10.4155/fmc.11.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun H, Pang YP, Lockridge O, Brimijoin S. Molecular Pharmacology. 2002;62:220–224. doi: 10.1124/mol.62.2.220. [DOI] [PubMed] [Google Scholar]
- 12.Hamza A, Cho H, Tai HH, Zhan CG. The Journal of Physical Chemistry B. 2005;109:4776–4782. doi: 10.1021/jp0447136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Y, Gao D, Yang W, Cho H, Yang G, Tai HH, Zhan CG. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16656–16661. doi: 10.1073/pnas.0507332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao D, Cho H, Yang W, Pan Y, Yang G, Tai HH, Zhan CG. Angewandte Chemie. 2006;118:669–673. doi: 10.1002/anie.200503025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan Y, Gao D, Yang W, Cho H, Zhan CG. Journal of the American Chemical Society. 2007;129:13537–13543. doi: 10.1021/ja073724k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng F, Yang W, Ko MC, Liu J, Cho H, Gao D, Tong M, Tai HH, Woods JH, Zhan CG. Journal of the American Chemical Society. 2008;130:12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun H, Shen ML, Pang YP, Lockridge O, Brimijoin S. Journal of Pharmacology and Experimental Therapeutics. 2002;302:710–716. doi: 10.1124/jpet.302.2.710. [DOI] [PubMed] [Google Scholar]
- 18.Duysen EG, Bartels CF, Lockridge O. Journal of Pharmacology and Experimental Therapeutics. 2002;302:751–758. doi: 10.1124/jpet.102.033746. [DOI] [PubMed] [Google Scholar]
- 19.Blong RM, Bedows E, Lockridge O. Biochem J. 1997;327:747–757. doi: 10.1042/bj3270747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lockridge O, Bartels CF, Vaughan TA, Wong CK, Norton SE, Johnson LL. Journal of Biological Chemistry. 1987;262:549–557. [PubMed] [Google Scholar]
- 21.Geyer BC, Kannan L, Garnaud PE, Broomfield CA, Cadieux CL, Cherni I, Hodgins SM, Kasten SA, Kelley K, Kilbourne J, Oliver ZP, Otto TC, Puffenberger I, Reeves TE, Robbins N, Woods RR, Soreq H, Lenz DE, Cerasoli DM, Mor TS. Proceedings of the National Academy of Sciences. 2010;107:20251–20256. doi: 10.1073/pnas.1009021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang YJ, Huang Y, Baldassarre H, Wang B, Lazaris A, Leduc M, Bilodeau AS, Bellemare A, Côté M, Herskovits P, Touati M, Turcotte C, Valeanu L, Lemée N, Wilgus H, Bégin I, Bhatia B, Rao K, Neveu N, Brochu E, Pierson J, Hockley DK, Cerasoli DM, Lenz DE, Karatzas CN, Langermann S. Proceedings of the National Academy of Sciences. 2007;104:13603–13608. doi: 10.1073/pnas.0702756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saxena A, Ashani Y, Raveh L, Stevenson D, Patel T, Doctor BP. Molecular Pharmacology. 1998;53:112–122. doi: 10.1124/mol.53.1.112. [DOI] [PubMed] [Google Scholar]
- 24.Sundy J, Ganson N, Kelly S, Scarlett E, Rehrig C, Huang W, Hershfield M. Arthritis Rheum. 2007;56:1021–1028. doi: 10.1002/art.22403. [DOI] [PubMed] [Google Scholar]
- 25.Yue C, Huang W, Alton M, Maroli A, Waltrip R, Wright D, Marco M. J Clin Pharmacol. 2008;48:708–718. doi: 10.1177/0091270008317589. [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Yuan Y, Zhan CG, Liao F. Drug Development Research. 2012;73:66–72. doi: 10.1002/ddr.20493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altamirano CV, Lockridge O. Biochemistry. 1999;38:13414–13422. doi: 10.1021/bi991475+. [DOI] [PubMed] [Google Scholar]
- 28.Altamirano CV, Bartels CF, Lockridge O. Journal of Neurochemistry. 2000;74:869–877. doi: 10.1046/j.1471-4159.2000.740869.x. [DOI] [PubMed] [Google Scholar]
- 29.Biberoglu K, Schopfer LM, Tacal O, Lockridge O. FEBS Journal. 2012;279:3844–3858. doi: 10.1111/j.1742-4658.2012.08744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parikh K, Duysen EG, Snow B, Jensen NS, Manne V, Lockridge O, Chilukuri N. Journal of Pharmacology and Experimental Therapeutics. 2011;337:92–101. doi: 10.1124/jpet.110.175646. [DOI] [PubMed] [Google Scholar]
- 31.Li H, Schopfer LM, Masson P, Lockridge O. Biochem J. 2008;411:425–432. doi: 10.1042/BJ20071551. [DOI] [PubMed] [Google Scholar]
- 32.Korkegian A, Black ME, Baker D, Stoddard BL. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao D, Narasimhan DL, Macdonald J, Ko MC, Landry DW, Woods JH, Sunahara RK, Zhan CG. Mol Pharmacol. 2009;75:318–323. doi: 10.1124/mol.108.049486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fei B, Xu H, Cao Y, Ma S, Guo H, Song T, Qiao D, Cao Y. J Ind Microbiol Biotechnol. 2013;40:457–464. doi: 10.1007/s10295-013-1260-z. [DOI] [PubMed] [Google Scholar]
- 35.Joo JC, Pack SP, Kim YH, Yoo YJ. J Biotechnol. 2011;151:56–65. doi: 10.1016/j.jbiotec.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Kim SJ, Lee JA, Joo JC, Yoo YJ, Kim YH, Song BK. Biotechnol Prog. 2010;26:1038–1046. doi: 10.1002/btpr.408. [DOI] [PubMed] [Google Scholar]
- 37.Lockridge O, Adkins S, La Du BN. Journal of Biological Chemistry. 1987;262:12945–12952. [PubMed] [Google Scholar]
- 38.Pan Y, Muzyka JL, Zhan CG. The Journal of Physical Chemistry B. 2009;113:6543–6552. doi: 10.1021/jp8114995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Case DA, Darden TA, Cheatham TE, CLS, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. Amber. Vol. 9. University of California; San Francisco: 2006. [Google Scholar]
- 40.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. Journal of Computational Chemistry. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. Journal of Computational Chemistry. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. The Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- 43.Darden T, York D, Pedersen L. The Journal of Chemical Physics. 1993;98:10089–10092. [Google Scholar]
- 44.Ryckaert JP, Ciccotti G, Berendsen HJC. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- 45.Braman J, Papworth C, Greener A. The Nucleic Acid Protocols Handbook. 2000. pp. 835–844. [Google Scholar]
- 46.Masson P, Xie W, Froment MT, Levitsky V, Fortier PL, Albaret C, Lockridge O. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology. 1999;1433:281–293. doi: 10.1016/s0167-4838(99)00115-6. [DOI] [PubMed] [Google Scholar]
- 47.Karnovsky MJ, Roots L. Journal of Histochemistry & Cytochemistry. 1964;12:219–221. doi: 10.1177/12.3.219. [DOI] [PubMed] [Google Scholar]
- 48.Pan Y, Gao D, Zhan CG. Journal of the American Chemical Society. 2008;130:5140–5149. doi: 10.1021/ja077972s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kronman C, Chitlaru T, Elhanany E, Velan B, Shafferman A. Journal of Biological Chemistry. 2000;275:29488–29502. doi: 10.1074/jbc.M004298200. [DOI] [PubMed] [Google Scholar]
- 50.Bourne Y, Grassi J, Bougis PE, Marchot P. Journal of Biological Chemistry. 1999;274:30370–30376. doi: 10.1074/jbc.274.43.30370. [DOI] [PubMed] [Google Scholar]