

Abstract
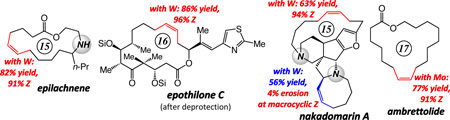
The first broadly applicable set of protocols for efficient and highly Z-selective formation of macrocyclic disubstituted alkenes through catalytic ring-closing metathesis (RCM) is described. Cyclizations are performed in the presence of 1.2–7.5 mol % of a Mo- or W-based mono-aryloxide pyrrolide (MAP) complex at 22 °C and typically proceed to complete conversion within two hours. The utility of the catalytic strategy is demonstrated by stereoselective synthesis of representative macrocyclic alkenes, including natural products yuzu lactone (13-membered ring: 73% Z) epilachnene (15-membered ring: 91% Z), ambrettolide (17-membered ring: 91% Z), an advanced precursor to epothilones C and A (16-membered ring: up to 97% Z) and nakadomarin A (polycyclic 15-membered ring: up to 97% Z). We demonstrate the complementary nature of the Mo-based catalysts, which deliver high activity but can be more prone to causing post-RCM stereoisomerization, versus W-based variants, which furnish lower activity but are less inclined towards causing loss of kinetic Z selectivity; a number of catalytic Z-selective cases are provided to elucidate which catalyst class is best suited for which substrate and particular type of alkene RCM process. Mechanistic models that rationalize the origin and the trends in Z selectivity as a function of alterations in the catalyst structure (i.e., Mo vs W and different imido and aryloxide or alkoxide ligands) are provided; we show that reaction time can be critical in retaining the Z selectivity attained not only with MAP complexes but with the original Mo-based bis-alkoxides as well. The W-based catalysts are sufficiently stable to be manipulated in air even with humidity levels of up to 80%; the catalytic Z-selective cyclizations can be performed on gram scale with complex molecule starting materials.
Introduction
Few transformations have had as palpable an impact on organic chemistry as catalytic alkene ring-closing metathesis (RCM).1 During the last two decades, the routes to countless molecules, natural products or otherwise, with one or more cyclic moieties have contained an RCM reaction that generates a small, medium or large ring alkene.2 There are several reasons for such strong predilection. Alkenes are relatively robust and yet can be readily modified in a variety of ways once the cyclization products are in hand; olefins do not typically require protection and unmasking in the course of a synthesis route; by comparison, and as an example, the alcohol and carboxylic acid (or equivalents thereof) required to bring about a lactonization must often be differentiated from other polar functional groups at the time of the ring closure. Reliability is another factor – an attribute that receives increasing credence with every new total synthesis that utilizes this remarkable set of processes.
One arena where catalytic RCM has played a prominent role is in connection with the synthesis of macrocyclic alkenes, a structural motif found in an array of biologically active molecules.3 It was the pioneering disclosures by Villemin,4 and Tsuji,5 which appeared in 1980 and concerned non-stereoselective macrocyclic RCM reactions promoted by (ill-defined) W-based complexes, that first made evident the potential value of catalytic RCM. It is, ironically, in the same arena that a glaring deficiency in catalytic alkene metathesis has persisted for decades: the absence of catalysts that reliably deliver kinetic control of stereoselectivity in the formation of large ring olefins. Dependence on substrate control – attaining stereoselectivity that largely originates from the thermodynamic preference for one alkene isomer – does not typically lead to the selective formation of the targeted isomer; often, E and Z olefins are generated in near equal ratios or, even less favorably, it is the undesired product that is obtained predominantly. A catalyst class that furnishes dependable control of the stereochemical outcome in a macrocyclization would enhance the utility of this elite collection of catalytic processes. In spite of such a shortcoming, it is catalytic RCM that is commonly viewed as perhaps the most suitable approach to macrocyclic ring formation; such is true even in the precarious situations where the cyclization must be implemented after a multi-step sequence is expended to obtain the requisite precursor – when a non-selective RCM can reduce the yield of the desired alkene isomer and the entire synthesis route by 50% or more (see below for examples).
Development of effective complexes that preferentially provide the often higher energy Z alkenes constitutes a hard challenge. One principal complication arises from the reversible nature of catalytic olefin metathesis, which threatens the long-term survival of the energetically less favored Z alkene. There are, therefore, two distinct but equally significant requirements for successful design of efficient Z-selective catalysts that promote macrocyclic RCM: not only must such species facilitate the formation of the typically higher energy alkene isomer with exceptional selectivity, they must then refrain from reacting with it to cause adventitious Z-to-E isomerization. The latter complication regarding chemoselectivity grows increasingly daunting as substrate conversion escalates and the concentration of the desired Z olefin product surpasses that of the starting terminal olefins.
Herein, we detail the outcome of our investigations regarding the development of the first class of catalytic olefin metathesis reactions that provide high efficiency and Z selectivity in the synthesis of macrocyclic disubstituted alkenes.6 We show that an assortment of large-ring Z alkenes can be accessed through transformations promoted by Mo- or W-based mono-aryloxide pyrolide (MAP) complexes.7 The study utilizes the total synthesis of several macrocyclic natural products as the framework for underlining different subtleties of catalyst selection; cyclizations of starting materials ranging from sparsely to amply functionalized dienes have been examined, generating insight about the relationship between the optimal catalyst activity and nuances of substrate structure. Syntheses of yuzu lactone, epilachnene, ambrettolide, as well as anti-cancer and anti-bacterial agents epothilones A and C8 nakadomarin A9 (Chart 1) have been accomplished through catalytic Z-selective RCM reactions. In all cases, reactions with previously available catalysts either afford a near equal mixture of alkene isomers (≤65:35) or furnish the undesired E alkene predominantly. Our investigations indicate that the more conformationally mobile substrates frequently require Mo-based complexes; in contrast, with the relatively pre-organized unsaturated chains, use of the less active W catalysts is often preferable. We demonstrate that with the relatively robust W-based alkylidenes, which can be handled in air without rigorous exclusion of air and moisture, gram-scale RCM reaction of a structurally complex diene can be carried out efficiently and with effective kinetic control of stereoselectivity. We present data regarding the basis of the high chemoselectivity furnished by W-based catalysts; such complexes can catalyze RCM but exhibit minimal tendency to react with the resulting and relatively strained macrocyclic Z olefin and cause isomerization to the E isomer. The latter attribute is highlighted through a total synthesis of nakadomarin A, where the RCM process used to install the cyclooctene moiety proceeds without significant erosion of Z:E ratio at the sensitive fifteen-membered ring alkene site.
chart 1.

Natural Products with a Z Macrocyclic Alkene Examined in this Study.
Results and Discussion
1. Mechanistic Models for Z Selectivity in Macrocyclic RCM Promoted by MAP Complexes
We begin by an analysis of the origin of Z selectivity in the projected macrocyclic RCM reactions and some of the structural features that lead to effective catalysis by Mo- and W-based MAP complexes. Such an examination helps elucidate the nature of some of the difficulties faced in the course of development of the present class of transformations.
Based on the Z-selective ring-opening/cross-metathesis10 as well as cross-metathesis reactions studied before,11 we projected that control of olefin stereochemistry originates from sufficient size differential between the imido (blue; Figure 1) and aryloxide ligands of the catalyst. Thus, as shown in Figure 1, the bulky and freely rotating aryloxide can force the alkene, tethered to the metal-alkylidene, to coordinate with the metal center so that the resulting metallacyclobutane substituents are oriented towards the smaller imido unit (cf. III, Figure 1). Productive decomposition of III would furnish alkene complex IV and subsequent release of macrocyclic Z olefin produces methylidene complex V, which can react with another diene molecule to re-generate I/II.
Figure 1.

Key structural features of MAP alkylidenes responsible for high Z selectivity in macrocyclic RCM reactions.
Catalytic olefin metathesis is a rearrangement process and therefore inherently reversible;12 as depicted by the general pathway in Figure 2 (box), the kinetically generated Z alkene can be catalytically isomerized to the (often) lower energy E isomer. Consequently, the same factors that lead to high Z selectivity, make available a pathway for catalytic post-RCM isomerization. The steric repulsion between the alkylidene substituent and the sizeable aryloxide, shown in complex VI (Figure 2), can culminate in a strong preference for modes of reaction represented by I/II (Figure 1). It follows that re-association of an E macrocyclic alkene with the complex, as featured in VII (Figure 2), would be similarly unfavorable compared to the corresponding system containing the Z isomer (cf. IV, Figure 1). That is, the Z olefin products are more susceptible towards undergoing ring-opening reactions that lead to Z-to-E isomerization (cf. Figure 2), while any E alkene formed is less prone to re-enter the catalytic cycle. The above considerations further emphasize the central challenge in designing olefin metathesis catalysts that efficiently and selectively deliver the higher energy stereoisomers: the metal complex must be sufficiently active and discriminating to generate the Z alkene with a strong preference but not too potent so that the fragile kinetic selectivity can be preserved.
Figure 2.

A general scheme for catalytic post-RCM isomerization; structural attributes that result in high Z selectivity can also lead to facile post-RCM isomerization.
2. Z-Selective Macrocyclic RCM of Sparsely Substituted Dienes
We commenced by examining reactions of several relatively unfunctionalized diene precursors as the means to investigate several fundamental aspects of the cyclization process. The influence of ring size and different types of linking units (e.g., carboxylic esters or amides) on the efficiency and stereoselectivity of macrocyclization reactions would thus be probed. We surmised that the requirements for an optimal catalyst for a more conformationally mobile substrate versus one that is more restricted might be distinct; accordingly, such studies, together with subsequent investigations involving the more highly functionalized dienes (i.e., precursors to epothilone C and nakadomarin A), would help delineate factors regarding the suitability of different catalyst classes and various substrate types.
2.1. Stereoselective synthesis of epilachnene
We chose the fifteen-membered ring macrolactone epilachnene (cf. Scheme 1),13 secreted by the Mexican bean beetle as part of its pupae defense mechanism,14 to serve as the initial platform for our investigations. The basis for this selection was the fact that the aza-macrolide natural product has been synthesized through catalytic diene RCM as well as alkyne RCM/catalytic hydrogenation strategies. Any limitation in the state-of-the-art in connection with either synthesis route could therefore be evaluated and addressed. A catalytic stereoselective RCM furnishing epilachnene would represent a more efficient approach than those formerly outlined,15 and would likely be applicable to the synthesis of an assortment of other sparingly substituted macrocycles.
scheme 1.

Previous Approaches to Epilachnene Involving Catalytic Ring-Closing Metathesisa
a As noted, some findings have been previously reported; other data are from the present investigation.
2.1.1. Previous syntheses of epilachnene
The results of extant investigations regarding synthesis of epilachene are summarized in Scheme 3. In the presence of Mo-based bis-alkoxide 1,16 Ru-based bis-phosphines 2a17 and 2b18 or those that bear an N-heterocyclic carbene (2c–d),19,20 complexes commonly used in olefin metathesis, diene 3 is preferentially converted to the undesired E isomer (67–75%). To address this problem, as shown in Scheme 1, an alternative stereoselective route, involving W- and Mo-catalyzed alkyne metathesis,21 was introduced.22 Catalytic RCM with the diyne substrate delivers the macrocyclic alkyne in approximately 70% yield; subsequent partial hydrogenation, promoted by Lindlar’s catalyst (i.e., deposited Pd containing Pb salts) leads to the formation of the Z olefin. The high stereoselectivity of the latter indirect route notwithstanding, several attributes detract from it:
The basic amine must be masked in order to achieve high catalyst activity, leading to the addition of two steps to the synthesis route (protection and removal of the Fmoc group); the deprotection step was reported to proceed in 62% yield (Scheme 1).22
Preparation of methyl-substituted internal alkyne, typically involving deprotonation of the corresponding acetylene and alkylation with iodomethane, is needed (vs terminal C–C triple bonds); otherwise, oligomerization processes dominate.23
In general, synthesis of alkyne containing substrates tends to be less concise than that of dienes. Indeed, synthesis of diyne precursor to 4 (before amine protection) was accomplished in eleven steps (longest linear sequence of eight steps);22 in contrast, diene 3 is prepared in four steps from inexpensive materials. Such discrepancy is due to several factors, among which is the substantially larger number of inexpensive commercially available olefinic compounds. Alkynes are generally more reactive than alkenes (e.g., they can be partially hydrogenated) and carry a relatively acidic proton, which might necessitate precautions that can diminish synthesis efficiency; for instance, depronation/metylation of a terminal alkyne might require proper masking of other functional groups (e.g., an alcohol).
Catalytic alkyne RCM may need elevated temperatures (Scheme 1). More recent advances have led to the development of catalysts that do not call for the use of chlorinated solvents and/or an additive and operate at ambient temperature; rigorously dried 5 Å molecular sieves must be used to ensure high conversions.24
scheme 3.

Reactivity vs Z-Selectivity Furnished by W-Based Complex 13
2.1.2. Z-Selective diene RCM with MAP complexes
Treatment of unsaturated amine 3 with 1.2 mol %25 Mo-based adamantylimido alkylidene 6 or W-based metallacyclobutane arylimido complex 7 (Scheme 2) delivers epilachnene in 70% and 82% yield and 91% Z selectivity. The suitability of the aforementioned complexes was established through an initial screening of a simpler model system, details of which were disclosed in the preliminary account of this investigation.6 Several additional aspects of the above findings are noteworthy:
Metallacyclobutane 7 is sufficiently robust that it can be weighed in air and various manipulations can be performed in a fume hood with standard glassware. The W-catalyzed cyclization, performed at 0.1 gram scale, entailed handling of the W-based complex in air at nearly 80% humidity level.
Metallacyclobutane 726 is prepared by subjection of a solution of the corresponding neophylidene complex to an atmosphere of ethylene; this complex can be viewed as a more attractive option for catalyzing olefin metathesis reactions, compared to its more sterically congested alkylidene precursor, perhaps due to a faster rate of initiation [i.e., release of ethylene in solution affords the active methylidene (cf. V, Figure 1)].
The Z-selective synthesis of epilachnene by catalytic diene RCM does not demand protection and deprotection of the amine unit, underscoring the stability of the Mo- and W-based alkylidenes towards this commonly occurring class of functional groups (in contrast to the corresponding alkylidynes; cf. Scheme 1).
scheme 2.

A Practical Catalytic Method for Z-Selective Macrocyclic RCM
2.2. Stereoselective synthesis of yuzu lactone, ambrettolide and other relatively unfunctionalized Z-macrocyclic olefins
Macrocyclic esters or amides of different ring sizes can be synthesized by the use of Mo or W alkylidenes 6 and 7 with unprecedented Z selectivity and with efficiency levels should render the method of notable utility; relevant results are summarized in Table 1. The disparity between the percent conversion and yield is largely due to adventitious oligomerization (as judged by 1H NMR analysis).
Table 1.
Synthesis of Macrocyclic Esters and Amides through Catalytic Z-Selective RCMa
| entry | macrocyclic Z alkene | complex;b loading | condition | conv (%);c yield (%)d |
Z:Ec |
|---|---|---|---|---|---|
| 1 | 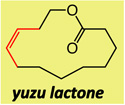 |
1; 5.0 mol % | ambient | 92; 30 | 17:83 |
| 2 | 2c; 5.0 mol % | ambient | 93; 46 | 15:85 | |
| 3 | 6; 3.0 mol % | 7.0 torr | 63; 49 | 69:31 | |
| 4 | 7; 5.0 mol % | 7.0 torr | 74; 46 | 73:27 | |
| 5 | 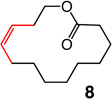 |
1; 5.0 mol % | ambient | 97; 67 | 7:93 |
| 6 | 2c; 5.0 mol % | ambient | 98; 74 | 7:93 | |
| 7 | 6; 3.0 mol % | 7.0 torr | 74; 50 | 80:20 | |
| 8 | 7; 5.0 mol % | 7.0 torr | 82; 54 | 82:18 | |
| 9 | 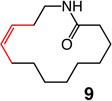 |
1; 5.0 mol % | ambient | 96; 56 | 21:79 |
| 10 | 2c; 5.0 mol % | ambient | 98; 68 | 15:85 | |
| 11 | 6; 3.0 mol % | 7.0 torr | 72; 50 | 95:5 | |
| 12 | 7; 5.0 mol % | 7.0 torr | <20; nd | nd | |
| 13 | 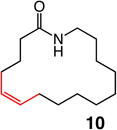 |
1; 5.0 mol % | ambient | 93; 53 | 56:44 |
| 14 | 2c; 5.0 mol % | ambient | 98; 60 | 66:34 | |
| 15 | 6; 3.0 mol % | 7.0 torr | 81; 69 | 93:7 | |
| 16 | 7; 5.0 mol % | 7.0 torr | <20; nd | nd | |
| 17 | 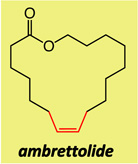 |
1; 5.0 mol % | ambient | 95; 65 | 23:77 |
| 18 | 2c; 5.0 mol % | ambient | 95; 61 | 24:76 | |
| 19 | 6; 3.0 mol % | 7.0 torr | 85:77 | 91:9 | |
| 20 | 7; 5.0 mol % | 7.0 torr | 90:78 | 88:12 |
Reactions were carried out in purified toluene (5.0 mM) at 22 °C for one h under an atmosphere of N2 or under vacuum, as noted; see the Supporting Information for details.
Complexes 1, 2c and 7 were prepared prior to use, whereas alkylidene 6 was synthesized in situ from the corresponding bis-pyrrolide and aryl alcohol, proceeding in ~60% yield (3.0 mol % effective catalyst loading). See the Supporting Information for details.
Conversion and Z:E ratios measured by analysis of 400 MHz 1H NMR spectra of unpurified mixtures; the variance of values are estimated to be ±2%.
Yield of isolated products (isomeric mixtures) after purification; the variance of values are estimated to be ±5%. nd = not determined.
Two of the macrocycles are natural products: the thirteen-membered camphor- and minty-smelling yuzu lactone (entries 1–4, Table 1), and seventeen-membered musk-odored ambrettolide (entries 17–20). For comparison, and to underline the uniqueness of the MAP complexes, the data regarding the RCM promoted by Mo alkylidene 1 and Ru-based carbene 2c are presented in Table 1; with the latter two complexes, in all cases, substantial amounts of the E alkenes are formed and, at times, with a significant preference (93% E in entries 5–6, Table 1). With Mo- and W-based alkylidenes 6 and 7, there is 69–73% Z selectivity in the formation of thirteen-membered yuzu lactone whereas a higher 93:7 Z:E ratio can be obtained with sixteen-membered ring 10 (entry 15), the identity of which was further confirmed through X-ray crystallography.27 The above-mentioned Z selectivity variations as a function of ring size suggest that a comparatively strained ring (e.g., yuzu lactone) undergoes ring-opening more readily (cf. Figures 1 and 2) and alkene isomerization proceeds at a faster rate. In this vein, when the RCM in the presence of complex 6 leading to yuzu lactone is analyzed after 10 minutes (20% conv), 82% Z alkene is found in the mixture (vs 69% conv after one hour). It can therefore be concluded that, at least in certain cases, post-RCM isomerization might be more efficient and therefore E selectivity higher when 1 is used (83% E, entry 1, Table 1) than when the same catalysts promote formation of the less strained 10 (44% E, entry 13).28 That there is effective catalyst-control in the formation of Z-8 is supported by calculations indicating that the E isomer is 1.9 kcal/mol lower in energy,27 predicting ~96% E selectivity; the latter findings compare favorably with 93:7 E:Z ratio in entries 5–6, and yet RCM with 6 or 7 provide 80–82% of the Z macrocyclic alkene (entries 7–8, Table 1).
3. Z-Selective Macrocyclic RCM en Route to Epothilone C
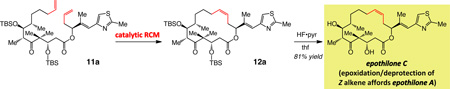
The next phase of our studies relates to examining the catalytic RCM reaction that have served as precursors to epothilones C and A.29 In such instances, lack of stereoselectivity in the RCM reactions is costly particularly since it arrives at the end of a multi-step route to the requisite diene;30 as reported previously29c and in our experience, the desired macrolactone is virtually inseparable from the E isomer. Furthermore, it has been demonstrated that olefin stereochemistry impacts the level of biological activity.31 Finally, access to the appropriate macrocyclic alkene precursors are required if the subsequent functionalizations are to proceed with the desired sense of stereochemical control (e.g., epoxidation29 or cyclopropanation32).
One goal of this segment of our studies was to showcase the practical utility of the catalytic RCM approach. A highly Z-selective RCM leading to epothilone A – particularly if performed on gram scale and in a practical manner – would bear notable implications regarding the efficiency with which macrocyclic natural products and their corresponding analogues, including those that are not accessible through fermentation procedures, can be accessed in meaningful quantities. Additionally, and as stated previously, the substrate for the cyclization is significantly more functionalized than those probed in the initial phase of our studies and can therefore be more conformationally rigid; accordingly, we thought it important to establish whether the requirements for optimal Z-selective macrocyclic RCM could prove to be distinct from those that can best effect cyclizations that furnish the less substituted macrocycles.
3.1. Effect of catalyst structure on Z selectivity
The RCM-based approach towards the sixteen-membered ring moiety of epothilone C has been investigated by several research groups;29 the desired Z olefin was formed as the minor isomer in most cases and in equal amounts to the undesired E alkene in others;30,30 the examples in entries 1–3 of Table 2 are representative (see below for a more detailed analysis of these earlier findings). As with epilachnene (cf. Scheme 1), the indirect route of utilizing catalytic alkyne RCM/partial hydrogenation strategy has been applied to this problem;33 similar analysis regarding the two strategies applies here as well.
Table 2.
Catalytic RCM for Stereoselective Total Syntheses of Epothilones C and Aa
 | |||||
|---|---|---|---|---|---|
| entry | complex; loadingb | conditions | time (h); temp (°C) | conv (%);c yield (%)d | Z:Ec |
| 1e | 1; 20 mol % | ambient; 1.0 mM (C6H6) | 1.0; 22 | >98; 86 | 33:67 |
| 2f | 2b; 10 mol % | ambient; 1.5 mM (CH2Cl2) | 20; 25 | >98; 85 | 54:46 |
| 3 | 2d; 5.0 mol % | ambient; 1.0 mM (C7H8) | 16; 40 | 96; nd | 34:66 |
| 4 | 6; 10 mol % | ambient; 1.0 mM (C6H6) | 1.5; 22 | 87; nd | 85:15 |
| 5 | 7; 10 mol % | ambient; 1.0 mM (C6H6) | 2.5; 22 | 72; nd | 79:21 |
| 6 | 13; 10 mol % | ambient; 1.0 mM (C6H6) | 2.5; 22 | 77; nd | 96:4 |
| 7 | 13; 10 mol % | 1.0 torr; 1.0 mM (C6H6) | 2.5; 22 | 98; 86 | 96:4 |
| 8 | 13; 5.0 mol % | 1.0 torr; 0.01 M (MeC6H5) | 2.0; 22 | 98; 86 | 96:4 |
| 9 | 13; 3.0 mol % | 1.0 torr; 0.05 M (MeC6H5) | 3.0; 22 | 97; 63 | 97:3 |
Reactions were carried out under an atmosphere of N2 or vacuum; see the Supporting Information for details.
Complex 2d, 7 and 13 were prepared prior to use, whereas alkylidene 6 was synthesized in situ from the corresponding bis-pyrrolide and aryl alcohol, proceeding in ~60% yield of the MAP complex (~3.0 mol % effective catalyst loading). See the Supporting Information for details.
Conversion and Z:E ratios measured by analysis of 500 MHz 1H NMR spectra of unpurified mixtures; the variance of values are estimated to be ±2%.
Yield of isolated products after purification (isomeric mixture); the variance of values are estimated to be ±5%. nd = not determined.
See ref 29b.
See ref 29a.
Use of Mo-based adamantylimido alkylidene 6 and W-based di(i-Pr)imido complex 7 (entries 4–5 of Table 2) gives rise to a significant reversal of selectivity in favor of the desired Z-12a, delivering 85% and 79% stereoselectivity, respectively. We attribute the improved stereochemical control with the Mo complex (6 vs 7) to the larger size difference between an adamantylimido and the aryloxide ligands (vs a 2,6-(i-Pr)2phenylimido in 7; cf. Figure 1). Support for the above proposal is found in the exceptional selectivity observed with W-based alkylidene 13 (structure shown in Scheme 3) even at a relatively high concentration for a macrocyclization (0.05 M),34 when reaction of the active complex with the Z macrocycle is more likely to occur; under the latter conditions, macrolactone 12a is isolated in up to 86% yield and with only 3% contamination with the undesired E isomer (entry 8, Table 2). The positive influence of reduced pressure (entries 6–7, Table 2),35 as detailed previously,11 is tied to enhancing catalyst longevity by minimization of the concentration of the relatively unstable methylidene complexes (cf. V, Figure 1), which can be generated by reaction of an alkylidene intermediate with the volatile ethylene (formed as byproduct). When the W-catalyzed RCM is performed with 3.0 mol % 13 at 0.05 M concentration, 12a is formed in 63% yield and 97% Z selectivity (entry 9, Table 2).

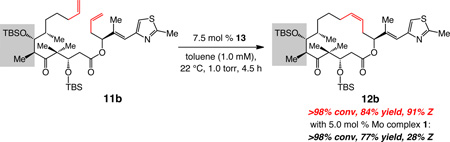
In a similar fashion, diene 11b, a diastereoisomer of 11a, can be converted to macrocyclic olefin 12b in 84% yield and with 91% Z selectivity (eq 1). The relatively lower degree of stereochemical control, versus RCM of 11a (96% Z), might be because there is a stronger inherent preference for the formation of the undesired E alkene in the case of 11b (i.e., substrate- vs catalyst-control are in stronger opposition); this is reflected in the slightly higher percentage of E isomer formed when the reaction is carried out with Mo bis-alkoxide 1 (28% Z vs 33% Z with 11a). The finding in eq 1 bodes well for the utility of MAP complexes in the preparation of analogues of this class of naturally occurring molecules.36
 |
(1) |
The above findings, collectively, confirm the notion that it is the capacity to generate high kinetic Z selectivity together with possessing the appropriate balance of sufficient (but not too high) reactivity levels that render W complex 13 the most attractive choice for macrocyclic RCM of the heavily functionalized dienes such as 11a (another relevant case will be presented below). The validity of such reactivity/Z selectivity relationships finds additional support in the variations in the conversion values and Z selectivities obtained for RCM of dienes leading to sparsely functionalized macrocyclic lactones 8, 14 and ambrettolide, respectively (Scheme 3). Compared to the transformations with complexes 6 and 7, W-based alkylidene 13 provides generally lower conversion as a result of its subordinate activity (vs Mo- based MAP complexes). On the other hand, higher Z:E values are observed in reactions with the corresponding conformationally more flexible dienes when 13 is employed (e.g., vs 11a): the less effective complex does not promote post-RCM isomerization or no more than to a minimal degree.
3.2. Study of catalyst-induced post-RCM isomerization in reactions with 12a
Considering the significant role that olefin metathesis-based stereoisomeric interconversion can play in determining the stereoisomeric purity with which a macrocyclic product is isolated, we chose to examine the extent of the influence of such processes, vis-à-vis the formation of macrolactone 12a in greater detail.
3.2.1. Studies with Mo-based bis-alkoxide 1
We first probed the ring closure performed in the presence of Mo complex 1, a transformation originally disclosed in 1997 (entry 1, Table 2).29b The highly reactive complex (1) generates 67% of the E alkene; based on our investigations described above, we suspected that post-RCM isomerization might be partly responsible for such preference. To probe this possibility, we investigated the degree of stereochemical control in the RCM of 11a promoted with bis-alkoxide 1 as a function of time. These studies led us to determine that, remarkably, as illustrated in Scheme 4, within only ten minutes, there is 92% conversion to 12a, which is isolated in 89% yield and 72% Z selectivity; the latter finding is significantly superior to 33% Z reported previously for the same reaction being performed with 20 mol % 1 for the duration of one hour (86% yield).29b These findings point to the importance of the need for careful determination of the optimal catalyst loading and/or reaction time when carrying out catalytic RCM that generate disubstituted macrocyclic alkenes; vigilant analysis of changes in the olefin stereochemistry as the cyclization progresses, regardless of which class of catalysts is being employed, might be necessary for achieving the best results.
scheme 4.

Z Selectivity as a Function of Time in RCM Promoted by Complex 1
Two additional points regarding this aspect of our studies are worthy of note:
The reduced size differential between the arylimido and hexa-fluoro-tert-butoxide ligands within Mo complex 1 conspire to furnish relatively moderate Z selectivity compared to that delivered with W-based alkylidene 13 (cf. Table 2). Further, the higher activity of the Mo bis-alkoxide, versus a MAP complex such as 13, manifested by a more efficient post-RCM isomerization, leads to a more facile erosion of kinetic selectivity.
Whereas there is no detectable difference in selectivity during the first ten minutes of the Mo-catalyzed transformation (Scheme 4), there is substantial Z-to-E isomerization within 90 minutes (71:29→37:63 Z:E). In stark contrast, in the cyclization with Ru-based carbene 2d, the E isomer is favored early on (Scheme 4), probably suggesting that the cyclization process might be kinetically E selective. The relatively low level of activity by the Ru complex makes it unlikely that olefin isomerization is occurring.
3.2.2. Studies with MAP complexes
We then turned to a more detailed analysis of the possible impact of post-RCM isomerization in reactions with Mo- and W-based complexes 6 and 13. Due to the relatively high Z selectivities observed with the latter two alkylidenes (≥85:15), we chose to investigate the extent of loss of stereoselectivity which could occur upon resubjection of a sample of 92% Z-12a to a solution containing the activated forms of alkylidenes 6 and 13, respectively. Thus, as depicted in Scheme 5, a sample of Mo complex 6 was pre-treated with diallylether to generate methylidene 15, the species released upon RCM and responsible for initiation of a new catalytic cycle (cf. V, Figure 1). Subsequent in vacuo removal of residual diallylether and the ethylene generated was followed by the addition of ten equivalents of 12a (corresponding to 10 mol % catalyst loading). After 90 minutes, the macrolactone was recovered as an 81:19 mixture of Z and E isomers (vs the initial 92:8). In contrast, when an identical procedure is performed with W alkylidene 13, there is no detectable loss of stereoselectivity (Scheme 5). The above experiments demonstrate that, whereas some loss of Z selectivity can take place in the course of RCM when Mo complex 6 is used, the methylidene derived W-based alkylidene 13 reacts with exceptional chemoselectivity with the terminal alkenes of 11a in preference to the cyclic disubstituted olefin in 12a. The small amount of E-12a, formed in the reactions with 13, is probably due to a lack of perfection in kinetic selectivity (i.e., reaction via complex related to VI, Figure 2).
scheme 5.

Catalyst-Induced Post-RCM Isomerization of Macrocyclic Epothilone C Precursor 12a with a Mo- and a W-Based Complexa
3.3. Practical aspects of W-catalyzed macrocyclic RCM
Handling of the W-based MAP complexes
As outlined in connection with the use of tungstacyclobutane 7 in the context of stereoselective synthesis of epilachnene, one of the notable discoveries made in the course of these investigations is the relative robustness of the W-based complexes. Thus, similar to 7, dichlorophenylimido MAP alkylidine 13, optimal in effecting the formation of macrolactone 12a, is sufficiently stable such that it can be handled and weighed in open air; the requisite apparatus can be set up and reactions performed in a typical fume hood without the need for strict exclusion of air and moisture.27 When complex 13, which must be stored under inert atmosphere (e.g., N2) for long terms storage, is exposed to air, there is ≤10% decomposition within ten minutes (as judged by spectroscopic analysis), more than sufficient time to set up a reaction.
Gram scale Z-selective macrocyclic RCM reaction
To challenge further the practical aspects of the W-catalyzed protocol, we chose to establish whether the W-catalyzed RCM of the relatively complex diene 11a can be performed on gram scale with MAP alkylidene 13 in an efficient and stereoselective fashion. We surmised that the reliability of the catalytic protocol would be more convincingly illustrated if the Z-selective RCM were to be carried out with a starting material prepared by a relatively protracted sequence (i.e., 16 steps for 11a).30,29a To accomplish this task, however, first, a significant revision of the originally reported route to 11a had to be implemented; otherwise, access to sufficient quantities of the diene proved to be too costly and labor intensive. A modified synthesis scheme was accordingly developed,37 allowing us to secure nearly 12 grams of 11a after two runs. Some of the notable attributes of the revised route for preparation of enantiomerically pure 11a include a highly diastereoselective aldol addition (96:4 vs 60:40 dr reported previously), the need for less equivalents of enantiomerically pure intermediates and smaller number of purifications through silica gel chromatography.27
We subsequently determined that in the presence of 6.5 mol % W complex 13, RCM can be effected with gram quantities of 11a to afford macrocycle 12a, precursor to epothilones C and A, in 83% yield and with 95% Z selectivity (Scheme 6). The transformation reaches 95% conversion within four hours at 22 °C and proceeds readily when performed in a typical laboratory fume hood with humidity levels reaching approximately the 80% level.
scheme 6.

A Practical Catalytic Method for Z-Selective Macrocyclic RCM
4. Z-Selective RCM Reactions en Route to Nakadomarin A
The final stage of our investigations corresponds to the formation of two ring structures, one macrocyclic and the other a cyclooctenyl moiety, en route to stereoselective total synthesis of nakadomarin A. The earlier approaches utilized olefin metathesis to prepare the larger ring structure of nakadomarin A, albeit with relatively low efficiency (15–30 mol % Ru complex) and minimal or stereoselectivity in favor of the undesired isomer (~2:1–1:2 E:Z).38 Due to the ineffective stereochemical control in the macrocyclic ring formation, as with epilachnene and epothilone C, strategies involving alkyne RCM/partial hydrogenation sequence have been introduced.22,39 We demonstrated in our initial disclosure6 that in the presence of W alkylidene 13, macrocyclic RCM reactions with tetracyclic substrate 16 as well as the more strained pentacyclic 18 proceed efficiently and with exceptional Z selectivity (90% yield and 97% Z and 63% yield and 94% Z, respectively); these advances are summarized in Scheme 7.
scheme 7.

W-Catalyzed Z-Selective RCM en route to Nakadomarin A
Attempts to effect alkyne RCM of diyne 19, shown in Scheme 8, bearing two Lewis basic tertiary amines, with either Mo- or W-based alkylidynes, including the more recently developed variations,24 led to <5% conversion even with 30–50 mol % of a metal complex and at 80 °C (up to 18 h). Use of tetracyclic diyne 20 (Scheme 8) was thus required in the latter approach (100 mol % Mo(CO)6, 500 mol % 2-fluorophenol, 0.26 mM, C6H5Cl, reflux, 2.5 h; 39% yield).39c
scheme 8.

Catalytic Alkyne RCM for Synthesis of Nakadomarin Aa
aSee ref 39c for details.
Development of olefin metathesis-based strategies towards nakadomarin A offers a distinct framework for outlining some of the advantages offered by high-oxidation metal complexes (Mo- and W-based) versus Ru-based carbenes. This part of our investigation allowed us to probe other unique attributes of the W-catalyzed method as well. The role of adventitious olefin-metathesis-based isomerization on the observed stereoselectivity can be further probed in the context of another relatively complex but structurally distinct polycyclic molecule (vs epothilone C precursor 11a). What is more, the polycyclic constitution of nakadomarin A provides the opportunity for investigation of whether the alkene within the eight-membered ring can be formed through Mo- or W-catalyzed RCM after the relatively sensitive macrocyclic olefin has been generated and without significant diminution in the stereochemical purity of the latter. Such studies would illustrate whether the RCM catalysts can be used for stereoselective preparation of polycyclic structures where the stereochemical integrity of one or more Z alkene units must be preserved while additional rings are being generated.
4.1. Comparison of macrocyclic RCM reactions with Ru- vs W-based complexes
Efficiency and stereoselectivity
In stark contrast to the findings in Scheme 7, the most selective formerly reported procedure for conversion of 18 to nakadomarin A involved the use of 20 mol % Ru carbene 2b; three equivalents of a strong BrØnsted acid was also required so that 63% Z selectivity could be achieved.38d The difficult nature of the latter RCM process originates largely from the relatively high ring strain associated with the Z alkene-containing macrocycle. Hence, it is the less active first-generation Ru carbene (vs the more active 2c and 2d), which must be added at a slow rate to the reaction mixture, that preserves the small kinetic preference for the Z alkene isomer.
Ease of product isolation/purification
In addition to the substantially higher efficiency (5.0 mol % vs ≥20 mol % or higher) and stereoselectivity (94% Z vs ≤63% Z), the W-based catalyst delivers another crucial practical advantage. Generally, purification of organic molecules prepared through the use of high oxidation-state catalysts, such as Mo- or W-based alkylidenes, is easier than those that contain a late transition metal (e.g., Ru-based carbenes); the former are more easily separated from the desired product through simple silica gel chromatography. With Ru-based complexes such as 2b, the reaction mixture contains significant amounts of tricyclohexylphosphine oxide, which cannot be easily removed chromatographically: treatment with 1.0 N HCl, to double-protonate the amine sites of the alkaloid and render it soluble in the aqueous phase, is therefore needed. Subsequent separation of the phosphine oxide-containing organic layer and basification of the aqueous phase regenerates nakadomarin A, which is then purified further. With W alkylidene 13, routine chromatography furnishes the desired macrocycle (94–95% Z); there is no need for acid treatment or aqueous extraction.
4.2. Study of catalyst-induced post-RCM isomerization in catalytic RCM of diene 16
We subjected samples of fifteen-membered ring structure 17, consisting of 97% of the Z alkene to solutions of Mo-based methylidene 15 and that derived from W-based complex 13 (Scheme 9). As in the case of 12a, substantial loss of Z selectivity is detected within one hour in the case of Mo complex 6 (from 3% to 18% E). In contrast to the macrocycle precursor to epothilone C where alkene isomerization is not detected (cf. Scheme 5), however, the presence of the less reactive methylidene derived from 13 leads to recovery of a sample of 17 that is of slightly lower Z:E ratio (93:7 vs 97:3; Scheme 9). It is likely that, as a result of diminished steric hindrance and a higher degree of angle strain within the macrocyclic unit of 17, in contrast to 12a, there is a stronger sensitivity towards ring-opening/ring-closing sequence (cf. Figure 2), culminating in lowering of stereochemical purity at the olefinic site. Considering that the reaction time for the conversion of 16 to 17 is two hours (cf. Scheme 7; vs 1.0 h in Scheme 9), it is probably that at least part of the 3% E-17 obtained is the result of some post–RCM isomerization, rather than any imperfection in kinetic selectivity delivered by W complex 13. The above findings emphasize the difficulty posed by an efficient and highly Z-selective macrocyclic RCM route to the polycyclic natural product.
scheme 9.

Examination of Catalyst-Induced Post-RCM Isomerization of Nakadomarin A Precursor 17 with a Mo- and a W-Based Complexa
4.3. Synthesis of the cyclooctene ring through catalytic RCM; impact on macrocyclic alkene stereochemistry
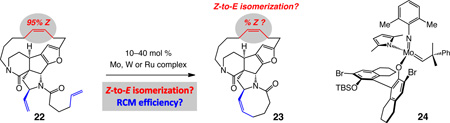
Studies with di-amide 22
As mentioned earlier, an intricate RCM reaction leading to nakadomarin A involves conversion of pentacyclic diamide 22 to hexacyclic 23 (Table 3), which can be synthesized from 17 in four steps and 66% overall yield. The resulting hexacyclic product would subsequently be converted to the target molecule through reduction of the amide functional groups (e.g., dibal–H). Can the strained hexacyclic natural product be synthesized without significant isomerization of the macrocyclic Z alkene?
Table 3.
Synthesis of Nakadomarin A Cyclooctene Ring through Catalytic RCM of a Diamide Precursor. Efficiency and Competitive Z-to-E Isomerization of the Macrocyclic Alkenea
 | |||||
|---|---|---|---|---|---|
| entry | complex; loadingb | solvent | temp (°C); time (h) |
conv (%)c |
Z:Ec (macrocycle) |
| 1 | 1; 30 | toluene | 22; 6 | 60 | 90:10 |
| 2 | 2b; 100 | CH2Cl2 | 40; 24 | >98 | 72:28 |
| 3 | 13; 30 | toluene | 22; 24 | <5% | na |
| 4 | 24; 10 | toluene | 80; 24 | 72 | 95:5 |
Reactions were carried out in purified toluene (5.0 mM) at 22 °C an atmosphere of N2 or under vacuum; see the Supporting Information for details.
Complex 1, 2b and 13 were prepared prior to use, whereas alkylidene 24 was synthesized in situ from the corresponding bis-pyrrolide and aryl alcohol. See the Supporting Information for details.
Conversion and Z:E ratios measured by analysis of 400 MHz 1H NMR spectra of unpurified mixtures. na = not applicable.
Conversion of bis-amide diene 22 to eight-membered ring lactam 23 proceeds to 60% conversion in the presence of 30 mol % Mo complex 1 after six hours at 22 °C (entry 1, Table 3); the desired product is obtained with minor loss of macrocyclic alkene stereoisomeric purity. With the less active Ru carbene 2b (entry 2, Table 3), 100 mol % loading, heating to 40 °C and 24 hours of reaction time are required for achieving >98% conversion, but formation of the cyclooctene ring proves costly: there is substantial reduction in the macrocyclic Z:E ratio (72% vs 95% Z).40 There is no detectable conversion to 23 with W-based alkylidene 13, even when 30 mol % of the starting complex is utilized (entry 3, Table 3). When 10 mol % of Mo-based MAP alkylidene 24 is employed (entry 4), ring closure occurs without loss of stereochemistry in spite of the elevated temperature and extended reaction time (80 °C, 24 h); nevertheless, a large portion of 22 remains unreacted (28%).41
There are several possible reasons for the comparative lack of efficiency in catalytic RCM reactions for converting 22 to 23. The angle strain associated with the formation of the desired hexacyclic diene is compounded by the presence of the two rigidifying amide groups; a related rationale has been put forth vis-à-vis cyclooctene formation through RCM with a macrocyclic diyne substrate.39b The sterically hindered vinyl group bearing a substitution at its allylic site can discourage facile ring closure. Finally, there is the possibility that the alkylidenes and carbenes derived from initial reaction at the either of the terminal alkenes might be partially deactivated due to intramolecular chelation with a neighboring Lewis basic amide carbonyl.42
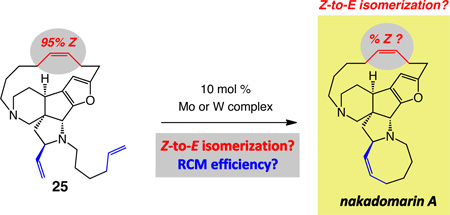
Studies with diamine 25
Based on the above-mentioned impediments that we synthesized diamine 25 (dibal–H, 75% yield) and examined the possibility of converting it to nakadomarin through catalytic RCM. As shown in entry 1 of Table 4, the presence of 10 mol % bis-alkoxide 1 gives rise to complete disappearance of excess Z macrocyclic alkene within one hour (95:5→55:45 Z:E). With the sterically more demanding Mo alkylidene 6 or tungstacyclobutane 7, substrate 25 is consumed at the same rate, but formation of the eight-membered ring amine is accompanied by significantly less diminution in Z:E ratio (entries 2–3, Table 4: 95:5→85:15 and 82:18 Z:E, respectively); it is noteworthy that W complex 7 delivers similar degrees of Z-to-E olefin isomerization as is observed with Mo species 6 (see Scheme 2 for another example). There is minimal RCM when Mo-based MAP complex 24 is used (entry 4, Table 4). The higher activity obtained with alkylidene 6 as opposed to 24 can be attributed to the smaller size of the adamantylimido ligand (vs the 2,6-di-alkyl-phenylimido). In all instances, where complete disappearance of triene 25 is observed (entries 1–3, Table 4), a relatively wide gap exists between the conversion and isolated yield values (>98% conv vs 32–43% yield). The origin of the latter discrepancy is the formation of a considerable amount of unidentifiable byproducts that likely arise from oligomerization reactions involving the two terminal olefins in 25 or those generated through ring-opening of the macrocyclic moiety (determined by spectroscopic analysis). It is similarly feasible that the terminal alkenes that serve as precursors to either the large or medium rings, some generated through a ring-opening process, can participate in RCM reactions with one another, leading to the formation of the alternative polycyclic products. Parallel issues regarding byproduct formation were observed in Mocatalyzed reactions with diamide 22 (Table 3, entries 1 and 4).
Table 4.
Synthesis of Nakadomarin A Cyclooctene Synthesis through Catalytic RCM of a Diamine Precursor. Efficiency and Competitive Z-to-E Isomerization of the Macrocyclic Alkenea
 | |||||
|---|---|---|---|---|---|
| entry | complexb | conv (%);c yield (%)d |
Z:Ec (macrocycle) |
||
| 1 | 1 | >98; 32 | 55:45 | ||
| 2 | 6 | >98; 38 | 85:15 | ||
| 3 | 7 | >98; 43 | 82:18 | ||
| 4 | 24 | 17; nd | nd | ||
Reactions were carried out in purified toluene at 22 °C for 6.0 h under 1.0 torr of under vacuum; see the Supporting Information for details.
Complexes 1, 7 and 27 were prepared prior to use; other alkylidenes were synthesized in situ from the corresponding bis-pyrrolide and aryl alcohol. See the Supporting Information for details.
Conversion and Z:E ratios measured by analysis of 400 MHz 1H NMR spectra of unpurified mixtures.
Yield of isolated products after purification; the variance of values are estimated to be <±5%. nd = not determined.
Considering the outcome of the experiments summarized in Schemes 5 and 9 in connection with post-RCM isomerization, it follows that with W-based MAP complex 13 formation of the cyclooctene ring occurs with the least degree of loss in the stereoselectivity at the macrocyclic alkene site. Specifically, as shown in Scheme 10, with 5.0 mol % of W complex 13, hexacyclic diamine 25 can be transformed to nakadomarin A in 56% yield (64% based on recovered substrate) with 91:9 Z:E ratio for the larger ring olefin. With 10 mol % 13, under otherwise identical conditions, the natural product is obtained in 48% yield and 88:12 Z:E ratio (>98% conversion); thus, with lower catalyst loading, a better balance between efficiency and preservation of kinetic selectivity can be achieved, resulting in 12% of recovered 25 and less product decomposition and post-RCM isomerization. The relative facility of catalytic RCM with diene 25 and the strong resistance of diyne 19 towards cyclization, shown in Scheme 8, point to comparative ease with which a less strained macrocyclic alkene can be synthesized (vs a large ring alkyne).
scheme 10.

Synthesis of Nakadomarin A through Cyclooctene Formation by W-Catalyzed RCM. Minimal Isomerization at Macrocyclic Alkene Site
Comparison with previously reported approaches
Conversion of pentacyclic diamine 25 to nakadomarin A demands 40 mol % of Ru carbene 2b and eight hours of reaction time, as shown in Scheme 10. Moreover, significantly more dilute conditions (0.3 vs 5.0 mM) and 300 mol % of camphorsulfonic acid are needed (60–70% conv, 27% yield and 60% E, otherwise). It is worthy of note that, in spite of the fact that formation of the cyclooctene moiety in the majority of the formerly reported total syntheses of nakadomarin A was implemented in the absence of the rigidifying macrocyclic alkene,38,39c 20–100 mol % of a Ru-based carbene proved necessary for achieving a reasonably efficient cyclization.38a–c,e
Conclusions
The ability of Mo- and W-based MAP species to promote Z-selective macrocyclic ring formation substantially enhances the effectiveness of one of the most critical and commonly employed transformations in chemistry. Successful applications to total syntheses of epilachene, epothilone C and nakadomarin A, leads to a near doubling of the overall efficiency with which these notable biologically active natural products can be accessed through an RCM-containing route. Our findings confirm that in planning a multi-step pathway for the preparation of a complex molecule, the Mo as well as the W complexes can be relied upon to deliver the desired outcome at the late stages of an extended synthesis scheme. As the result of the appropriate reactivity exhibited by the Mo or W catalysts, which are tolerant of basic amines, RCM reactions can be carried out often with minimal or no alkene isomerization. Similarly notable is that tungsten complexes 6 and 13 can be handled in air and reactions carried out in a common fume hood.
A critical outcome of the present studies is the complementarity of the Mo- and W-based catalysts. In situations where the substrate dienes do not enjoy a higher degree of pre-organization, the high activity of Mo-based alkylidenes might be needed; in cases where the relatively rigid starting materials are involved, and in particular where post-RCM isomerization can be detrimental, the relatively less active W alkylidenes might emerge as the catalysts of choice (Chart 2). The elucidation of the finely balanced activity profile furnished by the Mo and W mono-pyrrolide complexes and the significance of reaction time to the preservation of kinetic selectivity in catalytic olefin metathesis reactions are two of the noteworthy lessons learned in the course of our inquiries.
chart 2.

Highly Z-Selective and Complementary Moand W-Catalyzed Macrocyclic RCM.
The impact of catalytic Z-selective macrocyclic RCM is not limited to the target molecules examined here; routes leading to a number of other of complex and biologically active molecules43 can be made more efficient by the catalysts and strategies detailed above. The advances put forth in this report are expected to exert a wide-ranging and immediate impact on the synthesis of an assortment of organic molecules.
Supplementary Material
ACKNOWLEDGEMENTS
This research was funded by the NIH (GM-59426). M. Y. was a John LaMattina and AstraZeneca Graduate Fellow; A. F. K. was the recipient of an EPSRC-GlaxoSmithKline Synthesis Studentship, and P. J. of an EPSRC Postdoctoral Fellowship. D. J. D. acknowledges support through an EPSRC Leadership Fellowship. We thank B. Li for assistance in securing X-ray structures and S. J. Meek, S. J. Malcolmson, D. L. Silverio, R. V. O’Brien, T. J. Mann, E. T. Kiesewetter and S. Torker for valuable discussions. We are grateful to D. L. Silverio for assistance in computational analysis and K. Wu for experimental assistance and to Boston College for providing access to computational facilities
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures and spectral data for substrates and products (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.For relatively recent reviews on various aspects of catalytic olefin metathesis, see: Schrock RR, Hoveyda AH. Angew. Chem. Int. Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576. Grubbs RH, editor. Handbook of Metathesis. Weinheim, Germany: Wiley–VCH; 2003. Diver ST, Giessert AJ. Chem. Rev. 2004;104:1317–1382. doi: 10.1021/cr020009e. Hoveyda AH, Gillingham DG, Van Veldhuizen JJ, Kataoka O, Garber SB, Kingsbury KS, Harrity JPA. Org. Biomol. Chem. 2004;2:8–23. doi: 10.1039/b311496c. Hoveyda AH, Zhugralin AR. Nature. 2007;450:243–251. doi: 10.1038/nature06351. Donohoe TJ, Fishlock LP, Procopiou PA. Chem.–Eur. J. 2008;14:5716–5726. doi: 10.1002/chem.200800130. Samojlowicz C, Bieniek M, Grela K. Chem. Rev. 2009;109:3708–3742. doi: 10.1021/cr800524f. Vougioukalakis GC, Grubbs RH. Chem. Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424. Lozano-Vila AM, Monsaert S, Bajek A, Verpoort F. Chem. Rev. 2010;110:4865–4909. doi: 10.1021/cr900346r. Hoveyda AH, Malcolmson SJ, Meek SJ, Zhugralin AR. Angew. Chem. Int. Ed. 2010;49:34–44. doi: 10.1002/anie.200904491.
- 2.For reviews regarding applications of catalytic olefin metathesis in natural product synthesis, see: Love JA. In: Handbook of Metathesis. Grubbs RH, editor. Vol. 2. Wiley–VCH; Weinheim, Germany: 2003. pp. 296–322. Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. Cossy J, Arsenyadis S, Meyer C, editors. Metathesis in Natural Product Synthesis. Wiley–VCH; Weinheim, Germany: 2010. Fürstner A. Chem. Commun. 2011;47:6505–6511. doi: 10.1039/c1cc10464k.
- 3.For general overviews regarding macrocyclic ring-closing metathesis reactions, see: Gradillas A, Perez-Castells J. Angew. Chem. Int. Ed. 2006;45:6086–6101. doi: 10.1002/anie.200600641. Gradillas A, Pérez-Castells J. In: Metathesis in Natural Product Synthesis. Cossy J, Arsenyadis S, Meyer C, editors. Weinheim, Germany: Wiley–VCH; 2010. pp. 149–182.
- 4.Villemin D. Tetrahedron Lett. 1980;21:1715–1718. [Google Scholar]
- 5.Tsuji J, Hashiguchi S. Tetrahedron Lett. 1980;21:2955–2958. [Google Scholar]
- 6.For the preliminary account of this work, see: Yu M, Wang C, Kyle AF, Jakubec P, Dixon DJ, Schrock RR, Hoveyda AH. Nature. 2011;479:88–93. doi: 10.1038/nature10563.
- 7. Singh R, Schrock RR, Müller P, Hoveyda AH. J. Am. Chem. Soc. 2007;129:12654–12655. doi: 10.1021/ja075569f. Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008;456:933–937. doi: 10.1038/nature07594. Sattely ES, Meek SJ, Malcolmson SJ, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:943–953. doi: 10.1021/ja8084934. For an overview regarding the potential utility of this catalyst class in chemical synthesis, see: Ref 1j.
- 8. Höfle G, Bedorf N, Steinmetz H, Schomburg D, Gerth K, Reichenbach H. Angew. Chem. Int. Ed. 1996;35:1567–1569. Kowalski RJ, Giannakakou P, Hamel E. J. Biol. Chem. 1997;272:2534–2541. doi: 10.1074/jbc.272.4.2534. Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325–2333. For an overview on biological chemistry of epothilones, see: (d) Nicolaou KC, Roschangar F, Vourloumis D. Angew. Chem. Int. Ed. 1998;37:2014–2045. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2014::AID-ANIE2014>3.0.CO;2-2.
- 9.Kobayashi J, Watanabe D, Kawasaki N, Tsuda M. J. Org. Chem. 1997;62:9236–9239. [Google Scholar]
- 10.(a) Ibrahem I, Yu M, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3844–3845. doi: 10.1021/ja900097n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu M, Ibrahem I, Hasegawa M, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2012;134:2788–2799. doi: 10.1021/ja210946z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meek SJ, O’Brien RV, Llaveria J, Schrock RR, Hoveyda AH. Nature. 2011;471:461–466. doi: 10.1038/nature09957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For an early report regarding the significance reversible nature of catalytic olefin metathesis in the context of natural product synthesis, see: Xu Z, Johannes CW, Houri AF, La DS, Cogan DA, Hofilena GE, Hoveyda AH. J. Am. Chem. Soc. 1997;119:10302–10316. For a notable application, see: (b) Smith AB, III, Adam CM, Kozmin SA, Paone DV. J. Am. Chem. Soc. 2001;123:5925–5937. doi: 10.1021/ja0106164.
- 13.Attygalle AB, McCormick KD, Blankespoor CL, Eisner T, Meinwald J. Proc. Natl. Acad. Sci. USA. 1993;90:5204–5208. doi: 10.1073/pnas.90.11.5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossini C, González A, Farmer J, Meinwald J, Eisner T. J. Chem. Ecol. 2000;26:391–397. [Google Scholar]
- 15.Fürstner A, Langemann K. Synthesis. 1997:792–803. [Google Scholar]
- 16.Schrock RR, Murdzek JS, Bazan GC, Robbins J, DiMare M, O’Regan M. J. Am. Chem. Soc. 1990;112:3875–3886. [Google Scholar]; (b) Bazan G, Oskam JH, Cho H-N, Park LY, Schrock RR. J. Am. Chem. Soc. 1991;113:6899–6907. [Google Scholar]; (c) Bazan G, Schrock RR, Cho H-N, Gibson VC. Macromolecules. 1991;24:4495–4502. [Google Scholar]
- 17.(a) Fu GC, Grubbs RH. J. Am. Chem. Soc. 1992;114:7324–7325. [Google Scholar]; (b) Zuercher WJ, Hashimoto M, Grubbs RH. J. Am. Chem. Soc. 1996;119:6634–6640. [Google Scholar]
- 18.(a) Schwab P, France MB, Ziller JW, Grubbs RH. Angew. Chem. Int. Ed. Engl. 1995;34:2039–2041. [Google Scholar]; (b) Schwab P, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1996;118:100–110. [Google Scholar]
- 19.Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 20. Kingsbury JS, Harrity JPA, Bonitatebus PJ, Hoveyda AH. J. Am. Chem. Soc. 1999;121:791–799. Kingsbury JS, Garber SB, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168–8179. (c) Ref 1d.
- 21.For reviews on catalytic alkyne metathesis reactions, see: Fürstner A, Davis PW. Chem. Commun. 2005:2307–2320. doi: 10.1039/b419143a. Zhang W, Moore JS. Adv. Synth. Catal. 2007;349:93–120.
- 22.Fürstner A, Guth O, Rumbo A, Seidel G. J. Am. Chem. Soc. 1999;121:11108–11113. [Google Scholar]
- 23. Coutelier O, Mortreux A. Adv. Synth. Catal. 2006;348:2038–2042. and references cited therein.
- 24.Heppekausen J, Stade R, Goddard R, Fürstner A. J. Am. Chem. Soc. 2010;132:11045–11057. doi: 10.1021/ja104800w. [DOI] [PubMed] [Google Scholar]
- 25.Mo-based MAP complexes are routinely prepared in situ through reaction of the corresponding bis-pyrrolide and the requisite alcohol. In the case of alkylidene 6, the formation of the complex proceeds in ~60% yield; thus, use of 2.0 mol % of the precursors leads to 1.2 mol % catalyst loading.
- 26.Jiang AJ, Simpson JH, Müller P, Schrock RR. J. Am. Chem. Soc. 2009;131:7770–7780. doi: 10.1021/ja9012694. [DOI] [PubMed] [Google Scholar]
- 27.See the Supporting Information for details.
- 28.For example, when the RCM reaction, catalyzed by Mo-based bis-alkoxide 1 in entries 1 and 5 are examined after one minute, the product mixture contains 28% (vs 17% after one h) and 25% (vs 7% after one h) of the Z alkene, respectively (23% and 35% conv). For a study of post-RCM isomerization with reactions that are catalyzed by Ru-based carbenes and afford macrocyclic alkenes, see: Lee CW, Grubbs RH. Org. Lett. 2000;2:2145–2147. doi: 10.1021/ol006059s.
- 29.(a) Nicolaou KC, He Y, Vourloumis D, Vallberg H, Roschangar F, Sarabia F, Ninkovic S, Yang Z, Trujillo JI. J. Am. Chem. Soc. 1997;119:7960–7973. [Google Scholar]; (b) Meng D, Bertinato P, Balog A, Su D-S, Kamenecka T, Sorensen EJ, Danishefsky SJ. J. Am. Chem. Soc. 1997;119:10073–10092. [Google Scholar]; (c) Schinzer D, Bauer A, Böhm OM, Limberg A, Cordes M. Chem. Eur. J. 1999;5:2483–2491. [Google Scholar]; (d) Sinha SC, Sun J, Miller GP, Wartmann M, Lerner RA. Chem. Eur. J. 2001;7:1691–1702. doi: 10.1002/1521-3765(20010417)7:8<1691::aid-chem16910>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 30.For reviews and overviews regarding the use of catalytic RCM approaches to total synthesis of epothilones, including some biological activity studies, see: Harris CR, Danishefsky SJ. J. Org. Chem. 1999;64:8434–8456. Mulzer J. Monat. Chem. 2000;131:205–238. Nicolaou KC, Ritzen A, Namoto K. Chem. Commun. 2001:1523–1535. doi: 10.1039/b104949f. Rivkin A, Cho YS, Gabarda AE, Yoshimura F, Danishefsky SJ. J. Nat. Prod. 2004;67:139–143. doi: 10.1021/np030540k. Rivkin A, Chou T-C, Danishefsky SJ. Angew. Chem. Int. Ed. 2005;44:2838–2850. doi: 10.1002/anie.200461751. Altmann K-H, Pfeiffer B, Arseniyadis S, Pratt BA, Nicolaou KC. ChemMedChem. 2007;2:396–423. doi: 10.1002/cmdc.200600206. Mulzer J, Altmann K-H, Höfle G, Müller R, Prantz K. Chimie. 2008;11:1336–1368.
- 31.Altmann KH, Bold G, Caravatti G, Denni D, Flörsheimer A, Schmidt A, Rihs G, Wartmann M. Helv. Chim. Acta. 2002;85:4086–4110. [Google Scholar]
- 32.Johnson J, Kim S-H, Bifano M, DiMarco J, Fairchild C, Gougoutas J, Lee F, Long B, Tokarski J, Vite G. Org. Lett. 2000;2:1537–1540. doi: 10.1021/ol0058240. [DOI] [PubMed] [Google Scholar]
- 33.Fürstner A, Mathes C, Lehmann CW. Chem. Eur. J. 2001;7:5299–5317. doi: 10.1002/1521-3765(20011217)7:24<5299::aid-chem5299>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 34.The lower yield of 12 at higher concentration is (at least partly) due to larger amounts of adventitious homocoupling products.
- 35.When RCM of 11a is performed under the same conditions as in entry 6 of Table 2 but at 1.0 torr of vacuum, 97% conversion to 12a is observed (85% yield, 96% Z).
- 36.For representative studies in connection with epothilone analogues, see: (a) Ref 8d Nicolaou KC, He Y, Roschangar F, King NP, Vourloumis D, Li T. Angew. Chem. Int. Ed. 1998;37:84–87. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2014::AID-ANIE2014>3.0.CO;2-2.
- 37.See the Supporting Information for details.
- 38.(a) Nagata T, Nakagawa M, Nishida A. J. Am. Chem. Soc. 2003;125:7484–7485. doi: 10.1021/ja034464j. [DOI] [PubMed] [Google Scholar]; (b) Ono K, Nakagawa M, Nishida A. Angew. Chem. Int. Ed. 2004;43:2020–2023. doi: 10.1002/anie.200453673. [DOI] [PubMed] [Google Scholar]; (c) Young IS, Kerr MA. J. Am. Chem. Soc. 2007;129:1465–1469. doi: 10.1021/ja068047t. [DOI] [PubMed] [Google Scholar]; (d) Jakubec P, Cockfield DM, Dixon DJ. J. Am. Chem. Soc. 2009;131:16632–16633. doi: 10.1021/ja908399s. [DOI] [PubMed] [Google Scholar]; (e) Cheng B, Wu F, Yang X, Zhou Y, Wan X, Zhai H. Chem. Eur. J. 2011;17:12569–12572. doi: 10.1002/chem.201102101. [DOI] [PubMed] [Google Scholar]
- 39.(a) Nilson MG, Funk RL. Org. Lett. 2010;12:4912–4915. doi: 10.1021/ol102079z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kyle AF, Jakubec P, Cockfield DM, Cleator E, Skidmore J, Dixon DJ. Chem. Commun. 2011;47:10037–10039. doi: 10.1039/c1cc13665h. [DOI] [PubMed] [Google Scholar]; (c) Jakubec P, Kyle AF, Calleja J, Dixon DJ. Tetrahedron Lett. 2011;52:6094–6097. [Google Scholar]
- 40.A similar RCM reaction involving a bis-amide derivative has been reported under identical conditions as shown in entry 2 of Table 3 (see ref 39a); however, there is no mention of any changes in the stereosiomeric purity of the macrocyclic alkene.
- 41.When Mo-based MAP complex is used at 22 °C to effect the conversion of 22 to 23, only ~30% conv is observed (24 h, toluene); Z:E was not determined due to formation of byproducts. Since adamantylimido complex 6 tends to be less robust than the corresponding arylimido variants (e.g. 24 in entry 4, Table 3), RCM at elevated temperatures was not examined.
- 42.For an example and spectroscopic examination of catalyst deactivation by a resident Lewis basic functional group in an metathesis reaction involving a Mo-based alkylidene, see: Sattely ES, Cortez GA, Moebius DC, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2005;127:8526–8533. doi: 10.1021/ja051330s.
- 43.For representative examples where a Z-selective catalyst for macrocyclic RCM would significantly improve the overall efficiency of the total synthesis see: Humphrey JM, Liao Y, Ali A, Rein T, Wong Y-L, Chn HJ, Courtney AK, Martin SF. J. Am. Chem. Soc. 2002;124:8584–8592. doi: 10.1021/ja0202964. Fürstner A, Stelzer F, Rumbo A, Krause H. Chem. Eur. J. 2002;8:1856–1871. doi: 10.1002/1521-3765(20020415)8:8<1856::AID-CHEM1856>3.0.CO;2-R. She J, Lampe JW, Polianski AB, Watson PS. Tetrahedron Lett. 2009;50:298–301. Smith BJ, Sulikowski GA. Angew. Chem. Int. Ed. 2010;49:1599–1602. doi: 10.1002/anie.200905732.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.