

Abstract
Multiple myeloma is a malignant proliferation of monoclonal plasma cells leading to clinical features that include hypercalcaemia, renal dysfunction, anaemia, and bone disease (frequently referred to by the acronym CRAB) which represent evidence of end organ failure. Recent evidence has revealed myeloma to be a highly heterogeneous disease composed of multiple molecularly-defined subtypes each with varying clinicopathological features and disease outcomes. The major division within myeloma is between hyperdiploid and nonhyperdiploid subtypes. In this division, hyperdiploid myeloma is characterised by trisomies of certain odd numbered chromosomes, namely, 3, 5, 7, 9, 11, 15, 19, and 21 whereas nonhyperdiploid myeloma is characterised by translocations of the immunoglobulin heavy chain alleles at chromosome 14q32 with various partner chromosomes, the most important of which being 4, 6, 11, 16, and 20. Hyperdiploid and nonhyperdiploid changes appear to represent early or even initiating mutagenic events that are subsequently followed by secondary aberrations including copy number abnormalities, additional translocations, mutations, and epigenetic modifications which lead to plasma cell immortalisation and disease progression. The following review provides a comprehensive coverage of the genetic and epigenetic events contributing to the initiation and progression of multiple myeloma and where possible these abnormalities have been linked to disease prognosis.
1. Overview of Myeloma Genetics
Myeloma is a genetically complex disease which develops via a multistep process whereby plasma cells are driven towards malignancy through the accumulation of genetic “hits” over time. This multistep process permits myeloma to have various recognisable clinical phases, distinguished by biological parameters, along its development (Table 1). The earliest of these phases is termed monoclonal gammopathy of undetermined significance (MGUS) and is an indolent, asymptomatic, premalignancy phase characterized by a small clonal population of plasma cells within the bone marrow of <10% [1]. MGUS has a prevalence of >5% in adults aged over 70 and a progression risk to myeloma quantified at 1% per year [2, 3]. Following MGUS is smouldering multiple myeloma (SMM), another asymptomatic phase distinguished from MGUS by a greater intramedullary tumour cell content of >10% and an average risk of progression to myeloma of 10% per year for the first five years [4]. Next, myeloma itself is recognised, whereby malignant clones cause clinically relevant end-organ damage including the features of CRAB. The final phase is plasma cell leukemia (PCL), an aggressive disease end-point characterised by the existence of extramedullary clones and rapid progression to death. The basic premise of this disease progression is that the accumulation of genetic “hits” across different cellular pathways drives malignant change through deregulation to the intrinsic biology of the plasma cell. With advancements in molecular biology, many of these disrupted genes and pathways have now been characterised and the current challenge is therefore how to correctly interpret these molecular findings and develop them into clinically useful advances.
Table 1.
Diagnostic criteria for myeloma of undetermined significance (MGUS), smouldering multiple myeloma (SMM), myeloma, and plasma cell leukemia (PCL). Reproduced from international myeloma working group, 2003 [37].
| MGUS | SMM | Myeloma | PCL |
|---|---|---|---|
| Serum M-protein <30 g/L | Serum M-protein ≥30 g/L AND/OR Bone marrow clonal plasma cells ≥10% | M-protein in serum and/or urine.* No specific concentration required | Presence of ≥20% circulating plasma cells Absolute level of >2.0 × 109/L |
| Bone marrow clonal plasma cells <10%. If done—low level of plasma cell infiltration in a trephine biopsy | Confirmed clonal plasma cells in bone marrow | ||
| Absence of end-organ disease and symptoms | Absence of end-organ disease and symptoms | Presence of myeloma-related organ or tissue impairment (ROTI)** |
*1-2% of patients have no detectable M-protein in serum or urine but do have myeloma-related organ or tissue impairment (ROTI) and increased intramedullary plasma cells; this is termed nonsecretory myeloma. **ROTI: corrected serum calcium >0.25 mmol/L above the upper limit of normal or >2.75 mmol/L, creatinine >173 mmol/L, Hb 2 g/dL below the lower limit of normal or <10 g/dL, lytic bone lesions or osteoporosis with compression fractures (may be clarified by CT or MRI), symptomatic hyperviscosity, amyloidosis, recurrent bacterial infections (>2 episodes in 12 months).
1.1. Myeloma Intraclonal Heterogeneity
Alongside aiding the characterisation of genes and pathways disrupted in myeloma, molecular studies have also revealed that intraclonal heterogeneity is a common feature of the malignancy [5, 6]. This heterogeneity adds an extra layer of complexity to myeloma progression as it is apparent that genetic “hits” are not acquired in a linear fashion but rather through nonlinear branching pathways synonymous to Darwin's evolution of the species [7, 8]. A model of myeloma development through branching pathways is represented in Figure 1. This model however is designed as an oversimplification and should be viewed as a gross overview of disease progression as the process is highly complex with multiple progression pathways possible [8]. This analogy to Darwin's work explains that plasma cell clones acquire genetic lesions randomly and that these aberrations are then selected out based on their survival advantage. Consideration of intraclonal heterogeneity is important for disease understanding, as it is likely that the findings from many genomic studies represent the genetic aberrations in the predominant clonal population at the time of sampling and that these results may not be applicable to all subclonal populations. This has particular therapeutic relevance, as the genes and pathways deregulated in the predominant clonal population are unlikely to be uniform across the many subclones allowing drug resistance and relapse to occur through the evolution and progression of these minority populations.
Figure 1.
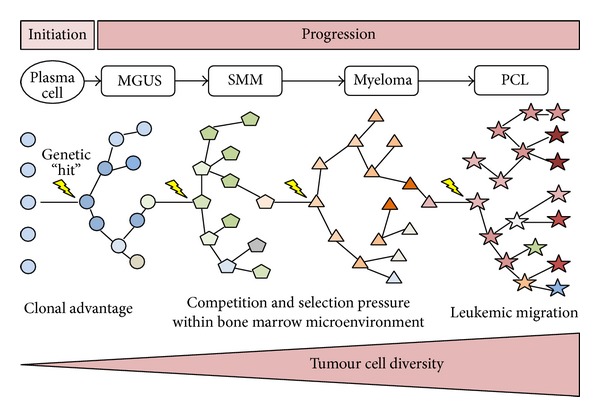
Initiation and progression of myeloma. A postgerminal centre B cell receives a genetic “hit” which immortalizes the cell and initiates transition to the indolent phase of monoclonal gammopathy of undetermined significance (MGUS). MGUS clones may then transition through the other disease phases of smouldering multiple myeloma (SMM), myeloma, and plasma cell leukemia (PCL) as genetic “hits”, which confer a survival advantage and are acquired over time. Clonal evolution develops through branching pathways whereby numerous ecosystems composed of multiple subclones exist at each disease phase, as represented by the differing shapes. At the end of this process, proliferative clones no longer become confined to the bone marrow and expand rapidly as a leukemic phase. At each disease phase, the precursor clones are present only at a low level as they have been outcompeted by more advantageous clones. It should be noted that the above figure represents an oversimplification of myeloma initiation and progression, as the process is highly complex with multiple pathways possible at any one time (adapted from Morgan et al., 2012 [8]).
1.2. Nonhyperdiploid and Hyperdiploid Myeloma
Along the progression from MGUS to PCL, genetic aberrations can be classified as primary events, contributing to plasma cell immortalisation, or secondary events, contributing to disease progression. This classification facilitates the division of myeloma into two broad groups, nonhyperdiploidy myeloma and hyperdiploidy myeloma, based on one of two genetic aberrations observed in the primary phase [9, 10], a distinction originally suggested by Smadja et al., supported by the work of others, who put forward the idea of myeloma representing two closely related diseases [11–13]. Nonhyperdiploidy myeloma involves the translocation (t) of immunoglobulin heavy chain alleles (IGH@) at 14q32 with various partner chromosomes including 4, 6, 11, 16, and 20. These primary translocations occur due to aberrant class switch recombination (CSR) in lymph node germinal centres and act to juxtapose the partner chromosome oncogenes under the influence of the IGH@ enhancer region. Hyperdiploidy myeloma is generally associated with better survival and involves trisomies of the odd numbered chromosomes 3, 5, 7, 9, 11, 15, 19, and 21 coupled to a low prevalence of IGH@ translocations [14, 15]. Either directly, or indirectly, one consequence of hyperdiploid and nonhyperdiploid events is to result in deregulation of the G1/S cell cycle transition point via the overexpression of cyclin D genes, an event shown to be a key early molecular abnormality in myeloma (Figure 2) [16]. For completion, it should be stated that exceptions to the hyperdiploidy and nonhyperdiploidy divisions do exist and that cases with primary translocations and multiple trisomies are detected in a minority.
Figure 2.
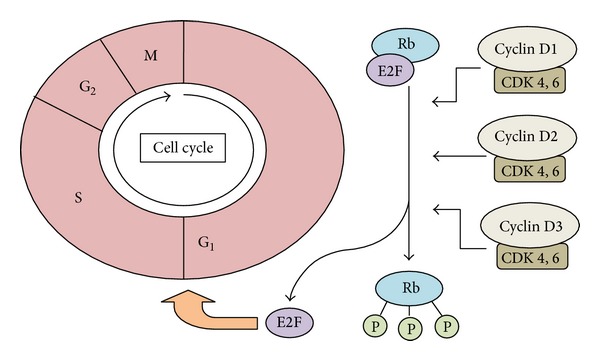
Overexpression of cyclin D genes influence cell cycle progression at the G 1 /S transition point in myeloma. Increased cyclin D gene expression through hyperdiploid or nonhyperdiploid events in myeloma facilitates activation of a cyclin-dependent kinase (CDK 4 or 6). The respective CDK then phosphorylates Rb (retinoblastoma protein), which subsequently resides from its role inhibiting E2F transcription factors allowing these to facilitate cell cycle progression at the G1/S transition. G1: Gap-1 phase; S: synthesis phase; G2: Gap-2 phase; M: mitosis; P: phosphate group.
1.3. Secondary Genetic Events and the Bone Marrow Microenvironment
Secondary genetic events drive disease progression and are generally found at higher frequencies in SMM, myeloma, and PCL. These secondary events cooperate with primary events to produce the malignant phenotype of myeloma and include secondary translocations, copy number variations (CNV), loss of heterozygosity (LOH), acquired mutations, and epigenetic modifications. Coupled to the development of these secondary events, clonal cells require a specialised relationship with bone marrow stromal cells for growth and survival. Studies have shown that this microenvironment interaction is highly complex, involving positive and negative interactions between the many cell types mediated through a variety of adhesion molecules, receptors, and cytokines [17, 18]. Furthermore, the derangement of these stromal-clone interactions has been shown to have important consequences in facilitating plasma cell homing to the bone marrow [18], promoting plasma cell immortalisation, and helping spread to secondary bone marrow sites [19, 20]. This stromal-clone relationship is relatively poorly understood at present but represents an area where investigation is ongoing and treatments are likely to be developed [21, 22].
1.4. Inherited Variation
Several studies have demonstrated that the majority of, if not all, myeloma cases pass through the MGUS phase [23, 24]. Therefore, in order to gain a fuller understanding of the disease, it is important to consider the genetic and environmental factors influencing transition to this indolent phase. From familial studies on index cases of myeloma, it is apparent that inherited genetic variation can predispose to the development of MGUS as these families have a two- to four-fold increased risk of developing the premalignant condition [25]. By investigating these families further, molecular epidemiology studies identified three genetic loci with associated gene pairs (2p: DNMT3A and DTNB, 3p: ULK4 and TRAK1, 7p: DNAH11 and CDCA7L) which incur a modest but increased risk of developing myeloma [26]. The complete functional role of these gene pairs is currently unknown, although deregulation of the proto-oncogene MYC encoding a transcription factor which regulates genes involved in DNA replication, cell proliferation, and apoptosis, has been implicated [26]. From these initial studies, it is likely that more susceptibility loci will be identified in the future and that these may be correlated to specific myeloma subtypes. The reliable identification of those at risk of developing myeloma would be an important advancement as it may facilitate comprehensive disease monitoring and early disease detection. Furthermore, it could be postulated that future targeted therapies or gene knockdown interventions may be developed against these susceptibility loci to restrict progression to myeloma altogether.
2. Chromosomal Translocations in Myeloma
Chromosomal translocations account for 40–50% of primary events in myeloma and strongly influence disease phenotype [9]. Secondary translocations, not associated with aberrant CSR, occur later in disease and are likely to represent progression events. The key primary and secondary translocations occurring in myeloma are highlighted in Figure 3.
Figure 3.
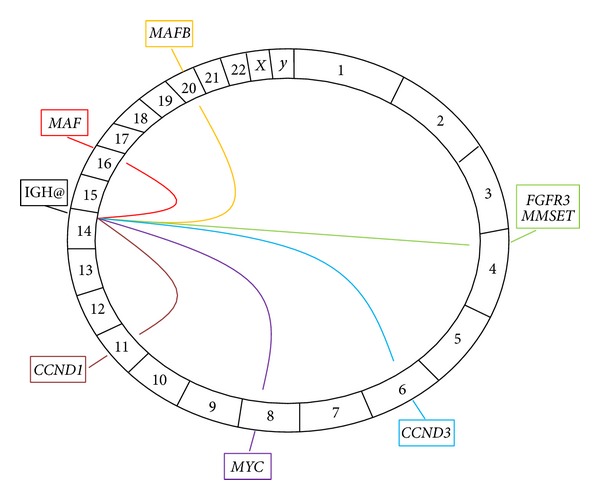
The key chromosomal translocations in myeloma. A Circos plot, with the chromosomes arranged in a clockwise direction, demonstrating the key translocations in myeloma. The translocations are represented as lines emerging from the immunoglobulin heavy chain (IGH@) locus on chromosome 14 to their respective partner chromosomes. The genes involved in each translocation are represented in boxes outside the plot. All translocations represent primary events except t(8; 14) involving MYC which is a secondary translocation.
2.1. t(4; 14) in Myeloma
The t(4; 14) is observed in 15% of myeloma cases and has been associated with an adverse prognosis in a variety of clinical settings such as those receiving high dose therapy with autologous stem cell transplant (ASCT) [27–30]. Pathologically, t(4; 14) results in the overexpression of two genes, FGFR3 and MMSET, by juxtaposition next to the IGH@ enhancers [31]. The upregulation of FGFR3 results in the ectopic expression of the FGFR3 tyrosine kinase receptor, an aberration with a currently unclear role in myelomagenesis. Interestingly, the pathogenic role of FGFR3 is somewhat in question, as approximately 30% of t(4; 14) tumours are imbalanced and lack FGFR3 expression due to loss of the derivative 14 chromosome [28, 32]. Furthermore, in these 30% lacking FGFR3 expression, the adverse prognosis of t(4; 14) remains [28], lending support for the role of the second gene MMSET. MMSET is overexpressed in all t(4; 14) tumours and encodes a chromatin-remodelling factor with histone methyltransferase (HMT) activity [27]. As for FGFR3, the exact role MMSET plays in pathogenesis is unclear although epigenetic regulation and a role in DNA repair have been suggested [33, 34]. In keeping with the unifying event of cyclin D deregulation, t(4; 14) with MMSET and/or FGFR3 overexpression have been shown to upregulate CCND2, and in some instances CCND1, through an unknown mechanism [9]. It is interesting to note that despite the poor prognosis associated with t(4; 14) a clear survival advantage in these tumours has recently been demonstrated through early treatment with the proteasome inhibitor bortezomib [35, 36], with a suggestion that prolonged bortezomib treatment can overcome the adverse prognosis altogether [35, 36]. This point demonstrates that future myeloma prognostication is likely to be determined by the success of therapeutically targeting high-risk lesions through a personalised approach.
2.2. t(6; 14) and t(11; 14) in Myeloma
The t(6; 14) is a rare translocation present in 2% of myeloma patients which results in the direct upregulation of the CCND3 gene via juxtaposition to the IGH@ enhancers [27, 38]. t(11; 14) is more common, occurring in approximately 17% of myeloma patients and also directly upregulates a cyclin D gene in the form CCND1 [27, 39]. Gene expression studies have shown that the overexpression of CCND3 and CCND1 results in a clustering of downstream gene expression suggesting that activation of these two genes results in the deregulation of common downstream transcriptional events [27]. Due to the seeming importance of cyclin D gene deregulation in myeloma, cyclin D inhibitors with a variety of specificities have shown promise targeting myeloma in vitro [40, 41], with many of these inhibitors now entering early human trials. Unlike t(4; 14), the overall prognostic impact of these two translocations is neutral [42], although t(11; 14) patients do show considerable heterogeneity and in some instances the translocation may manifest with an aggressive phenotype such as PCL.
2.3. t(14; 16) and t(14; 20) in Myeloma
The t(14; 16) and t(14; 20) both result in increased expression of a MAF family oncogene and combined are identified in 5–10% of presenting myeloma cases [27]. Specifically, t(14; 16) results in overexpression of the MAF gene splice variant c-MAF, a transcription factor which upregulates a number of genes including CCND2 by binding directly to its promoter [43]. t(14; 16) has been associated with a poor prognosis in a number of clinical series [29, 44], although this concept has recently been challenged by retrospective multivariate analysis on 1003 newly diagnosed myeloma patients which showed t(14; 16) not to be prognostic [45]. t(14; 20) is the rarest translocation involving the IGH@ and results in upregulation of the MAF gene paralog MAFB. Microarray studies have demonstrated that MAFB overexpression results in a very similar gene expression profile (GEP) to that seen with c-MAF [27], suggesting that common downstream targets, including CCND2, are deregulated by each. Interestingly, t(14; 20) is associated with a poor prognosis when present in myeloma but correlates to long-term stable disease when found in MGUS and SMM [46]. This suggests that the translocation alone is not responsible for the poor prognosis but that additional genetic events are required.
2.4. Secondary Translocations in Myeloma
As opposed to primary translocations, secondary translocations are CSR-independent events occurring later in disease. Furthermore, although the most frequent secondary translocation is t(8; 14), they do not always involve the IGH@ at 14q32 with approximately 40% linking different partner genes [47]. The gene typically deregulated by secondary translocations is MYC, the overexpression of which is linked directly to late disease stages and indirectly to a poor prognosis via a strong correlation to high levels of serum β 2-microglobulin (Sβ 2M) [48], an established indicator of a poor prognosis [49]. The frequency of MYC overexpression from secondary translocations supports its role as a progression event, as it is infrequently witnessed in MGUS but seen in 15% of myelomas and 50% of advanced disease [48, 50]. In opposition to this, a mouse model has previously demonstrated that the sporadic activation of a MYC transgene in germinal centre B cells of MGUS-prone mice results in the universal development of myeloma [51], whereas as previously discussed, an association also exists between MYC deregulation and certain genetic loci linked with myeloma susceptibility [26]. From these conflicting findings, it appears that MYC may play a role in both early and late disease phases and that further studies are required to elucidate an exact role for the gene.
3. Copy Number Variations in Myeloma
Copy number variations result from gains and losses of DNA and are common events in myeloma. These gains and losses can be both focal or of an entire chromosome/chromosome arm. In general, losses of DNA contribute to malignancy through loss of tumour suppressor genes, whereas gains are pathogenic through oncogene overexpression/activation.
3.1. Hyperdiploidy
Hyperdiploidy involves trisomies of the odd numbered chromosomes and is an event witnessed in approximately 50% of myeloma cases [14]. More common in elderly patients and associated with a high incidence of bone disease, hyperdiploidy confers a relatively favourable prognosis in the majority of cases [14], a factor held particularly true in instances where amplification 5q31.3 is concurrently present [52]. The underlying mechanism to generate hyperdiploidy is unknown, although one hypothesis, based on what is suggested to occur in hyperdiploid acute lymphoblastic leukemia, is that a single catastrophic mitosis results in the gain of whole chromosomes rather than their serial accumulation over time [53]. Along with the underlying mechanism, the consequence of hyperdiploidy towards myelomagenesis is poorly understood. However, alongside the known dysfunction of cyclin D genes, recent GEP studies have demonstrated that a high proportion of protein biosynthesis genes, specifically ribosomal protein genes representing end-points in MYC, NF-κB, and MAPK signalling pathways, are also concurrently overexpressed in hyperdiploid tumours [54, 55]. One explanation for this is that these genes are overexpressed due to rapid cell proliferation. This however is unlikely, as myeloma has a distinctively low proliferation rate. Instead, it is proposed that the overexpression is driven by gene copy number increases, with hyperdiploid cells then possessing more ribosomes and translational initiation factors to promote myelomagenesis through the overexpression of cellular growth genes [54].
3.2. Gain of 1q
Gain of the chromosome 1q arm (+1q) is an event observed in 35–40% of presenting myeloma cases and one which is frequently observed along with loss of 1p [56–59]. +1q is associated with a poor prognosis in patients treated both intensively and nonintensively and is an observation which remains when other adverse cytogenetic lesions which frequently coexist are removed [57, 60, 61]. Despite this knowledge, the relevant genes on 1q are not fully explored. One region of the chromosome arm which has been identified as a frequently minimally amplified region; however, 1q21 does contain many candidate oncogenes in the form of CKS1B, ANP32E, BCL-9, and PDZK1 [57, 60, 62]. The importance of this region is supported by the demonstration of a strong association between +1q21 and an adverse prognosis using both fluorescence in situ hybridization (FISH) and GEP techniques [56, 57, 61]. Of these genes, ANP32E, a protein phosphatise 2A inhibitor with a role in chromatin remodelling and transcriptional regulation, is of particular interest as it has been shown to be independently associated with shortened survival [57]. These findings support the importance of +1q in myeloma pathogenesis and suggest that patients in this group may benefit from specific inhibitors of the candidate genes and pathways identified.
3.3. Loss of 1p
Whole arm deletion or interstitial deletions of the 1p chromosome arm are observed in approximately 30% of myeloma patients and are associated with a poor prognosis in a range of treatment settings [57, 63, 64]. Molecular genetics has revealed that two regions of 1p, 1p12, and 1p32.3 are particularly important in myeloma pathogenesis when deleted. 1p12 may be hemi- or homozygously deleted and contains the candidate tumour suppressor gene FAM46C [5]. The function of FAM46C is unknown, although recent sequencing and homology studies have shown that its expression is correlated to both that of ribosomal proteins and eukaryotic initiation/elongation factors involved in protein translation [5]. FAM46C is considered a gene of significance as it has been shown to be frequently mutated in myeloma whilst also being independently correlated to a poor prognosis [5, 57, 58, 63]. 1p32.3 may also be hemi- and homozygously deleted and contains the two target genes, FAF1 and CDKN2C. CDKN2C is a cyclin-dependent kinase 4 inhibitor involved in negative regulation of the cell cycle, whereas FAF1 encodes a protein involved in initiation and/or enhancement of apoptosis through the Fas pathway. Homozygous deletion of 1p32.3 is associated with a poor prognosis in those receiving ASCT whereas in those receiving nonintensive treatment its prognostic impact is neutral [63]. Significant evidence points to CDKN2C as being the influential gene lost through homozygous 1p32.3 deletion [63, 65], although as CDKN2C and FAF1 lie in such close proximity, the vast majority of deletions lose both genes and therefore the importance of FAF1 relative to CDKN2C is difficult to delineate.
3.4. Loss of Chromosome 13/13q
Chromosome 13 deletion is observed in approximately 50% of myeloma cases and is commonly associated with nonhyperdiploid tumours [66–68]. In approximately 85% of cases, deletion of chromosome 13 constitutes a monosomy or loss of the q arm, whereas in the remaining 15% various interstitial deletions occur [66, 69]. With this, the identification of key genes contributing to myeloma pathogenesis is challenging as often a level of gene function remains from the residual allele(s). Despite this, molecular studies have shown that the tumour suppressor gene RB1 is significantly underexpressed in del(13/13q) and may therefore result in inferior negative cell cycle regulation [57]. To establish the prognostic impact of del(13/13q) is challenging due to its frequent association with other high-risk lesions, such as that of t(4; 14) where it is concurrently present in approximately 90% of cases [59]. When del(13/13q) is detected via conventional cytogenetics a link to poor survival exists [70, 71]. However, when detected via FISH, and in the absence of coexisting high-risk lesions, its significance towards survival is lost [42, 72]. This finding suggests that the historical link between del(13/13q) and a poor prognosis is therefore a surrogate of its association with high-risk lesions. One caveat to this statement however is that few long term follow-up studies comparing patient outcomes with or without these high-risk lesions have been completed whereas several long-term studies comparing the presence or absence of del(13/13q) do exist. In one of these studies, conducted by Gahrton et al. [73], a 96-month followup of 357 myeloma patients treated with either autologous transplantation or tandem autologous/reduced intensity conditioning allogenic transplantation (auto/RICallo) showed that whilst del(13/13q) acted as a poor prognostic marker for those receiving autologous transplantation this factor was apparently overcome for patients with del(13/13q) receiving auto/RICallo. This therefore suggests that del(13/13q) may have value as a poor prognostic marker for long-term outcomes in those receiving autologous transplantation.
3.5. Loss of 17p
The majority of chromosome 17 deletions are hemizygous and of the whole p arm, a genetic event observed in approximately 10% of new myeloma cases with this frequency increasing in later disease stages [29, 74]. The relevant gene deregulated in del(17p) is thought to be the tumour suppressor gene TP53, as GEP has shown that myeloma samples with monoallelic 17p deletions express significantly less TP53 compared to nondeleted samples [57]. Furthermore, in cases without del(17p) the rate of TP53 mutation is <1%, whereas in cases with del(17p) this rises to 25–37% [75]; a finding providing some evidence that monoallelic 17p deletion contributes to disruption of the remaining allele. The TP53 gene has been mapped to 17p13 and is known to function as a transcriptional regulator influencing cell cycle arrest, DNA repair, and apoptosis in response to DNA damage. In myeloma, del(17p) is the most important molecular finding for prognostication as it linked to an aggressive disease phenotype, a greater degree of extramedullary disease, and shortened survival [29, 42, 76]. It is hypothesised that PCL is largely a consequence of TP53 dysfunction, as the majority of these cases have abnormalities in the gene [74]. Furthermore, most, if not all, human myeloma cell lines which survive in laboratory cell culture have TP53 deficiency, further suggesting its importance in extramedullary disease. Despite the consensus that TP53 is the relevant gene disrupted in del(17p); however, it should be stated that no direct biological evidence exists to support this hypothesis and that further exploration of the genetic consequences of the deletion is required.
3.6. Other Chromosomal Losses
Many other chromosomal deletions, focal copy number losses, and regions of LOH are seen in myeloma, and as with the deletions of 1p, 13/13q, and 17p, the relatively high frequencies of these events in regions containing tumour suppressor genes suggest they are “driver” lesions contributing to myelomagenesis. Chromosome 11q deletion is observed in 7% of myeloma cases and harbours the tumour suppressor genes BIRC2 and BIRC3 [57]. del(14q) is a common event found in 38% of cases and includes the tumour suppressor gene TRAF3 [57]. 16q deletion is another common event, seen in 35% of myeloma cases, and contains the tumour suppressor genes CYLD and WWOX [57]. All of these genes, except WWOX which is implicated in apoptosis [77], are involved in the NF-κB pathway and demonstrate that activation of this signalling pathway is important in myeloma pathogenesis [57, 78, 79]. del(12p) is another lesion of interest in myeloma, as a recent single-nucleotide polymorphism (SNP) assay found it to be an independent adverse prognostic marker in 192 newly diagnosed patients [52]; a finding however not repeated in other studies [57]. Two further common chromosomal arm deletions frequently witnessed in myeloma are del(6q) and del(8p), observed 33% and 19–24% of cases, respectively [57, 80–82]. The relevance of del(6q) towards survival is as yet not clear. For del(8p) however, it has been shown that this aberration acts as an independently poor prognostic factor for both progression free survival (PFS) and overall survival (OS) [80, 81]. Furthermore, it has been shown that the tumour necrosis factor-related apoptosis-inducing ligand (TRALI) receptor gene is located on 8p, and that during del(8p) a consequential downregulation of TRALI occurs [83]. As TRALI is associated with TNF-induced apoptosis, it is proposed that with reduced receptor expression in del(8p) the sensitivity of tumour cells to TRAIL-medicated apoptosis may be decreased providing an advantage for the immune escape of malignant clones from surveillance by natural killer cells and cytotoxic T lymphocytes [84].
4. Deregulation of Myeloma Cellular Pathways and Processes
A range of signalling pathways are deregulated in myeloma and contribute towards pathogenesis through associations with proliferation, survival, apoptosis, migration, and drug resistance (Figure 4) [85]. Other cellular processes such as DNA repair, RNA editing, protein homeostasis, and cell differentiation may also contribute towards myelomagenesis through aberrant functioning.
Figure 4.
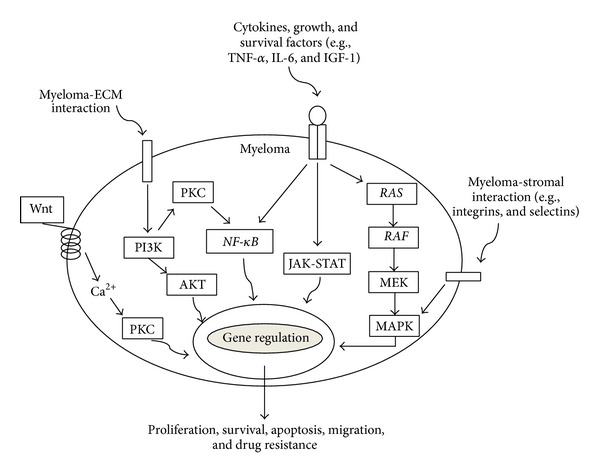
Signalling pathways involved in myeloma pathogenesis. The various pathways involved in myeloma pathogenesis may be stimulated via exogenous factors, such as Wnt proteins, myeloma-stromal interactions, cytokines, growth and survival factors, and myeloma-extracellular matrix (ECM) interactions, or the pathways may be aberrantly activated endogenously through genetic abnormalities such as activating mutations in RAS, RAF, and NF-κB genes.
4.1. NF-κB
NF-κB comprises a family of structurally related transcription factors which are upregulated during cellular stress to mediate gene responses. Salient to cancer, aberrant activation of NF-κB contributes to cell survival, proliferation and adhesion pathways. In myeloma, the NF-κB pathway is constitutively active in at least 50% of cases and is likely to represent a “driver” event due to its differing activation frequency between MGUS and later disease phases [78, 79]. Interestingly, NF-κB may be upregulated in both plasma cells and surrounding bone marrow stromal cells. In these supporting cells, NF-κB stimulates the release of key cytokines such as IL-6, BAFF and APRIL resulting in paracrine stimulation and critical survival signals to malignant clones [86, 87]. Activation of NF-κB within myeloma cells occurs through a range of mechanisms, including the inactivation of pathway suppressors through gene deletions and/or mutations, and pathway hyperactivity due to translocations and copy number gains [78, 79]. Furthermore, a recent whole genome sequencing (WGS) and whole exome sequencing (WES) study expanded the possible mechanisms through which the pathway may be activated by demonstrating 14 novel mutations/rearrangements affecting 11 NF-κB pathway genes [5]. The frequency with which NF-κB is deregulated in myeloma supports its importance in pathogenesis, although the prognostic impact for many of the implicated genes are yet to be fully elucidated. As the pathway involves the proteasome, inhibitors of this protein complex have been developed, with evidence suggesting that tumours “addicted” to the NF-κB pathway are particularly sensitive to these drugs [79]. Any adverse prognosis of NF-κB activation may therefore be potentially therapeutically ameliorated in the future.
4.2. Cell Proliferation
Of the pathways highlighted in Figure 4, three of them, the MAPK pathway, the JAK-STAT pathway, and the PI3K pathway, are particularly implicated in myelomagenesis through influences on cell proliferation.
4.2.1. The Mitogen Activated Protein Kinase (MAPK) Pathway
The MAPK pathway is a highly conserved cellular signalling cascade involved in cell differentiation, proliferation, and survival. The pathway may be stimulated via a range of inflammatory cytokines, such as TNF-a, IL-6, and IGF-1, which in turn activate the downstream kinase cascades RAS, RAF, MEK, and MAPK ultimately influencing gene expression. Two dominant oncogenes in the MAPK pathway, deregulated in many cancers, are NRAS and KRAS. These genes are frequently mutated in myeloma with a combined prevalence of 20–35% [88]. RAS mutations are likely to represent progression events as they are rarely found in MGUS but occur more frequently in later disease [89]. Additionally, RAS mutations are a poor prognostic marker, frequently being associated to a more aggressive disease phenotype and shortened survival times [88]. Recently however, it has been suggested that KRAS, and not NRAS, is the more influential gene impacting on prognosis [88], a finding which may have important consequences if genetic lesions are used to define risk. Due to the importance of RAS mutations and the MAPK pathway across many cancers, therapeutic inhibitors within this area are a key focus of research.
Showing further importance of the MAPK pathway, a recent study by Chapman et al. identified that seven out of 161 (4%) myeloma patients harboured a previously unobserved mutation in the BRAF gene [5]. BRAF encodes a serine/threonine-protein kinase in which activating mutations are known to be important in many cancers including melanoma and hairy cell leukemia [90]. This has particular clinical relevance, as myeloma patients with BRAF mutations may benefit from newly developed BRAF inhibitors, drugs which in some instances have shown marked clinical activity [91]. The premise to perform genome analysis for BRAF mutations on 161 samples arose from an original WGS/WES study on 38 myeloma samples which revealed the mutation in one patient [5]. This highlights the advantage of WGS/WES, in that the technique can be used as a screening tool to identify unknown genetic aberrations across the whole genome/exome, a benefit which may prove paramount in identifying novel therapeutic targets and disease biomarkers.
4.2.2. The JAK-STAT Pathway
The JAK-STAT pathway is constitutively activated in 50% of myeloma samples as well as a proportion of surrounding bone marrow stromal cells [92, 93]. The principal method thought to induce JAK-STAT activation is through autocrine and paracrine stimulation with IL-6, a cytokine shown to be important in myelomagenesis through the regulation of growth and survival [94, 95]. One key consequence of JAK-STAT activation is overactivity of STAT3, a STAT family transcription factor which results in high expression of the antiapoptotic protein Bcl-xL [94], a protein correlated to chemoresistance in myeloma patients [96]. With this, inhibition of STAT3 with compounds such as curcumin, atiprimod, and the JAK2 kinase inhibitor AG490 are associated with inhibition of IL-6-induced myeloma survival in vitro [97–99]. Furthermore, inhibition of STAT3 has been shown to sensitize the U266 myeloma cell line to apoptosis induced through conventional chemotherapy agents [100]. Development of STAT3 inhibitors may therefore facilitate improved results with conventional chemotherapy agents in the future.
4.2.3. The Phosphatidylinositol-3 Kinase (PI3K) Pathway
A range of molecular signals, such as IL-6 and IGF-1, acting on tyrosine kinase receptors can activate the PI3K pathway leading to phosphorylation of the serine-threonine-specific kinase AKT. AKT then subsequently activates several downstream targets including mTOR, GSK-3B and FKHR which influence many processes including cell proliferation and apoptosis resistance. Deregulation of the PI3K pathway is thought to be important in myeloma as phosphorylated AKT, an indicative marker of pathway activity, is observed in approximately 50% of cases [101]. Additionally, DEPTOR, a positive regulator of the PI3K pathway is commonly upregulated in myeloma, especially in those with MAF translocations [102], further demonstrating pathway activity. Of interest, unlike the MAPK pathway, the PI3K pathway is rarely mutated in myeloma [5]. However, as the pathway is known to be active, therapeutic targeting of PI3K is of interest within myeloma research.
4.3. Cell Cycle Deregulation
Alongside the overexpression of cyclin D genes in myeloma, the loss of function to negative cell cycle regulatory genes also proves to be a key event which destabilises cell cycle regulation. For example, the downregulation of CDKN2C through del(1p), or the inactivation of CDKN2A via DNA methylation changes may both deregulate the G1/S transition as these genes encode cyclin-dependent kinase inhibitors [65, 103]. Inactivation of the tumour suppressor gene RB1, a negative cell cycle regulator, also affects the G1/S transition and may occur frequently due to monosomy 13 or infrequently due to homozygous deletion or mutational inactivation [57]. The disruption of RB1 is known to be a key pathological event in many cancers and the development of anti-cancer drugs targeting cell cycle regulators, including RB1, is a rapidly growing field.
4.4. Abnormal DNA Repair
Chromosomal instability is a defining feature of myeloma and contributes to the perpetual accumulation of genetic aberrations during disease progression. Despite this, consistent mutations of DNA repair genes have not been demonstrated in the disease, with loss of TP53 function through del(17p), found in 10% of cases, the most common finding. Another gene of emerging importance however is PARP1, a gene encoding the PARP1 enzyme which contributes to repairing ssDNA breaks. A recent GEP study has demonstrated that increased expression of PARP1 is associated with shortened survival in myeloma patients [104], whereas another GEP study identified the gene as one of 15 which may be used as an expression signature to define high-risk disease [105]. Investigations in this area may prove pivotal for myeloma, as PARP inhibitors have shown promising activity in clinical trials [106]. This activity is especially prominent in cancers with defective homologous recombination (HR)-mediated DNA repair mechanisms, as cells with defects in this system are sensitized to PARP inhibitors [107]. Although not a recognised de novo finding for myeloma, it has been shown that bortezomib can induce a HR-mediated DNA repair defective state, a so called “BRCAness”, through interference of BRCA1 and RAD51 recruitment to the sites of dsDNA breaks in vitro [104]. Thus, bortezomib and a PARP inhibitor may induce synthetic lethality and be utilised as a future combined treatment for myeloma.
4.5. Abnormal RNA Editing
A recent study revealed that nearly half of 38 myeloma samples contained mutations in genes involved in RNA processing, protein translation and the unfolded protein response (UPR) [5]. Four different mutations of DIS3, a gene encoding an exonuclease serving as the catalytic component of the exosome complex involved in regulating the abundance of RNA species [108, 109], were observed in 11% of samples [5]. The DIS3 gene has been mapped to 13q22.1, and in three out the four mutations identified loss of function was exhibited by monoallelic mutation coupled to deletion of the remaining allele [5]. This demonstrates the key contribution del(13) is likely to play to DIS3 mutations and suggests another implication of this genetic aberration alongside RB1 haploinsufficiency. Furthermore, two of the four mutations in DIS3 have been functionally characterised in microorganisms where they result in loss of enzymatic activity with consequential accumulation of RNA targets [110, 111]. As it has been shown that the exosome plays a vital role in regulating the available pool of mRNAs for translation [112], these mutational findings indicate that loss of DIS3 activity may contribute to myelomagenesis through deregulation of protein translation. Another gene, FAM46C, implicated in del(1p) and previously discussed, gives further support for the role of translational control in myeloma pathogenesis, as WGS/WES found this gene to be mutated in 13% of samples [5]. This frequency supports the implication of FAM46C in myeloma pathogenesis as recurrently mutated genes are likely to be of biological significance.
4.6. Protein Homeostasis: The Unfolded Protein Response
The UPR is essential for the normal functioning of plasma cells as it serves a critical function in the efficient production of immunoglobulin by regulating cellular responses to unfolded/misfolded protein in the endoplasmic reticulum. Of interest, sequencing has revealed mutations of the LRRK2 gene at a frequency of 8% [5]. LRRK2 encodes a serine-threonine kinase responsible for phosphorylating the eukaryotic translation initiation factor 4E-binding protein 1 (EIF4EBP1), a protein which functions in regulating protein translation. LRRK2 is predominantly known for its association with Parkinson's disease, where mutations in the gene are linked to a predisposition for the condition [113, 114]. As with other neurodegenerative conditions, Parkinson's disease is in part characterised by a dysfunctional UPR and abrogated protein, and as myeloma has a vastly increased rate of immunoglobulin production [115, 116], any changes in protein homeostasis are likely to be pathogenically important. Of related interest, sequencing data has also revealed mutations in the UPR gene XBP1, although at a low frequency of 3% [5]. When over-expressed in transgenic mice, a splice form of XBP1 has been shown to induce a myeloma-like syndrome [117], whereas in mice deficient of XBP1 B cells are able to proliferate and construct germinal centres but are unable to differentiate into immunoglobulin secreting plasma cells [118]. The exact role of XBP1 in human myeloma pathogenesis is unclear, although a recent study has shown that finding a high ratio between un-spliced and spliced variants of XBP1 in myeloma samples is linked to a poor outcome and serves as an independent prognostic factor [119].
4.7. Abnormal Plasma Cell Differentiation
One method to establish biologically significant gain-of-function changes in cancer genomes is to use WGS/WES to search for recurrent identical mutations in candidate oncogenes. Utilising this method, Chapman et al. found two myeloma patients from 38 harboured an identical mutation (K123R) in the DNA-binding domain of the interferon regulatory factor 4 (IRF4) [5]. As its name suggests, IRF4 is involved in regulating the transcription of interferon's whilst it also plays an important role in B cell proliferation and differentiation. Interestingly, a recent RNA-inference-based genetic screen revealed that IRF4 function is required for myeloma cell line survival as inhibition of the gene proved toxic to the malignant cells [120], an in vitro finding supporting the genes role in pathogenesis. One way in which IRF4 acts is as a transcription factor for BLIMP1, also a transcription factor itself which plays a key role in plasma cell differentiation. The Chapman et al. study identified two mutations in the BLIMP1 gene from their 38 samples, and as loss-of-function mutations in BLIMP1 are known to occur in diffuse large B-cell lymphoma [121], this suggests BLIMP1 mutations may be of pathogenic importance to myeloma. Of consideration, as myeloma is a malignancy of terminally differentiated plasma cells, the importance of dysfunction within differentiation pathways may be of less importance than in cancers of immature cells, further studies are however required to investigate this.
4.8. Myeloma Bone Disease
Bone disease occurs in 80–90% of patients with myeloma and can be either focal or diffuse resulting in pain, pathological factures, cord compression and hypercalcaemia. A recent GEP study aimed to identify the molecular basis of patients presenting with bone disease in order to elucidate whether a gene expression signature could identify those at high-risk of skeletal-related events after randomization into one of two bisphosphonate arms [122]. The study identified that 50 genes were significantly associated with presenting bone disease, mostly from pathways involved in growth factor signalling, apoptosis and transcription regulation. The two most significantly differently expressed genes were the Wnt pathway inhibitors DKK1 and FRZB. The Wnt pathway is known to be important in regulating bone turnover and DKK1 has been shown to both inhibit osteoblast differentiation and increase bone resorption through an increase in the RANKL/OPG ratio [123, 124]. An antibody against DKK1 is now being tested in clinical trials after showing promise by improving bone disease and inhibiting myeloma cell growth in a murine model [125]. The GEP study was also able to make more generalised observations of which some have been previously been reported [72, 126]. For example, bone disease is more prevalent in those with a hyperdiploidy signature and less associated with t(4; 14) and MAF translocations. Interestingly, this study found that DKK1 and FRZB were more highly expressed in hyperdiploid tumours, providing a potential explanation for this finding. Secondly, patients with bone disease have shorter OS compared to those without [127], a finding which suggests bone disease significantly contributes to the impaired outcome of these patients, or, alternatively, that disease biology in myeloma with bone disease is distinctly different.
5. Epigenetic Changes in Myeloma
The study of epigenetics is an emerging field in myeloma and one which is demonstrating an increasing amount of influence on pathogenesis [128]. As outlined in Figure 5, the three main areas of epigenetic regulation include histone modification, RNA interference and DNA methylation.
Figure 5.
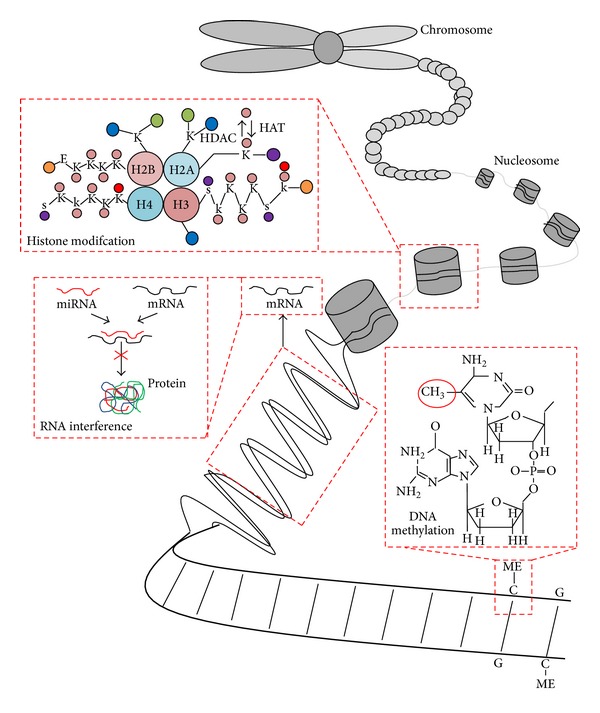
Mechanisms of epigenetic regulation. Three main forms of epigenetic modification include histone modification, RNA interference, and DNA methylation. Histone (chromatin) modification refers to the covalent posttranslational modifications to the N-terminal tails of the four core histone proteins; this modification is commonly acetylation/deacetylation changes at lysine residues mediated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). RNA interference is predominantly mediated through microRNAs, which inhibit the translation of mRNA into protein. DNA methylation occurs at cytosine residues of CpG dinucleotides and acts to regulate gene expression. Pink circle = acetyl group, purple circle = phosphate group, red circle = methyl group, blue circle = carboxyl terminus, green circle = ubiquitin, orange circle = amino terminus, k = lysine, E = glutamic acid, S = serine. H2A, histone 2A; H2B, histone 2B; H3, histone 3; H4, histone 4.
5.1. DNA Methylation
DNA Methylation changes occur at CpG dinucleotides which are generally found at higher frequencies in promoter regions, repeat sequences and transposable elements. Changes in DNA methylation act to regulate gene expression and are known to be important in contributing to cell development and differentiation as well as the progression of many cancers. Myeloma genomes, as for many other cancers, often follow a recognised pattern of methylation represented by global DNA hypomethylation and gene-specific hypermethylation [129]. A recent study using a genome-wide methylation microarray built on this knowledge to demonstrate that a marked loss methylation occurred at the transition from MGUS to myeloma [129]. Furthermore, gene-specific hypo and hypermethylation was demonstrated at this transition with the genes affected involved in the cell cycle, transcriptional and cell development pathways [129]. During progression from myeloma to PCL, rather than finding global DNA hypomehtylation, gene-specific hypermethylation was found in genes involved in cell adhesion and cell signalling [129]. This finding suggests these methylation changes may contribute to destabilisation of the stromal-clone relationship and promotion of clonal transition into the circulation and a proliferating leukaemic phase. The most significant DNA methylation changes, influencing cell survival, cell cycle progression and DNA repair, are seen in t(4; 14) tumours [33, 130], presumably as they over-express the MMSET gene which encodes a HMT transcription repressor.
5.2. Histone Modification
Other genes involved in methylation and chromatin modification are also deregulated in myeloma, including KDM6A, MLL genes and HOXA9 [5]. Recent sequencing observed that HOXA9 was ubiquitously expressed across 38 myeloma samples and hypothesised whether this gene represented a candidate oncogene [5]. The HOXA9 gene is primarily regulated by HMTs and encodes a DNA-binding transcription factor which contributes to regulating gene expression, morphogenesis and differentiation. As the majority of cases over-expressing HOXA9 in the study exhibited bi-allelic expression, consistent with deregulation of an upstream HMT event, genes involved in regulating HOXA9 were evaluated for mutations with findings revealing mutations in several genes: MLL, MLL2, MLL3, and MMSET [5]. To establish the functional importance of HOXA9 expression in myeloma, gene knockdown studies in a range of myeloma cell lines was performed and demonstrated that HOXA9-depleted cells incurred a competitive disadvantage against those with remaining HOXA9 function [5]. These findings indicate that the expression of HOXA9 has a role in myeloma pathogenesis and that these epigenetic changes may represent new therapeutic targets in myeloma.
5.3. MicroRNA Changes
MicroRNA (miRNA) genes encode a class of small RNAs (17–25 base pairs) which function to regulate the translation of other proteins by forming complementary base parings to specific mRNA transcripts. Studies have shown that miRNAs can act as both tumour suppressors and oncogenes in a range of cancers and that their transcriptional control is regulated by promoter methylation changes [131]. A substantial amount of work has been completed to investigate which miRNAs are differentially expressed in myeloma [132–134], and it has been shown that miRNA changes can deregulate genes and pathways relevant to myeloma pathogenesis including cell cycle progression, TP53 and MYC [135–137]. Although there is some discrepancy between which miRNAs are differentially expressed, and when, in myeloma, the overall conclusion is that miRNA deregulation is likely to be an important contributor to the malignancy and that further investigation is warranted to improve understanding and to highlight potential treatment targets.
6. Conclusion
Myeloma is a highly heterogeneous disease and one which may progress from an indolent, asymptomatic phase, to an aggressive extramedullary phase as genetic “hits” are acquired over time. The primary genetic events contributing towards plasma cell immortalisation can be broadly divided into a hyperdiploid group, characterised by trisomies of odd numbered chromosomes, and a nonhyperdiploid group, characterised by IGH@ translocations to various partner chromosomes; it appears that overexpression of the cyclin D family of genes is an almost universal sequelae of primary events. Secondary genetic events, contributing to disease progression, are complex and involve secondary translocations, CNVs, acquired mutations, LOH, and epigenetic modification. From the use of molecular techniques to investigate myeloma, many of these primary and secondary events are now well characterised, and from this characterisation, it is apparent that disease behaviour can be correlated to the genetic makeup of a patient's disease. With this, it is therefore essential that genetic research remains focused in translating molecular characterisation into clinically useful advances.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Kyle RA, Durie BG, Rajkumar SV, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24(6):1121–1127. doi: 10.1038/leu.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. The New England Journal of Medicine. 2006;354(13):1362–1369. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 3.Kyle RA, Therneau TM, Vincent Rajkumar S, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. The New England Journal of Medicine. 2002;346(8):564–569. doi: 10.1056/NEJMoa01133202. [DOI] [PubMed] [Google Scholar]
- 4.Kyle RA, Remstein ED, Therneau TM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. The New England Journal of Medicine. 2007;356(25):2582–2590. doi: 10.1056/NEJMoa070389. [DOI] [PubMed] [Google Scholar]
- 5.Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egan JB, Shi CX, Tembe W, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120(5):1060–1066. doi: 10.1182/blood-2012-01-405977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356–361. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 8.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nature Reviews Cancer. 2012;12(5):335–348. doi: 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 9.Kuehl WM, Bergsagel PL. Early genetic events provide the basis for a clinical classification of multiple myeloma. Hematology. 2005:346–352. doi: 10.1182/asheducation-2005.1.346. [DOI] [PubMed] [Google Scholar]
- 10.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. Journal of Clinical Oncology. 2005;23(26):6333–6338. doi: 10.1200/JCO.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 11.Smadja N-V, Fruchart C, Isnard F, et al. Chromosomal analysis in multiple myeloma: cytogenetic evidence of two different diseases. Leukemia. 1998;12(6):960–969. doi: 10.1038/sj.leu.2401041. [DOI] [PubMed] [Google Scholar]
- 12.Gould J, Alexanian R, Goodacre A, Pathak S, Hecht B, Barlogie B. Plasma cell karyotype in multiple myeloma. Blood. 1988;71(2):453–456. [PubMed] [Google Scholar]
- 13.Sawyer JR, Waldron JA, Jagannath S, Barlogie B. Cytogenetic findings in 200 patients with multiple myeloma. Cancer Genetics and Cytogenetics. 1995;82(1):41–49. doi: 10.1016/0165-4608(94)00284-i. [DOI] [PubMed] [Google Scholar]
- 14.Smadja NV, Bastard C, Brigaudeau C, Leroux D, Fruchart C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood. 2001;98(7):2229–2238. doi: 10.1182/blood.v98.7.2229. [DOI] [PubMed] [Google Scholar]
- 15.Dewald GW, Kyle RA, Hicks GA, Greipp PR. The clinical significance of cytogenetic studies in 100 patients with multiple myeloma, plasma cell leukemia, or amyloidosis. Blood. 1985;66(2):380–390. [PubMed] [Google Scholar]
- 16.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J., Jr. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106(1):296–303. doi: 10.1182/blood-2005-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. Journal of Clinical Investigation. 2012;122(10):3456–3463. doi: 10.1172/JCI61188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neri P, Ren L, Azab AK, et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. 2011;117(23):6202–6213. doi: 10.1182/blood-2010-06-292243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brennan SK, Wang Q, Tressler R, et al. Telomerase inhibition targets clonogenic multiple myeloma cells through telomere length-dependent and independent mechanisms. PLoS ONE. 2010;5(9) doi: 10.1371/journal.pone.0012487.12487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nature Reviews Cancer. 2002;2(3):175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- 21.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nature Reviews Cancer. 2007;7(8):585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 22.Roodman GD. Targeting the bone microenvironment in multiple myeloma. Journal of Bone and Mineral Metabolism. 2010;28(3):244–250. doi: 10.1007/s00774-009-0154-7. [DOI] [PubMed] [Google Scholar]
- 23.Rasmussen T, Haaber J, Dahl IM, et al. Identification of translocation products but not K-RAS mutations in memory B cells from patients with multiple myeloma. Haematologica. 2010;95(10):1730–1737. doi: 10.3324/haematol.2010.024778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–5417. doi: 10.1182/blood-2008-12-194241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altieri A, Chen B, Bermejo JL, Castro F, Hemminki K. Familial risks and temporal incidence trends of multiple myeloma. European Journal of Cancer. 2006;42(11):1661–1670. doi: 10.1016/j.ejca.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 26.Broderick P, Chubb D, Johnson DC, et al. Common variation at 3p22.1 and 7p15.3 influences multiple myeloma risk. Nature Genetics. 2012;44(1):58–61. doi: 10.1038/ng.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keats JJ, Reiman T, Maxwell CA, et al. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003;101(4):1520–1529. doi: 10.1182/blood-2002-06-1675. [DOI] [PubMed] [Google Scholar]
- 29.Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569–4575. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- 30.Chang H, Sloan S, Li D, et al. The t(4;14) is associated with poor prognosis in myeloma patients undergoing autologous stem cell transplant. British Journal of Haematology. 2004;125(1):64–68. doi: 10.1111/j.1365-2141.2004.04867.x. [DOI] [PubMed] [Google Scholar]
- 31.Chesi M, Nardini E, Lim RSC, Smith KD, Michael Kuehl W, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92(9):3025–3034. [PubMed] [Google Scholar]
- 32.Santra M, Zhan F, Tian E, Barlogie B, Shaughnessy J., Jr. A subset of multiple myeloma harboring the t(4;14)(p16;q32) translocation lacks FGFR3 expression but maintains an IGH/MMSET fusion transcript. Blood. 2003;101(6):2374–2376. doi: 10.1182/blood-2002-09-2801. [DOI] [PubMed] [Google Scholar]
- 33.Martinez-Garcia E, Popovic R, Min D-J, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117(1):211–220. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pei H, Zhang L, Luo K, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470(7332):124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. The New England Journal of Medicine. 2008;359(9):906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 36.Avet-Loiseau H, Leleu X, Roussel M, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p) Journal of Clinical Oncology. 2010;28(30):4630–4634. doi: 10.1200/JCO.2010.28.3945. [DOI] [PubMed] [Google Scholar]
- 37.Kyle RA, Child JA, Anderson K, et al. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. British Journal of Haematology. 2003;121(5):749–757. [PubMed] [Google Scholar]
- 38.Shaughnessy J, Jr., Gabrea A, Qi Y, et al. Cyclin D3 at 6p21 is dysregulated by recurrent chromosomal translocations to immunoglobulin loci in multiple myeloma. Blood. 2001;98(1):217–223. doi: 10.1182/blood.v98.1.217. [DOI] [PubMed] [Google Scholar]
- 39.Chesi M, Bergsagel PL, Brents LA, Smith CM, Gerhard DS, Kuehl WM. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood. 1996;88(2):674–681. [PubMed] [Google Scholar]
- 40.Baughn LB, di Liberto M, Wu K, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Research. 2006;66(15):7661–7667. doi: 10.1158/0008-5472.CAN-06-1098. [DOI] [PubMed] [Google Scholar]
- 41.Mao X, Cao B, Wood TE, et al. A small-molecule inhibitor of D-cyclin transactivation displays preclinical efficacy in myeloma and leukemia via phosphoinositide 3-kinase pathway. Blood. 2011;117(6):1986–1997. doi: 10.1182/blood-2010-05-284810. [DOI] [PubMed] [Google Scholar]
- 42.Avet-Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007;109(8):3489–3495. doi: 10.1182/blood-2006-08-040410. [DOI] [PubMed] [Google Scholar]
- 43.Hurt EM, Wiestner A, Rosenwald A, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5(2):191–199. doi: 10.1016/s1535-6108(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 44.Ross FM, Ibrahim AH, Vilain-Holmes A, et al. Age has a profound effect on the incidence and significance of chromosome abnormalities in myeloma. Leukemia. 2005;19(9):1634–1642. doi: 10.1038/sj.leu.2403857. [DOI] [PubMed] [Google Scholar]
- 45.Avet-Loiseau H, Malard F, Campion L, et al. Translocation t(14;16) and multiple myeloma: is it really an independent prognostic factor? Blood. 2011;117(6):2009–2011. doi: 10.1182/blood-2010-07-295105. [DOI] [PubMed] [Google Scholar]
- 46.Ross FM, Chiecchio L, Dagrada G, et al. The t(14;20) is a poor prognostic factor in myeloma but is associated with long-term stable disease in monoclonal gammopathies of undetermined significance. Haematologica. 2010;95(7):1221–1225. doi: 10.3324/haematol.2009.016329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dib A, Gabrea A, Glebov OK, Bergsagel PL, Kuehl WM. Characterization of MYC translocations in multiple myeloma cell lines. Journal of the National Cancer Institute. 2008;(39):25–31. doi: 10.1093/jncimonographs/lgn011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harousseau J-L, Bataille R. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001;98(10):3082–3086. doi: 10.1182/blood.v98.10.3082. [DOI] [PubMed] [Google Scholar]
- 49.Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. Journal of Clinical Oncology. 2005;23(15):3412–3420. doi: 10.1200/JCO.2005.04.242. [DOI] [PubMed] [Google Scholar]
- 50.Gabrea A, Martelli ML, Qi Y, et al. Secondary genomic rearrangements involving immunoglobulin or MYC loci show similar prevalences in hyperdiploid and nonhyperdiploid myeloma tumors. Genes Chromosomes and Cancer. 2008;47(7):573–590. doi: 10.1002/gcc.20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chesi M, Robbiani DF, Sebag M, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13(2):167–180. doi: 10.1016/j.ccr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Avet-Loiseau H, Li C, Magrangeas F, et al. Prognostic significance of copy-number alterations in multiple myeloma. Journal of Clinical Oncology. 2009;27(27):4585–4590. doi: 10.1200/JCO.2008.20.6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onodera N, McCabe NR, Rubin CM. Formation of a hyperdiploid karyotype in childhood acute lymphoblastic leukemia. Blood. 1992;80(1):203–208. [PubMed] [Google Scholar]
- 54.Chng WJ, Kumar S, VanWier S, et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Research. 2007;67(7):2982–2989. doi: 10.1158/0008-5472.CAN-06-4046. [DOI] [PubMed] [Google Scholar]
- 55.Agnelli L, Bicciato S, Mattioli M, et al. Molecular classification of multiple myeloma: a distinct transcriptional profile characterizes patients expressing CCND1 and negative for 14q32 translocations. Journal of Clinical Oncology. 2005;23(29):7296–7306. doi: 10.1200/JCO.2005.01.3870. [DOI] [PubMed] [Google Scholar]
- 56.Boyd KD, Ross FM, Chiecchio L, et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: analysis of patients treated in the MRC Myeloma IX trial. Leukemia. 2012;26(2):349–355. doi: 10.1038/leu.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walker BA, Leone PE, Chiecchio L, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010;116(15):e56–e65. doi: 10.1182/blood-2010-04-279596. [DOI] [PubMed] [Google Scholar]
- 58.Chang H, Qi X, Jiang A, Xu W, Young T, Reece D. 1p21 deletions are strongly associated with 1q21 gains and are an independent adverse prognostic factor for the outcome of high-dose chemotherapy in patients with multiple myeloma. Bone Marrow Transplantation. 2010;45(1):117–121. doi: 10.1038/bmt.2009.107. [DOI] [PubMed] [Google Scholar]
- 59.Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210–2221. doi: 10.1038/leu.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shaughnessy J. Amplification and overexpression of CKS1B at chromosome band 1q21 is associated with reduced levels of p27Kip1 and an aggressive clinical course in multiple myeloma. Hematology. 2005;10(supplement 1):117–126. doi: 10.1080/10245330512331390140. [DOI] [PubMed] [Google Scholar]
- 61.Shaughnessy JD, Jr., Zhan F, Burington BE, et al. Avalidated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 62.Shi L, Wang S, Zangari M, et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget. 2010;1(1):22–33. doi: 10.18632/oncotarget.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boyd KD, Ross FM, Walker BA, et al. Mapping of chromosome 1p deletions in myeloma identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as being genes in regions associated with adverse survival. Clinical Cancer Research. 2011;17(24):7776–7784. doi: 10.1158/1078-0432.CCR-11-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang H, Jiang A, Qi C, Trieu Y, Chen C, Reece D. Impact of genomic aberrations including chromosome 1 abnormalities on the outcome of patients with relapsed or refractory multiple myeloma treated with lenalidomide and dexamethasone. Leukemia and Lymphoma. 2010;51(11):2084–2091. doi: 10.3109/10428194.2010.524325. [DOI] [PubMed] [Google Scholar]
- 65.Leone PE, Walker BA, Jenner MW, et al. Deletions of CDKN2C in multiple myeloma: biological and clinical implications. Clinical Cancer Research. 2008;14(19):6033–6041. doi: 10.1158/1078-0432.CCR-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fonseca R, Oken MM, Harrington D, et al. Deletions of chromosome 13 in multiple myeloma identified by interphase FISH usually denote large deletions of the q arm or monosomy. Leukemia. 2001;15(6):981–986. doi: 10.1038/sj.leu.2402125. [DOI] [PubMed] [Google Scholar]
- 67.Avet-Loiseau H, Li J-Y, Morineau N, et al. Monosomy 13 is associated with the transition of monoclonal gammopathy of undetermined significance to multiple myeloma. Intergroupe Francophone du Myélome. Blood. 1999;94(8):2583–2589. [PubMed] [Google Scholar]
- 68.Chiecchio L, Protheroe RKM, Ibrahim AH, et al. Deletion of chromosome 13 detected by conventional cytogenetics is a critical prognostic factor in myeloma. Leukemia. 2006;20(9):1610–1617. doi: 10.1038/sj.leu.2404304. [DOI] [PubMed] [Google Scholar]
- 69.Avet-Loiseau H, Daviet A, Saunier S, Bataille R. Chromosome 13 abnormalities in multiple myeloma are mostly monosomy 13. British Journal of Haematology. 2000;111(4):1116–1117. doi: 10.1046/j.1365-2141.2000.02488.x. [DOI] [PubMed] [Google Scholar]
- 70.Tricot G, Barlogie B, Jagannath S, et al. Poor prognosis in multiple myeloma is associated only with partial or complete deletions of chromosome 13 or abnormalities involving 11q and not with other karyotype abnormalities. Blood. 1995;86(11):4250–4256. [PubMed] [Google Scholar]
- 71.Pérez-Simón JA, García-Sanz R, Tabernero MD, et al. Prognostic value of numerical chromosome aberrations in multiple myeloma: a FISH analysis of 15 different chromosomes. Blood. 1998;91(9):3366–3371. [PubMed] [Google Scholar]
- 72.Chng WJ, Santana-Dávila R, van Wier SA, et al. Prognostic factors for hyperdiploid-myeloma: effects of chromosome 13 deletions and IgH translocations. Leukemia. 2006;20(5):807–813. doi: 10.1038/sj.leu.2404172. [DOI] [PubMed] [Google Scholar]
- 73.Gahrton G, Iacobelli S, Björkstrand B, et al. Autologous/reduced-intensity allogeneic stem cell transplantation vs autologous transplantation in multiple myeloma: long-term results of the EBMT-NMAM2000 study. Blood. 2013;121(25):5055–5063. doi: 10.1182/blood-2012-11-469452. [DOI] [PubMed] [Google Scholar]
- 74.Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22(5):1044–1052. doi: 10.1038/leu.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lodé L, Eveillard M, Trichet V, et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica. 2010;95(11):1973–1976. doi: 10.3324/haematol.2010.023697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Drach J, Ackermann J, Fritz E, et al. Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood. 1998;92(3):802–809. [PubMed] [Google Scholar]
- 77.Jenner MW, Leone PE, Walker BA, et al. Gene mapping and expression analysis of 16q loss of heterozygosity identifies WWOX and CYLD as being important in determining clinical outcome in multiple myeloma. Blood. 2007;110(9):3291–3300. doi: 10.1182/blood-2007-02-075069. [DOI] [PubMed] [Google Scholar]
- 78.Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sutlu T, Alici E, Jansson M, et al. The prognostic significance of 8p21 deletion in multiple myeloma. British Journal of Haematology. 2009;144(2):266–268. doi: 10.1111/j.1365-2141.2008.07454.x. [DOI] [PubMed] [Google Scholar]
- 81.Neben K, Jauch A, Bertsch U, et al. Combining information regarding chromosomal aberrations t(4;14) and del(17pl3) with the international staging system classification allows stratification of myeloma patients undergoing autologous stem cell transplantation. Haematologica. 2010;95(7):1150–1157. doi: 10.3324/haematol.2009.016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gmidène A, Saad A, Avet-Loiseau H. 8p21.3 deletion suggesting a probable role of TRAIL-R1 and TRAIL-R2 as candidate tumor suppressor genes in the pathogenesis of multiple myeloma. Medical Oncology. 2013;30(2):p. 489. doi: 10.1007/s12032-013-0489-8. [DOI] [PubMed] [Google Scholar]
- 83.Gazitt Y. TRAIL is a potent inducer of apoptosis in myeloma cells derived from multiple myeloma patients and is not cytotoxic to hematopoietic stem cells. Leukemia. 1999;13(11):1817–1824. doi: 10.1038/sj.leu.2401501. [DOI] [PubMed] [Google Scholar]
- 84.Takeda K, Smyth MJ, Cretney E, et al. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. Journal of Experimental Medicine. 2002;195(2):161–169. doi: 10.1084/jem.20011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu H, Tamashiro S, Baritaki S, et al. TRAF6 activation in multiple myeloma: a potential therapeutic target. Clinical Lymphoma Myeloma and Leukemia. 2012;12(3):155–163. doi: 10.1016/j.clml.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chauhan D, Uchiyama H, Akbarali Y, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-κB. Blood. 1996;87(3):1104–1112. [PubMed] [Google Scholar]
- 87.Tai Y-T, Li X-F, Breitkreutz I, et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Research. 2006;66(13):6675–6682. doi: 10.1158/0008-5472.CAN-06-0190. [DOI] [PubMed] [Google Scholar]
- 88.Chng WJ, Gonzalez-Paz N, Price-Troska T, et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia. 2008;22(12):2280–2284. doi: 10.1038/leu.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rasmussen T, Kuehl M, Lodahl M, Johnsen HE, Dahl IMS. Possible roles for activating RAS mutations in the MGUS to MM transition and in the intramedullary to extramedullary transition in some plasma cell tumors. Blood. 2005;105(1):317–323. doi: 10.1182/blood-2004-03-0833. [DOI] [PubMed] [Google Scholar]
- 90.Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. The New England Journal of Medicine. 2011;364(24):2305–2315. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Patrawala S, Puzanov I. Vemurafenib, (RG67204, PLX4032): a potent, selective BRAF kinase inhibitor. Future Oncology. 2012;8(5):509–523. doi: 10.2217/fon.12.31. [DOI] [PubMed] [Google Scholar]
- 92.Bharti AC, Shishodia S, Reuben JM, et al. Nuclear factor-κB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103(8):3175–3184. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 93.Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 94.Kawano M, Hirano T, Matsuda T, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332(6159):83–85. doi: 10.1038/332083a0. [DOI] [PubMed] [Google Scholar]
- 95.Klein B, Zhang X-G, Lu Z-Y, Bataille R. Interleukin-6 in human multiple myeloma. Blood. 1995;85(4):863–872. [PubMed] [Google Scholar]
- 96.Tu Y, Renner S, Xu F-H, et al. BCL-X expression in multiple myeloma: possible indicator of chemoresistance. Cancer Research. 1998;58(2):256–262. [PubMed] [Google Scholar]
- 97.Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. Journal of Immunology. 2003;171(7):3863–3871. doi: 10.4049/jimmunol.171.7.3863. [DOI] [PubMed] [Google Scholar]
- 98.Amit-Vazina M, Shishodia S, Harris D, et al. Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. British Journal of Cancer. 2005;93(1):70–80. doi: 10.1038/sj.bjc.6602637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.de Vos J, Jourdan M, Tarte K, Jasmin C, Klein B. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. British Journal of Haematology. 2000;109(4):823–828. doi: 10.1046/j.1365-2141.2000.02127.x. [DOI] [PubMed] [Google Scholar]
- 100.Alas S, Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin’s lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clinical Cancer Research. 2003;9(1):316–326. [PubMed] [Google Scholar]
- 101.Aronson L, Davenport E, Giuntoli S, et al. Autophagy is a key myeloma survival pathway that can be manipulated therapeutically to enhance apoptosis. ASH Annula Meeting Abstracts. 2010;116(21):p. 4083. [Google Scholar]
- 102.Boyd KD, Walker BA, Wardell CP, et al. High expression levels of the mammalian target of rapamycin inhibitor DEPTOR are predictive of response to thalidomide in myeloma. Leukemia and Lymphoma. 2010;51(11):2126–2129. doi: 10.3109/10428194.2010.509893. [DOI] [PubMed] [Google Scholar]
- 103.Dib A, Barlogie B, Shaughnessy JD, Jr., Kuehl WM. Methylation and expression of the p16INK4A tumor suppressor gene in multiple myeloma. Blood. 2007;109(3):1337–1338. doi: 10.1182/blood-2006-09-049510. [DOI] [PubMed] [Google Scholar]
- 104.Neri P, Ren L, Gratton K, et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood. 2011;118(24):6368–6379. doi: 10.1182/blood-2011-06-363911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Decaux O, Lodé L, Magrangeas F, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myélome. Journal of Clinical Oncology. 2008;26(29):4798–4805. doi: 10.1200/JCO.2007.13.8545. [DOI] [PubMed] [Google Scholar]
- 106.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature Reviews Molecular Cell Biology. 2012;13(7):411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 107.Kummar S, Chen A, Parchment RE, et al. Advances in using PARP inhibitors to treat cancer. BMC Medicine. 2012;10, article 25 doi: 10.1186/1741-7015-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dziembowski A, Lorentzen E, Conti E, Séraphin B. A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nature Structural and Molecular Biology. 2007;14(1):15–22. doi: 10.1038/nsmb1184. [DOI] [PubMed] [Google Scholar]
- 109.Schmid M, Jensen TH. The exosome: a multipurpose RNA-decay machine. Trends in Biochemical Sciences. 2008;33(10):501–510. doi: 10.1016/j.tibs.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 110.Schneider C, Anderson JT, Tollervey D. The exosome subunit Rrp44 Plays a direct role in RNA substrate recognition. Molecular Cell. 2007;27(2):324–331. doi: 10.1016/j.molcel.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barbas A, Matos RG, Amblar M, López-Viñas E, Gomez-Puertas P, Arraiano CM. Determination of key residues for catalysis and RNA cleavage specificity. One mutation turns RNase II Into a ‘super-enzyme’. Journal of Biological Chemistry. 2009;284(31):20486–20498. doi: 10.1074/jbc.M109.020693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ibrahim H, Wilusz J, Wilusz CJ. RNA recognition by 3′-to-5′ exonucleases: the substrate perspective. Biochimica et Biophysica Acta. 2008;1779(4):256–265. doi: 10.1016/j.bbagrm.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 114.Paisán-Ruíz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 115.Masciarelli S, Fra AM, Pengo N, et al. CHOP-independent apoptosis and pathway-selective induction of the UPR in developing plasma cells. Molecular Immunology. 2010;47(6):1356–1365. doi: 10.1016/j.molimm.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cenci S, Sitia R. Managing and exploiting stress in the antibody factory. FEBS Letters. 2007;581(19):3652–3657. doi: 10.1016/j.febslet.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 117.Carrasco DR, Sukhdeo K, Protopopova M, et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell. 2007;11(4):349–360. doi: 10.1016/j.ccr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412(6844):300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 119.Bagratuni T, Wu P, Gonzalez de Castro D, et al. XBP1s levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood. 2010;116(2):250–253. doi: 10.1182/blood-2010-01-263236. [DOI] [PubMed] [Google Scholar]
- 120.Shaffer AL, Emre NCT, Lamy L, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226–231. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mandelbaum J, Bhagat G, Tang H, et al. BLIMP1 Is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell. 2010;18(6):568–579. doi: 10.1016/j.ccr.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu P, Walker BA, Brewer D, et al. A gene expression-based predictor for myeloma patients at high risk of developing bone disease on bisphosphonate treatment. Clinical Cancer Research. 2011;17(19):6347–6355. doi: 10.1158/1078-0432.CCR-11-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tian E, Zhan F, Walker R, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. The New England Journal of Medicine. 2003;349(26):2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 124.Qiang Y-W, Chen Y, Stephens O, et al. Myeloma-derived dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: a potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood. 2008;112(1):196–207. doi: 10.1182/blood-2008-01-132134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fulciniti M, Tassone P, Hideshima T, et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood. 2009;114(2):371–379. doi: 10.1182/blood-2008-11-191577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Robbiani DF, Chesi M, Bergsagel PL. Bone lesions in molecular subtypes of multiple myeloma. The New England Journal of Medicine. 2004;351(2):197–198. doi: 10.1056/NEJM200407083510223. [DOI] [PubMed] [Google Scholar]
- 127.Morgan GJ, Davies FE, Gregory WM, et al. First-line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC Myeloma IX): a randomised controlled trial. The Lancet. 2010;376(9757):1989–1999. doi: 10.1016/S0140-6736(10)62051-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sawan C, Vaissière T, Murr R, Herceg Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutation Research. 2008;642(1-2):1–13. doi: 10.1016/j.mrfmmm.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 129.Walker BA, Wardell CP, Chiecchio L, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. 2011;117(2):553–562. doi: 10.1182/blood-2010-04-279539. [DOI] [PubMed] [Google Scholar]
- 130.Brito JLR, Walker B, Jenner M, et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica. 2009;94(1):78–86. doi: 10.3324/haematol.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rouhi A, Mager DL, Humphries RK, Kuchenbauer F. MiRNAs, epigenetics, and cancer. Mammalian Genome. 2008;19(7-8):517–525. doi: 10.1007/s00335-008-9133-x. [DOI] [PubMed] [Google Scholar]
- 132.Zhoua Y, Chena L, Barlogiea B, et al. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(17):7904–7909. doi: 10.1073/pnas.0908441107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lionetti M, Biasiolo M, Agnelli L, et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood. 2009;114(25):e20–e26. doi: 10.1182/blood-2009-08-237495. [DOI] [PubMed] [Google Scholar]
- 134.Gutiérrez NC, Sarasquete ME, Misiewicz-Krzeminska I, et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia. 2010;24(3):629–637. doi: 10.1038/leu.2009.274. [DOI] [PubMed] [Google Scholar]
- 135.Pichiorri F, Suh S-S, Rocci A, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18(4):367–381. doi: 10.1016/j.ccr.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 136.Pichiorri F, Suh S-S, Ladetto M, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(35):12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chang T-C, Yu D, Lee Y-S, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nature Genetics. 2008;40(1):43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]