

Abstract
Uncoupling protein 2 (UCP2) is a mitochondrial membrane protein that regulates energy metabolism and reactive oxygen species (ROS) production. We generated mouse carboxy- and amino-terminal green fluorescent protein (GFP)-tagged UCP2 constructs to investigate the effect of UCP2 expression on cell proliferation and viability. UCP2-transfected Hepa 1–6 cells did not show reduced cellular adenosine triphosphate (ATP) but showed increased levels of glutathione. Flow cytometry analysis indicated that transfected cells were less proliferative than nontransfected controls, with most cells blocked at the G1 phase. The effect of UCP2 on cell cycle arrest could not be reversed by providing exogenous ATP or oxidant supply, and was not affected by the chemical uncoupler carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP). However, this effect of UCP2 was augmented by treatment with genistein, a tyrosine kinase inhibitor, which by itself did not affect cell proliferation on control hepatocytes. Western blotting analysis revealed decreased expression levels of CDK6 but not CDK2 and D-type cyclins. Examination of cell viability in UCP2-transfected cells with Trypan Blue and Annexin-V staining revealed that UCP2 transfection led to significantly increased cell death. However, characteristics of apoptosis were absent in UCP2-transfected Hepa 1–6 cells, including lack of oligonucleosomal fragmentation (laddering) of chromosomal DNA, release of cytochrome c from mitochondria, and cleavage of caspase-3. In conclusion, our results indicate that UCP2 induces cell cycle arrest at G1 phase and causes nonapoptotic cell death, suggesting that UCP2 may act as a powerful influence on hepatic regeneration and cell death in the steatotic liver.
Introduction
Uncoupling proteins (UCPs) are a family of mitochondrial inner membrane proteins. Five UCP homologs have been described so far. UCP1, mainly expressed in brown adipose tissue,1 was the first uncoupling protein characterized with proton transport activity.2 It is involved in adaptive thermoregulation through uncoupling of the electron transport chain from oxidative phosphorylation by dissipating the proton gradient between the mitochondrial intermembrane space and matrix.3 The later identified isoforms 2–4 include UCP3, which is predominately expressed in skeletal muscles and heart,4 and UCPs 4 and 5 [also called brain mitochondrial carrier protein-1 (BMPC1)], which are mostly expressed in the brain.5,6 UCP2 is the only uncoupling protein ubiquitously distributed in various tissues.7 Expression of UCP2 occurs in a wide variety of organs and tissues, including adipose tissue, muscle, heart, lung, kidney, and liver. Action of UCP2 reduces adenosoine triphosphate (ATP) production through thermogenesis or a futile cycle.8,9 Yeast expression of UCP210,11 and UCP311,12 results in increased respiration and decreased ability to maintain normal mitochondrial potential. Similar effects have been observed in mammalian cells.13,14 Recent literature suggests that the physiological roles of UCP2 may not be limited to uncoupling of oxidative phosphorylation and reduced ATP production. In addition to the effect on reduced ATP production, mitochondrial uncoupling proteins have been proposed to play a role in other physiological processes including: (1) Regulation of fatty acid and glucose oxidation,15 (2) regulation of reactive oxygen species (ROS) production,16,17 (3) body weight regulation,18 and (4) fever and thermoregulation.8,10 Mitochondria are the predominant energy supply of the cell and are the key regulators of apoptotic cell death.10 Located in the inner membrane of the mitochondria, increased expression of UCP2 has been reported to either positively20–23 or negatively24–26 regulate programmed cell death.
Recently, mitochondria have drawn attention as being potential regulators of cell proliferation and tumor suppression.27,28 In the present study, we investigate and report the effects of UCP2 overexpression on cell proliferation and viability using Hepa 1–6 cells. Our results, using this cell culture system, demonstrate that UCP2 negatively regulates cell proliferation and increases cell death in a liver cell line. Coupled with our observations that UCP2 is increased during steatosis and during ischemia reperfusion,29 these are important observations that have implications in the development of steatohepatitis, liver regeneration following surgical resection, and hepatic ischemia/reperfusion injury.
Experimental Procedures
Cell culture
Hepa 1–6 cells, Hela cells, 293 cells, and MG63 cells were cultured at 37°C in a 5% CO2 incubator with high-glucose Dulbecco modified Eagle medium (DMEM; Invitrogen), supplemented with 10% fetal bovine serum (FBS; Hyclone), 50 IU/mL penicillin, and 50 μg/mL streptomycin. Cells were passaged every 5–7 days after rinsing with phosphate-buffered saline (PBS) and trypsinization.
Subcloning of UCP2 fusion protein constructs and transfection
To examine the effect of UCP2 overexpression in hepatocytes, we constructed mouse UCP2–green fluorescent protein (GFP) fusion protein constructs with both coding and noncoding sequences. To make mouse UCP2–GFP fusion proteins, PCR primers (5′ primer, gccgctcgagAAATCAGAATCATGGTT; 3′ primer, gccgctcgagGAAAGGTGCCTCCCGAG; lowercase bold characters indicate added XhoI sites) were synthesized and used to make the PCR product of mouse UCP2 from total RNA of mouse liver that contains a full coding sequence of mouse UCP2 and has XhoI sites at both ends. This mouse UCP2 PCR product was subcloned into pEGFP-N1 (Clontech) for sense mouse UCP2 expression with a GFP tag at the carboxyl terminus (construct N-UCP2) and into pEGFP-C1 (Clontech) for the sense mouse UCP2 expression with a GFP tag at the amino terminus (construct C-UCP2). The UCP2 PCR product was also subcloned into pEGFP-C2 (Clontech) for noncoding mouse UCP2 expression with a GFP tag at the amino terminus (construct noncoding UCP2). All constructs were checked by DNA sequencing. Hepa 1–6 cells were transfected with UCP2 fusion protein constructs using Lipofectamine 2000 (Invitrogen), according to supplier's instructions. Cells were split the day before transfection so that cells would become 50%–70% confluent on the day of transfection. For each 35-mm culture plate transfected, 5 μg of plasmid DNA was mixed with 4 μL of Lipofectamine 2000 in 500 μL of Opti-MEM (Invitrogen), and the mixture was allowed to sit for 30 min at room temperature. For cell transfection in 24-well or eight-well culture plates, all reagents were downsized proportionally. Cells were washed twice with sterile PBS and were then loaded with transfection mixture without serum. Ten hours later, cells were switched back to normal culture medium. At 48 or 72 hr after transfection, cells were processed for gene expression and functional assays.
Immunohistochemistry
Cells were cultured in eight-well Lab-Tek culture slides at 5×104/well. After transfection, standard immunohistochemical techniques were employed. Briefly, cells were fixed in 2% paraformaldehyde (phosphate buffered) for about 10 min at room temperature, washed three times with Tris-buffered saline (TBS), permeablized with TBSA-BSAT [TBS containing 0.25% sodium azide, 1% bovine serum albumin (BSA), and 0.1% Triton-100] for 2 hr before staining. Immunostaining was performed with goat anti-UCP2 antibody (Santa Cruz, 1:200 dilution in PBS containing 1% BSA and 0.05% Triton-100) and then TRITC-labeled rabbit anti-goat antibody (Sigma). Fluorescent signals were detected with confocal microscopy.
Flow cytometry analysis of cell cycle
Cell proliferation activities were analyzed by flow cytometry with propidium iodide nuclear staining. At 48 hr after transfection, HEP6–16 liver cells were trypsinized and collected in 5 mL of culture medium. Cells were spun down at 200× g and were then fixed in 70% ethanol and immediately vortexed. After 1 hr of fixation, cells were washed twice with 2 mL of PBS and resuspended in 100 μL of PBS, 100 μL of 1 mg/mL RNase A, and 200 μL propidium iodide (100 μg/mL) and incubated for 30 min in the dark. Samples were then analyzed by flow cytometry (Becton Dickinson FACSCaliber).
ATP assay
Cellular ATP content was analyzed in triplicate as described previously.30 For normalization between samples, the total cellular protein of the tissue homogenate was determined by bicinchoninic acid assay (BCA) assay.
Annexin-V staining
Hepa 1–6 cells were cultured in eight-well Lab-Tek culture slides. Forty-eight hours after transfection, 1 μL of red fluorescent-tagged annexin-V (Molecular Probes) was added directly into each well of culture medium and incubated for 15 min at room temperature. Cells were then viewed immediately under fluorescent microscope.
Western blot analysis
Total cellular proteins were prepared using radioimmunoprecipitation assay (RIPA) buffer containing 5% mammalian proteinase inhibitor (Sigma). Protein electrophoresis (50 g for each well) and blotting were performed using premade 4%–12% NuPage polyacrylamide gel (Invitrogen) according to suggested protocols. After blocking with TBS containing 0.05% Tween-20 and 5% milk for 30 min, blots were incubated with primary antibody overnight at 4°C. The blots were then washed and incubated with peroxidase-tagged goat anti-rabbit or anti-mouse secondary antibodies (Sigma) for 30 min. After washing, the blots were incubated with Lumi-Light Western Blotting substrate solution (Roche Diagnostics, for detection of peroxidase) or CDP-Star Nitro-Block II (Fisher, for detection of alkaline phosphatase) and exposed to X-ray film.
Glutathione assay
A microtiter plate assay for the measurement of glutathione (GSH) and glutathione disulfide (GSSG) was done as described previously.31 All reagents for this assay were from Sigma. Cellular GSH contents were normalized with total cellular protein.
Statistics
All experiments were repeated at least three times or each experimental group included at least three samples. Data obtained from each experiment are shown as the mean±standard deviation (SD) and were analyzed for statistical significance using a two-tailed Student t-test. A P value<0.05 was regarded as statistically significant.
Results
Expression patterns of UCP2 fusion protein in Hepa 1–6 cells
Hepa 1–6, a mouse hepatoma cell line, was used for this study. Hepa 1–6 cells express modest levels of UCP2, which has been shown to be partially regulated by the intracellular energy state.30 Forty-eight hours after transfection with UCP2 constructs, cells were examined for gene expression patterns. Cells were fixed in 2% paraformaldehyde for 20 min at room temperature, followed by immunostaining with goat anti-UCP2 antibody and then TRITC-labeled rabbit anti-goat antibody. A representative result is presented in Fig. 1. Transfected Hepa 1–6 cells with C-UCP2 or N-UCP2 expressed GFP signals mainly in the cytoplasm; some were in a filamentous state, which might indicate mitochondrial localization (Fig. 1, top left). The intracellular localization of the UCP2 fusion protein seems to be not affected by GFP tag at either the amino or carboxyl terminus, suggesting that the mitochondrial localization sequence was not masked. In contrast, cells transfected with control noncoding UCP2 or vector alone had GFP expressed across the whole cell (especially in the nuclei) without any pronounced localization pattern (Fig. 1, top right). Fluorescent immunocytochemical staining using a UCP2 antibody revealed that in Hepa 1–6 cells transfected with C-UCP2 GFP-positive cells were also UCP2 positive, and a UCP2 immuno-signal was in the cytoplasm corresponding to GFP signals (Fig. 1, middle and bottom left), whereas UCP2 immunostaining is negative in nontransfected cells (Fig. 1, middle and bottom right). Western blots of transfected cells confirmed UCP2 fusion protein expression in both cell types (Fig. 1B).
FIG. 1.

Expression patterns of the uncoupling protein 2 (UCP2) fusion protein in Hepa 1–6 cells. (A) Green fluorescent protein (GFP) and fluorescent UCP2 immunocytochemistry (IC) signals of Hepa 1–6 cells transfected with either C-sense UCP2 or noncoding UCP2 show a UCP2 expression pattern in hepatocytes. GFP+IC shows merged pictures of GFP and immunostaining. (B) Western blot analysis of total proteins from transfected Hepa 1–6 cells using alkaline phosphatase–labeled anti-GFP antibody (Clontech, 1:2000 dilution). The figure shows ∼60-kD protein bands in sense UCP2-transfected cells, and ∼30-kD protein bands in noncoding UCP2 and vector-transfected cells. Western blot samples from left to right: transfections with C-UCP2, N-UCP2, noncoding-UCP2, and vector alone, respectively. Color images available online at www.liebertpub.com/met
UCP2 expression is associated with reduced intracellular ATP production in hepatocytes.21,29 We performed ATP assays to examine cellular energy status in UCP2-transfected cells. Hepa 1–6 cells were cultured in 24-well culture plates, and 48 hr after transfection cellular ATP concentrations were determined and normalized with total cellular protein. We found that cellular ATP concentrations in sense UCP2-transfected cells were approximately 10% lower than other groups, but did not reach the level of statistical significance (Fig. 2A). Cellular GSH levels are correlated with resistance to oxidative stress. UCP2-transfected cells were evaluated by examination of cellular GSH levels in Hepa 1–6 cells 48 hr after UCP2 transfection. Significantly higher levels of GSH were found in sense UCP2-transfected cells than other groups (Fig. 2B), suggesting that robust ROS defense mechanisms are present in UCP2-overexpressing cells.
FIG. 2.

Uncoupling protein 2 (UCP2) expression has minimal effect on intracellular adenosine triphosphate (ATP) content (A), but slightly increases glutathione (GSH) contents (B) in transfected cells. From left to right: transfection with C-UCP2, N-UCP2, noncoding-UCP2, EGFP-C1 vector alone, and nontransfected Hepa 1–6 cells. (*) P<0.05 and represents statistical analysis versus noncoding and vector alone–transfected cells.
Overexpression of UCP2 inhibits cell division
We attempted to clone a stably transfected Hepa 1–6 cell line with UCP2. Two days after transfection, about 40% of the cells became GFP positive. G418 selection (1 μg/mL) started 2 days after transfection and resulted in 95% of cell death within 1 week when approximately 10% of all remaining cells were GFP positive. In our effort to clone stable cell lines expressing a UCP2 fusion protein, we were able to eliminate most nontransfected cells by G418 selection and obtained high percentages of transfected cells in the first week. However, we found that these transfected cells did not divide. Upon long-term incubation (4–6 weeks), we repeatedly found that nontransfected Hepa 1–6 cells soon outnumbered GFP-positive cells, and GFP-positive cells were eventually eradicated. Then we cultured transfected Hepa 1–6 cells without G418 selection, and a similar phenomenon was observed: GFP-positive cells were soon found dead and detached from culture dishes. Cell cycle analysis by flow cytometry indicated that cells transfected with sense UCP2 had much less proliferative activity (Fig. 3). In GFP-positive transfected cells, the percentage of cells at S phase was decreased by as much as 50%–55% compared with vector alone or noncoding UCP2 transfected cells and was accompanied by a significant increase in cells at G0/G1 phase. GFP-negative nontransfected cells from the same preparations were not affected. Generally, the cell number in S phase was 37% in vector-transfected cells, 34% in noncoding UCP2 transfected cells, and only 17% in sense UCP2 transfected Hepa 1–6 cells (whereas nontransfected cells from the same culture dish of sense UCP2 transfection had 33% of cells in S phase). Cell numbers in G2 phase were also decreased significantly: 12% in vector transfected cells, 9% in noncoding UCP2-transfected cells, and only 1.5% in sense UCP2-transfected Hepa 1–6 cells (nontransfected cells from all other preparations had about 15% of cells in G2 phase). Flow cytometry analysis also suggested that with longer UCP2 expression times (72 hr), some degree of cell death began to develop (Fig. 3).
FIG. 3.

Uncoupling protein 2 (UCP2) expression inhibits cell proliferation with G1 phase arrest in Hepa 1–6 cells. This figure shows cell cycle analysis of UCP2-transfected Hepa 1–6 cells by flow cytometry, comparing cell cycle patterns of green fluorescent protein (GFP)-positive versus GFP-negative cells from the same sample transfected with different UCP2 constructs or vector alone, with a posttransfection time of either 48 hr (first 2 rows) or 72 hr (last 2 rows).
To investigate whether the effect of UCP2 expression on cell proliferation is an isolated incident or a universal phenomenon, we examined UCP2 transfection in other cell lines, including HeLa, 293, and MG63 cells. Similar phenomena were observed in these cells as in Hepa 1–6 cells (Table 1).
Table 1.
Effect of Uncoupling Protein 2 (UCP2) Expression on the Cell Cycle in Hela, MG63, and 293 Cells
| G1 phase % in | S phase % in | G2-M phase % in | |||||
|---|---|---|---|---|---|---|---|
| Cell type | UCP2 | GFP+ cells | GFP− cells | GFP+ cells | GFP− cells | GFP+ cells | GFP− cells |
| Hela | Sense | 95.6% | 58.7% | 2.2% | 28.8% | 2.2% | 12.5% |
| Noncoding | 86.7% | 61.4% | 13.3% | 26.2% | 0.0% | 12.4% | |
| MG63 | Sense | 84.39% | 68.19% | 11.01% | 19.88% | 4.60% | 11.92% |
| Noncoding | 65.43% | 63.39% | 21.01% | 21.23% | 13.56% | 15.37% | |
| 293 cell | Sense | 67.81% | 50.05% | 19.24% | 32.93% | 12.95% | 17.01% |
| Noncoding | 41.33% | 44.30% | 37.18% | 36.97% | 21.50% | 18.73% | |
Forty-eight hours after transfection with sense or noncoding mouse UCP2 constructs, cell proliferation activity was analyzed by flow cytometry.
GFP, green fluorescent protein.
Having shown that these results were robust across multiple hepatic cell lines, we focused our further experiments on Hepa 1–6 cells. Because intracellular ATP and ROS were slightly altered in UCP2-transfected cells, we set out to examine if the effect of UCP2 overexpression on cell division was related to decreased cellular ATP or ROS content. To exclude the role of this subtle change of cellular ATP on cell division, we performed an “ATP restoration” experiment. One day after transfection, cells were cultured in a medium containing 5 mM phosphor(enol)pyruvate (pH balanced at 7.4), which is a direct source of ATP and increases cellular ATP content.32,33 Forty-eight hours after transfection, cells were analyzed for proliferation activity. We found that addition of phosphor(enol)pyruvate in cell culture medium did not change the cell population in each phase. Similarly, to examine whether the change in cellular GSH content was associated with G1 arrest in UCP2-transfected cells, we did a “ROS restoration” experiment. One day after transfection, cells were cultured in a medium containing 200 nM or 1 μM hydrogen peroxide (H2O2). Addition of H2O2 masked the effect of UCP2 on cellular GSH levels by providing the cell with saturated oxidants. However, with 24 hr of treatment of H2O2, cell proliferation activity was not affected in either UCP2-transfected or nontransfected Hepa 1–6 cells. We then set out to examine if the effect of UCP2 on cell proliferation was due to its uncoupling effect on mitochondrial membrane potential. We had previously shown that carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, a mitochondrial uncoupling reagent) treatment in Hepa 1–6 cells decreases cellular ATP content and reduces UCP2 expression.30 When we examined FCCP-treated Hepa 1–6 cells for cell proliferation activity and found that 24 hr of FCCP treatment, with or without co-treatment with 2-deoxy-D-glucose, did not significantly affect cell proliferation activity. There was no observed difference with either 8 μM or 80 μM FCCP treatment. In UCP2-transfected Hepa 1–6 cells, the effect of UCP2 overexpression was not changed by FCCP treatment (data not shown).
Proteins such as cyclin-dependent kinases are important regulators of the cell cycle by phosphorylation of other proteins involved in regulation of cell cycle progression, such as the retinoblastoma (Rb) protein. We examined the effect of protein phosphorylation on UCP2 overexpression in Hepa 1–6 cells. We found that the effect of UCP2 in Hepa 1–6 cells was enhanced by treatment with genistein, a tyrosine kinase inhibitor. Treatment with 200 μM genistein for 24 hr further and significantly decreased the ratio of cells in S phase in UCP2-transfected Hepa 1–6 cells that was already low with an accompanying increase in cells in G1 phase (Fig. 4). Interestingly, such a phenomenon was not observed in nontransfected Hepa 1–6 cells. In GFP-negative Hepa 1–6 cells, genistein actually increased the ratio of cells in S phase in nontransfected cells with less cells in G1 phase. This was not observed in samples treated with other reagents that modulate protein kinase activities. Protein kinase A and C inhibitor (H7, 30 μM), protein kinase C activator [12-O-tetradecanoylphorbol-13-acetate (TPA), 200 nM], SB 203580 [p38 mitogen-activated protein kinase (MAPK) inhibitor], cycle adenosine monophosphate (cAMP) activator (forskolin at 10 μM, 8-Br-cAMP at 20 μM), did not show any effect on cell division in Hepa 1–6 cells (data not shown). Most interestingly, okadaic acid, an inhibitor of type 1 and type 2A protein phosphatases but not an inhibitor of tyrosine phosphatases, did not significantly affect the cell cycle in Hepa 1–6 cells, either transfected or not (Fig. 4). These findings suggest a specific role for protein tyrosine phosphorylation in the regulation of cell proliferation activity by UCP2. To elucidate regulatory mechanisms that may be involved in this increase in G0/G1 cells after UCP2 transfection, we examined expression levels of some cell cycle regulatory proteins. Western blots revealed that the protein level of CDK6 but not CDK4 was remarkably decreased in UCP2-transfected Hepa 1–6 cells, whereas cyclins D1 and D3 were not changed (Fig. 5).
FIG. 4.

The inhibitory effect of uncoupling protein 2 (UCP2) expression on the cell cycle is enhanced by genistein treatment. This figure shows cell populations in G1, S, and G2 phases in green fluorescent protein (GFP)-positive transfected cells (left) and GFP-negative nontransfected cells from the same transfection preparations: (1) C-UCP2-transfected cells; (2) C-UCP2-transfected cells with 24 hr of treatment with 200 μM genistein; (3) C-UCP2-transfected cells with 24 hr of treatment with okadaic acid; (4) noncoding UCP2-transfected cells; (5) vector alone–transfected cells. Data represent three repeated experiments. (*) Statistical analysis versus noncoding and/or vector alone–transfected cells (as indicated); (#) statistical analysis versus C-UCP2 transfected cells. (**) or (##) P<0.01.
FIG. 5.
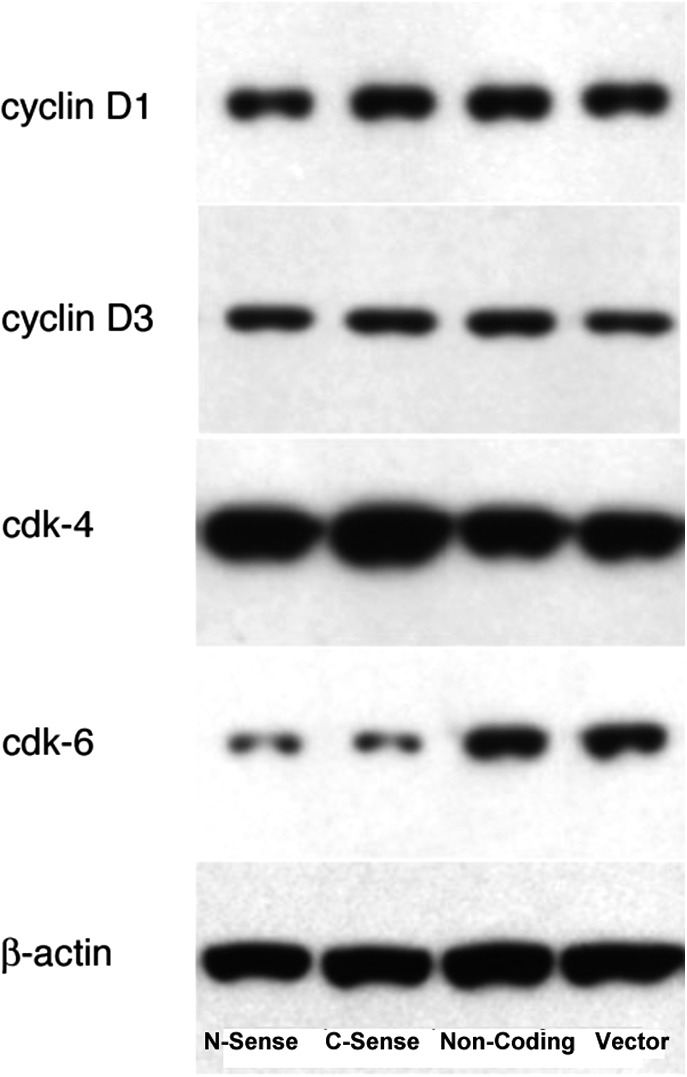
CDK6 is decreased in uncoupling protein 2 (UCP2)-transfected cells. Western blot analysis of cell division–related proteins from total cellular protein preparations of Hepa 1–6 cells 48 hr after transfection shows a remarkable decrease of Cdk6 but not Cdk4; cyclins D1 and D3 remain constant. Mouse monoclonal antibodies against cyclin D1 (1:1000 dilution); cyclin D3 (1:1000 dilution), Cdk4 (1:2000 dilution), Cdk6 (1:1000 dilution) were all from cell signaling, and anti-β-actin antibody (1:2000 dilution) was from Sigma.
Overexpression of UCP2 leads to nonapoptotic cell death
Another phenomenon we observed was that, upon transfection, some Hepa 1–6 cells become detached from the culture dish. Direct observation under a fluorescent microscope revealed that more sense UCP2-transfected cells become morphologically unhealthy and detached, a phenomenon not observed in cells transfected with noncoding constructs or vector alone. Cell cycle analysis by flow cytometry also suggested increased cell death with prolonged (72 hr) UCP2 transfection (Fig. 3). We investigated the effect of UCP2 overexpression on cell viability 72 hr after transfection. Trypan Blue staining and counting revealed that the number of dead cells with sense UCP2 transfection was higher than nontransfected cells. In nontransfected cells, less than 1% cells were stained with Trypan Blue. About 3% cells became Trypan Blue positive in preparations of Lipofectamine, noncoding UCP2, and vector-transfected cells. This increased to approximately 11% in sense UCP2-transfected cells (Fig. 6A). To further differentiate cell death between transfected versus nontransfected cells, fluorescent-tagged Annexin-V live staining was employed 72 hr after transfection, and cells were examined directly under fluorescent microscope. We found that the majority of dead cells were transfected with the sense construct, whereas nontransfected cells from the same preparation were not affected (Fig. 6B); only a few cells died in cells transfected with noncoding construct or vector alone (Fig. 6C). Flow cytometry analysis was employed to differentiate dead cells in transfected versus nontransfected cells and to confirm this finding. Seventy-two hours after transfection, live Hepa 1–6 cells were stained with propidium iodide (Fig. 6D). Approximately 26.11% of C-UCP2-transfected cells were propidium iodide–positive dead cells, whereas only 10.48% of nontransfected cells in the same preparations were dead. Transfection with a noncoding UCP2 construct or vector alone did not show a difference in cell viability between transfected and nontransfected cells (Fig. 6D).
FIG. 6.

Uncoupling protein 2 (UCP2) expression induces cell death. (A) Trypan Blue count. Viability of transfected cells: 3% in Lipofectamine and noncoding and vector-transfected cells, 11% in sense UCP2-transfected cells. (B) Live staining with Annexin-V in C-UCP2-transfected cells (left panel) and noncoding UCP2-transfected cells (right panel). Green is the green fluorescent protein (GFP) signal and red is Annexin-V. (C) Flow cytometry analysis with propidium iodide in C-UCP2-transfected cells (black) and noncoding UCP2-transfected cells (white). (D) Apoptosis-related gene expression analyzed by western blot from total cellular protein preparations (except for released cytochrome c, which was cytoplasmic protein preparation without mitochondria) of Hepa 1–6 cells 72 hr after transfection. (E and F) Rabbit polyclonal antibodies against Bcl-2 (sc-492; 1:1000 dilution) and Bax (sc-6236, 1:1000 dilution) were from Santa Cruz; rabbit polyclonal antibody against caspase 3 (1:1000 dilution) was from Cell Signaling; mouse monoclonal antibody against cytochrome c (1:300); and anti-bcl-x (1:500 dilution) was from BD Biosciences. Western blot samples from left to right: Transfections with C-UCP2, N-UCP2, noncoding UCP2, and vector alone, respectively. Color images available online at www.liebertpub.com/met
To clarify whether cell death is apoptotic, we examined whether some apoptotic characters were present in UCP2-transfected Hepa 1–6 cells. Agarose gel analysis of chromosome DNA from UCP2-transfected Hepa 1–6 cells did not show a characteristic DNA ladder pattern of apoptotic cells (data not shown). Western blot analysis of caspase-3 demonstrated a limited amount of cleaved caspase-3 signals, but there was no difference among sense, noncoding, or vector-transfected cells (Fig. 6E). Western blot analysis of cytochrome c from total cellular proteins showed uniform levels of this protein, but this was absent from cytosolic protein preparations without mitochondria, suggesting no cytochrome c release from mitochondria. Western blotting analysis of expression levels of some pro- and antiapoptotic proteins revealed only moderate decrease of bcl-2 expression in sense UCP2-transfected cells, whereas bax and bcl-xl expression was not changed in all preparations of Hepa 1–6 cells (Fig. 6F).
Discussion
Mitochondria are involved in many critical cellular processes and are essential to cell viability. Although the association between mitochondria and cell death is well established, the relationship between mitochondrial function and cell proliferation has been largely unexplored.28 The size and shape of mitochondria are highly variable during a cell cycle of physiologically normal cells34,35; however, it is not until recently that the role of mitochondria acting as a regulator of cell proliferation and as a tumor suppressor was proposed.28 In several tumor cell lines, the antiproliferative effects of tyrphostin AG17 were related to disruption of mitochondrial function, decreased ATP content, and loss of the mitochondrial membrane potential.27 This raises the possibility that mitochondria, mediated by their multiple life-critical functions, could be a major player in the regulation of cell proliferation. In the present study, we investigated the effect of UCP2 expression on cell proliferation and cell death in Hepa 1–6 cells. We provide here, for the first time, direct evidence that UCP2, a mitochondrial inner membrane protein, causes cell cycle arrest at the G1 phase with accompanied alteration in CDK6 expression, a phenomenon that is boosted by tyrosine phosphorylation inhibition. We further demonstrate that overexpression of UCP2 also leads to loss of cell viability through nonapoptotic cell death.
The inhibitory effect of UCP2 on cell proliferation is strong. Within 48 hr, UCP2-transfected hepatocytes showed more than 50% decrease in the cell subpopulation in S phase, and most cells were blocked at G1 phase. This was in clear contrast to other control groups, including noncoding UCP2 or vector-transfected hepatocytes, lipofectamine-treated hepatocytes, and especially nontransfected hepatocytes from the same preparation of the UCP2-transfected hepatocytes. Because cells were accumulated at the G1 phase and reduced at S phase, cells at the G2 phase were almost depleted. This, together with cell death caused by UCP2 overexpression, led to gradual loss of UCP2-transfected cells in culture.
Several different cell types are similarly affected by UCP2 expression, suggesting that the effect of UCP2 on cell proliferation could be a widespread phenomenon. It is not known how UCP2 regulates cell proliferation. Previous studies show that cell cycle progression may be altered in response to cellular energy supply or the response to ROS stress. When treated with oligomycin, a potent inhibitor of ATP production, HL-60 cells show a significant increase in the proportion of cells in G1 at low doses of the agent and in G2–M at higher doses.36 Cells arrested in G1 phase have a modest decrease in cellular ATP, whereas cells arrested in G2 have more severe ATP depletion.36 Separately, the redox state has multiple effects on cell proliferation. ROS stimulates human hepatoma cell proliferation via cross-talk between phosphoinositide 3-kinase (PI3-K)/PKB and Jun amino-terminal kinase (JNK) signaling pathways.37,38 ROS induce rat hepatic stellate cell activation, proliferation, and collagen gene expression in vitro.39 Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu–transformed rat-1 fibroblasts through the induction of a stress response.40 Because the production of ROS is directly proportional to mitochondrial potential,41,42 UCP2 is believed to reduce the production of free radicals and ROS in some pathological states,8,43 which may explain the effect of UCP2 on GSH levels and cell cycle arrest.
We did find that cellular ATP and GSH levels are affected by UCP2 transfection, but only to a limited extent. Whereas UCP2 dramatically suppressed cell division in Hepa 1–6 cells, intracellular ATP contents were reduced by only approximately 10% and did not always reach a statistically significant level. Cellular GSH levels were altered similarly. One explanation of this modest change in cellular ATP levels in UCP2-overexpressing cells is that transformed cells use more energy from glycolysis,44,45 which compensates for decreased ATP production due to UCP2 overexpression. Phosphoenolpyruvate is an endogenous substance and an ATP precursor that can cross cell membranes via an anion exchanger.32,33 Treatment with phosphoenolpyruvate in UCP2-transfected Hepa 1–6 cells failed to reverse the effect of UCP2 overexpression on the cell cycle. Similarly, addition of H2O2 to the culture medium of UCP2-transfected cells did not change the fate of UCP2 expression on cell cycle. To further elucidate the role of mitochondrial uncoupling in the regulation of cell division by UCP2, we examined the effect of FCCP on Hepa 1–6 cells. Treatment with FCCP, which reduces mitochondrial membrane potential and ROS production, did not affect the cell cycle by itself and did not block or reduce the effect of UCP2 on cell division. These data suggest that changes in cellular ATP content or ROS production do not contribute significantly to cell cycle arrest by UCP2 overexpression. Rather, UCP2 may act in a separate manner, perhaps serving as a signaling molecule in the cell division regulatory cascade.
In mammalian cells, the regulation of cell cycle progression is tightly controlled through a series of checkpoints, mainly by an intricate network of two protein families—the cyclins and the cyclin-dependent protein kinases (CDKs).46 CDKs control cell cycle progression in all eukaryotes and are active only at the appropriate time in the cell cycle through phosphorylation, dephosphorylation, and binding to cell cycle–specific cyclins.47,48 The G1 phase is mainly controlled by the cyclin D proteins and their associated kinases, including CDK4 and CDK6.49,50 In response to extracellular signals and growth factor stimulation, D-type cyclins assemble with CDK4 and CDK6 to form holoenzymes, and direct interaction of CDKs with cyclins is required for their full enzymatic activity to allow progression through the different phases of the cell cycle. In the present study, we found that UCP2 overexpression in Hepa 1–6 cells did not affect expression of D-type cyclins. However, the CDK6 level was markedly decreased when UCP2 was overexpressed. This was observed consistently with transfection of different UCP2 fusion protein constructs in Hepa 1–6 cells and was not affected by the amino- or carboxy-terminal positions of the GFP tag. It has been documented that CDK6 decreases transit time through the G1 phase of the cell cycle and thus enhances cell proliferation, and that this effect is not dependent on co-transfection of the kinase-activating partner, cyclin D.51 Accordingly, reduced CDK6 expression causes cell growth arrest in different cell types.52,53 This is supported by our present study in Hepa 1–6 cells. It is possible that overexpression of UCP2 reduces CDK6 expression level or activity, thus leading to cell cycle arrest by an unknown mechanism. Treatment with genistein, a specific tyrosine kinase inhibitor, significantly enhanced the effect of UCP2 on cell cycle arrest, suggesting a synergistic effect of protein tyrosine phosphorylation and UCP2 expression. This was a phenomenon not observed with treatments of other protein kinase inhibitors or activators. It is noteworthy that genistein by itself has no effect on cell proliferation activity in nontransfected or vector alone–transfected Hepa 1–6 cells. Taken together, these data raise the possibility that reduced levels of tyrosine phosphorylation contribute to G1 arrest in Hepa 1–6 cells and that this effect of tyrosine phosphorylation could be downstream of UCP2 expression.
Cell proliferation and cell death are highly interrelated and coordinated during normal development of an organism and in many other physiological and pathological conditions in which it is essential to identify and eliminate inappropriately proliferating cells.47,54 Changes in the expression and activity of CDKs and cyclins are also observed during apoptosis of many different cell types,54,55–57 suggesting that UCP2 may regulate cell death/apoptosis via altering mitochondrial function or by altering the expression and activity of other cell cycle regulators. The idea of UCP2 in the regulation of apoptosis is supported by Voehringer et al.; using gene microarray analysis, they reported that UCP2 mRNA levels increased significantly in LYas cells in response to apoptogenic radiation treatment.23 Upon exposure to radiation, UCP2 message levels peaked at 1 hr, followed by mitochondrial changes at 1.5–2 hr in LYas cells, well before apoptosis.23 In HeLa cells, but not in normal diploid fibroblasts, increased UCP2 expression leads to a dramatic fall in mitochondrial membrane potential, a reduction of intracellular ATP, and a form of cell death that is not inhibited by Bcl2.20
In the present study, we observed an association of UCP2 expression with decreased cell viability. However, our data suggest that overexpression of UCP2 leads to nonapoptotic cell death, which is consistent with the report by Mills et al.20 We observed significant cell death in sense UCP2-transfected cells, with simultaneously lower expression of Bcl2. However, characteristic apoptotic markers such as DNA ladder patterns upon chromosome DNA electrophoresis, cytochrome c release from mitochondria, and increased cleavage of caspase-3 were not observed. We speculate that because of an uncoupling protein located in the inner membrane of mitochondria, overexpression of UCP2 may interrupt multiple functions of mitochondria, and that disruption of mitochondrial function by UCP2 may lead to cell death other than apoptosis. However, controversies exist regarding the role of UCP2 in the regulation of cell viability. Especially in neural tissues24,26 or with ROS insults,25 expression of UCP2 shows a protective effect on tissue damage and apoptosis. In nonneural tissues, it has been reported that UCP2 overexpression protects cardiomyocytes from oxidative stress–induced apoptosis by reducing ROS production.58 These data suggest that UCP2 could be neuroprotective and antiapoptotic in response to some environmental insults, especially with ROS excess. Thus, the exact role of UCP2 expression on cell viability is very complex and depends on tissue type, expression level, exposure time, and other experimental conditions.
In summary, UCP2 overexpression induced cell cycle arrest on the G1 phase as well as nonapoptotic cell death in Hepa 1–6 cells, accompanied by CDK6 and bcl-2 downregulation. This effect on cell cycle arrest is enhanced by inhibition of protein tyrosine phosphorylation, and these elements suggest a role for UCP2 in the balance among proliferation, quiescence, and cell death, the basic cellular processes that must be strictly maintained to avoid unwanted tissue atrophy or tissue overgrowth. Tumor cells have increased cell proliferation and decreased cell death, and both of these are inhibited by UCP2 overexpression. Therefore, our data suggest the possibility that UCP2 may be a cell cycle regulator and a tumor suppressor and may constitute a novel target in the development of drugs designed to modulate cell proliferation and viability.
Acknowledgment
This work was funded by a grant from the National Institutes of Health (grant no. DK069369).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Stuart JA, Cadenas S, Jekabsons MB, et al. Mitochondrial proton leak and the uncoupling protein 1 homologues. Biochim Biophys Acta 2001;1504:144–158 [DOI] [PubMed] [Google Scholar]
- 2.Nicholls DG, Bernson VS, Heaton GM. The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Experientia Suppl 1978;32:89–93 [DOI] [PubMed] [Google Scholar]
- 3.Nicholls DG, Locke RM. Thermogenic mechanisms in brown fat. Physiol Rev 1984;64:1–64 [DOI] [PubMed] [Google Scholar]
- 4.Boss O, Samec S, Paoloni-Giacobino A, et al. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett 1997;408:39–42 [DOI] [PubMed] [Google Scholar]
- 5.Mao W, Yu XX, Zhong A, et al. UCP4, a novel brain-specific mitochondrial protein that reduces membrane potential in mammalian cells. FEBS Lett 1999;443:326–330 [DOI] [PubMed] [Google Scholar]
- 6.Sanchis D, Fleury C, Chomiki N, et al. BMCP1, a novel mitochondrial carrier with high expression in the central nervous system of humans and rodents, and respiration uncoupling activity in recombinant yeast. J Biol Chem 1998;273:34611–34615 [DOI] [PubMed] [Google Scholar]
- 7.Fleury C, Sanchis D. The mitochondrial uncoupling protein-2: Current status. Int J Biochem Cell Biol 1999;31:1261–1278 [DOI] [PubMed] [Google Scholar]
- 8.Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J 2000;345(Pt 2):161–179 [PMC free article] [PubMed] [Google Scholar]
- 9.Rousset S, Alves-Guerra MC, Mozo J, et al. The biology of mitochondrial uncoupling proteins. Diabetes 2004;53(Suppl 1):S130–S135 [DOI] [PubMed] [Google Scholar]
- 10.Fleury C, Neverova M, Collins S, et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat Genet 1997;15:269–272 [DOI] [PubMed] [Google Scholar]
- 11.Larkin S, Mull E, Miao W, et al. Regulation of the third member of the uncoupling protein family, UCP3, by cold and thyroid hormone. Biochem Biophys Res Commun 1997;240:222–227 [DOI] [PubMed] [Google Scholar]
- 12.Hinz W, Gruninger S, De Pover A, et al. Properties of the human long and short isoforms of the uncoupling protein-3 expressed in yeast cells. FEBS Lett 1999;462:411–415 [DOI] [PubMed] [Google Scholar]
- 13.Fink BD, Hong YS, Mathahs MM, et al. UCP2-dependent proton leak in isolated mammalian mitochondria. J Biol Chem 2002;277:3918–3925 [DOI] [PubMed] [Google Scholar]
- 14.Hong Y, Fink BD, Dillon JS, et al. Effects of adenoviral overexpression of uncoupling protein-2 and −3 on mitochondrial respiration in insulinoma cells. Endocrinology 2001;142:249–256 [DOI] [PubMed] [Google Scholar]
- 15.Lameloise N, Muzzin P, Prentki M, et al. Uncoupling protein 2: A possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes 2001;50:803–809 [DOI] [PubMed] [Google Scholar]
- 16.Negre-Salvayre A, Hirtz C, Carrera G, et al. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 1997;11:809–815 [PubMed] [Google Scholar]
- 17.Simonyan RA, Skulachev VP. Thermoregulatory uncoupling in heart muscle mitochondria: Involvement of the ATP/ADP antiporter and uncoupling protein. FEBS Lett 1998;436:81–84 [DOI] [PubMed] [Google Scholar]
- 18.Simoneau JA, Kelley DE, Neverova M, et al. Overexpression of muscle uncoupling protein 2 content in human obesity associates with reduced skeletal muscle lipid utilization. FASEB J 1998;12:1739–1745 [DOI] [PubMed] [Google Scholar]
- 19.Green DR, Amarante-Mendes GP. The point of no return: Mitochondria, caspases, and the commitment to cell death. Results Probl Cell Differ 1998;24:45–61 [DOI] [PubMed] [Google Scholar]
- 20.Mills EM, Xu D, Fergusson MM, et al. Regulation of cellular oncosis by uncoupling protein 2. J Biol Chem 2002;277:27385–27392 [DOI] [PubMed] [Google Scholar]
- 21.Rashid A, Wu TC, Huang CC, et al. Mitochondrial proteins that regulate apoptosis and necrosis are induced in mouse fatty liver. Hepatology 1999;29:1131–1138 [DOI] [PubMed] [Google Scholar]
- 22.Tsuboyama-Kasaoka N, Tsunoda N, Maruyama K, et al. Overexpression of GLUT4 in mice causes up-regulation of UCP3 mRNA in skeletal muscle. Biochem Biophys Res Commun 1999;258:187–193 [DOI] [PubMed] [Google Scholar]
- 23.Voehringer DW, Hirschberg DL, Xiao J, et al. Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis. Proc Natl Acad Sci USA 2000;97:2680–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bechmann I, Diano S, Warden CH, et al. Brain mitochondrial uncoupling protein 2 (UCP2): A protective stress signal in neuronal injury. Biochem Pharmacol 2002;64:363–367 [DOI] [PubMed] [Google Scholar]
- 25.Diano S, Matthews RT, Patrylo P, et al. Uncoupling protein 2 prevents neuronal death including that occurring during seizures: A mechanism for preconditioning. Endocrinology 2003;144:5014–5021 [DOI] [PubMed] [Google Scholar]
- 26.Mattiasson G, Shamloo M, Gido G, et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med 2003;9:1062–1068 [DOI] [PubMed] [Google Scholar]
- 27.Burger AM, Kaur G, Alley MC, et al. Tyrphostin AG17, [(3,5-Di-tert-butyl-4-hydroxybenzylidene)-malononitrile], inhibits cell growth by disrupting mitochondria. Cancer Res 1995;55:2794–2799 [PubMed] [Google Scholar]
- 28.Rustin P. Mitochondria, from cell death to proliferation. Nat Genet 2002;30:352–353 [DOI] [PubMed] [Google Scholar]
- 29.Evans ZP, Ellett JD, Schmidt MG, et al. Mitochondrial uncoupling protein-2 mediates steatotic liver injury following ischemia/reperfusion. J Biol Chem 2008;283:8573–8579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng G, Polito CC, Haines JK, et al. Decrease of intracellular ATP content downregulated UCP2 expression in mouse hepatocytes. Biochem Biophys Res Commun 2003;308:573–580 [DOI] [PubMed] [Google Scholar]
- 31.Baker MA, Cerniglia GJ, Zaman A. Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem 1990;190:360–365 [DOI] [PubMed] [Google Scholar]
- 32.Golbidi S, Moriuchi H, Yang C, et al. Preventive effect of phosphoenolpyruvate on hypoxemia induced by oleic acid in Guinea pigs. Biol Pharm Bull 2003;26:336–340 [DOI] [PubMed] [Google Scholar]
- 33.Saiki S, Yamaguchi K, Chijiiwa K, et al. Phosphoenolpyruvate prevents the decline in hepatic ATP and energy charge after ischemia and reperfusion injury in rats. J Surg Res 1997;73:59–65 [DOI] [PubMed] [Google Scholar]
- 34.Dewey WC, Fuhr MA. Quantification of mitochondria during the cell cycle of Chinese hamster cells. Exp Cell Res 1976;99:23–30 [DOI] [PubMed] [Google Scholar]
- 35.Posakony JW, England JM, Attardi G. Mitochondrial growth and division during the cell cycle in HeLa cells. J Cell Biol 1977;74:468–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sweet S, Singh G. Accumulation of human promyelocytic leukemic (HL-60) cells at two energetic cell cycle checkpoints. Cancer Res 1995;55:5164–5167 [PubMed] [Google Scholar]
- 37.Han MJ, Kim BY, Yoon SO, et al. Cell proliferation induced by reactive oxygen species is mediated via mitogen-activated protein kinase in Chinese hamster lung fibroblast (V79) cells. Mol Cells 2003;15:94–101 [PubMed] [Google Scholar]
- 38.Liu SL, Lin X, Shi DY, et al. Reactive oxygen species stimulated human hepatoma cell proliferation via cross-talk between PI3-K/PKB and JNK signaling pathways. Arch Biochem Biophys 2002;406:173–182 [DOI] [PubMed] [Google Scholar]
- 39.Svegliati-Baroni G, Saccomanno S, van Goor H, et al. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver 2001;21:1–12 [DOI] [PubMed] [Google Scholar]
- 40.Preston TJ, Muller WJ, Singh G. Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu-transformed rat-1 fibroblasts through the induction of a stress response. J Biol Chem 2001;276:9558–9564 [DOI] [PubMed] [Google Scholar]
- 41.Skulachev VP. Possible role of reactive oxygen species in antiviral defense. Biochemistry (Mosc) 1998;63:1438–1440 [PubMed] [Google Scholar]
- 42.Yang S, Zhu H, Li Y, et al. Mitochondrial adaptations to obesity-related oxidant stress. Arch Biochem Biophys 2000;378:259–268 [DOI] [PubMed] [Google Scholar]
- 43.Boss O, Hagen T, Lowell BB. Uncoupling proteins 2 and 3: Potential regulators of mitochondrial energy metabolism. Diabetes 2000;49:143–156 [DOI] [PubMed] [Google Scholar]
- 44.Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem 1981;256:8699–8704 [PubMed] [Google Scholar]
- 45.Nakashima RA, Paggi MG, Pedersen PL. Contributions of glycolysis and oxidative phosphorylation to adenosine 5′-triphosphate production in AS-30D hepatoma cells. Cancer Res 1984;44:5702–5706 [PubMed] [Google Scholar]
- 46.Morgan DO. Principles of CDK regulation. Nature 1995;374:131–134 [DOI] [PubMed] [Google Scholar]
- 47.King KL, Cidlowski JA. Cell cycle regulation and apoptosis. Annu Rev Physiol 1998;60:601–617 [DOI] [PubMed] [Google Scholar]
- 48.Sherr CJ. Growth factor-regulated G1 cyclins. Stem Cells 1994;12(Suppl 1):47–55; discussion 55–47. [PubMed] [Google Scholar]
- 49.Bates S, Parry D, Bonetta L, et al. Absence of cyclin D/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene 1994;9:1633–1640 [PubMed] [Google Scholar]
- 50.Sherr CJ. D-type cyclins. Trends Biochem Sci 1995;20:187–190 [DOI] [PubMed] [Google Scholar]
- 51.Grossel MJ, Baker GL, Hinds PW. cdk6 can shorten G(1) phase dependent upon the N-terminal INK4 interaction domain. J Biol Chem 1999;274:29960–29967 [DOI] [PubMed] [Google Scholar]
- 52.Dai C, Chung IJ, Jiang S, et al. Reduction of cell cycle progression in human erythroid progenitor cells treated with tumour necrosis factor alpha occurs with reduced CDK6 and is partially reversed by CDK6 transduction. Br J Haematol 2003;121:919–927 [DOI] [PubMed] [Google Scholar]
- 53.Veiga-Fernandes H, Rocha B. High expression of active CDK6 in the cytoplasm of CD8 memory cells favors rapid division. Nat Immunol 2004;5:31–37 [DOI] [PubMed] [Google Scholar]
- 54.Meikrantz W, Schlegel R. Apoptosis and the cell cycle. J Cell Biochem 1995;58:160–174 [DOI] [PubMed] [Google Scholar]
- 55.Freeman RS, Estus S, Johnson EM., Jr Analysis of cell cycle-related gene expression in postmitotic neurons: Selective induction of Cyclin D1 during programmed cell death. Neuron 1994;12: 343–355 [DOI] [PubMed] [Google Scholar]
- 56.Gao C, Negash S, Wang HS, et al. Cdk5 mediates changes in morphology and promotes apoptosis of astrocytoma cells in response to heat shock. J Cell Sci 2001;114:1145–1153 [DOI] [PubMed] [Google Scholar]
- 57.Hoang AT, Cohen KJ, Barrett JF, et al. Participation of cyclin A in Myc-induced apoptosis. Proc Natl Acad Sci USA 1994;91:6875–6879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teshima Y, Akao M, Jones SP, et al. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res 2003;93:192–200 [DOI] [PubMed] [Google Scholar]