

Abstract
Retinal ischemia-reperfusion (I/R) involves extensive increase in reactive oxygen species as well as pro-inflammatory changes that result in significant histopathologic damage, including neuronal and vascular degeneration. Nrf2 has a well-known cytoprotective role in many tissues, but its protective function in the retina is unclear. We investigated the possible role of Nrf2 as a protective mechanism in retinal ischemia-reperfusion injury using Nrf2 −/− mice. I/R resulted in an increase in retinal levels of superoxide and pro-inflammatory mediators, as well as leukocyte infiltration of the retina and vitreous, in Nrf2 +/+ mice. These effects were greatly accentuated in Nrf2 −/− mice. With regard to histopathologic damage, Nrf2 −/− mice exhibited loss of cells in the ganglion cell layer and markedly accentuated retinal capillary degeneration, as compared to wild-type. Treatment with the Nrf2 activator CDDO-Me increased antioxidant gene expression and normalized I-R induced superoxide in the retina in wild-type but not Nrf2 −/− mice. CDDO-Me treatment abrogated retinal capillary degeneration induced by I/R in wild-type, but not Nrf2 −/− mice. These studies indicate that Nrf2 is an important cytoprotective mechanism in the retina in response to ischemia-reperfusion injury and suggest that pharmacologic induction of Nrf2 could be a new therapeutic strategy for retinal ischemia-reperfusion and other retinal diseases.
Keywords: Apoptosis, Capillary degeneration, Cytokines, Inflammation, Ischemia-reperfusion, Knockout mice, Nuclear factor erythroid-2 related factor 2, Reactive oxygen species, Retina, Triterpenoids
Introduction
Retinal ischemia is a major contributor to tissue damage in diseases including acute angle-closure glaucoma, retinal vascular occlusions, diabetic retinopathy, and retinopathy of prematurity [1]. An important aspect of this injury is the condition of ischemia-reperfusion (I/R), in which blood supply returns to a tissue bed after a period of ischemia. The absence of oxygen and nutrients during ischemia creates a condition in which restoration of circulation results in the generation of reactive oxygen species [2, 3], leading to inflammation [4]. Animal models of retinal ischemia-reperfusion have been developed and have been widely used to study the effects of I/R on neuronal injury in the retina, especially ganglion cell loss [5, 6]. More recently, it has been demonstrated that injury to the retinal microvasculature, including capillary degeneration, is another major sequela in this retinal ischemia/reperfusion model [7]. This is consistent with ischemia-reperfusion in systemic diseases, which is known to have a major impact on the vasculature [8].
Similar to other tissue beds, retinal I/R injury involves extensive increase in oxidative stress as well as pro-inflammatory changes. The generation of excessive free radicals is known to be an important primary event in retinal I-R. Indeed, modulation of oxidative stress reduces histopathologic changes in retinal I-R injury, including leukocyte infiltration [9] and capillary degeneration [10]. Modulation of inflammation has also been found to be beneficial in retinal IR [11]. . Overall, the retinal I/R model serves as a useful approach for studying the regulation of oxidative stress and inflammation in the retina by various genes and molecules, including iNOS, manganese superoxide dismutase, and catalase [7, 10–12]. This model is especially valuable in evaluating the importance of various molecules in protecting the retina against cellular damage, particularly ganglion cell loss and retinal capillary degeneration, which are critical endpoints in retinal diseases such as glaucoma and diabetic retinopathy, respectively,
Nrf2 (NF-E2-related factor 2) is a transcription factor that plays a significant role in protecting cells from endogenous and exogenous stresses [13]. In a wide range of tissues, Nrf2 acts as a master regulator of the antioxidant response, serving as one of the most important cellular pathways protecting against oxidative stress [14]. Nrf2 also plays an important role as a negative regulator of inflammation [15, 16]. Nrf2 normally resides in the cytoplasm, bound by its cytosolic inhibitor, Keap1, which targets Nrf2 for proteosomal degradation. Various endogenous or exogenous inducing agents such as reactive oxygen species can disrupt the association of Nrf2 with Keap1, leading to nuclear translocation of Nrf2 and the transcriptional activation of an array of cytoprotective genes.
Although Nrf2 is known to play a critical role in many organs including the liver, lung, and brain [13], its role in the retina has been less investigated. In a model of oxygen-induced retinopathy, Nrf2 was found to have a beneficial effect on the retinal vasculature at postnatal day 9, although this effect was not seen at postnatal day 12 [17]. In a model of uveitis in which ocular inflammation is experimentally induced by administration of LPS, Nrf2 was found to modulate the inflammatory response and associated oxidative stress in the retina [18]. However, a retinal cytoprotective role for Nrf2 has not yet been established. In this study, we sought to determine whether Nrf2 prevents cellular injury in retinal ischemia-reperfusion, specifically neuronal and capillary degeneration. We found that Nrf2 is indeed a cytoprotective mechanism in the retina after I/R, and that pharmacologic activation of Nrf2 is a promising strategy for protecting the retina from damage in I/R injury.
Materials and methods
Mice
Nrf2−/− and wild-type (Nrf2+/+) mice were generated as previously described [19] and backcrossed into C57BL/6 background. The animals were maintained on an AIN-76A diet and water ad libitum and housed at a temperature range of 20–23°C under 12:12-h light-dark cycles. Mice were used in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University School of Medicine, and mouse studies adhered to the ARVO (Association of Research in Vision & Ophthalmology) Statement for the Use of Animals in Ophthalmic and Vision Research.
Model of retinal ischemia-reperfusion (I/R)
Retinal ischemia was induced as previously described [7]. Adult mice were anesthetized with a cocktail of 50 mg/kg ketamine, 10 mg/kg xylazine and 2 mg/kg acepromazine. The anterior chamber of one eye was cannulated with a 30-gauge needle attached to a line infusing sterile saline. The intraocular pressure (IOP) was raised to 80–90 mmHg by elevating the saline reservoir. The retina was monitored for blanching, indicating loss of blood flow. After 90 min of ischemia, the needle was withdrawn and the IOP was normalized to allow reperfusion. The other eye of the same mouse was set up as the control.
CDDO-Me Treatment
Some mice were pre-treated with three intraperitoneal injections of 1 μmol/kg CDDO-Me (Reata Pharmaceuticals, Irving, TX, USA; dissolved in 10% DMSO, 10% cremophor-EL, 80% PBS) or vehicle at 48, 24 and 0 hr before being subjected to I/R, similar to a previous dosing scheme used for LPS-induced uveitis [18] and based on a previous study showing that the effect of CDDO-Me on Nrf2 activation lasts for at least 24 hours [20]. After I/R, the mice were treated with 1 μmol/kg CDDO-Me or vehicle every 48 hr. For gene expression analysis, the mice were treated with one intraperitoneal injection of 1 μmol/kg CDDO-Me or vehicle.
Superoxide measurement
Dihydroethidium (DHE; Invitrogen-Molecular Probes, Eugene, IR, USA) was used for histochemical evaluation of superoxide production. DHE is a cell-permeable compound that oxidized on reaction with superoxide to 2-hydroxyethidium, which binds to DNA in the nucleus and fluoresces red [21, 22]. Serial cryosections (5 μm) were incubated with 2.5 μM DHE in DMSO at room temperature for 15 min. After fixation with 2% paraformaldehyde in phosphate buffered saline (PBS; pH 7.4) for 1 min, the cryosections were rinsed twice in PBS for 5min, and visualized using a Zeiss Axiovert 200M microscope (Zeiss Microimaging, Thornwood, NY, USA) under the condition of absorption/emission: 518/605 nm.
Superoxide anion in retina was quantified by lucigenin assay as previously described [23] with minor modification. Briefly, fresh retinas were put into 0.2 mL Krebs/HEPES buffer and incubated in the dark at 37 °C under 85%O2/5%CO2 for 45 min. Lucigenin (Sigma, St. Louis, MO) was added up to a final concentration of 0.5 mM and the plate was incubated for 10 min at room temperature. Photon emission was measured over 10 s by a luminometer (BMG Labtech, Inc., Durham, NC). Repeated measurements were made over a 30 min period for 15 measurements. After averaging the measurements, the values were then subtracted the background from blank and normalized by the protein concentration for each retina.
Histopathology and analysis of cells in retina
Following enucleation, eyes were immediately fixed with 10% formalin overnight at room temperature. The fixed eyes were then dehydrated with graded series of ethanol, embedded in paraffin, cross-sectioned (4μm), and stained with hematoxylin and eosin (H&E). For ganglion cell layer counting, pictures were taken of three sections, randomly selected from each eye in the region of the optic nerve. The nuclei in the ganglion cell layer (excluding the nuclei in the vessels) were counted in each entire section. Counting was performed in a masked fashion. For quantitation of inflammatory cells, H&E stained sections of eyes were scored by an ophthalmic pathologist (CGE) who was masked with respect to animal genotype. Inflammatory cells in the vitreous and below the internal limiting membrane (ILM) were counted in 10 random 400X high-powered fields for each eye.
Apoptotic DNA Cleavage ELISA
Apoptotic DNA cleavage was assayed using a Cell Death Detection ELISA plus Kit (Roche Applied Science, Indianapolis, IN) as previously described [12]. Briefly, fresh retina was immediately put into 200 μL chilled lysis buffer supplied in kit and vortex at the highest speed for 2 min. The tube with retina was incubated for 30 min at room temperature with gentle shaking. The supernatant was collected after centrifugation for 10 min at 10,000g at 4 °C. For each sample, 20 μL supernatant was subjected to the ELISA assay according to the manufacturer’s instructions. Following the color reaction to detect captured DNA fragments, the samples were measured at 405 nm (with 490 nm as reference). The values were normalized by the weight of retina.
Quantitative PCR
Total RNA from retina was isolated using the RNeasy kit (Qiagen, Maryland, MD), then treated with DNase (Qiagen, Hilden, Germany). Single-stranded cDNA was synthesized from 0.5 μg total RNA using oligo (dT)12–18 primer (Invitrogen, Carlsbad, CA), and MMLV Reverse Transcriptase (Invitrogen, Carlsbad, CA) in a final reaction volume of 25 μL. Real-time PCR was performed using SYBR Premix Ex Taq (Takara, Dalian, China) with Stratagene Mx3005P qPCR system (Angilent Technologies, Santa Clara, CA). The primers were NQO-1 sense (5′-TTCTCTGGCCGATTCAGAGT-3′) and antisense (5′-TCCAGACGTTTCTTCCATCC -3′); GCLM sense (5′-TGGAGCAGCTGTATCAGTGG -3′) and antisense (5′-AGAGCAGTTCTTTCGGGTCA-3′); GCLC sense (5′-GTCTCAAGAACATCGCCTCC-3′) and antisense (5′-CTGCACATCTACCACGCAGT -3′); HO-1 sense (5′-GAGCCTGAATCGAGCAGAAC-3′) and antisense (5′-CTTCCAGGGCCGTGTAGATA -3′); TNF-α sense (5′-GACAAGGCTGCCCCGACTA-3′) and antisense (5′-AGGGCTCTTGATGGCAGAGA-3′); COX-2 sense (5′-TGCAGAATTGAAAGCCCTCT -3′) and antisense (5′-CCCAATGAGTAGGCTGGAGA -3′); MCP-1 sense (5′-CCCAATGAGTAGGCTGGAGA -3′) and antisense (5′-TCTGGACCCATTCCTTCTTG -3′); ICAM-1 sense (5′-CTTCCAGCTACCATCCCAAA -3′) and antisense (5′-CTTCAGAGGCAGGAAACAGG -3′); IL-6 sense (5′-CCGGAGAGGAGACTTCACAG -3′) and antisense (5′-TCCACGATTTCCCAGAGAAC -3′)’); iNOS sense (5′-TGGTGGTGACAAGCACATTT-3′) and antisense (5′-AAGGCCAAACACAGCATACC-3′). Cyclophilin A (sense: 5′-AACGACCCCTTCATTGAC-3′; antisense: 5′-TCCACGACATACTCAGCAC-3′) was used for normalization. Each cDNA sample was run in duplicate.
Quantitation of acellular capillaries
Isolation of retinal vasculature and quantification of acellular capillaries were performed as previously described [24, 25] with minor modification. Briefly, the enucleated eyes were fixed with 10% formalin for 1 day. After removing the cornea, the eye-cups were fixed with 10% formalin for 24hrs and dissected to isolate the retina. The retina were rinsed by tap-water overnight and then incubated with 3% Difco trypsin (BD Biosciences, Sparks, MD) containing 0.2 M NaF at 37 °C for 1h. Nonvascular tissues were removed by gentle brushing, and the isolated vasculature was mounted on the slide. After drying overnight, the slides with the retinal blood vessels were subjected to the periodic acid-schiff and hematoxylin stain. To quantify the acellular capillaries, the slides were analyzed for 20~25 fields covering nearly the entire retina in masked fashion (200× magnification). Acellular capillaries were identified as capillary-sized vessel tubes without nuclei along their length. The numbers were normalized by the counting area.
Statistical analysis
Results were expressed as mean ± SE. Data were analyzed by Student t-test for Fig. 1–3, and one-way ANOVA for Fig. 4–8. P values less than 0.05 were considered statistically significant.
Fig. 1. Nrf2-deficient mice exhibit accentuated induction of superoxide levels after retinal I/R injury.
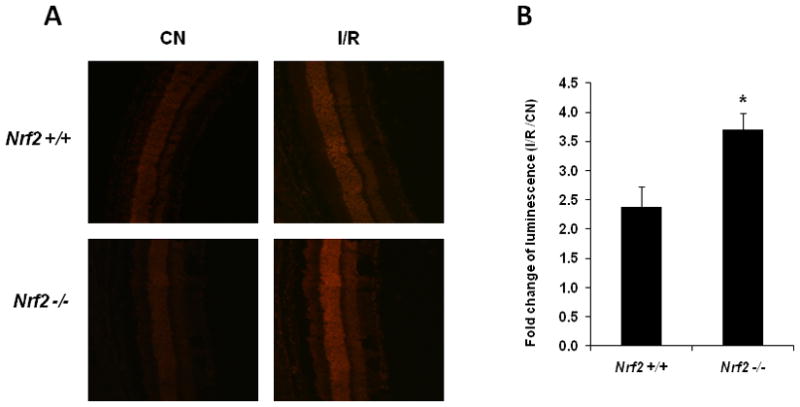
Retinas were obtained and analyzed 24 hrs after I/R. (A) DHE fluorescence was significantly increased in Nrf2 +/+ retinas, but were even greater in retinas from Nrf2 −/− mice following I/R injury. Representative images from each group are shown (n = 4 – 5). (B) Retinal superoxide levels were determined by biochemical assay using lucigenin. Data are expressed as fold-change of luminescence in the retina subjected to I/R vs. control retina (contralateral eye, not subjected to I/R) (n = 4 – 5). The increase in luminescence induced by I/R was significantly greater in Nrf2 −/− mice. CN, control retinas without I/R. * p < 0.05.
Fig. 3. Deletion of Nrf2 promotes more extensive inflammation after retinal I/R injury.

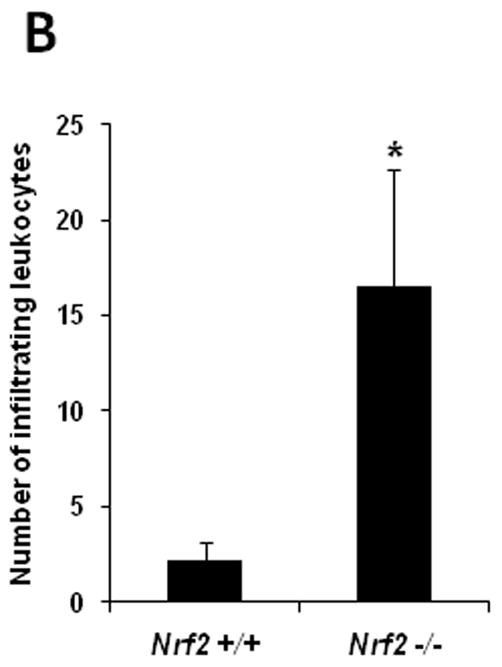
Eyes were harvested and subjected to H&E staining for histological analysis 2 days after I/R. (A) The histological features of the retina from control (non-I/R) wild-type and Nrf2 −/− mice were similar and showed no significant histological changes (a and d). Wild-type mice exhibited leukocyte (predominantly neutrophil) infiltration in the vitreous (short arrow) after I/R injury (b and c). The severity of inflammation was far more extensive after I/R in Nrf2 −/− mice (e and f), with significantly greater numbers of neutrophils (short arrow and inset of panel f). There was detachment of the internal limiting membrane (ILM) (long arrow) after I/R in all the Nrf2 −/− mice, and many neutrophils accumulated in this space (short arrow). Fig. 3c and 3f are higher magnification images of the enclosed area in Fig. 3b and 3e, respectively. Scale bar, 50 μm. (B) Infiltrating leukocytes were quantitated in 10 random high-powered fields for each eye, as described in Methods. The number of infiltrating leukocytes was significantly higher in Nrf2 −/− than wild-type retina after I/R injury. n = 5, * p < 0.05.
Fig. 4. Nrf2-deficient mice exhibit more pronounced neuronal cell loss after retinal I/R injury.
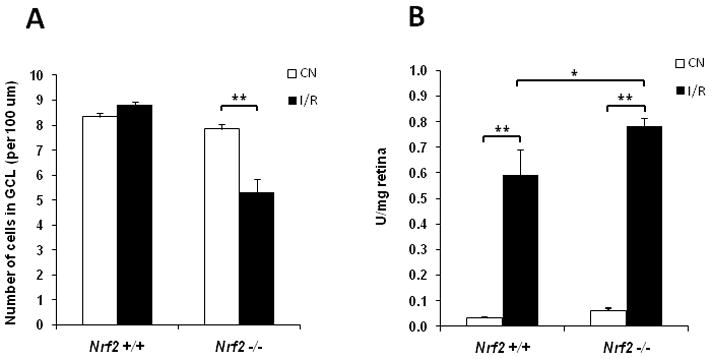
(A) Ganglion cell layer counting was performed as described in Methods. The number of cells in ganglion cell layer (GCL) of the I/R retina was significantly less than that of control retina (CN) in Nrf2 −/−mice 2 days after I/R injury, while no significant change was observed in the wild-type mice (n = 5, ** p < 0.01). (B) Apoptotic DNA Cleavage ELISA was performed 48 hrs after I/R injury. Nucleosomal DNA fragmentation was significantly increased in wild-type mice after I/R injury. There was a further increase in apoptotic DNA cleavage in Nrf2 −/− mice after I/R injury, beyond what was observed in wild-type mice. n = 5, * p < 0.05; ** p < 0.01.
Fig. 8. CDDO-Me inhibits retinal capillary degeneration in wild-type but not Nrf2 −/− retinas following I/R injury.
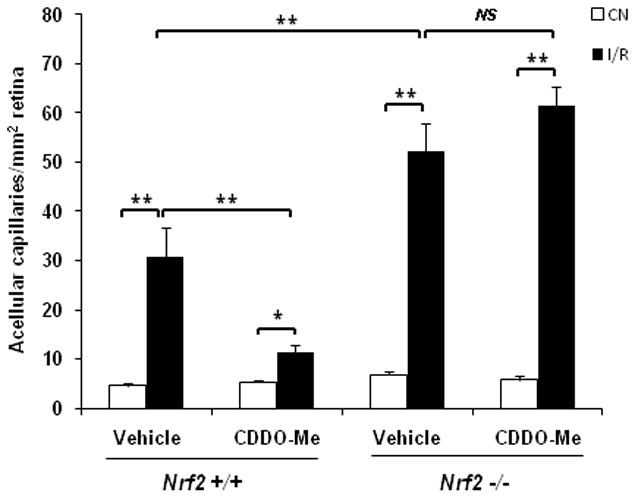
Mice were pre-treated with three intraperitoneal injections of 1 μmol/kg CDDO-Me or vehicle at 48, 24 and 0 hr before being subjected to I/R. After I/R, the mice were treated with 1 μmol/kg CDDO-Me or vehicle every 48 hours. 8 days after I/R, retinas were subjected to trypsin digest to visualize the microvasculature and quantify acellular capillaries. Treatment with CDDO-Me markedly reduced the number of acellular capillaries in wild-type retina, but showed no beneficial effect on Nrf2 −/− retinas. CN, control retinas without I/R. n = 4 – 5, * p < 0.05; ** p < 0.01; NS: not significant.
Results
Increase in retinal ROS by ischemia-reperfusion is accentuated in Nrf2-deficient mice
The generation of excessive free radicals is well-recognized as a critical primary event in retinal ischemia-reperfusion [6, 7]. . We investigated the effect of retinal ischemia-reperfusion on reactive oxygen species in the retina using both a histochemical and biochemical assay. We compared wild-type and Nrf2 −/− mice to determine if Nrf2 is a protective mechanism in the retina in I/R. Dihydroethidium (DHE) or hydroethidine (HE), a cell-permeable, redox-sensitive probe, has been widely used to detect intracellular superoxide anion in histologic imaging studies [26]. We investigated dihydroethidium (DHE) staining for ROS using cryosections, 24 hours after reperfusion. Control (non-I/R) retinas in both wild-type and Nrf2 −/− mice exhibited a low level of detectable ROS (Fig. 1A). DHE fluorescence was significantly increased in wild-type retinas after 24 hours of I/R (Fig. 1A), consistent with a previous report [10]. However, DHE fluorescence was even greater in retinas from Nrf2 −/− mice, indicating an accentuation in retinal superoxide levels in these mice.
As a complementary approach, we also employed a biochemical assay of retinal samples using lucigenin (bis-N-methylacridinium nitrate), an acridylium dinitrate compound which emits light on interaction with superoxide anion [27]. This approach was previously used to detect increased retinal superoxide in diabetes [23]. In wild-type mice, retinas subjected to I/R produced significantly more luminescence (about 2.4-fold) compared to control (non-I/R) retinas (Fig. 1B), consistent with the DHE fluorescence studies. This increase in luminescence induced by I/R was significantly greater Nrf2 −/− mice (about 3.7-fold; Fig. 1B).
Exaggerated retinal expression of inflammatory mediators in Nrf2-deficient mice after ischemia-reperfusion
Retinal I/R injury is also known to be associated with the upregulation of multiple cytokines and inflammatory mediators [7, 11]. We compared the induction of inflammatory genes in the retina by ischemia-reperfusion in wild-type and Nrf2 −/− mice. There was a significant induction of TNF-α, COX-2, MCP-1, ICAM-1, and IL-6 by I/R in wild-type mice, consistent with previous reports (Fig. 2). The induction of these genes by I/R was markedly greater in Nrf2 −/− mice. Retinal iNOS levels were not induced by I/R in wild-type mice, but were significantly increased in Nrf2 −/− mice (Fig. 2).
Fig. 2. Deletion of Nrf2 increases mRNA expression of inflammatory mediators in the retina after I/R injury.
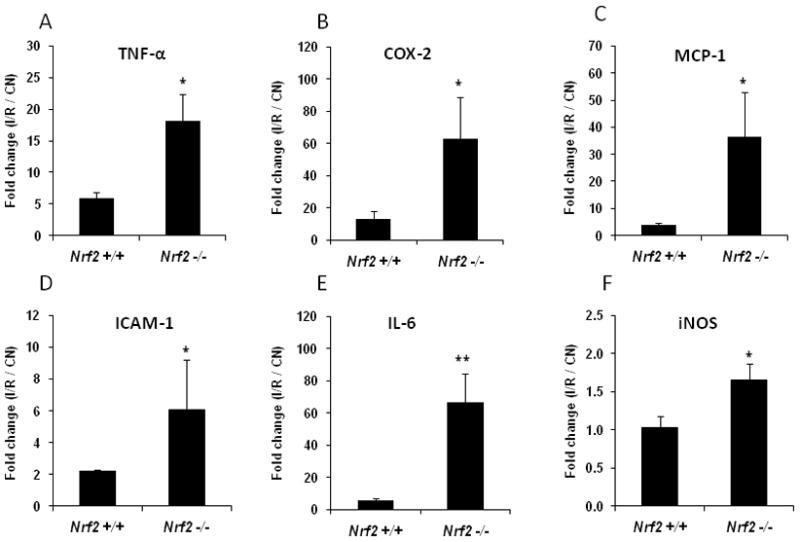
Real-time PCR analysis of inflammatory mediators was performed on retinas that were harvested 6 hours after I/R. Data are expressed as fold-change in the retina subjected to I/R vs. control (CN) retina (contralateral eye, not subjected to I/R). n = 5, * p < 0.05; ** p < 0.01.
More pronounced inflammation and neuronal cell loss in Nrf2-deficient mice after ischemia-reperfusion
Our results above indicate that Nrf2 is operative in the retina and acts as a protective mechanism against oxidative stress and inflammation in ischemia-reperfusion. Although Nrf2 has previously been demonstrated to be operative in the retina in the setting of inflammation [18], it is unclear whether Nrf2 is protective against cellular damage in the retina. Retinal inflammation and neurodegeneration are well-described consequences of ischemia-reperfusion injury. Neurodegeneration occurs via apoptotic cell death, and retinal ganglion cells are particularly susceptible [5–7, 11]. We evaluated the effect of retinal I/R injury on these endpoints in wild-type and Nrf2 −/− mice. The histological features of the retina from control (non-I/R) wild-type and Nrf2 −/− mice were similar and showed no significant histological changes (Fig. 3A). Microscopic examination 2 days after I/R disclosed the presence of scattered leukocytes infiltrating the inner retina and vitreous in wild-type mice (Fig. 3A). The severity of inflammation was far more extensive after I/R in Nrf2 −/− mice, with significantly greater numbers of inflammatory cells, predominantly neutrophils, in the retina and vitreous (Fig. 3A, B). There was detachment of the internal limiting membrane after I/R in all the Nrf2 −/− mice, and many neutrophils accumulated in this space. Serous fluid was present underlying the detached ILM in some eyes. In contrast, the internal limiting membrane remained intact in all wild-type mice after I/R (Fig. 3A).
In addition to inflammation, we also analysed cell loss in the ganglion cell layer after I/R injury. Two days after I/R, we did not observe a significant effect on ganglion cell layer count in wild-type B6 mice (Fig. 4A), in contrast to a previous study [7]. However, in Nrf2 −/− mice, there was a statistically significant (33%) reduction in ganglion cell layer count, implicating a retinal neuroprotective role for Nrf2.
Retinal neuronal degeneration is associated with neuronal apoptosis following I/R injury [7]. We analysed this endpoint using an apoptotic DNA cleavage ELISA of retinal samples. Nucleosomal DNA fragmentation was significantly increased in wild-type mice following I/R injury. There was a further increase in apoptotic DNA cleavage in Nrf2 −/− mice, beyond what was observed in wild-type mice (Fig. 4B). In the absence of I/R, there was a trend toward increased apoptotic DNA cleavage in Nrf2 −/− compared to wild-type, but this was not statistically significant.
Marked accentuation of retinal vascular degeneration in Nrf2-deficient mice following ischemia-reperfusion injury
Capillary degeneration is a central feature of ischemic retinopathies including diabetic retinopathy. Capillary degeneration is known to be an important sequela of retinal ischemia-reperfusion injury [7, 10, 11]. We investigated whether Nrf2 protects against capillary degeneration in retinal I/R injury. As demonstrated in Fig. 5, there was significant (2-fold) increase in retinal capillary degeneration 8 days following I/R injury, consistent with previous reports in mice. Strikingly, Nrf2 −/− mice exhibited a dramatic accentuation of I/R-induced retinal capillary degeneration (about 20-fold increase; Fig. 5).
Fig. 5. Retinal capillary degeneration following I/R injury is markedly accentuated in Nrf2-deficient mice.
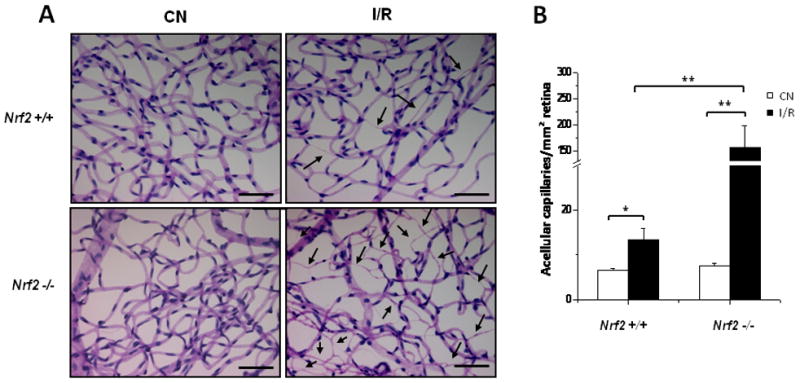
Retinas were subjected to trypsin digest to visualize the microvasculature and quantify acellular capillaries 8 days after I/R. (A) Following I/R injury, Nrf2 +/+ retinas exhibited increased number of acellular capillaries (arrows). Nrf2 −/− retinas exhibited a greatly accentuated increase in acellular capillaries following I/R injury. Scale bar, 100 μm. (B) Quantification of acellular capillaries normalized by the counting area. CN, control retinas without I/R. n = 4 – 5, * p < 0.05; ** p < 0.01.
Expression of Nrf2-responsive antioxidant genes are induced by CDDO-Me treatment
The experiments above with Nrf2 −/− mice indicate that Nrf2 is an important mechanism that confers retinal cytoprotection in ischemia-reperfusion injury, with especially profound effects on capillary degeneration. We were interested in determining whether pharmacologic activation of Nrf2 might have a therapeutic effect on the retina, particularly on capillary degeneration. For this purpose, synthetic triterpenoids are potent inducers of Nrf2 [28, 29], and have been used successfully to induce Nrf2 in the retina [18, 30]. We used the triterpenoid derivative, CDDO-Me [29], a synthetic triterpenoid that activates Nrf2 and attenuates LPS-induced inflammation in human peripheral blood mononuclear cells [31]. We first tested whether exogenous administration of CDDO-Me activates Nrf2 in the retina by evaluating the mRNA expression of four genes known to be targets of transcriptional activation by Nrf2 [32]. We used a dose of 1 micromol/kg body weight, based on a pilot dose-response experiment which demonstrated that this dose was sufficient to achieve maximal upregulation of Nrf2 target genes in the retina (data not shown). We found that intraperitoneal administration of CDDO-Me significantly induces the expression of the Nrf2 target genes in wild-type mice (Fig. 6). Importantly, CDDO-Me did not significantly affect the expression of these genes in Nrf2 −/− mice, confirming that this was an Nrf2-specific effect of CDDO-Me. We did not find any significant differences in the basal levels (i.e., before CDDO-Me treatment) of any of these four genes between Nrf2 +/+ and Nrf2 −/− mice (data not shown).
Fig. 6. CDDO-Me increases the expression of Nrf2-responsive antioxidant genes in wild-type, but not Nrf2 −/− retinas.

Mice were treated with one intraperitoneal injection of 1 μmol/kg CDDO-Me or vehicle 6 hrs before isolating the retina. n = 4, * p < 0.05; NS: not significant.
Treatment with CDDO-Me suppresses superoxide increase in wild-type but not Nrf2 −/− mice
Since excessive free radical generation is known to be a primary event in the pathogenesis of I/R injury in the retina and other tissues, we examined whether CDDO-Me treatment had a functional impact on oxidative stress in the retina following ischemia-reperfusion injury. As demonstrated in Fig. 7, CDDO-Me treatment completely abrogated the increase in retinal superoxide levels induced by I/R in wild-type mice. CDDO-Me treatment had no effect on this I/R-induced increase in retinal superoxide in Nrf2 −/− mice, indicating that the therapeutic effect of CDDO-Me is dependent on Nrf2.
Fig. 7. CDDO-Me suppresses superoxide increase in wild-type but not Nrf2 −/− retinas following I/R injury.
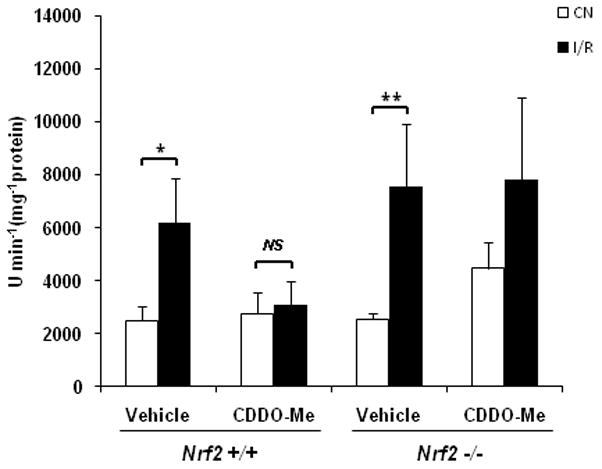
Mice were pre-treated with three intraperitoneal injections of 1 μmol/kg CDDO-Me or vehicle at 48, 24 and 0 hr before being subjected to I/R. Retinal superoxide levels were determined by biochemical assay using lucigenin 24 hrs after I/R. CDDO-Me treatment completely abrogated the increase in retinal superoxide levels induced by I/R in wild-type mice. CDDO-Me treatment had no effect on this I/R-induced increase in retinal superoxide in Nrf2 −/− mice. CN, control retinas without I/R. n = 5, * p < 0.05; ** p < 0.01; NS: not significant.
Treatment with CDDO-Me inhibits retinal vascular degeneration following ischemia-reperfusion injury
Finally, we investigated whether CDDO-Me treatment can have a therapeutic effect on capillary degeneration in the retina following I/R injury. As shown in Fig. 8, treatment with CDDO-Me markedly reduced capillary drop-out in the retina 8 days after ischemia reperfusion by 60%. CDDO-Me treatment had no beneficial effect on retinal capillary drop-out in Nrf2 −/− mice, confirming that this therapeutic benefit of CDDO-Me is mediated by Nrf2.
Discussion
Although Nrf2 is well-known to have a protective role in many organs including liver, lung, and brain [13], it has thus far not been demonstrated whether Nrf2 plays a cytoprotective role in the retina. In similarity to other tissue beds, retinal ischemia-reperfusion is characterized by the generation of excessive free radicals leading to inflammation and tissue damage, including both neuronal and capillary degeneration. Since Nrf2 is considered to be one of the most important cellular pathways protecting against oxidative stress in other tissues [13, 14], we were interested in determining whether Nrf2 serves a similar protective role in the retina in ischemia-reperfusion injury. Our studies indicate that Nrf2 does indeed serve as a protective mechanism against oxidative stress in the retina: Nrf2-deficient mice exhibited a marked accentuation of I-R induced superoxide in the retina, and pharmacologic induction of Nrf2 normalized I-R induced superoxide in wild-type mice. Importantly, our studies also indicate that Nrf2 protects against both neuronal and capillary degeneration in retinal I-R. To our knowledge, this is the first direct evidence for a cytoprotective role of Nrf2 in retinal disease.
Retinal neurodegeneration has been well-described after ischemia-reperfusion injury in rodents, with a particular susceptibility of the ganglion cell layer [5–7, 11]. This phenomenon has been demonstrated in both rats and mice. Interestingly, a study by one lab found that the time-course is faster in rats than mice. In rats, much of the GCL loss had already occurred at 2 days after I/R as compared to 5 days. In B6 mice, there was significantly greater GCL loss after 5 days, although some GCL loss was observed after 2 days [7]. In our study, we did not observe a reduction in GCL counts after 2 days in wild-type B6 mice, in contrast to this earlier study, perhaps reflecting a somewhat delayed time-course of GCL loss in our mice. Nevertheless, we did observe a significant reduction in GCL counts in the corresponding Nrf2-deficient mice. This suggests that Nrf2 plays a physiologic role in modulating retinal neurodegeneration following ischemia-reperfusion.
In addition to neuronal damage, retinal ischemia-reperfusion is known to have a significant injurious impact on the retinal vasculature [7, 10, 11]. We found that retinal I/R injury leads to significant increase in capillary degeneration (Fig. 5), consistent with published reports. This pathologic effect was markedly accentuated in mice deficient in Nrf2 (Fig. 5), indicating that Nrf2 is an important protective mechanism in the retina for both neurons and blood vessels.
Since our studies indicate that Nrf2 is operative in the retina and play a protective role, we were interested in exploiting Nrf2 as a therapeutic target. For this purpose, we utilized the synthetic triterpenoid, CDDO-Me, since triterpenoids have previously been demonstrated to induce Nrf2-regulated genes in the retina [30], and are beneficial in modulating pro-inflammatory retinal changes, including leukocyte adhesion, in an LPS-induced model of ocular inflammation [18]. Given the primary role of free radicals in mediating retinal ischemia-reperfusion injury, we first looked at the effect of CDDO-Me on retinal superoxide and found that this drug normalized I-R induced superoxide in wild-type but not Nrf2-deficient mice. This is consistent with previous studies demonstrating that the anti-oxidative effects of this triterpenoid class are largely mediated by Nrf2 [33, 34], likely via modulation of its inhibitor Keap1 [33]. We then found that CDDO-Me abrogated I-R induced capillary degeneration, the most striking effect of retinal ischemia-reperfusion in our study. Importantly, as with the effect on superoxide, this effect on capillary degeneration was observed only in wild-type and not Nrf2-deficient mice, indicating that the therapeutic effect of CDDO-Me is dependent on Nrf2. These results serve as a proof of principle that pharmacologic targeting of Nrf2 can be a useful approach for retinal protection, and suggest specifically that synthetic triterpenoids such as CDDO-Me are useful agents for this purpose. This builds on previous work suggesting that Nrf2 activation by pharmacologic agent (genipin derivative) [35] or environmental modulation (bright cyclic light) [36] are promising strategies for retinal neuronal protection.
Although free radical generation is a primary event in the initiation of tissue damage in IR in tissues including the retina, the inflammation that ensues also plays a very important role in causing tissue damage. Our studies demonstrated that the inflammatory response to retinal I/R is also modulated by Nrf2. Nrf2-deficient mice exhibited a marked accentuation of the induction of inflammatory mediators as well as neutrophil infiltration of the retina and vitreous. We observed detachment of the internal limiting membrane (ILM) in all eyes from Nrf2-deficient mice following I/R. This ILM detachment might have resulted from the vascular leakage and serous fluid “pushing up” on the ILM, excessive proteolysis by leukocytes digesting the tissue anchoring the ILM to the retina, vitreous inflammation promoting traction on the ILM, or a combination of these factors.
It will be of interest in future studies to investigate the global regulation of retinal gene expression by Nrf2, in order to determine the important genes which mediate its therapeutic effect. In this study, we specifically examined the expression of NQO-1, GCLM, and HO-1 because they are established targets of transcriptional activation by Nrf2 [32], thereby allowing us to confirm that systemic CDDO-Me administration was successful in activating Nrf2 in the retina. We also looked at GCLC, another Nrf2 target gene induced by triterpenoids in the retina [30]. All four genes were induced in the retina by CDDO-Me in an Nrf2-dependent fashion. GCLC and GCLM are the catalytic and modifier subunits, respectively, of the glutamate cysteine ligase (GCL) complex, which is the rate-limiting enzyme in glutathione (GSH) synthesis [37]. In light of glutathione’s role as an important cellular antioxidant, induction of GCLC and GCLM could be involved in the protective response of CDDO-Me. NQO1 is known to have multiple cytoprotective roles, including superoxide scavenging activity [38]. NQO1 also exhibits anti-inflammatory properties, such as the inhibition of inflammatory cytokine induction by LPS [39]. Heme oxygenase-1 (HO-1) is an enzyme induced ubiquitously in response to oxidative stress [40] and has both antioxidant and anti-inflammatory activity in multiple tissues, including the vasculature [41]. Each of these genes may well participate in the Nrf2 response in the retina. However, given the ability of Nrf2 to induce a wide array of cytoprotective genes, it is likely that multiple other genes participate in mediating the effect of Nrf2 in retinal ischemia-reperfusion.
In conclusion, our studies demonstrate that Nrf2 is an important factor in protecting the retina against injury in ischemia-reperfusion. Deficiency of Nrf2 magnified retinal superoxide and inflammation induced by retinal I/R injury and significantly exacerbated degeneration of both neurons and capillaries. Activation of Nrf2 by CDDO-Me increased antioxidant gene expression, decreased superoxide levels, and markedly reduced capillary degeneration in the retina. Together these results suggest that Nrf2 is an important mechanism used by the retina to protect against injury. It will therefore be important to consider the possibility that Nrf2 could be important in other retinal conditions such as glaucoma and ischemic retinopathies, including diabetic retinopathy. Pharmacologic activation of Nrf2 could be a therapeutic strategy to protect the retina against oxidative stress and inflammation in ischemia-reperfusion and possibly other conditions.
Acknowledgments
We are grateful to Shyam Biswal for sharing the Nrf2 −/− mice and thankful to Dr Masayuki Yamamoto who originally provided these mice to Shyam Biswal. We also thank Shyam Biswal and Rajesh Thimmulappa for many helpful suggestions during the study. The authors thank Timothy Kern, Jie Tang, and Yunpeng Du for invaluable technical assistance regarding the isolation of retinal vasculature and quantification of acellular capillaries and lucigenin assay for superoxide. We are grateful to Jiangxia Wang for providing advice about the data analysis, including ANOVA test. These studies were supported in part by research grants from the National Institutes of Health EY018138 (EJD) and EY019904 (JTH), the Juvenile Diabetes Research Foundation, Thome Foundation Grant, Robert Bond Welch Professorship, and Cindy and Jeong H. Kim, Ph.D. EJD is a recipient of an RPB Career Development Award.
List of abbreviations
- CDDO-Me
2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid-methyl ester
- COX-2
cyclooxygenase-2
- DHE
dihydroethidium
- GCLC
glutamate--cysteine ligase, catalytic subunit
- GCLM
glutamate-cysteine ligase, modifier subunit
- HO-1
heme oxygenase-1
- ICAM-1
intercellular adhesion molecule-1
- iNOS
inducible nitric oxide synthase
- IL-6
interleukin-6
- ILM
internal limiting membrane
- IOP
intraocular pressure
- I/R
ischemia-reperfusion
- LPS
lipopolysaccharide
- MCP-1
monocyte chemotactic protein-1
- NQO1
NAD(P)H dehydrogenase, quinone 1
- Nrf2
NF-E2-related factor 2
- ROS
reactive oxygen species
- TNF-α
tumor necrosis factor-α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 2.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 3.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci U S A. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korthuis RJ, Granger DN. Reactive oxygen metabolites, neutrophils, and the pathogenesis of ischemic-tissue/reperfusion. Clin Cardiol. 1993;16:I19–26. doi: 10.1002/clc.4960161307. [DOI] [PubMed] [Google Scholar]
- 5.Hangai M, Yoshimura N, Hiroi K, Mandai M, Honda Y. Inducible nitric oxide synthase in retinal ischemia-reperfusion injury. Exp Eye Res. 1996;63:501–509. doi: 10.1006/exer.1996.0140. [DOI] [PubMed] [Google Scholar]
- 6.Neufeld AH, Kawai S, Das S, Vora S, Gachie E, Connor JR, Manning PT. Loss of retinal ganglion cells following retinal ischemia: the role of inducible nitric oxide synthase. Exp Eye Res. 2002;75:521–528. doi: 10.1006/exer.2002.2042. [DOI] [PubMed] [Google Scholar]
- 7.Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007;48:361–367. doi: 10.1167/iovs.06-0510. [DOI] [PubMed] [Google Scholar]
- 8.Gourdin MJ, Bree B, De Kock M. The impact of ischaemia-reperfusion on the blood vessel. Eur J Anaesthesiol. 2009;26:537–547. doi: 10.1097/EJA.0b013e328324b7c2. [DOI] [PubMed] [Google Scholar]
- 9.Szabo ME, Droy-Lefaix MT, Doly M, Carre C, Braquet P. Ischemia and reperfusion-induced histologic changes in the rat retina. Demonstration of a free radical-mediated mechanism. Invest Ophthalmol Vis Sci. 1991;32:1471–1478. [PubMed] [Google Scholar]
- 10.Chen B, Caballero S, Seo S, Grant MB, Lewin AS. Delivery of antioxidant enzyme genes to protect against ischemia/reperfusion-induced injury to retinal microvasculature. Invest Ophthalmol Vis Sci. 2009;50:5587–5595. doi: 10.1167/iovs.09-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Portillo JA, Van Grol J, Zheng L, Okenka G, Gentil K, Garland A, Carlson EC, Kern TS, Subauste CS. CD40 mediates retinal inflammation and neurovascular degeneration. J Immunol. 2008;181:8719–8726. doi: 10.4049/jimmunol.181.12.8719. [DOI] [PubMed] [Google Scholar]
- 12.Abcouwer SF, Lin CM, Wolpert EB, Shanmugam S, Schaefer EW, Freeman WM, Barber AJ, Antonetti DA. Vascular Permeability and Apoptosis are Separable Processes in Retinal Ischemia-Reperfusion Injury: Effects of Ischemic Preconditioning, Bevacizumab and Etanercept. Invest Ophthalmol Vis Sci. 2010;51:5920–5933. doi: 10.1167/iovs.10-5264. [DOI] [PubMed] [Google Scholar]
- 13.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 14.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen XL, Kunsch C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: a new therapeutic approach for the treatment of inflammatory diseases. Curr Pharm Des. 2004;10:879–891. doi: 10.2174/1381612043452901. [DOI] [PubMed] [Google Scholar]
- 16.Li N, Nel AE. Role of the Nrf2-mediated signaling pathway as a negative regulator of inflammation: implications for the impact of particulate pollutants on asthma. Antioxid Redox Signal. 2006;8:88–98. doi: 10.1089/ars.2006.8.88. [DOI] [PubMed] [Google Scholar]
- 17.Uno K, Prow TW, Bhutto IA, Yerrapureddy A, McLeod DS, Yamamoto M, Reddy SP, Lutty GA. Role of Nrf2 in retinal vascular development and the vaso-obliterative phase of oxygen-induced retinopathy. Exp Eye Res. 2010;90:493–500. doi: 10.1016/j.exer.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagai N, Thimmulappa RK, Cano M, Fujihara M, Izumi-Nagai K, Kong X, Sporn MB, Kensler TW, Biswal S, Handa JT. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic Biol Med. 2009;47:300–306. doi: 10.1016/j.freeradbiomed.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 20.Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, Guilarte TR, Yamamoto M, Sporn MB, Kensler TW. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6:154–162. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- 21.Miller FJ, Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 22.Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci U S A. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du Y, Miller CM, Kern TS. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic Biol Med. 2003;35:1491–1499. doi: 10.1016/j.freeradbiomed.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Gubitosi-Klug RA, Talahalli R, Du Y, Nadler JL, Kern TS. 5-Lipoxygenase, but not 12/15-lipoxygenase, contributes to degeneration of retinal capillaries in a mouse model of diabetic retinopathy. Diabetes. 2008;57:1387–1393. doi: 10.2337/db07-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 26.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gyllenhammar H. Lucigenin chemiluminescence in the assessment of neutrophil superoxide production. J Immunol Methods. 1987;97:209–213. doi: 10.1016/0022-1759(87)90461-3. [DOI] [PubMed] [Google Scholar]
- 28.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 2005;65:4789–4798. doi: 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- 29.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–369. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 30.Pitha-Rowe I, Liby K, Royce D, Sporn M. Synthetic triterpenoids attenuate cytotoxic retinal injury: cross-talk between Nrf2 and PI3K/AKT signaling through inhibition of the lipid phosphatase PTEN. Invest Ophthalmol Vis Sci. 2009;50:5339–5347. doi: 10.1167/iovs.09-3648. [DOI] [PubMed] [Google Scholar]
- 31.Thimmulappa RK, Fuchs RJ, Malhotra D, Scollick C, Traore K, Bream JH, Trush MA, Liby KT, Sporn MB, Kensler TW, Biswal S. Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid Redox Signal. 2007;9:1963–1970. doi: 10.1089/ars.2007.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P, Tuder RM, Biswal S. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 2008;178:592–604. doi: 10.1164/rccm.200803-380OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Ichikawa T, Li J, Meyer CJ, Janicki JS, Hannink M, Cui T. Dihydro-CDDO-trifluoroethyl amide (dh404), a novel Nrf2 activator, suppresses oxidative stress in cardiomyocytes. PLoS ONE. 2009;4:e8391. doi: 10.1371/journal.pone.0008391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Calingasan NY, Thomas B, Chaturvedi RK, Kiaei M, Wille EJ, Liby KT, Williams C, Royce D, Risingsong R, Musiek ES, Morrow JD, Sporn M, Beal MF. Neuroprotective effects of the triterpenoid, CDDO methyl amide, a potent inducer of Nrf2-mediated transcription. PLoS ONE. 2009;4:e5757. doi: 10.1371/journal.pone.0005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koriyama Y, Chiba K, Yamazaki M, Suzuki H, Muramoto K, Kato S. Long-acting genipin derivative protects retinal ganglion cells from oxidative stress models in vitro and in vivo through the Nrf2/antioxidant response element signaling pathway. J Neurochem. 2010;115:79–91. doi: 10.1111/j.1471-4159.2010.06903.x. [DOI] [PubMed] [Google Scholar]
- 36.Tanito M, Agbaga MP, Anderson RE. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic Biol Med. 2007;42:1838–1850. doi: 10.1016/j.freeradbiomed.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 37.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 501:116–123. doi: 10.1016/j.abb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J Immunol. 2008;181:6730–6737. doi: 10.4049/jimmunol.181.10.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 41.Dulak J, Loboda A, Jozkowicz A. Effect of heme oxygenase-1 on vascular function and disease. Curr Opin Lipidol. 2008;19:505–512. doi: 10.1097/MOL.0b013e32830d81e9. [DOI] [PubMed] [Google Scholar]