

Abstract
Background
Fluphenazine is one of the first drugs to be classed as an ‘antipsychotic’ and has been widely available for five decades.
Objectives
To compare the effects of oral fluphenazine with placebo for the treatment of schizophrenia.
Search methods
We updated searches of the Cochrane Schizophrenia Group's trials register, which includes relevant randomised controlled trials from the bibliographic databases Biological Abstracts, CINAHL, The Central Register of Controlled Trials in The Cochrane Library, EMBASE, MEDLINE, PsycLIT, LILACS, PSYNDEX, Sociological Abstracts and Sociofile, 15 May, 2012. References of all identified studies were searched for further trial citations.
Selection criteria
We sought all randomised controlled trials comparing oral fluphenazine with placebo relevant to people with schizophrenia. Primary outcomes of interest were global state and adverse effects.
Data collection and analysis
We inspected citations and abstracts independently, ordered papers and re-inspected and quality assessed trials. We extracted data independently. Dichotomous data were analysed using fixed-effect risk ratio (RR) and the 95% confidence interval (CI). Continuous data were excluded if more than 50% of people were lost to follow-up, but, where possible, mean differences (MD) were calculated.
Main results
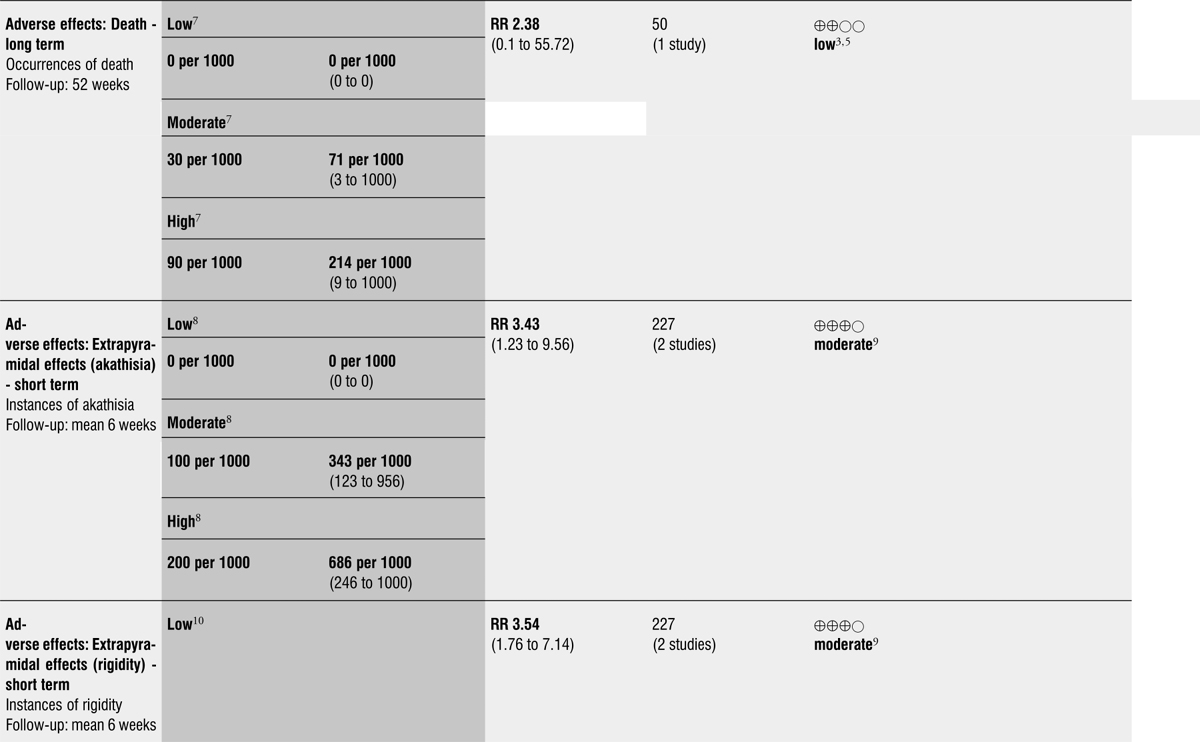
From over 1200 electronic records of 415 studies identified by our initial search and this updated search, we excluded 48 potentially relevant studies and included seven trials published between 1964 and 1999 that randomised 439 (mostly adult participants). No new included trials were identified for this review update. Compared with placebo, global state outcomes of ‘not improved or worsened’ were not significantly different in the medium term in one small study (n = 50, 1 RCT, RR 1.12 CI 0.79 to 1.58, very low quality of evidence). The risk of relapse in the long term was greater in two small studies in people receiving placebo (n = 86, 2 RCTs, RR 0.39 CI 0.05 to 3.31, very low quality of evidence), however with high degree of heterogeneity in the results. Only one person allocated fluphenazine was reported in the same small study to have died on long-term follow-up (n = 50, 1 RCT, RR 2.38 CI 0.10 to 55.72, low quality of evidence). Short-term extrapyramidal adverse effects were significantly more frequent with fluphenazine compared to placebo in two other studies for the outcomes of akathisia (n = 227, 2 RCTs, RR 3.43 CI 1.23 to 9.56, moderate quality of evidence) and rigidity (n = 227, 2 RCTs, RR 3.54 CI 1.76 to 7.14, moderate quality of evidence).
Authors’ conclusions
The findings in this review confirm much that clinicians and recipients of care already know, but they provide quantification to support clinical impression. Fluphenazine's global position as an effective treatment for psychoses is not threatened by the outcome of this review. However, fluphenazine is an imperfect treatment and if accessible, other inexpensive drugs less associated with adverse effects may be an equally effective choice for people with schizophrenia.
BACKGROUND
Description of the condition
One in every 10,000 people per year is diagnosed with schizophrenia, with a lifetime prevalence of about 1% (Jablensky 1992). It often runs a chronic course with acute exacerbations and often partial remissions. Over the past few decades, a large body of evidence has accumulated linking excessive dopamine transmission to psychosis and more direct evidence emerges from neuro-imaging studies which showed an increased dopamine synthesis (Hietala 1995; Lindstrom 1999; Meyer 2002), an exaggerated release of dopamine and a higher than normal levels of dopamine at baseline (Abi-Dargham 2000; Gjedde 2001). The antipsychotic group of drugs with its anti-dopaminergic effects is the mainstay treatment for this illness (Dencker 1980). These are generally regarded as highly effective, especially in controlling such symptoms as hallucinations and fixed false beliefs (delusions) (Kane 1986). Moreover, they seem to reduce the risk of acute relapse. A systematic review undertaken two decades ago also suggested that, for those with serious mental illness, stopping antipsychotics resulted in 58% of people relapsing, whereas only 16% of those who were still on the drugs became acutely ill within a one-year period (Davis 1986). Schizophrenia usually begins in young adulthood and has a lifetime prevalence of about 1% irrespective of culture, social class and race. Schizophrenia is a chronic relapsing mental illness, characterised by symptoms such as hallucinations, delusions, disordered thinking, and emotional withdrawal. Antipsychotic drugs are effective for controlling florid symptoms such as hallucinations and delusions but are less effective for treating emotional withdrawal. Antipsychotics are associated with adverse effects such as movement disorders, and the overall cost of the illness to the individual, their carers and the community is considerable.
Description of the intervention
Fluphenazine, a phenothiazine derivative, was one of the first drugs to be classed as an ‘antipsychotic’ and was approved by the FDA in 1959. In Britain it was first used for the relief of anxiety. The American reports, however, were the first to indicate its value in psychotic illness (Darling 1959; Holt 1960). Fluphenazine has trifluoromethyl and piperazine groups which bring about increase in potency that in many pharmacodynamic properties may be about 40 times as potent as chlorpromazine. This is associated with a rapid and prolonged action, relatively little sedative activity and little or no increase in autonomic and haemodynamic effects. Fluphenazine is an inexpensive and widely accessible antipsychotic drug that has been available to treat people with schizophrenia for five decades. In this review, for perhaps the first time, we objectively quantify the effects of oral administration of fluphenazine in comparison with placebo. It is indeed a potent antipsychotic but with considerable adverse effects. Other drugs may well be preferable.
How the intervention might work
Fluphenazine is thought to elicit its antipsychotic effects via interference with central dopaminergic pathways and blocking receptors, particularly D2, in the mesolimbic zone of the brain. Extrapyramidal side effects are a result of interaction with dopaminergic pathways in the basal ganglia. As fluphenazine is not specific to one action within the body, it is known to cause adverse effects ranging from orthostatic hypotension as a result of its alpha adrenergic blocking activity to anticholinergic and extrapyramidal symptoms (tardive dyskinesia, pseudo-parkinsonism, dystonia, dyskinesia, akathisia). In addition, the use of fluphenazine has been associated with a potentially fatal disturbance of blood pressure, temperature and muscle control (neuroleptic malignant syndrome). As with all antipsychotic medications, fluphenazine is characterised by inter-individual variability in pharmacokinetics, most marked with the oral preparation. It is extensively metabolised, undergoing ‘first pass’ metabolism by the liver and is excreted in both the urine and faeces. Fluphenazine is highly protein-bound (greater than 90%) in plasma. With oral fluphenazine, peak plasma/serum levels are attained within a few hours. The serum half-life of it is approximately 15 hours. Fluphenazine crosses the blood-brain barrier, crosses the placenta easily and cannot be removed by dialysis (Wikipedia 2006).
Why it is important to do this review
Fluphenazine is still one of the drugs commonly used for people with schizophrenia and is given by mouth or short-acting injection. Although we have not found precise data on how much fluphenazine is used worldwide, it is one of the World Health Organization's Essential Drugs (WHO 2005) and in the developing world, where non-proprietary preparations of fluphenazine are inexpensive, it may be one of the only drug treatments available. However, although it is still available in most of Europe and North America, the arrival of a newer generation of antipsychotic drugs has reduced its market share in the respective countries. This version of the review updates our past work (Matar 2007a; Matar 2007b). This is an update of a Cochrane Review first published in 2007 (Issue 1) of The Cochrane Database of Systematic Reviews.
OBJECTIVES
To compare the effects of oral fluphenazine with placebo for the treatment of schizophrenia.
METHODS
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials. We included trials described as ‘double-blind’ if it was implied that the study was randomised and we included these in a sensitivity analysis. If their inclusion did not result in a substantive difference, they remained in the analyses. If their inclusion did result in statistically significant differences, we did not add the data from these lower quality studies to the results of the better trials, but presented these within a subcategory. We excluded quasi-randomised studies, such as those allocating by alternate days of the week.
Types of participants
We included people diagnosed with schizophrenia or schizophrenia-like illnesses using any criteria, irrespective of age, sex or severity of illness.
Types of interventions
1. Fluphenazine: any dose of only oral administration
2. Placebo: (active or inactive) or no treatment
Types of outcome measures
Where possible, outcomes were made binary by dividing them into two categories - ‘clinically significant change’ and ‘no clinically significant change’.
We categorised outcomes as short term (0-8 weeks), medium term (9 to 26 weeks) and long term (27 weeks to 104 weeks).
Primary outcomes
1. Global state
1.1 Not improved or worsened
2. Adverse effects
2.1 General
2.2 Specific
2.2.1 Extrapyramidal symptoms (parkinsonian symptoms, dystonia, akathisia, and tardive dyskinesia)
2.2.2 Anticholinergic symptoms
2.2.3 Others
Secondary outcomes
1. Global state
1.1 Relapse
1.2 Time in exacerbated state
1.3 Leaving the study early
1.4 Length of stay in hospital
1.5 Satisfaction with treatment - participant/carer
1.6 Death
2. Mental state
2.1 General symptoms
2.2 Specific symptoms
2.2.1 Positive symptoms (delusions, hallucinations, disordered thinking)
2.2.2 Negative symptoms (avolition, poor self-care, blunted affect)
2.2.3 Mood - depression
3. Behaviour
3.1 General behaviour
3.2 Specific behaviours (e.g. aggressive or violent behaviour)
3.2.1 Social functioning
3.2.2 Employment status during trial (employed/unemployed)
3.2.3 Occurrence of violent incidents (to self, others or property)
4. Economic
4.1 Cost of care
5. ‘Summary of findings’ table
We used the GRADE approach to interpret findings (Schünemann 2008) and used the GRADE profiler to import data from Review Manager (RevMan) to create ‘Summary of findings’ table/s. These tables provide outcome-specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient-care and decision making. We selected the following main outcomes for inclusion in the ‘Summary of findings’ table.
Global state - not improved or worsened - medium term.
Relapse - long term.
Adverse effects: death - long term.
Adverse effects: extrapyramidal effects (akathisia) - short term.
Adverse effects: extrapyramidal effects (rigidity) - short term.
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia Group's trials register (May 2012)
The Trial Search Co-ordinator searched the Cochrane Schizophrenia Group's Trials Register 15th May 2012 using the same search criteria as our initial 2006 review.
For details of previous search please see Appendix 1. [((fluphen* or flufen* or modec* or moditen* or eutimax* or prolixin* or siqualon* or anaten* or dapotum* or decazate* or decafen* or decentan* or fludecate* or lyogen* or lyoridin* or mirenil*) in title, abstract and index fields in REFERENCE) OR (fluphenazin* in interventions field in STUDY)]
The Cochrane Schizophrenia Group's Trials Register is compiled by systematic searches of major databases, handsearches and conference proceedings (see group module).
Searching other resources
1. Reference searching
We inspected references of all identified studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Methods used in data collection and analysis for this update are below; for previous methods please see Appendix 2.
Selection of studies
For this 2012 update, review authors HEM and MQA inspected citations from the new electronic search and identified relevant abstracts. HEM and MQA also inspected full articles of the abstracts meeting inclusion criteria and carried out the reliability check of all citations from the new electronic search.
Data extraction and management
1. Extraction
For this update, HEM and MQA extracted data from included studies. We extracted data presented only in graphs and figures whenever possible. When further information was necessary, we contacted authors of studies in order to obtain missing data or for clarification. We encountered multi-centre trials, however, we were unable to extract data relevant to each component centre separately; this was because the study was published many years ago, and such data were unavailable (Goldberg 1964).
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale-derived data
We included continuous data from rating scales only if:
a. the psychometric properties of the measuring instrument have been described in a peer-reviewed journal (Marshall 2000); and b. the measuring instrument has not been written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should either be i. a self-report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly; we have noted whether or not this is the case in Description of studies.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between-person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. Had enough studies reported continuous data, we would have combined endpoint and change data in the analysis and used mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011, Chapter 9.4.5.2).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non-parametric data, we aimed to apply the following standards to all data before inclusion:
a) standard deviations (SDs) and means are reported in the paper or obtainable from the authors;
b) when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996));
c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), (Kay 1986)), which can have values from 30 to 210), we modified the calculation described above to take the scale starting point into account. In these cases skew is present if 2 SD > (S-S min), where S is the mean score and S min is the minimum score.
Endpoint scores on scales often have a finite start and end point and these rules can be applied. We entered skewed endpoint data from studies of fewer than 200 participants as ‘other data’ within the Data and analyses rather than into a statistical analysis. Skewed data pose less of a problem when looking at mean if the sample size is large; we would have entered such endpoint data into syntheses had we encountered such data.
When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not; therefore, we entered skewed change data into analyses regardless of size of study.
2.5 Common measure
Had we encountered such measures, in order to facilitate comparison between trials, we would have converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Had such data been available, we would have made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut-off points on rating scales and dividing participants accordingly into ‘clinically improved’ or ‘not clinically improved’. It is generally assumed that if there is a 50% reduction in a scale-derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986); this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we would have used the primary cut-off presented by the original authors.
2.7 Direction of graphs
We entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for oral fluphenazine. Where keeping to this made it impossible to avoid outcome titles with clumsy double-negatives (e.g. ‘Not improved’), we reported data where the left of the line indicates an unfavourable outcome. This was the case with outcome 1.10 Leaving the study early: 3. marked improvement/ hospital discharge (Analysis 1.8), which reflected a positive outcome.
Analysis 1.8.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 8 Leaving the study early: 3. Marked improvement/ hospital discharge.
Assessment of risk of bias in included studies
For this update, HEM and MQA worked independently by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This new set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain additional information. We have noted the level of risk of bias in both the text of the review and in the Summary of findings for the main comparison.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). For statistically significant results, we used ‘Summary of findings’ tables to calculate the number needed to treat to provide benefit (NNTB)/to induce harm (NNTH) statistic and its 95% CI.
2. Continuous data
For continuous outcomes, we estimated mean difference (MD) between groups.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ ‘cluster randomisation’ (such as randomisation by clinician or practice), but analysis and pooling of clustered data poses problems. Authors often fail to account for intra-class correlation in clustered studies, leading to a ‘unit of analysis’ error (Divine 1992) whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Had we encountered such studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intra-class correlation coefficients (ICCs) or their clustered data and to adjust for this by using accepted methods (Gulliford 1999). We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a ‘design effect’. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1+(m-1)*ICC] (Donner 2002). No cluster trials were identified in this review; however, if in future updates of this review cluster-randomised studies are identified, where the ICC is not reported it will be assumed to be 0.1 (Ukoumunne 1999).
2. Cross-over trials
A major concern of cross-over trials is the carry-over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash-out phase. For the same reason cross-over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data of the first phase of cross-over studies. This was the case in Millar 1963.
3. Studies with multiple treatment groups
Had we found studies that involved more than two relevant treatment arms, we would have presented the additional treatment arms in comparisons. However, we found no such studies; if, in future updates of this review, if such studies are identified, binary data will be simply added and combined within the two-by-two table. If data are continuous, we will combine data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011). Where the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow-up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 60% of data be unaccounted for, we would not reproduce these data or use them within analyses. If, however, more than 60% of those in one arm of a study were lost, but the total loss was less than 40%, we would have marked such data with (*) to indicate that such a result may well be prone to bias.
2. Binary
In the case where attrition for a binary outcome is between 0% and 60% and where these data are not clearly described, had we found such studies, we would have presented data on a ‘once-randomised-always-analyse’ basis (an intention-to-treat analysis). Those leaving the study early would be assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. For these outcomes, the rate of those who stay in the study - in that particular arm of the trial - would be used for those who did not. We undertook a sensitivity analysis to test how prone the primary outcomes were to change when data only from people who completed the study to that point were compared to the intention-to-treat analysis using the above assumptions.
3. Continuous
3.1 Attrition
In the case where attrition for a continuous outcome is between 0 and 60%, and data only from people who complete the study to that point are reported, we presented and used these data.
3.2 Standard deviations
Had standard deviations not been reported, we first would have tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error and confidence intervals available for group means, and either P value or T value available for differences in mean, we would have calculated them according to the rules described in the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011). Had only the standard error (SE) been reported, standard deviations (SDs) can be calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011) present detailed formula for estimating SDs from P values, T or F values, confidence intervals, ranges or other statistics. If these formula do not apply, we would have calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study's outcome and thus to lose information. Had we imputed data, and if future updates of this review employ use of imputed data, we will examine the validity of the imputations in a sensitivity analysis excluding the imputed values. Had we needed to, we would have imputed standard deviations using this model for Clark 1971.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, where LOCF data have been used in the trial, if less than 50% of the data have been assumed, we reproduced these data and indicated that they are the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, we fully discussed these.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose, we fully discussed these.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a CI for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic was interpreted as evidence of substantial levels of heterogeneity (Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small-study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes.
Data synthesis
We understand that there is no closed argument for preference for use of fixed-effect or random-effects models. The random-effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random-effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random-effects model: it puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the fixed-effect model for all analyses, but used of the random-effects model where heterogeneity was present.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses - only primary outcomes
1.1 Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of oral fluphenazine for people with schizophrenia in general. In addition, however, we tried to report data on subgroups of people in the same clinical state, stage and with similar problems.
2. Investigation of heterogeneity
If inconsistency was high, we have reported this. First, we investigated whether data had been entered correctly. Second, if data were correct, we visually inspected the graph and successively removed outlying studies to see if homogeneity was restored. For this review we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present data. If not, then we did not pool data and discussed issues. We know of no supporting research for this 10% cut-off, but we used prediction intervals as an alternative to this unsatisfactory state.
When unanticipated clinical or methodological heterogeneity was obvious, we simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Hypotheses
It was expected that several sensitivity analyses could be undertaken within this review. The following hypotheses were to be tested: When compared with placebo, for the primary outcomes of interest (see: Criteria for considering studies for this review), fluphenazine is differentially effective for:
1.1 Men and women.
1.2 People who are under 18 years of age, between 18 and 64, or over 65 years of age.
1.3 People who became ill recently (i.e. acute episode approximately less than one month's duration) as opposed to people who have been ill for a longer duration.
1.4 People who are given low doses (1- 5 mg/day), and those given high doses (over 5 mg/day).
1.5 People who have schizophrenia diagnosed according to any operational criterion i.e. a pre-stated checklist of symptoms/problems/time periods/exclusions) as opposed to those who have entered the trial with loosely defined illness.
1.6 People treated earlier (pre-1990) and people treated in recent years (1990 to 2006).
We additionally applied all sensitivity analyses to the primary outcomes of this review.
2. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way so as to imply randomisation. For the primary outcomes, we included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we entered all data from these studies.
3. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow-up (see Dealing with missing data), we compared the findings of the primary outcomes when we use our assumption/s and when we used data only from people who completed the study to that point. If there was a substantial difference, we reported results and discussed them but continued to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. A sensitivity analysis was undertaken to test how prone results are to change when completer-only data only are compared to the imputed data using the above assumption. If there was a substantial difference, we reported results and discussed them.
4. Risk of bias
We analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available): allocation concealment, blinding and outcome reporting for the meta-analysis of the primary outcome. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we included data from these trials in the analyses.
5. Imputed values
We also sought to undertake a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster-randomised trials.
If we noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
6. Fixed-effect and random-effects
We synthesised data using a fixed-effect model; however, we also synthesised data for the primary outcome using a random-effects model to evaluate whether this altered the significance of the results.
RESULTS
Description of studies
For substantive descriptions of studies please see the Included studies and Excluded studies tables.
Results of the search
The initial 2006 search yielded 1274 electronic records for 422 studies, of which we rejected 834 during the first inspection. We inspected the remaining 433 papers. Three hundred and forty-three were rejected, as they were clearly not relevant. The remaining 82 papers (55 studies) were considered to match the inclusion criteria closely enough to be mentioned in either the ‘Included studies’; ‘excluded studies’ and ‘awaiting classification’ tables. The 2012 update search yielded seven studies, each of which was excluded. The seven studies previously awaiting ‘classification’ were moved to ‘excluded studies’ for this review. Therefore, the current review cites 48 studies as ‘excluded studies’ and 16 reports of the seven ‘included studies’. There was over 90% agreement for trial selection and once we had investigated any disagreement and acquired and reassessed the papers, concordance was 100%. Initially, there was also over 90% agreement in the extracted data. We easily resolved any disagreement by discussion (see Figure 1 and Figure 2).
Figure 1.

Study flow diagram: 2006 search.
Figure 2.

Study flow diagram: 2012 update search (no additional studies).
Included studies
We included seven studies (total n = 439 participants).
1.1 Methods
In all included studies, randomisation was either reported or implied. The mean duration of treatment was about 170 days (~six months), but this was highly skewed (standard deviation (SD) 253). The most common study length was six weeks (Carpenter 1999; Clark 1971; Millar 1963) but the range was considerable with the longest lasting two years (Marder 1994).
1.2 Setting
Four studies were hospital-based (Clark 1971; Goldberg 1964; Hordern 1964; Millar 1963), while three were undertaken in the community (Carpenter 1999; Marder 1994; Rifkin 1976). Five studies were conducted in the United States of America (Carpenter 1999; Clark 1971; Goldberg 1964; Marder 1994; Rifkin 1976), one in Australia (Hordern 1964) and one in the United Kingdom (Millar 1963).
1.3 Participants
All trials included participants diagnosed with schizophrenia and two of the seven trials described the diagnostic criteria used (Diagnostic and Statistical Manual version III (DSM-III) or Research Diagnostic Criteria (RDC)). The other studies used a clinical diagnosis of schizophrenia. The mean age of the participants was about 38 years, range 16 to 75, and they were mostly chronic patients with a mean hospitalisation period of about 20 years. In Marder 1994, all participants were men, whilst in Hordern 1964 and Millar 1963 the participants were all women. In the remaining studies participants were of mixed sex.
1.4 Study size
The mean number of participants was about n = 60, ranging from 36 to 190.
1.5 Interventions
All trials compared oral fluphenazine with inactive placebo. The doses of oral fluphenazine in these studies ranged from 2.5 mg/day (Millar 1963) up to a potential of 20 mg/day (Rifkin 1976). The mean dose was 8.2 mg per day (SD 3.9). The standard oral dosage in minor disturbances 2 mg to 5 mg/day, or in the treatment of psychotic disorders up to 20 mg to 40 mg daily (www.psychotropics.dk).
1.6 Outcomes
1.6.1 General remarks
Most outcomes were dichotomous, and presented as such, or were continuous data. In many studies, outcomes were few and where data were reported they were rendered unusable. None of the included studies attempted to quantify levels of satisfaction or quality of life and there is no evidence of any direct economic evaluation of fluphenazine. However, we were able to measure some aspects of the global and mental state and adverse effects.
1.7 Outcome scales
The following scales were used and provided data for the analysis.
1.7.1 Global state
1.7.1.1 Clinical Global Impression (CGI) Guy 1976
A rating instrument commonly used in studies in schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement. A seven-point scoring system is usually used with low scores indicating decreased severity and/or greater recovery. Carpenter 1999 and Clark 1971 reported dichotomised data from this scale, measuring improvement scores. Continuous data from Clark 1971 were also used in our results, however SDs were imputed using the method described in Dealing with missing data.
1.7.1.2 The Multidimensional Scale for Rating Psychiatric Patients (MDRSP) Lorr 1953
The Multidimensional Rating Scale or Hamilton's schizophrenia scale is a modification of the Inpatient Multidimensional Psychiatric Scale. The MDRSP is completed after a psychiatric interview. It consists of 18 items, in the form of simple questions, to be rated along a four-point scale. The severity scores are defined by short behavioural descriptions on the form, thus avoiding interpretation problems. The scale is mainly designed for the evaluation of chronically hospitalised schizophrenic patients. Hordern 1964 reported dichotomised data from this scale for levels of improvement.
1.7.1.3 Brief Psychiatric Rating Scale (BPRS)
Overall 1962 The scale measures positive symptoms and quantifies factors such as thought disorder, activation, hostility. somatic, hallucinatory, and depressive states. The original scale had 16 items, but a revised 18-item scale is more commonly used, with scores ranging from 0 to 126. Each item is defined on a seven-point scale from 0 = not present to seven = extremely severe. Higher scores equate to severity of illness.
Excluded studies
We excluded 48 studies. Seven were not randomised, did not imply randomisation or did not describe the allocation procedure at all. In one study, participants were not suffering from schizophrenia. Another sizeable proportion of the trials did not compare oral fluphenazine with placebo, but with other treatments. A few were fluphenazine-withdrawal studies which are not relevant to this review. These withdrawal studies will be included in a future review. Three studies had no usable outcomes. Either data did not have clear clinical implications or genuinely relevant clinical data were not adequately reported. Frequently the numbers of participants in each group were not specified, means or SDs were not given or data were not reported from individual arms of cross-over studies. The seven studies identified in the 2012 search were all excluded, owing either to no randomisation (Kinross-Wright 1963; Matheu 1961), no diagnosis of schizophrenia (Hanlon 1970; Howell 1961), not the appropriate intervention (Marder 1993; Shafti 2009) or no usable data were presented (Zahn 1993).
Awaiting assessment
No studies are awaiting assessment.
Ongoing studies
We are not aware of any ongoing studies.
Risk of bias in included studies
Please see the relevant ‘Risk of bias’ tables in the Characteristics of included studies section and Figure 3; Figure 4.
Figure 3.

’Risk of bias’ graph: review authors’ judgements about each risk of bias item presented as percentages across all included studies.
Figure 4.

’Risk of bias’ summary: review authors’ judgements about each risk of bias item for each included study.
Allocation
None of the seven included studies described the methods used to generate random allocation, yet six of them were reported to be “randomly assigned” and readers are given little assurance that bias was minimised during the allocation procedure. Hordern 1964, did not mention “random assignment”, and reported that assignment of participants to fluphenazine or placebo groups was matched on age, length of illness, and severity of illness and the number of participants in each group was identical. Millar 1963 also obtained the same numbers in each group in a randomly assigned procedure with no further description.
Blinding
Only two studies (25%) gave a description of their attempts to make the investigation double-blind. Clark 1971 reported that identically appearing medication was administrated from a bottle labelled only with the participant's name and Millar 1963 reported that only the hospital pharmacist knew the composition of the groups. However neither of them actually tested how successful these attempts were. In the other six trials, it was indicated that attempts at blinding had been made, but without any further description.
Incomplete outcome data
Five studies reported that participants left the study early. Only two, Goldberg 1964 and Clark 1971, reported specifically the reasons for withdrawal.
Selective reporting
Studies frequently presented both dichotomous and continuous data in graphs, or just reported statistical measures of probability (P values). This often made it impossible to acquire raw data for synthesis. Continuous data were frequently poorly described; often no standard deviations/standard errors were presented or no data were presented at all. In this way a lot of potentially informative data were lost. In some studies it seemed that attempts had been made to use the original trials as vehicles for answering a host of other questions about schizophrenia. As a consequence, data from the randomised parts of the studies became buried beneath copious subgroup analyses.
Other potential sources of bias
The quality of trials has been assessed in individual ‘Risk of bias’ tables under the Characteristics of included studies section, using the guidance from the Cochrane Handbook for Systematic Reviews of Interventions categories (Higgins 2011). Due to poor reporting standards from particularly old studies, the majority of the risk of bias domains have been rated ‘unclear’ for the risk of bias.
Effects of interventions
See: Summary of findings for the main comparison ORAL FLUPHENAZINE versus PLACEBO for Schizophrenia
1. COMPARISON: ORAL FLUPHENAZINE versus PLACEBO
1.1 Global state
1.1.1 Not improved or worsened
We found no significant difference between oral fluphenazine and placebo for ‘not improved or worse’ (CGI) over short-term assessment (n = 125, 3 RCTs). Dichotomised multidimensional rating scale scores at 12 weeks ‘not improved or worse’ were also nonsignificant (n = 50, 1 RCT, Analysis 1.1).
Analysis 1.1.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 1 Global state: 1. Not improved or worsened.
1.1.2 Relapse
Only Millar 1963 reported on relapse up to six weeks (short term) with results indicating a trend favouring fluphenazine (n = 38, 1 RCT, risk ratio (RR) 0.25 95% confidence interval (CI) 0.06 to 1.03, P = 0.05). Two studies, Marder 1994 and Rifkin 1976, reported data for long-term relapse, which significantly favoured fluphenazine but data are heterogeneous (I2 92%). Using a random-effects model rendered data equivocal (n = 86, 2 RCTs, Analysis 1.2).
Analysis 1.2.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 2 Global state: 2. Relapse.
1.1.3 Percentage of time in prodrome state
Marder 1994 reported data at one- and two-year time points, but data are skewed (high SDs) and are best inspected outside of a forest plot, using an additional table (Analysis 1.3). The data suggest that a greater amount of time was spent in a prodromal state amongst placebo participants, at both one and two years.
Analysis 1.3.
Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 3 Global state: 3. Percentage of time in prodrome state (skewed data).
| Global state: 3. Percentage of time in prodrome state (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| one-year data | ||||
| Marder 1994 | Oral fluphenazine | 10.5 | 15.90 | 17 |
| Marder 1994 | Placebo | 19.4 | 22.30 | 19 |
| two-year data | ||||
| Marder 1994 | Oral fluphenazine | 2.80 | 3.80 | 14 |
| Marder 1994 | Placebo | 4.90 | 5.70 | 15 |
1.1.4 Percentage of time in exacerbated state
Again, Marder 1994 reported data at one- and two-year time points, but data are skewed and are best inspected outside of a forest plot, using an additional table (Analysis 1.4). Data suggest that, by one year, participants receiving oral fluphenazine spent more time in an exacerbated state that those on placebo. However, by two years, people receiving placebo spent longer in an exacerbated state.
Analysis 1.4.
Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 4 Global state: 4. Percentage of time in exacerbated state (skewed data).
| Global state: 4. Percentage of time in exacerbated state (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| one-year data | ||||
| Marder 1994 | Oral fluphenazine | 11.8 | 15.00 | 17 |
| Marder 1994 | Placebo | 7.20 | 10.70 | 19 |
| two-year data | ||||
| Marder 1994 | Oral fluphenazine | 5.50 | 10.40 | 14 |
| Marder 1994 | Placebo | 12.9 | 13.6 | 15 |
1.1.5 CGI severity of illness - average score
Data for global state using the CGI were reported in one study (Clark 1971), which demonstrated greater improvement in global state (n = 36, 1 RCT, mean difference (MD) -0.77 95% CI -1.39 to -0.15), this was a statistically significant result (P = 0.02); however, these data were imputed using the adjusted final mean and the P value between studies, and the significance of the results should be treated with caution (Analysis 1.5).
Analysis 1.5.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 5 Global state: 5. average score: CGI - severity of illness score (high = poor).
1.2 Leaving study early
1.2.1 Non-specific reasons
Although people allocated to oral fluphenazine left the study less often than participants who were given placebo in the short term, the data did not reach statistical significance (n = 227, 2 RCTs). For -medium-term assessment, we found only one study reporting on attrition (n = 50) and data were not significantly different. Long-term follow-up from two studies (n = 86) were also equivocal, and as a proxy measure for treatment acceptability the oral fluphenazine group did not find treatment any more acceptable than the placebo group when assessed over short-, medium- and long-term evaluation. Overall, across all time periods, only about 15% of people left these studies early (n = 363, 5 RCTs, Analysis 1.6).
Analysis 1.6.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 6 Leaving the study early: 1. Non-specific reasons.
1.2.2 Specific reasons
Leaving the study early due to court case transfers was significantly higher (P = 0.02) in the fluphenazine group (n = 190, 1 RCT, RR 10.65 95% CI 1.39 to 81.58). Other reasons for leaving the study early: ‘incorrect diagnosis’ (n = 190, 1 RCT), ‘marked early remission’ (n = 190, 1 RCT) and ‘serious complication of treatment’ (n = 190, 1 RCT) were not significantly different. We found the number of participants leaving the study early due to treatment failure favoured oral fluphenazine with significantly more people dropping out from the placebo group (n = 190, 1 RCT, RR 0.11 95% CI 0.03 to 0.35, Analysis 1.7).
Analysis 1.7.


Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 7 Leaving the study early: 2. Specific reason - short term.
1.2.3 Marked improvement/ hospital discharge
Data were equivocal for this positive outcome (forest plots are presented to display results right to the line of no effect, indicating a favourable outcome for oral fluphenazine). Only one participant receiving oral fluphenazine in one small study was discharged due to marked improvement (n = 36, 1 RCT, Analysis 1.8).
1.3 Adverse effects
1.3.1 Anticholinergic effects
There is some suggestion that fluphenazine increases a person's chance of experiencing anticholinergic effects such as constipation (n = 190, 1 RCT, RR 2.22 95% CI 1.19 to 4.15), dry mouth (n = 227, 2 RCTs, RR 3.62 95% CI 1.39 to 9.42) and increased salivation (n = 190, 1 RCT, RR 18.10 95% CI 1.06 to 309.15). Data for blurred vision, drooling, gastrointestinal distress, nasal congestion, urinary disturbance, and vomiting were not significantly different (Analysis 1.9).
Analysis 1.9.


Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 9 Adverse effects: 1. Anticholinergic effects - short term.
1.3.2 Cardiovascular effects
No significant differences were found between fluphenazine and placebo for dizziness/faintness/weakness (n = 190, 1 RCT), hypotension (n = 37, 1 RCT), syncope (n = 37, 1 RCT), or tachycardia (n = 37, 1 RCT, Analysis 1.10).
Analysis 1.10.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 10 Adverse effects: 2. Cardivascular effects - short term.
1.3.3 Central nervous system
There is also some suggestion that fluphenazine increases a person's chance of experiencing some neurological symptoms such as drowsiness (n = 190, 1 RCT, RR 3.91 95% CI 1.98 to 7.71). Reports of headache did not reveal any significant differences between fluphenazine and placebo groups (n = 190, 1 RCT). Other outcomes, anxiety, convulsion/seizures, depression, sedation/lethargy were equivocal (Analysis 1.11).
Analysis 1.11.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 11 Adverse effects: 3. CNS - short term.
1.3.4 Death
Only one study (Rifkin 1976) reported on the outcome of death, with one death occurring in the fluphenazine group during long-term follow-up (n = 50, 1 RCT, Analysis 1.12).
Analysis 1.12.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 12 Adverse effects: 4. Death - long term.
1.3.5 Endocrine
We found no statistically significant difference between fluphenazine and placebo for the outcomes of amenorrhoea (n = 190, 1 RCT), lactation (n = 190, 1 RCT) or swelling of the breasts (n = 190, 1 RCT) at short term (Analysis 1.13).
Analysis 1.13.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 13 Adverse effects: 5. Endocrine - short term.
1.3.6 Extrapyramidal effects
In the short term, there is evidence that fluphenazine increases a person's chances of experiencing akathisia (n = 227, 2 RCTs, RR 3.43 95% CI 1.23 to 9.56), facial rigidity (n = 190, 1 RCT, RR 2.77 95% CI 1.03 to 7.46), ‘loss of associated movements’ (n = 190, 1 RCT, RR 6.39 95% CI 1.95 to 20.98), rigidity (n = 227, 2 RCTs, RR 3.54 95% CI 1.76 to 7.14) and tremor (n = 227, 2 RCTs, RR 3.19 95% CI 1.25 to 8.11). We found measures of akinesia, associated movements, dystonia and restlessness/insomnia were not significantly different from those allocated to placebo. Evidence in the medium term indicates that fluphenazine increases the likelihood of having parkinsonism (n = 50, 1 RCT, RR 5.50 95% CI 1.36 to 22.32), but akathisia, akinesia and dystonia were equivocal (Analysis 1.14).
Analysis 1.14.


Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 14 Adverse effects: 6a. Extrapyramidal effects - short term.
1.3.7 Other
We did not find any statistically significant data for the outcomes of convulsion/seizures (n = 190, 1 RCT), diarrhoea (n = 190, 1 RCT), infection (n = 190, 1 RCT), or rash (n = 227, 2 RCTs, Analysis 1.16).
Analysis 1.16.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 16 Adverse effects: 7. Others - short term.
2. SENSITIVITY ANALYSES
2.1 Men and women
Three studies included either all male (Marder 1994) or all female (Hordern 1964; Millar 1963) participants. However, only Marder 1994 reported data for any of our primary outcomes; therefore, a sensitivity analysis was not possible for this comparison. Note HM: this section is repeated below.
2.2 People who are under 18 years of age, between 18 and 64, or over 65 years of age
The age ranges of all participants across the included studies were all similar (between 16 and 58 years old). Where participants younger than 18 were included in the relevant studies (the youngest being 16 years old in Goldberg 1964, and 17 years old in Rifkin 1976), it was not possible to extract their data from the individual reports. Therefore, a sensitivity analysis has not been undertaken.
2.3 Chronic versus acutely ill people (< one month in duration)
Limited data were available from only one study. Participants who were chronically ill did not leave the study early in greater numbers than acutely ill patients. We found relapse did occur more often in the placebo arm of those chronically ill compared with those acutely ill, but the sample sizes were small and uneven and more data are required to draw any inferences (Analysis 1.17).
Analysis 1.17.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 17 Sensitivity analysis: 1. CHRONIC versus ACUTE.
2.4 Low doses (1-5 mg/day) and high doses (over 5 mg/day)
Again, limited data were available, but it is suggested that there is little difference between levels of improvement between participants in studies that used either a high dose (15 mg/day used in Carpenter 1999; n = 38, 1 RCT) or a flexible dose regimen in the short term (2-10 mg/day in Clark 1971; or up to 14 mg/day in Hordern 1964; n = 87, 2 RCTs, Analysis 1.18).
Analysis 1.18.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 18 Sensitivity analysis: 2. LOWDOSES (1-5 mg/day) versus HIGH DOSES (5mg/day>).
2.5 Diagnosed according to any operational criterion versus those with loosely defined illness
It is indicated that there is again little difference in results for no improvement when participants have been diagnosed according to operational criteria (DSM-III-R, n = 38, 1 RCT) or with loose diagnostic criteria or definitions (n = 87, 2 RCTs, Analysis 1.19).
Analysis 1.19.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 19 Sensitivity analysis: 3. OPERATIONAL CRITERIA versus LOOSE DEFINITIONS.
2.6 Studies published before 1990 versus studies published between 1990 and the present
Data were available for the outcomes of ‘no global improvement’ in the short term. There were no clear differences between the results of earlier studies and those published in the last 16 years (Analysis 1.20).
Analysis 1.20.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 20 Sensitivity analysis: 4. BEFORE 1990 versus AFTER 1990.
2.7 Implication of randomisation
Only one study was rated as a low risk of bias for adequate randomisation (Clark 1971); results for the primary outcome of global state (not improved or worse) remain the same when all other studies that implied randomisation were removed from the meta-analysis (n = 37, 1 RCT, RR 0.59, 95% CI 0.24 to 1.42). For the other secondary outcomes of adverse effects; the effect of removing the other studies that were meta-analysed with Clark 1971 and implied as being randomised is that data are no longer statistically significant for short-term extrapyramidal akathisia (from P = 0.02 to P = 0.18), or tremor (P = 0.01 to P = 0.09).
2.8 Assumptions for lost binary data
There were no clear differences between the results when completer-only data were used compared to data assumed for those lost to follow-up.
2.9 Risk of bias
Each included study was rated as a ‘high’ risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available): allocation concealment, blinding and outcome reporting for the meta-analysis of the primary outcome. The result of excluding each study on this basis leaves us with no data to compare, therefore a sensitivity analysis was not possible.
2.10 Imputed values
We also sought to undertake a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster-randomised trials. However, we encountered no such studies.
2.11 Fixed-effect and random-effects
We found that there was no difference in the results when synthesising data for the primary outcome of global state (not improved or worse) using a random-effects model. However, for short-term extrapyramidal effects (tremor), when using a random-effects model, results were no longer statistically significant (from P = 0.01 to P = 0.07).
DISCUSSION
Summary of main results
1. COMPARISON ONE: ORAL FLUPHENAZINE versus PLACEBO
1.1 Global State
1.1.1 No improvement
There were few data available for global improvement. Data were only obtained from three studies, and both short-term and medium-term assessment did not reveal any significant differences between fluphenazine and placebo, although a trend may be evident from the graphical representations of the data, suggesting that oral fluphenazine is superior to placebo, and perhaps this would have clearly emerged if larger and longer studies were used.
1.1.2 Relapse
There are surprisingly little data regarding relapse but the strongest results come from a small study with a duration of one year (Rifkin 1976), however, there was substantial heterogeneity present, perhaps due to the difference between the participant population and stages (remitted, non-psychotic participants in Rifkin 1976, and participants with at least two episodes of acute schizophrenia or continuing psychotic symptoms in previous two years in Marder 1994). A larger sample size would have probably ensured more unequivocal data in favour of fluphenazine.
1.2 Leaving study early
The finding that using fluphenazine resulted in more people staying in the study could be seen as heartening. Perhaps a genuine decrease in the distressing symptoms of schizophrenia leads to an increased concordance with medication, despite the unpleasant adverse effects of this drug.
1.3 Mental state
In spite of more than five decades of research on this benchmark antipsychotic treatment, very little can be said from trials regarding its direct effect on mental state regarding general or specific symptoms of schizophrenia.
1.4 Adverse effects
Although we were able to include a few studies in this review, clinicians will not be surprised that fluphenazine produces acute extrapyramidal disorders; dystonia, parkinsonism, akathisia, tremor, rigidity, weakness and anticholinergic effects. This Cochrane review, however, is a rare report of the best available and quantitative data on a compound over half a century old. Estimates of the incidence of extrapyramidal disorders, however, are not available from this review, as these necessitate a long follow-up period that was only attempted in a few trials. It may be surprising that there was only one death incident reported among over 439 people with schizophrenia who were randomised to oral fluphenazine or placebo. The lifetime incidence of suicide for people suffering from schizophrenia is 10% to 13% (Caldwell 1992). The occur-rence of only one death indicates that either trial-care is more vigilant than routine care or that death is an under-reported outcome.
2. SENSITIVITY ANALYSES
As we knew would be likely from the start, the power to detect a real difference between studies in any one of the sensitivity analyses was very low. Only subsets of already limited lists of trials were available. The wide confidence intervals could be hiding true differences in effect between the acutely and chronically ill people, and early trials versus current studies.
Overall completeness and applicability of evidence
1. Generalisability
This work includes studies that span nearly four decades of evaluative studies within psychiatry. It is possible that the rigour of these experiments has changed over time, as have the participants. There is some empirical evidence that the quality of schizophrenia trial reporting has not changed much over time (Thornley 1998) or, if it has changed, it may even have deteriorated (Ahmed 1998). We have found no time-related differences in reporting of studies within this review and no suggestion of a change of the effect size over time. We identified trials by meticulous searching; nevertheless, for a compound formulated so long ago, publication biases may be difficult to avoid. The strength of this review is that it presents up-to-date quantitative data for a benchmark treatment for schizophrenia which is used throughout the world.
2. Applicability
The seven included studies involve many people who would be recognisable in everyday practice. There are those with strictly diagnosed illnesses, very likely to suffer from schizophrenia, and people whose illness was diagnosed using less rigorous criteria. The dose of fluphenazine in the studies included in this review could be considered standard (mean 8.2 mg/day SD 3.9). Although the outcomes that have been used in this review are accessible to both clinicians and recipients of care, generalising to treatment in community settings, could be problematic. Four studies were undertaken in hospital and three in the community, which is where most people with schizophrenia now reside.
3. Heterogeneity
Two outcomes were heterogeneous, but all other outcomes were homogeneous. However, no more than five studies were pooled so the chances of data being heterogeneous were always small. In addition, several methods were used to observe different adverse effects e.g. tremor and blurred vision. This made analyses and interpretation for the results even more difficult.
4. Limited data
Data were often inadequately reported and rendered many outcomes unusable. Most trials report only six- to 12-week outcomes for a mostly lifelong illness. No studies reported on service utilisation, economic outcomes, or on satisfaction with care.
Quality of the evidence
Generally, the quality of the available evidence was rated as either very low, low to moderate (See Summary of findings for the main comparison). This was largely due to the age of the included studies and poor reporting standards, some of which were published up to 50 years ago, pre-dating the CONSORT statement (Moher 2001; Schulz 2010). Missing or unreported outcomes were common, which restricted the amount of evidence available for use in this systematic review and data that may have been relevant in meta-analysis. Any future studies in this area must adhere to CONSORT principles, ensuring that all trials are transparently reported, that all tables and figures express what data they are presenting, so that no trials can reach publication without revelation of methodological inadequacies. A flow diagram should also be used in order to document the process in which participants are recruited to those that are ultimately followed-up, promoting completeness, clarity and transparency of reporting.
Potential biases in the review process
The review authors sought to adhere to the protocol, through the independent inspection of citations and full articles of potentially relevant studies. Furthermore, the review authors independently extracted data onto standard simple forms; however, we met for discussion where inconsistencies or disagreements arose regarding the available data.
Agreements and disagreements with other studies or reviews
We are not aware of other systematic reviews evaluating the effects of oral fluphenazine versus placebo in the treatment of schizophrenia.
AUTHORS ‘CONCLUSIONS
Implications for practice
1. For people with schizophrenia
Many people with schizophrenia and their non-professional carers recognise psychotic symptoms as phenomena generated by a damaging and pernicious illness and may see the effect of fluphenazine, as demonstrated within this review, as positive. Others may consider these data as supporting well-publicised objections to the use of drugs; drugs potent in their ability to cause unpleasant adverse effects, and to potentially erode a person's ability to make informed decisions.
2. For clinicians
This review will confirm much that clinicians already know, but it does provide some quantification to support clinical impression. Fluphenazine is an antipsychotic, prone to cause a variety of extrapyramidal and anticholinergic effects. Evidence about its short-term antipsychotic effect is weak. However, fluphenazine is a low-cost and widely available choice for the clinician. Despite its many adverse effects, fluphenazine is likely to remain one of the most widely used treatments for schizophrenia worldwide.
3. For managers or policy makers
Fluphenazine is widely available and inexpensive. It is understandable that it remains one of the many drugs used for treating people with serious mental illnesses. However, some of fluphenazine's adverse effects could be expensive in terms of human suffering and cost of treatment. It could, therefore, be more beneficial to use another drug if the latter was equally potent, but had a more favourable adverse-effect profile.
Implications for research
1. General
So much more could have been learnt about the effects of oral fluphenazine if the studies in this review had clearly described the method of allocation and the integrity of blinding; especially for the more subjective outcomes. Most included studies, however, predated the CONSORT statement (Moher 2001). Concrete and simple outcomes are of interest. For example, clearly reporting improvement, ‘number of violent incidents’, ‘relapse’ (giving some description of criteria), ‘hospital discharge or admission’, and ‘presence of delusions or hallucinations’ would have been helpful, and simple reporting of levels of satisfaction and quality of life would have been very informative.
2. Specific
Even though fluphenazine has been used as an antipsychotic drug for decades, there are still a surprisingly small number of well-conducted randomised, placebo-controlled trials measuring its efficacy and potential to cause adverse effects. The use of oral fluphenazine for millions of people is based on clinical experience rather than the poorly reported trials that involve, in total, only a few hundred participants. Clinicians and researchers are mainly satisfied with the current levels of understanding, and, therefore new studies evaluating oral fluphenazine versus placebo will be very rare. The fluphenazine story is, however, incomplete. Questions remain regarding the effect of this drug on mental state and long-term extrapyramidal effects. One or more large, methodologically sound, randomised, placebo-controlled trials could help answer these questions. With the advent of universally available effective, even moderately effective, antipsychotic drugs, the day for studies comparing oral fluphenazine with placebo has passed.
| Date | Event | Description |
|---|---|---|
| 9 July 2013 | New citation required but conclusions have not changed | No new conclusions made to the review after results of 2012 update search added |
| 18 March 2013 | New search has been performed | Review update completed: seven new studies identified in update search, each of which was excluded with reasons. ‘Summary of findings’ table added to grade the quality of the evidence. New format ‘Risk of bias’ tables have been constructed, with new ratings applied |
| Date | Event | Description |
|---|---|---|
| 15 May 2012 | Amended | Update search of Cochrane Schizophrenia Group's Trial Register (see Search methods for identification of studies). 7 studies added to awaiting assessment |
| 5 October 2011 | Amended | Contact details updated. |
| 4 August 2010 | Amended | Contact details updated. |
| 15 February 2010 | Amended | Contact details updated. |
| 13 August 2008 | Amended | Contact Author details updated |
| 25 April 2008 | Amended | Converted to new review format. |
| 14 November 2006 | New citation required and conclusions have changed | Substantive amendment |
PLAIN LANGUAGE SUMMARY.
Oral fluphenazine versus placebo for schizophrenia
Antipsychotic drugs are the first line and mainstay of treatment for schizophrenia. They help to effectively treat psychotic symptoms such as hearing voices and seeing things (hallucinations) and having strange beliefs (delusions). Fluphenazine was one of the first antipsychotics and has been available for around 50 years. Fluphenazine is inexpensive and in developing countries, may be one of the only drug treatments available. In most of Europe and North America, despite still being available, the arrival of newer antipsychotic drugs has reduced the use of fluphenazine and its market share. Fluphenazine has debilitating side effects, including: dizziness; movement disorders such as involuntary movements or spasms; shaking and tremors; inner restlessness and the inability to sit still; and problems with blood pressure, fever and muscle stiffness.
This review included seven studies and compared the effects of fluphenazine taken by mouth with placebo (‘dummy’ treatment). In the main, the findings of the review support the widespread view that fluphenazine is a potent and effective antipsychotic but has considerable side effects, other antipsychotic drugs may well be preferable. Fluphenazine is an imperfect treatment with serious side effects, so other inexpensive antipsychotic drugs with fewer side effects may be better for people with schizophrenia. Despite this, fluphenazine has a low cost and is widely available, so is likely to remain one of the most widely used treatments for schizophrenia worldwide. However, some of fluphenazine's side effects could be expensive in terms of human suffering and personal cost of treatment. Even though fluphenazine has been used as an antipsychotic drug for decades, there are still a surprisingly small number of well-conducted studies measuring its effectiveness and potential to cause side effects. Future large-scale research should report on important outcomes such as improvement in mental health, relapse, hospital discharge and admission, levels of satisfaction with treatment and quality of life.
This plain language summary has been written by a consumer Ben Gray from RETHINK.



Analysis 1.15.

Comparison 1 ORAL FLUPHENAZINE versus PLACEBO, Outcome 15 Adverse effects: 6b. Extrapyramidal effects - medium term.
Characteristics of included studies [ordered by study ID]
| Carpenter 1999 | |
|---|---|
| Methods | Allocation: random. Blinding: double. Duration: 6 weeks (4 weeks presented usable data).* Design: parallel. |
| Participants | Diagnosis: schizophrenia (DSM-III-R or RDC). N = 53 (38 to relevant interventions).** Age: mean 37 yrs. Sex: M 38 (26 relevant), F 15 (12 relevant). History: illness for ~13 yrs, clinically stable patients. Excluded: patients with concurrent drug abuse, alcoholism, organic brain disorders and mental retardation. Setting: community, Maryland Psychiatric Research Center Outpatient Program (US) Consent: written informed consent required. |
| Interventions | 1.Oral fluphenazine: dose 15 mg/day, N = 18. 2.Placebo, N = 20. [3. Diazepam: dose 30 mg/day, N = 15]. |
| Outcomes | Global state (CGI) - not improved or worsened. Unable to use - Mental state: BPRS (no usable data). Relapse (not given by each group). Sleep change ratings (no data). |
| Notes | * Data were given only for the first 4 weeks of the study. ** Demographic data relate to the total of 38 people. |
| Risk of bias | ||
|---|---|---|
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random - “a stratified randomization procedure...was used to assign drug treatment to balance study groups on gender, prior social function, and past duration of hospital care” (p300). No details as to randomisation methods |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes |
Low risk | Double blind - no further details. If participants experienced worsening or exacerbation of symptoms, they were removed from the study and treated on an open basis with fluphenazine Rating scales: raters not stated to be independent of treatment |
| Incomplete outcome data (attrition bias) All outcomes |
Unclear risk | No mention of participants lost to follow-up or leaving the study early |
| Selective reporting (reporting bias) | High risk | No scale data reported for BPRS or CGI. |
| Other bias | Unclear risk | Funding: supported in part by NIMH grants (MH-35996 and MH-40279) |
| Clark 1971 | |
|---|---|
| Methods | Allocation: random. Blinding: double “identically appearing medication administrated from a bottle labelled only with the patient's name”. Duration: 6 weeks. Design: parallel. |
| Participants | Diagnosis: chronic schizophrenia. N = 76 (37 to relevant interventions). Age: mean 33 yrs (range 18 to 45). Sex: M 23, F 53. History: 6 months preadmission period free of hospitalisation or shock treatment. Excluded: childhood schizophrenia or autism, brain syndrome, IQ < 70, alcoholism, recent hepatitis, chronic physical illness, epilepsy, drug addiction. Setting: inpatient, Central State Griffin Memorial Hospital (Oklahoma, US) Consent: not stated. |
| Interventions | 1.Oral fluphenazine: dose 2-10 mg/day. N = 18. 2.Placebo. N = 19. [3. Chlorpromazine: dose 100-1000 mg/day. N = 20]. [4. Thioridazine: dose 100-1000 mg/day. N = 19]. |
| Outcomes | Global state (using CGI): not improved or worsened; average score (CGI severity of illness*) Mental state: average score (BPRS*). Leaving the study early: any reason; administrative/hospital transfer; AWOL; marked improvement allowing discharge. Adverse effects: anticholinergic (dry mouth; blurred vision; nasal congestion; tachycardia; gastrointestinal distress); EPS (tremor; rigidity; associated movements; akinesia; akithisia; drooling; restlessness/ insomnia); CNS (anxiety/agitation/excitement/ confusion; sedation and lethargy; depression); cardiovascular (hypotension; syncope); others (rash) Unable to use - Global state: NOSIE (no SD). Toxicity (no usable data). |
| Notes | Unscheduled dose adjustments were permitted for toxicity or intolerance *SDs imputed ‘between groups’ using RevMan calculator. |
| Risk of bias | ||
|---|---|---|
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random: “patients were assigned to treatment randomly in blocks of four” (p404) - no further details |
| Allocation concealment (selection bias) | Unclear risk | Participants were “assigned to treatment randomly in blocks of four” (p404) - no further details |
| Blinding (performance bias and detection bias) All outcomes |
Low risk | Double blind: “double blind design was maintained throughout the study” (p404) - no further details. Identically-appearing capsules were dispensed from a bottle labelled only with the participant name Rating scales: raters not stated to be independent of treatment |
| Incomplete outcome data (attrition bias) All outcomes |
Low risk | Follow-up: 85% - n = 2 participants left the study early, but their final measures were obtained and used in the analysis (n = 1 receiving placebo due to behavioural deterioration and n = 1 receiving fluphenazine was discharged from the hospital as markedly improved after two weeks). Afurther n = 11 participants, however, were dropped without final measures being obtained (placebo: n = 1 went AWOL; n = 1 on convalescent leave; n = 1 transferred to another hospital. Thioridazine group: n = 2 AWOL; n = 1 medication intolerance. Chlorpromazine: n = 1 AWOL; n = 2 refused oral medication. Fluphenazine: n = 1 administrative transfer, n = 1 AWOL). Dichotomised data presented as ITT (only n = 1 missing from placebo). LOCF for CGI and BPRS |
| Selective reporting (reporting bias) | Unclear risk | No SDs reported for all scale data. |
| Other bias | Unclear risk | Funding: supported in part by Public Health Service Grant MH 11666 and Research Scientist Development award No. K135278 from NIMH. Medication supplied by Smith Kline and French Laboratories (chlorpromazine and placebo); Sandoz Inc (thioridazine) and ER Squibb & Sons (fluphenazine) |
| Goldberg 1964 | |
|---|---|
| Methods | Allocation: random. Blinding: double. Duration: 6 weeks. Design: multi-centre, parallel. |
| Participants | Diagnosis: schizophrenia. N = 463 (190 to relevant interventions). Age: 16-45 yrs. Sex: male and female (equal distribution stated, however no number given). History: newly admitted patients. Excluded: childhood autism, brain syndrome, IQ < 70, epilepsy, drug addiction. Setting: inpatient, Boston State Hospital (Massachusettes); District of Columbia General Hospital (Washington DC); Kentucky State Hospital (Kentucky); Malcolm Bliss Mental Health Center (Missouri); Mercy-Dougleaa Hosptial (Pennsylvania); Payne-Whitney CLinic (New York); Rochester State Hosptial (Rochester, New York); Springfield State Hospital (Maryland); Institute of Living (Conneticut, US) Consent: not stated. |
| Interventions | 1.Oral fluphenazine: dose 1-16 mg/day. N = 92. 2.Oral placebo. N = 98. [3. Paraenteral fluphenazine. N = 23]. [4. Chlorpromazine: dose 200 mg/day. N = 112]. [5. Thioridazine: dose 200 mg/day. N = 111]. [6. Paraenteral placebo. N = 27]. Additional medication: Anti-parkinsonian medications.* |
| Outcomes | Leaving the study early (any reason; treatment failure; serious complication of treatment; marked early remission; incorrect diagnosis; court cases, transfer, eloped). Adverse effects: CNS (headache; drowsiness; convulsions or seizures) cardiovascular effects (dizziness, faintness, weakness) anticholinergic effects (increased salivation; dry mouth/throat; gastrointestinal distress and nausea; urinary disturbance; constipation; vomiting) endocrine (lactation; amenorrhoea; swelling of breasts) extrapyramidal effects (loss of associated movements; facial rigidity; rigidity; restlessness/insomnia; tremor; akithisia; dystonia) others (convulsion or seizures; diarrhoea; intercurrent infection; rash) Unable to use - Global state (no SD).** Mental state: IMPS, WBRS (no SD).*** |
| Notes | *44% of fluphenazine group and 5% of placebo received anti-parkinsonian drugs. **Global rating of severity of metal illness, Global rating of improvement. ***Results were not broken down by each drug group. |
| Risk of bias | ||
|---|---|---|
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random: participants were “randomly assigned to one of four treatments on a double-blind basis” (p247) |
| Allocation concealment (selection bias) | Unclear risk | Stratified by sex with “randomized assignment to drug treatment within each sex group” (p247). In the three out of nine hospitals participating in the study that admitted approximately equal number of White and Black participants, this was taken into account and groups were further stratified by race |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Double blind described, no further details. Participants received individually numbered containers of medication. A flexile dosage schedule permitted the treating physician to adjust dosage according to individuals’ needs Rating scales: raters not stated to be independent of treatment |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | Follow-up: 74%. Reasons for removal of study included administrative removals (incorrect diagnoses; intercurrent medical illness; court cases, transfer, elopement, etc) , treatment-related removals (marked early remission; serious complication of treatment; treatment failure). Those lost were not included in the study report analysis |
| Selective reporting (reporting bias) | High risk | No SDs reported for continuous data. |
| Other bias | Unclear risk | Funding: supported by NIMH grants (MH 04661, 04663, 04667, 04673, 04674, 04675, 04679, 04803). Medications provided free of charge from Sandoz Pharmaceuticals (Hanover); Squibb Institute for Medical Research (New Brunswick); Smith Kline and French Laboratories (Philiadelphia) Rating scales: raters not stated to be independent of treatment |
| Hordern 1964 | |
|---|---|
| Methods | Allocation: unclear. Blinding: double. Duration: 12 weeks. Design: parallel. |
| Participants | Diagnosis: chronic schizophrenia. N = 75 (50 to relevant interventions). Age: mean 49 yrs. Sex: all female. History: hospitalisation > 2 yrs (mean ~20 yrs, ~SD 9), all have had previous unsuccessful phenothiazines treatment, and none leucotomised. Excluded: physical illness, epilepsy. Setting: inpatient, Mont Park Hospital, Victoria (Australia) Consent: not stated. |
| Interventions | 1.Oral fluphenazine: dose < 14 mg/day. N = 25. 2.Placebo. N = 25. [3. Thioproperazine (max dose of 140 mg/day). N = 25]. |
| Outcomes | Global state: MADRS* - not improved or worsened. Adverse effects - extrapyramidal effects: dystonia; akinesia; parkinsonism; akathisia. Leaving study early.** |
| Notes | *Rated as either ‘no change’, ‘clear worsening’ and ‘marked worsening’ using the Multidimensional Rating Scale of the Veterans’ Administration (Lorr 1953). **Two participants from fluphenazine group left the study early and they were considered to have the worst outcomes. The ward sister's blind ratings of change of behaviour at the end of the trial: 9 placebo participants deteriorated in contrast to 6 participants on fluphenazine. One placebo participant required additional nursing care because of severe negativism |
| Risk of bias | ||
|---|---|---|
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation unclear - participants were “divided into three groups of 25 matched on age, chronicity and severity of illness” (p532) |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes |
Low risk | Double blind (implied) - members of the ward staff were told that powerful phenothiazine drugs were to be administered, but were unaware of which participants were receiving the active medication. All medications were identical-looking tablets, dispensed by a medical officer who “took no part in the rating procedure” (p533). Maximum blindness preserved in evaluations claimed, as neither physicians entered the closed wards between ratings and did not observe side effects during period of treatment. A blind assessment of overall change was made at the end of the trial by the ward sister Rating scales: raters not stated to be independent of treatment |
| Incomplete outcome data (attrition bias) All outcomes |
Unclear risk | Follow-up: 95% - n = 4 participants were lost to follow-up by 12 weeks of treatment, only n = 2 were accounted for as having discontinued from active drugs (n = 1 from fluphenazine; n = 1 from thioproperazine). ITT used for medium-term data |
| Selective reporting (reporting bias) | Unclear risk | None detected. |
| Other bias | Unclear risk | Funding: all drugs and placebos were provided free of charge by May and Baker (Australia) Limited (‘Majeptil’) and ER Squibb and Sons (Australia) Limited (‘Anatensol’) |
| Marder 1994 | |
|---|---|
| Methods | Allocation: randomly assigned. Blindness: double. Duration: 2 years (preceded by 2 months stabilisation phase with low dose of fluphenazine decanoate 5-10 mg) Design: parallel. |
| Participants | Diagnosis: schizophrenia (DSM-III-R). N = 36. Age: mean 40 yrs. Sex: all male. History: at least two documented episodes of acute schizophrenic illness or at least 2 years of continuing psychotic symptoms, randomly assigned when getting prodromal symptoms using Idiosyncratic Prodromal Scale. Excluded: patients who could not be stabilised for 2 or more months with 10 mg or less of fluphenazine decanoate every 2 weeks. Setting: community, outpatients at Brentwood Division of West Los Angeles Verterans Affairs Medical Center (US) Consent: not stated. |
| Interventions | 1.Oral fluphenazine hydrochloride: dose 10 mg/day. N = 17. 2.Placebo. N = 19. Additional medication - Fluphenazine decanoate: dose 5-10 mg/2weeks for all patients. N = 36 Factored to: A.Behavioural skills training. B.Supportive group therapy.** |
| Outcomes | Relapse: defined as number of psychotic exacerbations.* Percentage of time in exacerbated state (skew). Percentage of time in prodrome (skew). Leaving the study early: non-specific reasons. Unable to use - Adverse effects (no data). |
| Notes | *Defined as worsening of four points or more on the sum of the BPRS clusters for thought disturbance and paranoia or increase of three or more points on either cluster |
| Risk of bias | ||
|---|---|---|
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random - possible participants were stabilised on low dose fluphenazine decanoate (5 to 10 mg every 14 days) for two months and monitored every week using an idiosyncratic prodromal rating scale. Participants were randomised to either oral fluphenazine or placebo when they met criteria for a prodromal episode |
| Allocation concealment (selection bias) | Unclear risk | Not described. |
| Blinding (performance bias and detection bias) All outcomes |
Unclear risk | Double blind (implied), as participants who never experienced prodromal episodes nor were assigned to either treatment were treated with 5 mg oral fluphenazine on an open label basis during exacerbations Rating scales: raters not stated to be independent of treatment |
| Incomplete outcome data (attrition bias) All outcomes |
High risk | Follow-up: 81% - n = 6 participants lost to follow-up by two years of treatment. Only two participants accounted for as being omitted from analysis (n = 1 who dropped-out during a prodromal episode, and n = 1 who reached the two-year end point during a prodrome) |
| Selective reporting (reporting bias) | Unclear risk | None detected. |
| Other bias | Unclear risk | Funding: study supported by the Medical Research Service of the Department of Veterans Affairs, Washington DC (grant MH-41573) from National Institute of Mental Health, Bethesda, Md; UCLA Mental Health Clinical Research Center for Schizophrenia (grant MH-30911) from the National Institute of Mental Health |
| Millar 1963 | |
|---|---|
| Methods | Allocation: random. Blindness: double. Duration: 6 weeks. Design: cross-over (after 3 weeks). |
| Participants | Diagnosis: chronic schizophrenia. N = 38. Age: 28-58 yrs (mean 46 yrs). Sex: all female. Excluded: advanced age, doubtful diagnostic classification, epilepsy, severe sub-normality. History: in hospital 2-35 yrs (mean 18 yrs). Setting: inpatient, UK. Consent: not stated. |
| Interventions | 1.Oral fluphenazine: dose 2.5 mg/day. N = 19. 2.Placebo. N = 19. |
| Outcomes | Relapse. Unable to use - Improvement: no better or worse (data not reported by group). Adverse effects (no data). Lorr psychiatric rating scale (no SD, mean only). Baker and Thorpe behaviour rating scale (no SD, mean only). |
| Notes | Participants had been receiving chlorpromazine three times a day and were “mostly stabilised” on a certain dose. They were then given doses of fluphenazine, with this drug substituted for the chlorpromazine at approx. one fortieth of the dose. A single daily dose was given of 2.5 mg (with the exception of two cases who received 20 mg) with dose increase of 2.5 mg/day (one tablet). After participants received fluphenazine for 2 months, they were rated and randomised into intervention groups, receiving dosages established in the stabilisation phase |
| Risk of bias | ||
|---|---|---|
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random - participants were divided into two random groups; no further details |
| Allocation concealment (selection bias) | Unclear risk | Only hospital pharmacist knew the composition of the groups. |
| Blinding (performance bias and detection bias) All outcomes |
Low risk | Double blind - only hospital pharmacist knew the composition of the groups; medication was administered with matching placebo Rating scales: raters not stated to be independent of treatment |
| Incomplete outcome data (attrition bias) All outcomes |
Unclear risk | No mention of participants lost to follow-up or leaving the study early |
| Selective reporting (reporting bias) | High risk | Data not clearly described as to the stage of trial and relevant groups (i.e. pre-cross-over or post-cross-over). Full data not reported for continuous outcomes (missing means and SDs) |
| Other bias | Unclear risk | Funding: fluphenazine tablets were provided by ER Squibb & Sons |
| Rifkin 1976 | |
|---|---|
| Methods | Allocation: random. Blindness: double. Duration: 1 year (preceded by several months transitional period during which all patients were treated exclusively with fluphenazine decanoate and oral fluphenazine) Design: parallel. |
| Participants | Diagnosis: schizophrenia (remitted). N = 73 (50 to relevant interventions). Sex: M 50, F 23. Age: 17-40 yrs. History: reached a stable remission while receiving FD and FPZ and showed no adverse effects. Excluded: history of severe drug abuse or chronic schizophrenics.* Setting: psychiatric aftercare clinic, Long Island Jewish-Hillside Medican Center (US) Consent: informed consent obtained. |
| Interventions | 1.Oral fluphenazine: dose 5-20 mg/day. N = 28. 2.Placebo. N = 22. [3. Fluphenazine decanoate: dose 0.5-2.0 mL/2weeks. N = 23]. Additional medication: Prophylactic procyclidine (5-15 mg/day).** Placebo procyclidine. Psychotherapy biweekly during the first 6 months and monthly thereafter |
| Outcomes | Death. Relapse (clinical judgement). Leaving the study early. Unable to use - Social and vocational functioning (results not broken down by individual drugs). Mental state: BPRS (no data). Global state: CGI, PER-C (no data). KAS (no usable data). |
| Notes | *Chronic patients > 3 previous hospitalisations. **Prophylactic procyclidine for patients receiving active treatment and in the first 2 months for patients receiving placebo to prevent the emergence of extrapyramidal side effects during the fluphenazine decanoate washout period |
| Risk of bias | ||
|---|---|---|
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random: “randomly assigned” (p44). 15% of patients referred schizophrenics were re-diagnosed using Kraepelinian Criteria as non schizophrenic and randomised separately but treated in the same manner |
| Allocation concealment (selection bias) | Unclear risk | “Randomly assigned” (p44) - no further details. |
| Blinding (performance bias and detection bias) All outcomes |
Low risk | Double blind: drugs were given in a “double blind fashion” (p44) Rating scales: raters not stated to be independent of treatment |
| Incomplete outcome data (attrition bias) All outcomes |
Unclear risk | Follow-up: 89%. Drop-outs mainly due to moving from the geographical area or refusing further care. None of these patient's conditions had deteriorated clinically |
| Selective reporting (reporting bias) | High risk | Not all adverse effects reported by group. No data reported for individual groups using the BPRS, CGI, PER-C, KAS scales |
| Other bias | Unclear risk | Funding: supported by NIMH grant MH 21337-03. |
RDC - Research Diagnostic Criteria for schizophrenia or schizoaffective disorders
DSM - Diagnostic and Statistical Manual
FD - Fluphenazine Decanoate
FPZ - Oral fluphenazine
Rating Scales:
KAS- Katz Adjustment Scale
Global state:
CGI - Clinical Global Impression
NOSIE - Nurse's Observation Scale for Inpatient Evaluation
Mental state:
BPRS - Brief Psychiatric Rating Scale
IMPS - Inpatient Multidimensional Psychiatric Scale
MADRS - modified Montgomery-Asberg Depression Rating Scale
PER-C - Periodic Evaluation Record-Community
WBRS - Burdock Ward Behaviour Rating Scale
Other:
CNS - central nervous system
EPS - extrapyramidal symptoms
ITT - intention-to-treat
LOCF - last observation carried forward
SD - standard deviation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adler 1994 | Allocation: randomised. Participants: people with non-organic psychosis (schizophrenia, schizoaffective disorder, unipolar disorder, bipolar disorder or a delusional disorder). Interventions: vitamin E versus placebo. |
| Baladini 1970 | Allocation: randomised. Participants: people with schizophrenia. Interventions: amitriptyline and fluphenazine (combination tablets) versus placebo |
| Boyer 1995 | Allocation: randomised. Participants: people with schizophrenia. Interventions: amisulpride versus fluphenazine versus placebo |
| Breier 1987 | Allocation: unclear. Participants: people with schizophrenia. Interventions: fluphenazine versus placebo (withdrawal study) |
| Carpenter 1992 | Allocation: randomised. Participants: people with schizophrenia. Interventions: diazepam versus depot fluphenazine versus placebo |
| Chacon 1972 | Allocation: randomised. Participants: people with schizophrenia. Interventions: depot fluphenazine versus chlorpromazine versus placebo |
| Chacon 1973 | Allocation: randomised. Participants: people with schizophrenia. Interventions: depot fluphenazine versus chlorpromazine versus placebo |
| Coffman 1987 | Allocation: unclear. Participants: people with schizophrenia. Interventions: depot fluphenazine versus placebo (oral fluphenazine as a background) |
| Del Giudice 1975 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral phenothiazine versus phenothiazine enanthate |
| Doran 1990 | Allocation: not randomised. |
| Dowing 1963 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine versus chlorpromazine versus thioridazine versus placebo. Outcomes: mental state (no SD). |
| Elman 1999 | Allocation: not randomised. |
| Haider 1968 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral fluphenazine versus fluphenazine enanthate |
| Hanlon 1970 | Allocation: unclear. Participants: newly admitted patients to psychiatric wards, alcoholics, drug addicts, psychosis |
| Held 1970 | Allocation: randomised. Participants: people with schizophrenia. Interventions: phenothiazines versus placebo. |
| Hogarty 1979 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral fluphenazine versus fluphenazine decanoate |
| Holden 1970 | Allocation: unclear. Participants: people with schizophrenia. Interventions: oral fluphenazine versus haloperidol. |
| Howell 1961 | Allocation: unclear. Participants: people with functional psychosis. |
| Itil 1971 | Allocation: unclear. Participants: people with schizophrenia. Interventions: oral fluphenazine low dose versus high dose. |
| Itil 1975 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine versus haloperidol versus thiothixene |
| Kane 1982 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral fluphenazine, fluphenazine decanoate versus placebo. Outcomes: results are not broken down by individual drug. |
| Kinross-Wright 1963 | Allocation: not randomised. |
| Kinross-Wright 1964 | Allocation: randomised. Participants: people with schizophrenia. Interventions: first stage - oral fluphenazine versus fluphenazine enanthate, second stage - fluphenazine enanthate versus placebo |
| Leff 1971 | Allocation: randomised. Participants: people with schizophrenia. Interventions: trifluoperazine versus chlorpromazine versus placebo |
| Litman 1994 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine versus clozapine. |
| Marder 1989 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral fluphenazine versus fluphenazine decanoate |
| Marder 1993 | Allocation: randomised. Participants: people with schizophrenia. Interventions: intensive behavioural skills training versus supportive group psychotherapy |
| Martin 1975 | Allocation: randomised. Participants: people with schizophrenia. Interventions: benzhexol versus placebo. |
| Matheu 1961 | Allocation: not randomised. |
| Mattes 1984 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral fluphenazine versus fluphenazine decanoate versus lithium |
| Pichot 1988 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine versus amisulpride and haloperidol versus amisulpride |
| Pickar 1986 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral fluphenazine versus placebo (withdrawal study) |
| Pickar 1992 | Allocation: not randomised. |
| Quitkin 1978 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine decanoate versus penfluridol. |
| Sampath 1992 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine decanoate versus placebo (withdrawal study) |
| Schlosberg 1978 | Allocation: randomised. Participants: people with schizophrenia. Interventions: pipotiazine palmitate versus fluphenazine decanoate versus placebo |
| Schooler 1976 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine decanoate versus oral fluphenazine, oral fluphenazine versus placebo (withdrawal study), fluphenazine decanoate versus placebo (withdrawal study) |
| Shafti 2009 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine decanoate (IM) versus placebo. |
| Shenoy 1981 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine decanoate versus placebo (withdrawal study) |
| Steingard 1994 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine versus placebo. Outcomes: no usable data. |
| Stevens 1976 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine decanoate versus placebo. |
| Turner 1966 | Allocation: randomised. Participants: people with schizophrenia. Interventions: chlorpromazine versus fluphenazine versus placebo. Outcomes: critical flicker fusion frequency (no usable data) |
| Van Praag 1970 | Allocation: randomised. Participants: people with schizophrenia. Interventions: oral fluphenazine versus fluphenazine decanoate |
| Vestre 1962 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine versus triflupromazine versus phenobarbital |
| Watt 1978 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine versus oral pimozide. |
| Wistedt 1981 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine decanoate or flupentixol decanoate versus placebo (withdrawal study) |
| Wistedt 1983 | Allocation: randomised. Participants: people with schizophrenia. Interventions: fluphenazine decanoate, flupenthixol decanoate versus placebo |
| Zahn 1993 | Allocation: randomised. Participants: people with schizophrenia. Interventions: clozapine versus conventional neuroleptics including oral fluphenazine and placebo Outcomes: no useable data - results not presented for individual groups |
IM - intramuscular
SD - standard deviation
Comparison 1.
ORAL FLUPHENAZINE versus PLACEBO
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Not improved or worsened | 3 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 1.1 short term (CGI/MDRS) | 3 | 125 | Risk Ratio (M-H, Fixed, 95% CI) | 0.80 [0.57, 1.12] |
| 1.2 medium term (MDRS) | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 1.12 [0.79, 1.58] |
| 2 Global state: 2. Relapse | 3 | 124 | Risk Ratio (M-H, Random, 95% CI) | 0.35 [0.07, 1.68] |
| 2.1 short term | 1 | 38 | Risk Ratio (M-H, Random, 95% CI) | 0.25 [0.06, 1.03] |
| 2.2 long term | 2 | 86 | Risk Ratio (M-H, Random, 95% CI) | 0.39 [0.05, 3.31] |
| 3 Global state: 3. Percentage of time in prodrome state (skewed data) | Other data | No numeric data | ||
| 3.1 one-year data | Other data | No numeric data | ||
| 3.2 two-year data | Other data | No numeric data | ||
| 4 Global state: 4. Percentage of time in exacerbated state (skewed data) | Other data | No numeric data | ||
| 4.1 one-year data | Other data | No numeric data | ||
| 4.2 two-year data | Other data | No numeric data | ||
| 5 Global state: 5. average score: CGI - severity of illness score (high = poor) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 short term | 1 | 36 | Mean Difference (IV, Fixed, 95% CI) | −0.77 [−1.39, −0.15] |
| 6 Leaving the study early: 1. Non-specific reasons | 5 | 363 | Risk Ratio (M-H, Fixed, 95% CI) | 0.73 [0.49, 1.10] |
| 6.1 short term | 2 | 227 | Risk Ratio (M-H, Fixed, 95% CI) | 0.68 [0.43, 1.07] |
| 6.2 medium term | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 5.0 [0.25, 99.16] |
| 6.3 long term | 2 | 86 | Risk Ratio (M-H, Fixed, 95% CI) | 0.69 [0.24, 1.97] |
| 7 Leaving the study early: 2. Specific reason - short term | 3 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 7.1 administrative/hospital transfer | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 1.06 [0.07, 15.64] |
| 7.2 AWOL | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 1.06 [0.07, 15.64] |
| 7.3 court cases, transfer, eloped | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 10.65 [1.39, 81.58] |
| 7.4 incorrect diagnosis | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 1.07 [0.07, 16.78] |
| 7.5 marked early remission | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 2.13 [0.20, 23.10] |
| 7.6 serious complication of treatment | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 11.71 [0.66, 208.85] |
| 7.7 severe extrapyramidal effects | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 3.0 [0.13, 70.30] |
| 7.8 treatment failure | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 0.11 [0.03, 0.35] |
| 8 Leaving the study early: 3. Marked improvement/ hospital discharge | 1 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 8.1 discharged due to marked improvement | 1 | 36 | Risk Ratio (M-H, Fixed, 95% CI) | 3.0 [0.13, 69.09] |
| 9 Adverse effects: 1. Anticholinergic effects - short term | 2 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 9.1 blurred vision | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 5.26 [0.27, 102.66] |
| 9.2 constipation | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 2.22 [1.19, 4.15] |
| 9.3 drooling | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 3.16 [0.14, 72.84] |
| 9.4 dryness mouth or throat | 2 | 227 | Risk Ratio (M-H, Fixed, 95% CI) | 3.62 [1.39, 9.42] |
| 9.5 gastrointestinal distress and nausea | 2 | 227 | Risk Ratio (M-H, Fixed, 95% CI) | 0.90 [0.30, 2.72] |
| 9.6 increased salivation | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 18.10 [1.06, 309.15] |
| 9.7 nasal congestion | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 3.16 [0.14, 72.84] |
| 9.8 urinary disturbance | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 3.20 [0.34, 30.17] |
| 9.9 vomiting | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 5.32 [0.26, 109.41] |
| 10 Adverse effects: 2. Cardivascular effects - short term | 2 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 10.1 dizziness, faintness, weakness | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 2.34 [0.85, 6.49] |
| 10.2 hypotension | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 3.16 [0.14, 72.84] |
| 10.3 syncope | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 3.16 [0.14, 72.84] |
| 10.4 tachycardia | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 3.16 [0.14, 72.84] |
| 11 Adverse effects: 3. CNS - short term | 2 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 11.1 anxiety, agitation, excitement and confusion | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 1.06 [0.17, 6.72] |
| 11.2 convulsion or seizures | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 0.35 [0.01, 8.60] |
| 11.3 depression | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 0.35 [0.02, 8.09] |
| 11.4 drowsiness | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 3.91 [1.98, 7.71] |
| 11.5 headache | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 1.17 [0.52, 2.63] |
| 11.6 sedation and lethargy | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 1.06 [0.31, 3.60] |
| 12 Adverse effects: 4. Death - long term | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 2.38 [0.10, 55.72] |
| 13 Adverse effects: 5. Endocrine - short term | 1 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 13.1 amenorrhea | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 1.07 [0.27, 4.14] |
| 13.2 lactation | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 7.45 [0.39, 142.32] |
| 13.3 swelling of breasts | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 5.32 [0.26, 109.41] |
| 14 Adverse effects: 6a. Extrapyramidal effects - short term | 2 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 14.1 akinesia | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 3.16 [0.14, 72.84] |
| 14.2 akathisia | 2 | 227 | Risk Ratio (M-H, Fixed, 95% CI) | 3.43 [1.23, 9.56] |
| 14.3 associated movements | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 7.37 [0.41, 133.37] |
| 14.4 dystonia | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 13.84 [0.79, 242.25] |
| 14.5 facial rigidity | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 2.77 [1.03, 7.46] |
| 14.6 loss of associated movements | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 6.39 [1.95, 20.98] |
| 14.7 restlessness, insomnia | 2 | 227 | Risk Ratio (M-H, Fixed, 95% CI) | 0.99 [0.69, 1.40] |
| 14.8 rigidity | 2 | 227 | Risk Ratio (M-H, Fixed, 95% CI) | 3.54 [1.76, 7.14] |
| 14.9 tremor | 2 | 227 | Risk Ratio (M-H, Fixed, 95% CI) | 3.19 [1.25, 8.11] |
| 15 Adverse effects: 6b. Extrapyramidal effects - medium term | 1 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 15.1 akathisia | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 1.2 [0.42, 3.43] |
| 15.2 akinesia | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 11.0 [0.64, 188.95] |
| 15.3 dystonia | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 1.09 [0.92, 1.29] |
| 15.4 parkinsonism | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 5.5 [1.36, 22.32] |
| 16 Adverse effects: 7. Others - short term | 2 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 16.1 convulsion or seizures | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 0.35 [0.01, 8.60] |
| 16.2 diarrhoea | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 5.32 [0.26, 109.41] |
| 16.3 intercurrent infection | 1 | 190 | Risk Ratio (M-H, Fixed, 95% CI) | 1.07 [0.22, 5.14] |
| 16.4 rash | 2 | 227 | Risk Ratio (M-H, Fixed, 95% CI) | 0.76 [0.15, 3.78] |
| 17 Sensitivity analysis: 1. CHRONIC versus ACUTE | 2 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 17.1 Acute: Global state - not improved - short term | 1 | 37 | Risk Ratio (M-H, Fixed, 95% CI) | 0.59 [0.24, 1.42] |
| 17.2 Chronic: global state - not improved - short term | 1 | 50 | Risk Ratio (M-H, Fixed, 95% CI) | 0.93 [0.56, 1.55] |
| 18 Sensitivity analysis: 2. LOW DOSES (1-5 mg/day) versus HIGH DOSES (5mg/day>) | 3 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 18.1 High dose: Global state - not improved - short term | 1 | 38 | Risk Ratio (M-H, Fixed, 95% CI) | 0.79 [0.48, 1.31] |
| 18.2 Flexible dose: Global state - not improved - short term | 2 | 87 | Risk Ratio (M-H, Fixed, 95% CI) | 0.80 [0.51, 1.25] |
| 19 Sensitivity analysis: 3. OPERATIONAL CRITERIA versus LOOSE DEFINITIONS | 3 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 19.1 DSM-III-R: Global state - not improved - short term | 1 | 38 | Risk Ratio (M-H, Fixed, 95% CI) | 0.79 [0.48, 1.31] |
| 19.2 Loose definition: global state - not improved - short term | 2 | 87 | Risk Ratio (M-H, Fixed, 95% CI) | 0.80 [0.51, 1.25] |
| 20 Sensitivity analysis: 4. BEFORE 1990 versus AFTER 1990 | 3 | Risk Ratio (M-H, Fixed, 95% CI) | Subtotals only | |
| 20.1 Before 1990: Global state - not improved - short term | 2 | 87 | Risk Ratio (M-H, Fixed, 95% CI) | 0.80 [0.51, 1.25] |
| 20.2 After 1990: Global state - not improved - short term | 1 | 38 | Risk Ratio (M-H, Fixed, 95% CI) | 0.79 [0.48, 1.31] |
ACKNOWLEDGEMENTS
We would like to thank members of the Cochrane Schizophrenia Group, Prof. Clive E Adams, Tessa Grant and Judy Wright for their continuing support. Thanks also go to Dr. Adib Essali who first introduced us to The Cochrane Collaboration.
The Cochrane Schizophrenia Group produce and maintain a template for the methods section of their reviews. We have used this template and adapted it for our requirements.
SOURCES OF SUPPORT
Internal sources
• No internal sources of support provided, Not specified.
External sources
• National Institute for Health Research (NIHR), UK.
UK Cochrane Collaboration Programme Grant 2011; Reference number: 10/4001/15
APPENDICES
Appendix 1. Previous search strategy
1. Cochrane Schizophrenia Group's trials register (September 2006)
We searched the Cochrane Schizophrenia Group's trials register (September 2006) using the phrase: [((fluphen* or flufen* or modec* or moditen* or eutimax* or prolixin* or siqualon* or anaten* or dapotum* or decazate* or decafen* or decentan* or fludecate* or lyogen* or lyoridin* or mirenil*) in title, abstract and index fields in REFERENCE) OR (fluphenazin* in interventions field in STUDY)]
The Cochrane Schizophrenia Group's Trials Register is compiled by systematic searches of major databases, handsearches and conference proceedings (see group module).
Appendix 2. Previous data collection and analysis section
1. Selection of trials
We (HEM, MQA) independently inspected the citations identified from the search. We identified potentially relevant abstracts and ordered full papers to reassess these for inclusion and methodological quality. We discussed and reported any disagreements. 2. Assessment of methodological quality
We assessed the methodological quality of included trials in this review using the criteria described in the Cochrane Handbook (Higgins 2005). These criteria are based on the evidence of a strong relationship between allocation concealment and direction of effect (Schulz 1995), and define the following categories: A. Low risk of bias (adequate allocation concealment) B. Moderate risk of bias (some doubt about the results) C. High risk of bias (inadequate allocation concealment). For the purpose of the analysis in this review, trials were included if they met the Cochrane Handbook criteria A or B.
Only trials falling in category A or category B were included in this review. 3. Data collection
3.1 Data extraction
We (HEM and MQA) independently extracted data and, where further clarification was needed, the authors’ of trials were contacted to provide missing data. Any disagreements were discussed and the decisions documented. 3.2 Intention to treat analysis
Where people were lost to follow-up at the end of the study, it was assumed that they had a poor outcome and once they were randomised they were included in the analysis (intention-to-treat (ITT) analysis).
4. Data synthesis
4.1 Data types: We assessed outcomes using continuous (e.g. average changes on a behaviour scale), categorical (e.g. one of three categories on a behaviour scale, such as ‘little change’, ‘moderate change’ or ‘much change’) or dichotomous measures, e.g. either ‘no important changes’ or ‘important changes’ in a person's behaviour. RevMan software does not currently support categorical data, so we only presented these in the text of the review.
4.2 Dichotomous - yes/no - data
Where the original authors of the studies gave outcomes such as ‘clinically improved’ or ‘not clinically improved’ we recorded this. If possible, we attempted to convert relevant outcome measures to dichotomous data by identifying cut off points on rating scales and dividing people accordingly into ‘clinically improved’ or ‘not clinically improved’. For example, the Brief Psychiatric Rating Scale (BPRS) (Overall 1962) is used frequently as a measure of change of symptoms in studies. We defined a 50% change on this particular scale as clinically important although it was recognised that for many people, especially those with chronic or severe illness, a less rigorous definition of important improvement (e.g. 25% on the BPRS) would be equally valid. If individual patient data were available, we used the 50% cut-off for the definition in the case of non-chronically ill people and 25% for those with chronic illness. For dichotomous data we estimated fixed-effect (FE) relative risk (RR) with a 95% confidence interval (CI), and calculated the number needed to treat/ harm (NNT/H) statistic. If heterogeneity was found (see section 5) we used a random-effects model.
4.3 Continuous data: We excluded continuous data if more than 50% of people were lost to follow-up. Continuous data were reported as presented in the original studies, without making any assumptions about those lost to follow-up.
4.3.1. Skewed data: Continuous data on mental health outcomes are often not ‘normally’ distributed. To avoid the pitfall of applying parametric tests to non-parametric data, we applied the following standards to all data before inclusion: (a) standard deviations (SD) and means were reported in the paper or were obtainable from the authors, (b) when a scale started from a finite number (such as zero), the SD, when multiplied by two, was less than the mean (as otherwise the mean was unlikely to be an appropriate measure of the centre of the distribution - (Altman 1996). Endpoint scores on scales often have a finite start and end point and this rule can be applied to them. However, we reported data not meeting these standards in the text of the results section if they had been analysed with appropriate nonparametric tests. Scale-derived change data were reported if no endpoint data were available. Their normality cannot be tested as above, despite the distinct possibility of skew.
4.3.2. Rating scales: a wide range of instruments are available to measure outcomes in mental health studies. These instruments vary in quality and many are not validated, or are even ad hoc. It is accepted generally that measuring instruments should have the properties of reliability (the extent to which a test effectively measures anything at all) and validity (the extent to which a test measures that which it is supposed to measure) (Rust 1989). Unpublished scales are known to be subject to bias in trials of treatments for schizophrenia (Marshall 2000). Therefore, we only included continuous data from rating scales if the measuring instrument had been described in a peer-reviewed journal. In addition, we set the following minimum standards for instruments: The instrument should either be (a) a self-report or (b) completed by an independent rater or relative (not the therapist) and (c) the instrument should be a global assessment of an area of functioning.
4.3.3 Summary statistic: For continuous outcomes we estimated the weighted mean difference (WMD) between groups. Again, if heterogeneity was found (see section 5) we used a random-effects model.
4.3.4 Cluster trials: studies increasingly employ ‘cluster randomisation’ (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a ‘unit of analysis’ error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra class correlation co-efficients of their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non-cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a ‘design effect’. This is calculated using the mean number of participants per cluster (m) and the intra-class correlation co-efficient (ICC) Design effect = 1+(m-1)*ICC (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
5. Investigation for heterogeneity
We judged clinical heterogeneity within all comparisons between included studies, and visually inspected graphs in order to investigate the possibility of statistical heterogeneity. This was supplemented by the I2 statistic which provides an estimate of the percentage of variability due to heterogeneity rather than to chance alone. An I2 estimate equal to or greater than or equal to 75% indicates the presence of high levels of heterogeneity (Higgins 2003). In such cases, we sought to remove outlying trial(s) and perform a sensitivity analyses both with and without these outlying trials. Where no obvious outlying trial(s) could be identified, we analysed and reported the result using a random-effects model, which takes into account that the effects being estimated are not identical.
6. Sensitivity analyses
We undertook several sensitivity analyses (see: Objectives).
7. General
If possible, we presented data in the graphs so that a result to the left of the line of no effect favoured oral fluphenazine.
Footnotes
CONTRIBUTIONS OF AUTHORS
Hosam E Matar screened the search results, organised the retrieval of papers, appraised the papers and extracted data, analysed and interpreted the data, managed the data and entered data in RevMan, and wrote the final report.
Muhammad Qutayba Almerie screened the search results, organised the retrieval of papers, appraised the papers and extracted data, managed the data and entered data in RevMan, analysed and interpreted the data and wrote the final report.
Stephanie Sampson updated the abstract, search and results section, constructed PRISMA diagram and Summary of findings table.
Citation: Matar HE, Almerie MQ, Sampson S. Fluphenazine (oral) versus placebo for schizophrenia. Cochrane Database of Systematic Reviews 2013, Issue 7. Art. No.: CD006352. DOI: 10.1002/14651858.CD006352.pub2.
DECLARATIONS OF INTEREST
None known.
INDEX TERMS
Medical Subject Headings (MeSH)
Administration, Oral; Antipsychotic Agents [*therapeutic use]; Fluphenazine [*therapeutic use]; Placebos [therapeutic use]; Randomized Controlled Trials as Topic; Schizophrenia [*drug therapy]
MeSH check words
Humans
REFERENCES
* Indicates the major publication for the study
- Carpenter WT, Jr, Buchanan RW, Kirkpatrick B, Breier AF. Diazepam treatment of early signs of exacerbation in schizophrenia. American Journal of Psychiatry. 1999;156(2):299–303. doi: 10.1176/ajp.156.2.299. [MEDLINE: 99178642] [DOI] [PubMed] [Google Scholar]
- Clark ML, Huber WK, Charalampous KD, Serafetinides EA, Trousdale W, Colmore JP. Drug treatment in newly admitted schizophrenic patients. Archives of General Psychiatry. 1971;25(5):404–9. doi: 10.1001/archpsyc.1971.01750170020004. [MEDLINE: 72081786] [DOI] [PubMed] [Google Scholar]
- Cole JO, Goldberg SC, Klerman GL. Phenothiazine treatment in acute schizophrenia. Archives of General Psychiatry. 1964;10:246–61. [MEDLINE: 71204770] [PubMed] [Google Scholar]
- Gibbons RD, Lewine RRJ, Davis JM, Schooler NR, Cole JO. An empirical test of a kraepelinian vs. a bleulerian view of negative symptoms. Schizophrenia Bulletin. 1985;11(3):390–5. doi: 10.1093/schbul/11.3.390. [MEDLINE: 71204770] [DOI] [PubMed] [Google Scholar]
- Goldberg SC, Klerman GL, Cole JO. Changes in schizophrenic psychopathology and ward behaviour as a function of phenothiazine treatment. British Journal of Psychiatry. 1965;111:120–33. doi: 10.1192/bjp.111.471.120. [MEDLINE: 5677325] [DOI] [PubMed] [Google Scholar]
- Goldberg SC, Mattsson N, Cole JO, Klerman GL. Prediction of improvement in schizophrenia under four phenothiazines. Archives of General Psychiatry. 1967;16:107–17. doi: 10.1001/archpsyc.1967.01730190109015. [MEDLINE: 6066689] [DOI] [PubMed] [Google Scholar]
- Goldberg SC, Mattsson NB. Schizophrenic subtypes defined by response to drugs and placebo. Diseases of the Nervous System. 1968;29(5):S153–8. [MEDLINE: 68399894] [PubMed] [Google Scholar]
- Klerman GL, Goldberg SG, Davis D. Relationship between the hospital milieu and the response to phenothiazines in the treatment of schizophrenics. Acta Psychiatrica Belgica. 1970;70(6):716–29. [MEDLINE: 71204770] [PubMed] [Google Scholar]
- Hordern A, King A, Holt NF, Collins J, Toussaint J. Thioproperazine in chronic schizophrenia. British Journal of Psychiatry. 1964;110:531–9. doi: 10.1192/bjp.110.467.531. [MEDLINE: 79181666] [DOI] [PubMed] [Google Scholar]
- Marder SR, Wirshing WC, van Putten T, Mintz J, McKenzie J, Johnston-Cronk K, et al. Fluphenazine vs placebo supplementation for prodromal signs of relapse in schizophrenia. Archives of General Psychiatry. 1994;51(4):280–7. doi: 10.1001/archpsyc.1994.03950040024003. [MEDLINE: 94213575] [DOI] [PubMed] [Google Scholar]
- Millar J. A trial of fluphenazine in schizophrenia. British Journal of Psychiatry. 1963;109:428–32. [MEDLINE: 769023] [Google Scholar]
- Rifkin A, Quitkin F, Kane J, Klein DF. Fluphenazine decanoate, oral fluphenazine, and placebo in the treatment of remitted schizophrenics. II. Rating scale data. Psychopharmacology Bulletin. 1977;13(2):40–50. [MEDLINE: 323910] [PubMed] [Google Scholar]
- Rifkin A, Quitkin F, Kane J, Klein DF, Ross D. The effect of fluphenazine upon social and vocational functioning in remitted schizophrenics. Biological Psychiatry. 1979;14(3):499–508. [MEDLINE: 224958; : PsycINFO 64–08562] [PubMed] [Google Scholar]
- Rifkin A, Quitkin F, Klein DF. Fluphenazine decanoate, oral fluphenazine, and placebo in treatment of remitted schizophrenics. II. Rating scale data. Archives of General Psychiatry. 1977;34(10):15–9. doi: 10.1001/archpsyc.1977.01770220097011. [MEDLINE: 78019044] [DOI] [PubMed] [Google Scholar]
- Rifkin A, Quitkin F, Rabiner CJ, Klein DF. Comparison of fluphenazine decanoate, oral fluphenazine, and placebo in remitted outpatient schizophrenics. Psychopharmacology Bulletin. 1976;12(2):24–6. [MEDLINE: 769022] [PubMed] [Google Scholar]
- Rifkin A, Quitkin F, Rabiner CJ, Klein DF. Fluphenazine decanoate, fluphenazine hydrochloride given orally, and placebo in remitted schizophrenics. I. Relapse rates after one year. Archives of General Psychiatry. 1977;34(1):43–7. doi: 10.1001/archpsyc.1977.01770130045004. [MEDLINE: 77111134] [DOI] [PubMed] [Google Scholar]
- Adler LA, Rotrosen J, Edson R, Lavori P, Lohr J, Hitzemann R. Vitamin E treatment for tardive dyskinesia. Archives of General Psychiatry. 1999;56(9):836–41. doi: 10.1001/archpsyc.56.9.836. [EMBASE 1999313441] [DOI] [PubMed] [Google Scholar]
- Adler LA, Rotrosen J, Lavori P, Edson R. Vitamin E treatment of TD: development of a VA cooperative study. Biological Psychiatry. 1994;35:730–1. [MEDLINE: 11760] [Google Scholar]
- Caligiuri MP, Lohr JB, Rotrosen J, Adler L, Lavori P, Edson R, et al. Reliability of an instrumental assessment of tardive dyskinesia: results from VA Cooperative Study 394. Psychopharmacology. 1997;132(1):61–6. doi: 10.1007/s002130050320. [EMBASE 1997216311] [DOI] [PubMed] [Google Scholar]
- Baldini JT, Neary ER. Controlled trials of an amitriptylinefluphenazine combination in depressive neuroses and psychoses: a collaborative study. Current Therapeutic Research, Clinical and Experimental. 1970;12(2):84–93. [MEDLINE: 70113586] [PubMed] [Google Scholar]
- Boyer P, Lecrubier Y, Puech AJ. Treatment of positive and negative symptoms: pharmacologic approaches. Modern Problems of Pharmacopsychiatry. 1990;24:152–74. doi: 10.1159/000418016. [MEDLINE: 11782241] [DOI] [PubMed] [Google Scholar]
- Boyer P, Lecrubier Y, Puech AJ, Dewailly J, Aubin F. Treatment of negative symptoms in schizophrenia with amisulpride. British Journal of Psychiatry. 1995;166(1):68–72. doi: 10.1192/bjp.166.1.68. [MEDLINE: 7894879] [DOI] [PubMed] [Google Scholar]
- Boyer P, Puech AJ. Determinants for clinical activity of neuroleptic drugs: chemical substances, doses, assessment tools [Modalities d’action clinique des neuroleptiques: substances, doses, instruments de mesure utilises]. Psychiatrie and Psychobiologie. 1987;2(4):296–305. [PsycINFO 76–12830] [Google Scholar]
- Boyer P, Puech AJ, Lecrubier Y. Double blind trial versus placebo of low dose amisulpride (Solian 50) in schizophrenia with exclusively negative symptoms. Preliminary analysis of results [Etude en double insu contre placebo de l’amisulpride (Solian (r) 50) a faible dose chez des schizophrenes purement deficitaires. Premiere analyse des resultats]. Annales de Psychiatrie. 1988;3(3):321–5. [EMBASE 1988242981] [Google Scholar]
- Lecrubier Y. 6th World Congress of Biological Psychiatry. Nice; France: Jun 22-27, 1997. 1997. Amisulpride in deficit schizophrenia. [MEDLINE: 7894879] [Google Scholar]
- Rein W, Turjanski S. Clinical update on amisulpride in deficit schizophrenia. International Clinical Psychopharmacology. 1997;12(Suppl 2):S19–27. doi: 10.1097/00004850-199705002-00005. [MEDLINE: 97361270] [DOI] [PubMed] [Google Scholar]
- Breier A, Wolkowitz OM, Doran AR, Roy A, Boronow J, Hommer DW, et al. Neuroleptic responsivity of negative and positive symptoms in schizophrenia. American Journal of Psychiatry. 1987;144:1549–55. doi: 10.1176/ajp.144.12.1549. [MEDLINE: 3688278] [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Buchanan RW, Breier A, Kirkpatrick B, Hanlon T, Levine J, et al. Novel neuroleptic dosage reduction strategies. Schizophrenia Research. 1992;6(2):107. [MEDLINE: 3688278] [Google Scholar]
- Chacon C, Harper P, Harvey GF. Work study in the assessment of the effects of phenothiazines in schizophrenia. Comprehensive Psychiatry. 1972;13(6):549–54. doi: 10.1016/0010-440x(72)90055-7. [MEDLINE: 73051431; : PsycINFO 1973–31686–001] [DOI] [PubMed] [Google Scholar]
- Chacon C, Harper P. Clinical and work performance variables in phenothiazine therapy of schizophrenia. Acta Psychiatrica Scandinavica. 1973;49(1):65–76. doi: 10.1111/j.1600-0447.1973.tb04399.x. [MEDLINE: 4572169] [DOI] [PubMed] [Google Scholar]
- Coffman JA, Nasrallah HA, Lyskowski J, McCalley-Whitters M, Dunner FJ. Clinical effectiveness of oral and parenteral rapid neuroleptization. Journal of Clinical Psychiatry. 1987;48(1):20–4. [EMBASE 1987061089] [PubMed] [Google Scholar]
- Del Giudice J, Clark WG, Gocka EF. Prevention of recidivism of schizophrenics treated with fluphenazine enanthate. Psychosomatics. 1975;16(1):32–6. doi: 10.1016/s0033-3182(75)71232-x. [MEDLINE: 76032528; : PsycINFO 54–12277] [DOI] [PubMed] [Google Scholar]
- Doran AR, Labarca R, Wolkowitz OM, Roy A, Douillet P, Pickar D. Circadian variation of plasma homovanillic acid levels is attenuated by fluphenazine in patients with Schizophrenia. Archives of General Psychiatry. 1990;47:558–63. doi: 10.1001/archpsyc.1990.01810180058009. [MEDLINE: 2350208] [DOI] [PubMed] [Google Scholar]
- Downing RW, Ebert JN, Shubrooks SJ. Effect of phenothiazines on the thinking of acute schizophrenics. Perceptual and Motor Skills. 1963;17(2):511–20. doi: 10.2466/pms.1963.17.2.511. [PsycINFO 1964–06498–001] [DOI] [PubMed] [Google Scholar]
- Elman I, Goldstein DS, Eisenhofer G, Folio J, Malhotra AK, Adler CM, et al. Mechanism of peripheral noradrenergic stimulation by clozapine. Neuropyschopharmacology. 1999;20(1):29–34. doi: 10.1016/S0893-133X(98)00047-5. [MEDLINE: 99103168] [DOI] [PubMed] [Google Scholar]
- Haider I. A controlled trial of fluphenazine enanthate in hospitalized chronic schizophrenics. British Journal of Psychiatry. 1968;114(512):837–41. doi: 10.1192/bjp.114.512.837. [MEDLINE: 4874164] [DOI] [PubMed] [Google Scholar]
- Hanlon TE, Ota KY, Kurland AA. Comparative effects of fluphenazine, fluphenazine-chlordiazepoxide and fluphenazine-imipramine. Diseases of the Nervous System. 1970;31(3):171–7. [MEDLINE: 4909632] [PubMed] [Google Scholar]
- Held JM, Cromwell RL, Frank ET, Jr, Fann WE. Effect of phenothiazines on reaction time in schizophrenics. Journal of Psychiatric Research. 1970;7(3):209–13. doi: 10.1016/0022-3956(70)90008-7. [MEDLINE: 70166858] [DOI] [PubMed] [Google Scholar]
- Hogarty GE, Schooler NR, Ulrich R, Mussare F, Ferro P, Herron E. Fluphenazine and social therapy in the aftercare of schizophrenic patients. Relapse analyses of a two-year controlled study of fluphenazine decanoate and fluphenazine hydrochloride. Archives of General Psychiatry. 1979;36(12):1283–94. doi: 10.1001/archpsyc.1979.01780120013001. [MEDLINE: 80041623] [DOI] [PubMed] [Google Scholar]
- Holden JM, Itil TM, Keskiner A. Assessment and significance of changes in laboratory values with haloperidol and fluphenazine hydrochloride therapy. Biological Psychiatry. 1970;2(2):173–82. [MEDLINE: 4918019] [PubMed] [Google Scholar]
- Howell RJ, Brown HM, Beaghler HE. A comparison of fluphenazine, trifluoperazine and a placebo in the context of an active treatment unit. Journal of Nervous and Mental Disease. 1961;132:522–30. doi: 10.1097/00005053-196106000-00007. [MEDLINE: 13716275] [DOI] [PubMed] [Google Scholar]
- Itil TM, Saletu B, Hsu W, Kiremitci N, Keskiner A. Clinical and quantitative EEG changes at different dosage levels of fluphenazine treatment. Acta Psychiatrica Scandinavica. 1971;47(4):440–51. doi: 10.1111/j.1600-0447.1971.tb03701.x. [MEDLINE: 4947806] [DOI] [PubMed] [Google Scholar]
- Itil TM, Marasa J, Saletu B, Davis S, Mucciardi AN. Computerized EEG: predictor of outcome in schizophrenia. Journal of Nervous and Mental Disease. 1975;160(3):188–203. [MEDLINE: 75116033] [PubMed] [Google Scholar]
- Kane JM, Rifkin A, Quitkin F, Nayak D, Ramos Lorenzi J. Fluphenazine vs placebo in patients with remitted, acute first-episode schizophrenia. Archives of General Psychiatry. 1982;39(1):70–3. doi: 10.1001/archpsyc.1982.04290010048009. [MEDLINE: 82112452] [DOI] [PubMed] [Google Scholar]
- Kinross-Wright J, Vogt AH, Charalampous KD. A new method of drug therapy. American Journal of Psychiatry. 1963;119:779–80. doi: 10.1176/ajp.119.8.779. [MEDLINE: 14032917] [DOI] [PubMed] [Google Scholar]
- Kinross-Wright J, Charalampous KD. A controlled study of a very long-acting phenothiazine preparation. International Journal of Neuropsychiatry. 1964;1:66–70. [PsycINFO 60–03426] [Google Scholar]
- Hirsch SR, Gaind R, Rohde PD, Stevens BC, Wing JK. Outpatient maintenance of chronic schizophrenic patients with long-acting fluphenazine: double-blind placebo trial. Report to the Medical Research Council Committee on Clinical Trials in Psychiatry. British Medical Journal. 1973;1(854):633–7. doi: 10.1136/bmj.1.5854.633. [MEDLINE: 4571196] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff JP, Wing JK. Trial of maintenance therapy in schizophrenia. British Medical Journal. 1971;3(775):599–604. doi: 10.1136/bmj.3.5775.599. [MEDLINE: 71287275] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litman RE, Hommer DW, Radant A, Clem T, Pickar D. Quantitative effects of typical and atypical neuroleptics on smooth pursuit eye tracking in schizophrenia. Schizophrenia Research. 1994;12(2):107–20. doi: 10.1016/0920-9964(94)90068-x. [MEDLINE: 94318574] [DOI] [PubMed] [Google Scholar]
- Marder SR, van Putten T, Aravagiri M, Hubbard JW, Hawes EM, McKay G, et al. Plasma levels of parent drug and metabolites in patients receiving oral and depot fluphenazine. Psychopharmacology Bulletin. 1989;25(3):479–82. [MEDLINE: 2626520] [PubMed] [Google Scholar]
- Marder SR, Wirshing WC, Eckman T. Psychosocial and pharmacological strategies for maintenance therapy: effects on two - year outcome. Schizophrenia Research. 1993;9:260. [Google Scholar]
- Martin IC. Implications of phenothiazine side effects: a study of antiparkinsonian agents in an older population. Acta Psychiatrica Scandinavica. 1975;51(2):110–8. doi: 10.1111/j.1600-0447.1975.tb00220.x. [MEDLINE: 235189] [DOI] [PubMed] [Google Scholar]
- Matheu H, Fogel EJ. Clinical effects of fluphenazine dihydrychloride in chronic schizophrenia. Journal of Neuropsychiatry. 1961;3:105–11. [MEDLINE: 14471099] [PubMed] [Google Scholar]
- Mattes JA, Nayak D. Lithium versus fluphenazine for prophylaxis in mainly schizophrenic schizo-affectives. Biological Psychiatry. 1984;19(3):445–9. [MEDLINE: 6722235; : PsycINFO 72–10290] [PubMed] [Google Scholar]
- Boyer P, Lecrubier Y, Puech AJ. Treatment of positive and negative symptoms: pharmacologic approaches. Modern Problems of Pharmacopsychiatry. 1990;24:152–74. doi: 10.1159/000418016. [MEDLINE: 11782241] [DOI] [PubMed] [Google Scholar]
- Boyer P, Puech AJ. Determinants for clinical activity of neuroleptic drugs: chemical substances, doses, assessment tools [Modalities d’action clinique des neuroleptiques: substances, doses, instruments de mesure utilises]. Psychiatrie and Psychobiologie. 1987;2(4):296–305. [PsycINFO 76–12830] [Google Scholar]
- Pichot P, Boyer P. A double blind, controlled, multicenter trial of low dose amisulpride (Solian(R) 50) versus low dose fluphenazine in the treatment of negative symptoms in chronic schizophrenia [Essai multicentrique controle, en double insu, amisulpride (solian(r) 50) contre fluphenazine a faibles doses dans le traitement du syndrome deficitaire des schizophrenies chroniques]. Annales de Psychiatrie. 1988;3(3):312–20. [EMBASE 1988242980] [Google Scholar]
- Pickar D, Labarca R, Doran AR, Wolkowitz OM, Roy A, Breier A, Linnoila M, Paul SM. Longitudinal measurement of plasma homovanillic acid levels in schizophrenic patients. Correlation with psychosis and response to neuroleptic treatment. Archives of General Psychiatry. 1986;43(7):669–76. doi: 10.1001/archpsyc.1986.01800070059008. [MEDLINE: 3718170] [DOI] [PubMed] [Google Scholar]
- Pickar D, Owen RR, Litman RE, Hsiao JK, Su TP. Predictors of clozapine response in schizophrenia. Journal of Clinical Psychiatry. 1994;(Suppl B):55, 129–32. [MEDLINE: 95050371] [PubMed] [Google Scholar]
- Pickar D, Owen RR, Litman RE, Konicki E, Gutierrez R, Rapaport MH. Clinical and biologic response to clozapine in patients with schizophrenia. Crossover comparison with fluphenazine. Archives of General Psychiatry. 1992;49(5):345–53. doi: 10.1001/archpsyc.1992.01820050009001. [MEDLINE: 92264872] [DOI] [PubMed] [Google Scholar]
- Quitkin F, Rifkin A, Kane J, Ramos Lorenzi JR, Klein DF. Long-acting oral vs injectable antipsychotic drugs in schizophrenics: a one-year double-blind comparison in multiple episode schizophrenics. Archives of General Psychiatry. 1978;35(7):889–92. doi: 10.1001/archpsyc.1978.01770310095007. [MEDLINE: 78234523] [DOI] [PubMed] [Google Scholar]
- Sampath G, Shah A, Krska J, Soni SD. Neuroleptic discontinuation in the very stable schizophrenic patient - relapse rates and serum neuroleptic levels. Human Psychopharmacology. 1992;7(4):255–64. [EMBASE 1992364197] [Google Scholar]
- Schlosberg A, Shadmi M. A comparative controlled study of two long-acting phenothiazines: pipotiazine palmitate and fluphenazine decanoate. Current Therapeutic Research. 1978;23(5):642–54. [PsycINFO 62–01680] [Google Scholar]
- Gelenberg AJ, Doller JC, Schooler NR, Mieske M, Severe J, Mandel MR. Acute extrapyramidal reactions with fluphenazine hydrochloride and fluphenazine decanoate. American Journal of Psychiatry. 1979;136:217–9. doi: 10.1176/ajp.136.2.217. [MEDLINE: 79101484] [DOI] [PubMed] [Google Scholar]
- Levine J, Schooler NR, Severe J, Escobar J, Gelenberg A, Mandel M, et al. Discontinuation of oral and depot fluphenazine in schizophrenic patients after one year of continuous medication: a controlled study. Advances in Biochemical Psychopharmacology. 1980;24:483–93. [MEDLINE: 80262441] [PubMed] [Google Scholar]
- Mandel MR, Severe JB, Schooler NR, Gelenberg AJ, Mieske M. Development and prediction of postpsychotic depression in neuroleptic-treated schizophrenics. Archives of General Psychiatry. 1982;39(2):197–203. doi: 10.1001/archpsyc.1982.04290020051010. [MEDLINE: 82159410] [DOI] [PubMed] [Google Scholar]
- Schooler NR, Levine J. Dosage and side effect comparisons between oral and depot fluphenazine. Psychopharmacology Bulletin. 1977;13(3):29–31. [MEDLINE: 329328] [PubMed] [Google Scholar]
- Schooler NR, Levine J. +NIMH-PRB Collaborative Fluphenazine Study Group. The initiation of long-term pharmacotherapy in schizophrenia: dosage and side effect comparisons between oral and depot fluphenazine. Pharmacopsychiatry. 1976;9(4):159–69. [MEDLINE: 77036977] [PubMed] [Google Scholar]
- Schooler NR, Levine J, Severe JB. Depot fluphenazine in the prevention of relapse in schizophrenia: evaluation of a treatment regimen. Psychopharmacology Bulletin. 1979;15(2):44–7. [MEDLINE: 373006] [PubMed] [Google Scholar]
- Schooler NR, Levine J, Severe JB, Brauzer B, DiMascio A, Klerman GL, et al. Prevention of relapse in schizophrenia. An evaluation of fluphenazine decanoate. Archives of General Psychiatry. 1980;37(1):16–24. doi: 10.1001/archpsyc.1980.01780140018002. [MEDLINE: 80108511] [DOI] [PubMed] [Google Scholar]
- Shafti SS. Augmentation of olanzapine by fluphenazine decanoate in poorly responsive schizophrenia. Clinical Schizophrenia and Related Psychoses. 2009;3(2):97–102. [Google Scholar]
- Shenoy RS, Sadler AG, Goldberg SC, Hamer RM, Ross B. Effects of a six-week drug holiday on symptom status, relapse, and tardive dyskinesia in chronic schizophrenics. Journal of Clinical Psychopharmacology. 1981;1(3):141–5. doi: 10.1097/00004714-198105000-00005. [MEDLINE: 82053610] [DOI] [PubMed] [Google Scholar]
- Steingard S, Allen M, Schooler NR. A study of the pharmacologic treatment of medication-compliant schizophrenics who relapse. Journal of Clinical Psychiatry. 1994;55(11):470–2. [PubMed] [Google Scholar]
- Stevens B. The social value of fluphenazine decanoate. Acta Psychiatrica Belgica. 1976;76(5):792–804. [MEDLINE: 77178598] [PubMed] [Google Scholar]
- Turner P. A comparison of fluphenazine and chlorpromazine on critical flicker fusion frequency. Journal of Pharmacy and Pharmacology. 1966;18:836. doi: 10.1111/j.2042-7158.1966.tb07825.x. [MEDLINE: 67163670] [DOI] [PubMed] [Google Scholar]
- van Praag HM, Breetveld J, van Mesdag Etty H, Westerhuis R, Pen A, Schut T. A controlled comparative study of fluphenazine and fluphenazine enanthate in acute and chronic psychotic patients. Psychiatria Neurologia Neurochirurgia. 1970;73(3):165–75. [MEDLINE: 4912332] [PubMed] [Google Scholar]
- Vestre ND, Hall WB, Schiele BC. A comparison of fluphenazine, triflupromazine, and phenobarbital in the treatment of chronic schizophrenic patients: a double-blind controlled study. Journal of Clinical and Experimental Psychopathology. 1962;23:149–59. [MEDLINE: 67163670] [PubMed] [Google Scholar]
- Watt DC. Maintenance drugs for schizophrenia. Lancet. 1978;2(8098):1045–6. doi: 10.1016/s0140-6736(78)92361-9. [MEDLINE: 79052029] [DOI] [PubMed] [Google Scholar]
- Wistedt B. A depot neuroleptic withdrawal study. A controlled study of the clinical effects of the withdrawal of depot fluphenazine decanoate and depot flupenthixol decanoate in chronic schizophrenic patients. Acta Psychiatrica Scandinavica. 1981;64(1):65–84. doi: 10.1111/j.1600-0447.1981.tb00761.x. [MEDLINE: 7032224] [DOI] [PubMed] [Google Scholar]
- Wistedt B. Neuroleptics and depression. Archives of General Psychiatry. 1982;39(6):745. doi: 10.1001/archpsyc.1982.04290060085019. [MEDLINE: 6124226] [DOI] [PubMed] [Google Scholar]
- Wistedt B, Jorgensen A, Wiles D. A depot neuroleptic withdrawal study. Plasma concentration of fluphenazine and flupenthixol and relapse frequency. Psychopharmacology. 1982;78(4):301–4. doi: 10.1007/BF00433729. [MEDLINE: 83118214] [DOI] [PubMed] [Google Scholar]
- Wistedt B, Wiles D, Jorgensen A. A depot neuroleptic withdrawal study neurological effects. Psychopharmacology. 1983;80(2):101–5. doi: 10.1007/BF00427950. [MEDLINE: 4874164] [DOI] [PubMed] [Google Scholar]
- Zahn TP, Pickar D. Autonomic effects of clozapine in schizophrenia - comparison with placebo and fluphenazine. Biological Psychiatry. 1993;34(1-2):3–12. doi: 10.1016/0006-3223(93)90250-h. [DOI] [PubMed] [Google Scholar]
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proceedings of the National Academy of Sciences. 2000;97(14):8104–9. doi: 10.1073/pnas.97.14.8104. [MEDLINE: 10884434] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed I, Soares K, Seifas R, Adams CE. Randomised controlled trials in Archives of General Psychiatry (1959-1995): a prevalence study. Archives of General Psychiatry. 1998;55(8):754–5. doi: 10.1001/archpsyc.55.8.754. [DOI] [PubMed] [Google Scholar]
- Altman DG, Bland JM. Detecing skewness from summary information. BMJ. 1996;313:1200. doi: 10.1136/bmj.313.7066.1200. [OLZ020600] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ. 1997;315:600. doi: 10.1136/bmj.315.7108.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d’efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d’Etude des Indices D’efficacite]. Therapie. 1999;54(4):405–11. [PUBMED: 10667106] [PubMed] [Google Scholar]
- Caldwell CB, Gottesman II. Schizophrenia - a high-risk factor for suicide: clues to risk reduction. Suicide and Life-Threatening Behavior. 1992;22:479–93. [PubMed] [Google Scholar]
- Darling HF. Fluphenazine: a preliminary study. Disease of the Nervous System. 1959;20(4):167–70. [MEDLINE: 13652801] [PubMed] [Google Scholar]
- Davis JM, Andriukatis S. The natural course of schizophrenia and effective maintenance drug therapy. Journal of Clincal Psychopharmacology. 1986;(1Suppl):6, 2S–10S. [PubMed] [Google Scholar]
- Deeks J. Issues in the selection for meta-analyses of binary data; Proceedings of the 8th International Cochrane Colloquium; Cape Town. Cape Town: The Cochrane Collaboration. 2000 Oct 25-28.2000. [Google Scholar]
- Dencker SJ, Lepp M, Malm U. Do schizophrenics well adapted in the community need neuroleptics? A depot neuroleptic withdrawal study. Acta Psychiatria Scandinavica. 1980;(supp 279):61, 64–76. doi: 10.1111/j.1600-0447.1980.tb07084.x. [DOI] [PubMed] [Google Scholar]
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians’ patient care behavior. Journal of General Internal Medicine. 1992;7:623–9. doi: 10.1007/BF02599201. [DOI] [PubMed] [Google Scholar]
- Donner A, Klar N. Issues in the meta-analysis of cluster randomized trials. Statistics in Medicine. 2002;21:2971–80. doi: 10.1002/sim.1301. [DOI] [PubMed] [Google Scholar]
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315:629–34. doi: 10.1136/bmj.315.7109.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta-analyses involving crossover trials: methodological issues. International Journal of Epidemiology. 2002;31(1):140–9. doi: 10.1093/ije/31.1.140. [DOI] [PubMed] [Google Scholar]
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta-analyses can provide accurate results. Journal of Clinical Epidemiology. 2006;59(7):7–10. doi: 10.1016/j.jclinepi.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Gjedde A, Wong DF. Quantification of neuroreceptors in living human brain v. endogenous neurotransmiter inhibition of haloperidol binding in psychosis. Journal of Cerebral Blood Flow Metabolism. 2001;21(8):982–94. doi: 10.1097/00004647-200108000-00011. [MEDLINE: 11487734] [DOI] [PubMed] [Google Scholar]
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community-based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology. 1999;149:876–83. doi: 10.1093/oxfordjournals.aje.a009904. [DOI] [PubMed] [Google Scholar]
- Guy U. ECDEU assessment manual for psychopharmacology. Revised. National Institute of Mental Health; Rockville: 1976. [Google Scholar]
- Hietala J, Syvalahti E, Vuorio K, Rakkolainen V, Bergman J, Haaparanta M, et al. Presynaptic dopamine function in striatum of neuroleptic-naive schizophrenic patients. Lancet. 1995;346(8983):1130–1. doi: 10.1016/s0140-6736(95)91801-9. [DOI] [PubMed] [Google Scholar]
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–60. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]. The Cochrane Library. issue 3. John Wiley & Sons, Ltd; Chichester: 2005. [Google Scholar]
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration. 2011 Available from www.cochrane-handbook.org.
- Holt JP, Wright ER. Preliminary results with fluphenazine (prolixin) in chronic psychotic patients. American Journal of Psychiatry. 1960;117:157–9. doi: 10.1176/ajp.117.2.157. [MEDLINE: 14402852] [DOI] [PubMed] [Google Scholar]
- Jablensky A, Sartorius N, Ernberg G, Anker M, Korten A, Cooper JE, et al. Schizophrenia: manifestations, incidence and course in different cultures. A World Health Organization ten-country study. Psychological Medicine Monograph Suppl. 1992;22(4):1092. doi: 10.1017/s0264180100000904. [MEDLINE: 1565705] [DOI] [PubMed] [Google Scholar]
- Kane JM, Woerner M, Sarantakos S. Depot neuroleptics: A comparison review of standard, intermediate and low-dose regimens. Journal of Clinical Psychiatry. 1986;47(suppl 5):30–3. [PubMed] [Google Scholar]
- Kay SR, Opler LA, Fiszbein A. North Tonawanda. Multi-Health Systems; NY: 1986. Positive and Negative Syndrome Scale (PANSS) Manual. [Google Scholar]
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research. 2005;79(2-3):231–8. doi: 10.1016/j.schres.2005.04.008. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry. 2005;187:366–71. doi: 10.1192/bjp.187.4.366. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin. 2007;33(1):183–91. doi: 10.1093/schbul/sbl025. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom LH, Gefvert O, Hagberg G, Lundberg T, Bergstrom M, Hartvig P, et al. Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L-(beta-11C) DOPA and PET. Biological Psychiatry. 1999;46(5):681–8. doi: 10.1016/s0006-3223(99)00109-2. [MEDLINE: 10472420] [DOI] [PubMed] [Google Scholar]
- Lorr M. The Multidimensional Scale for Rating Psychiatric Patients. U.S.V.I, , T.B; Washington D.C.: 1953. pp. 19–507. [Google Scholar]
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry. 2000;176:249–52. doi: 10.1192/bjp.176.3.249. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson M, Quarantelli M, et al. Reduced prefronatal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nature Neuroscience. 2002;5(3):267–71. doi: 10.1038/nn804. [MEDLINE: 11865311] [DOI] [PubMed] [Google Scholar]
- Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. JAMA. 2001;285(12):1987–91. doi: 10.1001/jama.285.15.1987. [DOI] [PubMed] [Google Scholar]
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports. 1962;10:799–812. [Google Scholar]
- Rust J, Golombok S. Modern Psychmetrics. Routledge; London: 1989. [Google Scholar]
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias: dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA. 1995;273:408–12. doi: 10.1001/jama.273.5.408. [DOI] [PubMed] [Google Scholar]
- Schulz KF, Altman DG, Moher D. For the CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. Annals of Internal Medicine. 2010 Jun 1;152(11):726–32. doi: 10.7326/0003-4819-152-11-201006010-00232. [DOI] [PubMed] [Google Scholar]
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration. 2008:359–83. [Google Scholar]
- Thornley B, Adams C. Content and quality of 2000 controlled trials in schizophrenia over 50 years. BMJ 1998. 317:1181–4. doi: 10.1136/bmj.317.7167.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area-wide and organisation-based interventions in health and health care: a systematic review. Health Technology Assessment. 1999;3(5):iii–92. [MEDLINE: 10982317] [PubMed] [Google Scholar]
- Essential Medicines. WHO Model List. 14th Edition. http://whqlibdoc.who.int/hq/2005/a87017.eng.pdf.
- Wikipedia (The Free Encyclopedia) http://en.wikipedia.org/wiki/Fluphenazine#Pharmacokinetics Accessed in Nov 2006.
- Psychotropics. http://www.psychotropics.dk/.
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El-Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin. 2009;33(7):254–7. [Google Scholar]
- Matar HE, Almerie MQ. Oral fluphenazine versus placebo for schizophrenia. Cochrane Database of Systematic Reviews. 2007;(Issue 1) doi: 10.1002/14651858.CD006352. [DOI: 10.1002/14651858.CD006352] [DOI] [PubMed] [Google Scholar]
- Matar HE-D, Almerie MQ, Giraldo AM, Adams CE. Oral fluphenazine versus placebo for schizophrenia: A Cochrane systematic review of 40 years of randomised controlled trials.. Revista Colombiana de Psiquiatria. 2007;36(1):8–17. doi: 10.1002/14651858.CD006352. [DOI] [PubMed] [Google Scholar]