

Abstract
Cells respond to environmental stressors and xenobiotic exposures using regulatory networks to control gene expression, and there is an emerging appreciation for the role of numerous postsynthetic chemical modifications of DNA, RNA, and proteins in controlling transcription and translation of the stress response. In this Perspective, we present a model for a new network that regulates the cellular response to xenobiotic exposures and other stresses in which stress-induced reprogramming of a system of dozens of post-transcriptional modifications on tRNA (tRNA) promotes selective translation of codon-biased mRNAs for critical response proteins. As a product of novel genomic and bioanalytical technologies, this model has strong parallels with the regulatory networks of DNA methylation in epigenetics and the variety of protein secondary modifications comprising signaling pathways and the histone code. When present at the tRNA wobble position, the modified ribonucleosides enhance the translation of mRNAs in which the cognate codons of the tRNAs are highly over-represented and that represent critical stress response proteins. A parallel system may also downregulate the translation of families of proteins. Notably, dysregulation of the tRNA methyltransferase enzymes in humans has also been implicated in cancer etiology, with demonstrated oncogenic and tumor-suppressive effects.
1. Introduction
Cells respond to environmental signals and stresses using a variety of mechanisms that link the external stimuli to changes in cell phenotype by myriad biochemical reactions that ultimately lead to changes in gene expression and protein activity. Well-defined pathways of signal transduction affect transcription, mRNA stability, protein levels, and protein secondary modification, with the altered protein function and metabolite levels defining a new cell state. Among the mechanisms of cell response, the link between translation and environmental changes is the least understood. Here, we describe the emerging evidence for a new system by which cells respond and adapt to environmental stresses by reprogramming dozens of modified ribonucleosides in tRNA, which leads to selective translation of codon-biased mRNAs. This system exploits features of both genetics, in the form of a code of codon use in families of genes, and postsynthetic DNA and protein modifications, in the idea that editable modifications affect gene expression, with signal transduction pathways linking the environmental stress to controlled changes in gene expression at the level of translational elongation.
2. Systems for Regulating Transcription by Reprogramming DNA and Histone Modifications
As a parallel to the system of RNA modifications in control of translation, epigenetics is classically defined as heritable changes in gene expression without changing the DNA sequence. The prime example involves formation of m5C by DNA methyltransferases (DNMT’s) as a well-established regulator of gene expression,1−4 with methylation patterns in promoter regions dramatically altered in response to environmental stimuli or in different cancers.5,6 Although DNA methylation patterns are heritable and the patterns previously presumed to be stable in a specific cell type, global reprogramming of m5C patterns in the genome has now been observed in response to exposure to drugs and toxicants,5 which illustrates a dynamic role for epigenetic signals in cellular response and adaptation. Methylation of histone tails by protein methyltransferases (PMT’s) functions in a similar manner to DNA methylation as a well-recognized regulator of gene expression.7 As part of an integrated system with DNA methylation, histone methylation is theorized to be part of a complicated histone code that is composed of a variety of other posttranslational modifications and that is altered by environmental signals and disease pathologies to control gene expression. For example, lysine N7-methylation (H3K4, H3K36) in histone H3 and subsequent demethylation are considered dueling signals that regulate transcription.7 At their simplest, both promoter and histone methylation affect gene expression by regulating how much of a transcript is made, with these signals altered in cancer pathogenesis and reprogrammed after some environmental exposures (Figure 1A). However, the complexity of epigenetic DNA marks has become more complicated by the emergence of 5-hydroxylmethylcytosine, 5-formylcytosine, and 5-carboxycytosine modifications of DNA8 and by the diversity of histone modifications, including acetylation and phosphorylation.7 In this context of transcriptional control by DNA and histone modification, we introduce the concept of a system of dozens of RNA modifications, including RNA methylation, that reprogram in response to environmental changes and control gene expression at the level of translation.
Figure 1.

Nucleic acid and protein modifications regulate gene expression in transcription and translation. (A) DNA and histone methylation marks regulate transcript levels to affect gene expression. Dynamic RNA methylation signals have recently been demonstrated to regulate how well a transcript is translated to affect gene expression. (B) Structures of 5-methylcytosine (m5C) in DNA (X = H) and RNA (X = OH) and the tRNA wobble modification mcm5U. (C) Structures of four of >120 modified ribonucleosides in prokaryotes and eukaryotes. The RNA modification database can be accessed to view more modification structures (http://mods.rna.albany.edu/). The nucleosome image was prepared by David S. Goodsell and the RCSB PDB (http://www.rcsb.org/pdb/101/motm.do?momID=7) and is used with permission.
3. Regulatory Potential of RNA Methyltransfereases and tRNA Modifications
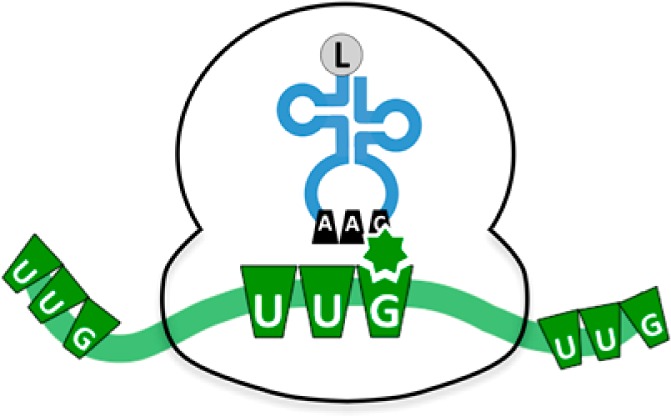
Similar to DNMT’s and PMT’s, RNA methytransferases (RNMT’s) have also been implicated in the pathophysiology of human disease,9,10 but a clear understanding of their mechanism of action in human cells has been elusive until recently. For example, the modification of mRNA with N6-methyladenosine (m6A) by the methyltransferase METTL3 and removal of m6A by the FTO demethylase are emerging as determinants of mRNA stability and translational efficiency.10 Here, we focus on tRNA, with recent work with the model eukaryote Saccharomyces cerevisiae demonstrating that RNA-methylation enzymes specific to tRNA (tRNA) are vital to cell viability after exposure to agents that generate reactive oxygen species (ROS) and DNA damage. Specifically, defects in the m5C tRNA methyltransferase 4 (Trm4, also called Ncl1) and mcm5U tRNA methyltransferase 9 (Trm9) lead to damage-induced growth and cell cycle phenotypes.11,12 This highlights an important connection between tRNA and stress response: modified ribonucleosides in tRNA. tRNA molecules are initially transcribed with U, A, C, and G bases, but the nucleobases and ribose sugars in a tRNA molecule are subject to chemical modification by a large system of enzymes. There are >100 chemical tRNA modifications throughout phylogeny, with ∼25–30 in all tRNA species in a cell, including S. cerevisiae and humans.13S. cerevisiae have an average of 11 modifications spread throughout the ∼70 bases in tRNA, whereas the average mammalian tRNA contains 13 modifications (Figure 1B,C).14 In general, tRNA methyltransferases transfer the methyl group from S-adenosyl methionine (SAM) to the 2′-OH of the ribose sugar, to the heterocyclic or exocyclic nitrogen atoms of the nucleobase, or to nucleophilic sites in modification intermediates. There are 18 known Trm enzymes in S. cerevisiae, with genomic analyses predicting 36 human Trms.9 In many cases, and for both Trm4 and Trm9, there are two or more human homologues for each yeast tRNA methyltransferases, which suggests diversification or specialization of Trm activity to new modifications in humans, modification of different tRNAs, or functions other than tRNA modification. Regardless of enzyme identify or regulation, modified ribonucleosides can promote tRNA structural stability and folding, translational fidelity, frame-shift prevention, and translation efficiency, with evidence for roles in tRNA quality control, cellular stress responses, and cell growth.15−21 The modifications are located in conserved positions throughout the four loops and termini of the tRNA molecule, with a large diversity of chemical structures occurring in the anticodon loop and the wobble position of the anticodon in particular.16 This is not surprising in light of the role of the wobble ribonucleotide in determining anticodon–codon interactions between tRNA and mRNA during translational elongation.
The diversity of both the chemical structures and locations of tRNA modifications suggests a role for modified ribonucleosides in controlling translation as part of a regulatory system. Simplistically, wobble base tRNA modifications can allow or prevent specific anticodon–codon interactions, which gives them great regulatory potential as a result of their ability to control the rate of translational elongation.17,22 Some wobble base modifications are only found on a subset of tRNAs for specific amino acids that interact with select codons, which supports the idea that regulation by tRNA modification can be very specific to a particular codon. If tRNA modifications are part of a such a regulatory system, then they must satisfy at least two criteria: (1) that they increase or decrease in response to specific changes in cell state and (2) that changes in the levels of the modifications alter the codon-reading properties of the associated tRNA and, in some cases, the selection of redundant codons. These behaviors transcend the chemical structure or location of individual ribonucleoside modifications and require a coordinated system with rules beyond the primary genetic code. Only recently have analytical and informatic technologies provided a means to define these transcendent properties of tRNA modifications.
4. Systems-Level Quantification of tRNA Modifications to Define Transcendent Properties
In the field of systems biology, the development of convergent technologies to quantify the thousands of individual components of the transcriptome, proteome, and metabolome has led to the discovery of regulatory networks and interactions that would not have been observed in single-molecule or -pathway analyses. The same has been true in the study of tRNA modifications. The power of liquid chromatography-coupled mass spectrometry (LC–MS) for identifying and quantifying modified ribonucleosides has recently been recognized by several groups.23−27 To explore the regulatory potential for tRNA modifications in cellular stress responses, we developed a systems-oriented LC–MS platform to measure changes in the relative quantities of all tRNA modifications in an organism (Figure 2).26,27 The platform involves artifact-free RNA isolation, purification of individual noncoding RNA species by HPLC,28 hydrolysis and HPLC resolution of individual ribonucleosides, and mass spectrometric identification and quantification of stress-induced changes in all modified ribonucleosides by quadrupole time-of-flight and tandem quadrupole mass spectrometry, respectively. The data set is subjected to bioinformatic and statistical analysis to define patterns of change and then to define pathways linked to altered ribonucleosides. As shown in Figures 2 and 3, our LC–MS method is capable of quantifying 23 of the ∼25 known ribonucleoside modifications in cytoplasmic tRNA in S. cerevisiae,29,30 with limited detection of two modifications (Ar(p) and ncm5Um) in positive ion mode. Of critical importance here is the sensitivity of detection, because low-level modifications are those most likely to be found at wobble positions of specific tRNA species, as opposed to more abundant modifications found in many tRNA species. LC–MS analysis reveals that modifications occur roughly at high (D, m5C, m1G, m22G, m1A, and Y), medium (ac4c, t6A, m5U, Cm, Gm, m7G, m2G, i6A, and Am), and low levels (ncm5Um, mcm5s2U, ncm5u, mcm5U, Um, m1I, I, and m3C),27 which generally reflects their presence in all or specific tRNA species as well as their presence at multiple or single positions in tRNA. These features make the sensitivity, precision, and accuracy of the analytical method particularly important in first-pass studies of stress-induced changes in tRNA modifications. For example, Trm4 catalyzes the formation of m5C in over 34 species of tRNA,30 yet tRNALeu(CAA) is the only tRNA with m5C at the anticodon wobble position 34 in addition to position 48 between the variable and TΨC loops.30 The observation of stress-dependent changes in m5C levels may thus depend on the ability to detect small changes in the total quantity of m5C in the tRNA population. Similarly, Trm9 catalyzes two modifications, mcm5s2U and mcm5U, at wobble positions in five tRNA species (tRNAArg-UCU, tRNAGly-UCC, tRNALys-UUU, tRNAGln-UUG, and tRNAGlu-UUC)31,32 such that changes could occur in any or all of the tRNA species. Ultimately, individual tRNA species must be isolated and analyzed for changes in tRNA-modification levels in an analysis of the regulatory properties of modified ribonucleosides, and this is accomplished by quantitative localization of modifications using combinations of RNase cleavage and oligonucleotide-based affinity purification along with LC–MS analysis.25
Figure 2.
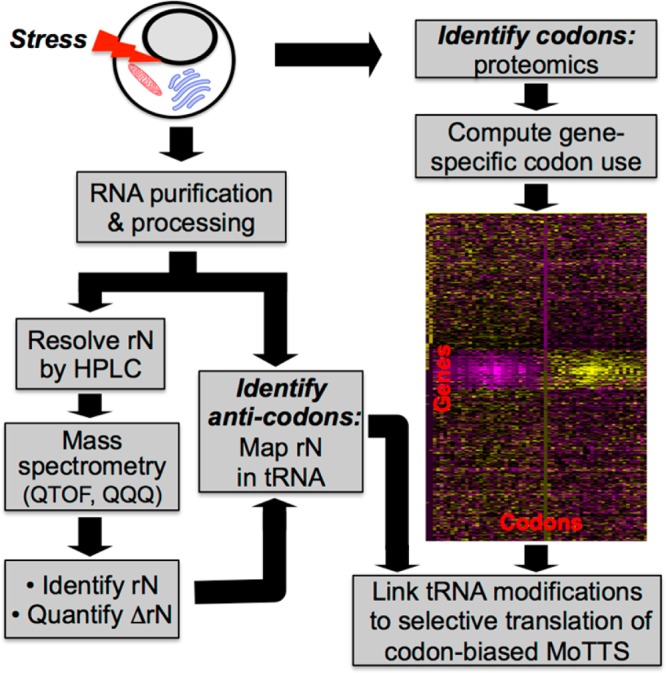
Platform for tRNA modification analytics and computational analysis of codon usage, which allows definition of the link between tRNA modifications and selective translation of MoTTs in the cell stress response. tRNA modifications are identified and quantified by HPLC-coupled mass spectrometry techniques to identify highly up- and downregulated ribonucleosides. Critical modifications are then mapped to wobble positions in specific tRNA species, the anticodon of which specifies a codon that is subjected to genomic analysis. Biased use of this codon in gene families specifies potential MoTTs. In parallel, proteomic analysis of stress-altered protein levels reveals codon-biased translation of MoTTs. Ultimately, the stress-altered tRNA reprogramming is linked to selective translation of codon-biased mRNAs, with patterns of gene expression unique to each stress.
Figure 3.
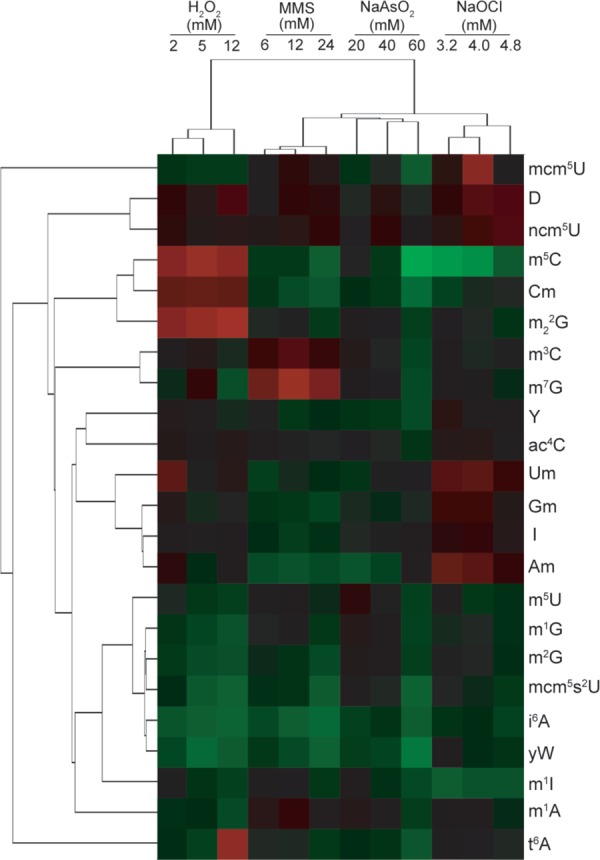
Hierarchical clustering of exposure- and genetic-induced changes in RNA modification levels. RNA modification data from wild-type cells exposed to different agents and mock-treated cells were identified and quantitated by mass spectrometry. Log-based fold-change values were determined relative to untreated, wild-type cells, and these data where hierarchically clustered. Image reproduced from ref (27).
5. Stress-Induced Tuning of tRNA Modifications: Biomarker Signatures of Exposure
As noted earlier, tRNA modifications fulfill a regulatory function in cell response if they increase or decrease following specific stresses. Application of the tRNA-modification analysis platform to yeast exposed to different chemical stresses revealed that this is indeed the case in yeast.12,27 Cells were exposed to three equitoxic doses of hydrogen peroxide (H2O2), methyl methanesulfonate (MMS), sodium arsenite (NaAsO2), and sodium hypochlorite (NaOCl), and changes in the levels of 23 modified ribonucleosides in total tRNA were quantified by LC–MS analysis. Application of multivariate statistical analysis to the fold-change data (e.g., hierarchical clustering) revealed that specific groups of tRNA modifications were uniquely up- or downregulated for each agent and for individual doses of each agent, as shown in Figure 3. The highly reproducible changes in tRNA-modification spectra demonstrate that the exposures promote reprogramming of the system of RNA modifications,27,33−36 which has been referred to as the ribonucleome.24 More recently, we quantitatively compared changes in the complete set of tRNA modifications in yeast exposed to four different oxidizing agents and five different alkylating agents. Multivariate statistical analysis revealed class-specific features that distinguished oxidizing agents from alkylating agents, with 14 modifications forming the basis for a data-driven model that predicted the chemical class of toxicant exposure with greater than 80% sensitivity and specificity (Chan et al., submitted). Furthermore, signature changes in tRNA modification spectra distinguished SN1 from SN2 alkylating agents (Chan et al., submitted). These systems-level changes in tRNA modifications are analogous to the stress-specific patterns of changes in mRNA levels in transcriptional profiling or to proteomic and metabolomic signatures of cell state, which suggests a role for RNA modifications in regulating gene expression after ROS stress and DNA damage. Recent evidence for codon biases across the genome has provided a basis for linking tRNA-modification reprogramming to selective translation of codon-biased transcripts as the regulatory mechanism in question.
6. Gene-Specific Biases in Codon Use: In Search of a Code of Codons in Translational Control of Cell Response
The second criterion for a translational regulatory role for tRNA modifications involves their ability to recognize information in mRNAs that is separate from the simple amino acid-specifying codon. More specifically, understanding how changes in the levels of specific tRNA modifications can affect gene expression requires insight into codon–anticodon interactions and the patterns of usage of so-called redundant codons in the genome. The general dogma in thinking about codons is that the rate by which they are translated by the ribosome is tightly linked to the concentration of the decoding tRNA, with reported correlations between genome-wide codon bias, tRNA copy number, and gene-expression levels in many model organisms.37 Simply put, current models correlate the most highly translated transcripts with possession of the most frequently used codons, which are specified and decoded by corresponding tRNA species whose genes have the highest number of copies in the genome and the highest number of tRNA copies in the pool.37 Although this model holds true for the expression of many genes, it suffers from being a static model: it cannot account for stress-induced regulation of translation. There are also many exceptions to the model, as revealed by transcripts showing clustering of low-frequency codons, distinct mRNA secondary structure, and internal ribosome binding sites.
So there is a need to better understand the information content of biased codon usage in genes and to identify a mechanistic link between codon-usage patterns and specific tRNA modifications. Developing rules, though, is a challenge because of the fact that there are 20 standard amino acids, 64 codons, 76 unique tRNA species, 300 tRNA genes, and >23 tRNA modifications in S. cerevisiae as a model organism. The complexity can be simplified by concentrating on wobble base modifications in specific tRNAs and then analyzing patterns of codon usage in specific transcripts, but an appreciation of the degeneracy of the genetic code is required to move the model to the next level. There are 64 standard codons possible from the four canonical bases found in mRNA, with several (i.e., synonymous) codons translated into the same amino acid. This degeneracy is illustrated well by leucine and arginine. Both amino acids have the maximum number of six degenerate codons in whole- (four) and split- (two) codon boxes (Figure 4A). In split-codon boxes, the wobble base tRNA modifications m5C and mcm5U can influence codon–anticodon affinity by dramatically enhancing interactions with one codon (i.e., TTG for Leu and AGA for Arg). Transcripts in which one codon from the split box is over-represented therefore have great potential for their translational efficiency to be tied to the levels of specific wobble base modifications. We can extend this idea further by proposing that specific transcripts may have over-representation of many specific codons from split boxes for multiple amino acids, which could lead to translational regulation by multiple tRNA modifications. We term the mRNAs from these codon-biased genes as modification tunable transcripts (MoTTs), and we have identified 425 of them in S. cerevisiae using a recently developed codon-analysis algorithm (Figure 4). These 425 genes contain statistically significant deviations in the usage of 29 codons when compared to all transcripts in the S. cerevisiae genome.11,38 Several recent studies have validated the use of the term MoTT by establishing a link between the dynamics of stress-induced tuning of tRNA wobble modifications and the selective translation of codon-biased mRNAs that represent critical stress response genes.12,27,39
Figure 4.

Calculation of biases in gene-specific codon usage. (A) Fold-difference in the average codon frequency of the 425 identified yeast MoTTs when compared to genome averages is noted. Those codons overused in the MoTTs are colored yellow (P < 10–5), whereas those under-represented are colored purple (P < 10–14), with the sum frequency that all degenerate codon are used for each amino acid = 1. (B) Run of 25 codons used at the C-terminal end of the YEF3 transcript is highly enriched (n = 24) for those over-represented in MoTTs. Notably, there are two (AAG)4 codon runs and one (AAG)5 codon run represented in this sequence, with 21 codons specific to Trm9 (AAG, GAA, and AGA) and Trm4 (UUG).
7. Stress Response Regulatory Mechanism That Links tRNA-Modification Reprogramming and Selective Translation of mRNAs with Biased Codon Use
The evidence for stress-induced reprogramming of tRNA modifications and for a link between specific tRNA modifications and biased codon used in MoTTs suggests that the system of tRNA modifications composes a mechanism for regulating cellular responses to environmental changes at the level of translation (Figure 5). Several recent studies confirm this model, with regulation of codon-biased transcripts demonstrated for MoTT’s linked to m5C and mcm5U modifications. Specifically, Trm4-catalyzed modification of C to m5C at the wobble position of anticodon of tRNALeu(CAA) has been shown to increase in response H2O2 exposure, with this increase driving increased translation of mRNAs (MoTTs) derived from the 38 genes in yeast in which 90% or more of the leucines are encoded by UUG.12 Among these UUG-enriched MoTTs is that for the ribosomal protein, Rpl22a.12 Of importance here is the fact that Rpl22a is one of two alternative proteins for Rpl22, with the gene for its paralogue, Rpl22b, lacking significant enrichment of UUG. H2O2 exposure did not increase the rate of translation of Rpl22b, and only loss of the gene for Rpl22a rendered the cells sensitive to killing by oxidative stress.12 These results provide a direct link between stress-induced increases in a specific wobble tRNA modification and selective translation of codon-biased mRNAs for critical stress response genes.
Figure 5.

RNA modifications and biased codon use form a system that controls cellular stress response at the level of translation. Emerging evidence supports a model in which stress induces a reprogramming of tRNA modifications that leads to selective translation of codon-biased mRNAs (i.e., MoTTs) representing critical stress response proteins.
Similar to Trm4, Trm9-specific mcm5U and mcm5s2U modifications have been demonstrated to be regulated in response to DNA damage and are required to increase the translation of codon-biased MoTTs.11,36 In S. cerevisiae, the mRNAs for yeast translation elongation factor 3 (YEF3) and ribonucleotide reductases 1 (RNR1) and 3 (RNR3) are over-represented with AGA, GAA, and AAG codons, and the basal translation of these proteins is dramatically decreased in trm9Δ cells lacking mcm5U and mcm5s2U.11,38 This again illustrates the concept of MoTTs. Proteins corresponding to transcripts with average codon usage (i.e., non-MoTTs) were found to occur at similar levels in wild-type and trm9Δ cells.11 Notably, mRNA levels for RPL22A, YEF3, RNR1, and RNR3 were identical in wild-type, trm4Δ (for RPL22A), and trm9Δ (for YEF3, RNR1, and RNR3) cells, which further demonstrates that tRNA-modification-dependent gene expression operates at the level of translational regulation.
Computational analysis of the MoTTs RPL22A, YEF3, RNR1, and RNR3 indicates that each of these transcripts is significantly over-represented in specific groups of codons, with protein analysis technologies clearly demonstrating that their protein levels can be regulated by specific tRNA modifications. The computed codon signature is an indicator of a MoTT and is limited at this point from the perspective of developing regulatory rules, as it is a trend for the whole gene. Notably, it does not provide any location-specific information detailing where these over-represented codons fall in the gene, and there will most likely be transcript regions that are more severely biased in certain codons and thus represent key regulatory motifs. As an example, the 3′/C-terminal region of YEF3 is shown in Figure 4B. In a span of 25 codons, it contains 21 codons whose regulation can be linked to Trm4 (UGU) and Trm9 (AGA, GAA, and AAG), thus representing a local transcript region that is predicted to be highly dependent on specific tRNA modifications for translation. Furthermore, development and testing of computational rules governing the precise mechanism of translational regulation of MoTTs is needed to identify the most significant regulatory regions where modifications regulate the translation of specific transcripts.
MoTTs are a new regulatory term, but they have been described before. There is an important precedent in the form of selenocysteine-containing proteins that also illustrates the concept of MoTTs, RNA modifications, and stress response proteins. Selenocysteine (Sec) is commonly called the 21st amino acid, and it is found in cellular detoxification and stress response proteins that include members of the glutathione peroxidase (GPX) and thioredoxin reductase (TRXR) families.40,41 Importantly, these Sec-containing proteins can detoxify H2O2 (GPX1 and GPX3) and lipid peroxidation products (GPX6) and contribute to the regulation of ribonucleotide reductase enzymes (TrxR1 and TrxR2). Sec lacks its own dedicated codon, and it is incorporated into proteins using a novel mechanism termed stop-codon recoding, which requires a number of key signals. The UGA codon (i.e., the stop codon) is normally used to signal the end of translation, but in stop-codon recoding, an internal UGA codon is used in conjunction with other factors to signal for the insertion of Sec. Importantly, the wobble base tRNA modification mcm5U, which is catalyzed by human and mouse ALKBH8, is required for efficient stop-codon recoding.40−43 As a side note, it has been proposed that the oxidative demethylation activity of ALKBH8 could serve as an off switch by reversing wobble methylation modifications, akin to DNA and histone demethylation, but no such activity has been demonstrated.8 Transcripts that encode Sec-containing proteins can be thought of as extreme MoTTs because they are over-represented with stop codons and need mcm5U for efficient translation. Transcripts for Sec-containing proteins also fit into the theme of MoTTs because they correspond to important stress response proteins, with specific GPX and TRXR activities well established as cellular detoxification enzymes.40−43
In conclusion, the connection between RNA modifications, biased codon use, and translational regulation of stress response protein highlights a complicated set of mechanisms to regulate gene expression. This parallels other methylation-based signals, as understanding regulation of transcription by m5C and histone methylation is also complicated, required new tools at their outset, and can have species-specific rules. It is important to note that the DNA, protein, and RNA modification activities and modifications specified by DNMT’s, PMT’s, and RNMT’s share a common theme of regulating gene expression by enzyme-catalyzed methylation, with all of them reprogrammable by environmental conditions and during some disease pathologies. There are significant challenges for better defining the roles and mechanisms of MoTTs and RNA modifications because codon usage and modification patterns change when studying different organisms and there are numerous, varied, and specialized instrumentation required for the study of RNA modifications. The study of MoTTs and RNA modifications is therefore required in multiple model systems and settings to define further and to develop general and then species-specific rules. We note that one possible path to make the study of RNA modifications simpler and more accessible is to follow the example set by researchers studying DNA and histone methylation signals: to develop antibodies for each modification.
Glossary
Abbreviations
- ac4C
N4-acetylcytidine
- Am
2′-O-methyladenosine
- Ar(p)
2′-O-ribosyladenosine phosphate
- Cm
2′-O-methylcytidine
- D
dihydrouridine
- DNMT
DNA methyltransferases
- Gm
2′-O-methylguanosine
- GPX
glutathione peroxidase
- H2O2
hydrogen peroxide
- HPLC
high-performance liquid chromatography
- I
inosine
- i6A
N6-isopentenyladenosine
- LC–MS
chromatography-coupled mass spectrometry
- m1A
N1-methyladenosine
- m3C
3-methylcytidine
- m5C
5-methylcytidine
- mcm5U
5-methoxycarbonylmethyluridine
- mcm5s2U
5-methoxycarbonylmethyl-2-thiouridine
- m1G
1-methylguanosine
- m2G
2-methylguanosine
- m22G
N2,N2-dimethylguanosine
- m7G
N7-methylguanosine
- m1I
N1-methylinosine
- MMS
methyl methanesulfonate
- MoTT
modification tunable transcript
- m5U
5-methyluridine
- NaAsO2
sodium arsenite
- NaOCl
sodium hypochlorite
- ncm5U
5-carbamoylmethyluridine
- ncm5Um
5-carbamoylmethyl-2′-O-methyluridine
- PMT
protein methyltransferases
- RNMT
RNA methyltransferases
- RNR1
ribonucleotide reductase 1
- RNR3
ribonucleotide reductase 3
- RPL22a
ribosomal protein large subunit 22A
- ROS
reactive oxygen species
- SAM
S-adenosyl methionine
- t6A
N6-threonylcarbamoyladenosine
- Trm
tRNA methyltransferases
- tRNA
transfer RNA
- TRXR
thioredoxin reductase
- Um
2′-O-methyluridine
- Y
pseudouridine
- YEF3
yeast elongation factor 3
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
NIH ES017010 (T.J.B.), and National Science Foundation CHE-1308839 and the National Research Foundation of Singapore through the Singapore-MIT Alliance for Research and Technology (P.C.D.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Esteller M. (2002) CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 21, 5427–5440. [DOI] [PubMed] [Google Scholar]
- Esteller M. (2005) Aberrant DNA methylation as a cancer-inducing mechanism. Annu. Rev. Pharmacol. Toxicol. 45, 629–656. [DOI] [PubMed] [Google Scholar]
- Kennedy B. K.; Liu O. W.; Dick F. A.; Dyson N.; Harlow E.; Vidal M. (2001) Histone deacetylase-dependent transcriptional repression by pRB in yeast occurs independently of interaction through the LXCXE binding cleft. Proc. Natl. Acad. Sci. U.S.A. 98, 8720–8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. Y.; Hayes J. J.; Pruss D.; Wolffe A. P. (1993) A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72, 73–84. [DOI] [PubMed] [Google Scholar]
- Thomson J. P., Moggs J. G., Wolf C. R., and Meehan R. R. (2013) Epigenetic profiles as defined signatures of xenobiotic exposure. Mutat. Res. [Online early access], DOI: 10.1016/j.mrgentox.2013.08.007, Published Online: August 31.. [DOI] [PubMed] [Google Scholar]
- Baylin S. B.; Jones P. A. (2011) A decade of exploring the cancer epigenome – biological and translational implications. Nat. Rev. Cancer 11, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P.; Allis C. D.; Wang G. G. (2010) Covalent histone modifications–miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; He C. (2012) Nucleic acid modifications with epigenetic significance. Curr. Opin. Chem. Biol. 16, 516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towns W. L.; Begley T. J. (2011) Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: Activities, predications, and potential roles in human health. DNA Cell Biol. 31, 434–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibbritt T.; Patel H. R.; Preiss T. (2013) Mapping and significance of the mRNA methylome. Wiley Interdiscip. Rev.: RNA 4, 397–422. [DOI] [PubMed] [Google Scholar]
- Begley U.; Dyavaiah M.; Patil A.; Rooney J. P.; Direnzo D.; Young C. M.; Conklin D. S.; Zitomer R. S.; Begley T. J. (2007) Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell 28, 860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. T. Y.; Pang Y. L. J.; Deng W.; Babu I. R.; Dyavaiah M.; Begley T. J.; Dedon P. C. (2012) Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka M. A.; Milanowska K.; Osman Oglou O.; Purta E.; Kurkowska M.; Olchowik A.; Januszewski W.; Kalinowski S.; Dunin-Horkawicz S.; Rother K. M.; Helm M.; Bujnicki J. M.; Grosjean H.. (2013) Modomics: A database of RNA modification pathways – a 2013 update. Nucleic Acids Res. 41, D262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl M.; Horn C.; Brown M.; Ioudovitch A.; Steinberg S. (1998) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 26, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yacoubi B.; Bailly M.; de Crecy-Lagard V. (2012) Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 46, 69–95. [DOI] [PubMed] [Google Scholar]
- Phizicky E. M.; Hopper A. K. (2010) tRNA biology charges to the front. Genes Dev. 24, 1832–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y.; Helm M. (2010) tRNA stabilization by modified nucleotides. Biochemistry 49, 4934–4944. [DOI] [PubMed] [Google Scholar]
- Alexandrov A.; Chernyakov I.; Gu W.; Hiley S. L.; Hughes T. R.; Grayhack E. J.; Phizicky E. M. (2006) Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96. [DOI] [PubMed] [Google Scholar]
- Thompson D. M.; Parker R. (2009) Stressing out over tRNA cleavage. Cell 138, 215–219. [DOI] [PubMed] [Google Scholar]
- Netzer N.; Goodenbour J. M.; David A.; Dittmar K. A.; Jones R. B.; Schneider J. R.; Boone D.; Eves E. M.; Rosner M. R.; Gibbs J. S.; Embry A.; Dolan B.; Das S.; Hickman H. D.; Berglund P.; Bennink J. R.; Yewdell J. W.; Pan T. (2009) Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 462, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V.; Naslund A. K.; Kurland C. G. (1992) Thiolation of transfer RNA in Escherichia coli varies with growth rate. Nucleic Acids Res. 20, 4499–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris P. F. (2004) Decoding the genome: A modified view. Nucleic Acids Res. 32, 223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J.; Pomerantz C.; Rozenski J.; Zhang Y.; McCloskey J. A. (1996) Interpretation of oligonucleotide mass spectra for determination of sequence using electrospray ionization and tandem mass spectrometry. Anal. Chem. 68, 1989–1999. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Ikeuchi Y.; Noma A.; Sakaguchi Y. (2007) Mass spectrometric identification and characterization of RNA-modifying enzymes. Methods Enzymol. 425, 211–229. [DOI] [PubMed] [Google Scholar]
- Meng Z.; Limbach P. A. (2006) Mass spectrometry of RNA: Linking the genome to the proteome. Briefings Funct. Genomics Proteomics 5, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. T.; Chionh Y. H.; Ho C. H.; Lim K. S.; Babu I. R.; Ang E.; Wenwei L.; Alonso S.; Dedon P. C. (2011) Identification of N6,N6-dimethyladenosine in transfer RNA from Mycobacterium bovis Bacille Calmette-Guerin. Molecules 16, 5168–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. T.; Dyavaiah M.; DeMott M. S.; Taghizadeh K.; Dedon P. C.; Begley T. J. (2010) A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 6, e1001247-1–e1001247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chionh Y. H.; Ho C. H.; Pruksakorn D.; Ramesh Babu I.; Ng C. S.; Hia F.; McBee M. E.; Su D.; Pang Y. L.; Gu C.; Dong H.; Prestwich E. G.; Shi P. Y.; Preiser P. R.; Alonso S.; Dedon P. C. (2013) A multidimensional platform for the purification of non-coding RNA species. Nucleic Acids Res. 41, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenski J.; Crain P. F.; McCloskey J. A. (1999) The RNA modification database: 1999 update.. Nucleic Acids Res. 196–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerwoniec A.; Dunin-Horkawicz S.; Purta E.; Kaminska K. H.; Kasprzak J. M.; Bujnicki J. M.; Grosjean H.; Rother K. (2009) Modomics: A database of RNA modification pathways: 2008 update. Nucleic Acids Res. 37, D118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M. J.; Esberg A.; Huang B.; Bjork G. R.; Bystrom A. S. (2008) Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 28, 3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M. J. O., and Bystrom A. S. (2005) Transfer RNA modification and modifying enzymes in Saccharomyces cerevisae, In Topics in Current Genetics: Fine-Tuning of RNA Functions by Modification and Editing (Grosjean H., Ed.) pp 87–120, Springer, Berlin. [Google Scholar]
- Begley U.; Sosa M. S.; Avivar-Valderas A.; Patil A.; Endres L.; Estrada Y.; Chan C. T.; Su D.; Dedon P. C.; Aguirre-Ghiso J. A.; Begley T. (2013) A human tRNA methyltransferase 9-like protein prevents tumour growth by regulating lin9 and hif1-alpha. EMBO Mol. Med. 5, 366–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. T.; Pang Y. L.; Deng W.; Babu I. R.; Dyavaiah M.; Begley T. J.; Dedon P. C. (2012) Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil A.; Chan C. T.; Dyavaiah M.; Rooney J. P.; Dedon P. C.; Begley T. J. (2012) Translational infidelity-induced protein stress results from a deficiency in trm9-catalyzed tRNA modifications. RNA Biol. 9, 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil A.; Dyavaiah M.; Joseph F.; Rooney J. P.; Chan C. T.; Dedon P. C.; Begley T. J. (2012) Increased tRNA modification and gene-specific codon usage regulate cell cycle progression during the DNA damage response. Cell Cycle 11, 3656–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iben J. R.; Maraia R. J. (2012) tRNAomics: tRNA gene copy number variation and codon use provide bioinformatic evidence of a new anticodon: Codon wobble pair in a eukaryote. RNA 18, 1358–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumu S.; Patil A.; Towns W. L.; Dyavaiah M.; Begley T. J. (2012) The gene-specific codon counting database: A genome-based catalog of one-, two-, three-, four- and five-codon combinations present in Saccharomyces cerevisiae genes. Database bas002-1–bas002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer F.; Hermand D. (2012) A coordinated codon-dependent regulation of translation by elongator. Cell Cycle 11, 4524–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa M. E.; Kumaraswamy E.; Zhong N.; Rao M.; Carlson B. A.; Hatfield D. L. (2003) Models for assessing the role of selenoproteins in health. J. Nutr. 133, 2494S–2496S. [DOI] [PubMed] [Google Scholar]
- Papp L. V.; Holmgren A.; Khanna K. K. (2010) Selenium and selenoproteins in health and disease. Antioxid. Redox Signaling 12, 793–795. [DOI] [PubMed] [Google Scholar]
- Carlson B. A.; Xu X. M.; Gladyshev V. N.; Hatfield D. L. (2005) Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine tRNA. J. Biol. Chem. 280, 5542–5548. [DOI] [PubMed] [Google Scholar]
- Songe-Moller L.; van den Born E.; Leihne V.; Vagbo C. B.; Kristoffersen T.; Krokan H. E.; Kirpekar F.; Falnes P. O.; Klungland A. (2010) Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol. Cell. Biol. 30, 1814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]