

Abstract
Background and Purpose
The objective of this study was to determine how the AMPK activating antidiabetic drug metformin affects the major activator of hepatic gluconeogenesis, PPARγ coactivator 1α (PGC-1α) and liver functions regulated by PGC-1α.
Experimental Approach
Mouse and human primary hepatocytes and mice in vivo were treated with metformin. Adenoviral overexpression, siRNA and reporter gene constructs were used for mechanistic studies.
Key Results
Metformin increased PGC-1α mRNA and protein expression in mouse primary hepatocytes. 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) (another AMPK activator) had the opposite effect. Metformin also increased PGC-1α in human primary hepatocytes; this effect of metformin was abolished by AMPK inhibitor compound C and sirtuin 1 siRNA. AMPK overexpression by AMPK-Ad also increased PGC-1α. Whereas metformin increased PGC-1α, it down-regulated gluconeogenic genes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase). Furthermore, metformin attenuated the increase in PEPCK and G6Pase mRNAs induced by PGC-1α overexpression, but did not affect PGC-1α-mediated induction of mitochondrial genes. Metformin down-regulated several key transcription factors that mediate the effect of PGC-1α on gluconeogenic genes including Krüppel-like factor 15, forkhead box protein O1 and hepatocyte NF 4α, whereas it increased nuclear respiratory factor 1, which is involved in PGC-1α-mediated regulation of mitochondrial proteins.
Conclusions and Implications
Down-regulation of PGC-1α is not necessary for suppression of gluconeogenic genes by metformin. Importantly, metformin selectively affects hepatic PGC-1α-mediated gene regulation and prevents activation of gluconeogenesis, but does not influence its regulation of mitochondrial genes. These results identify selective modulation of hepatic PGC-1α functions as a novel mechanism involved in the therapeutic action of metformin.
Keywords: PGC-1α, AMPK, metformin, gluconeogenesis, liver, SIRT1, PEPCK, G6Pase
Introduction
Metformin is a biguanide class drug widely used in the treatment of type 2 diabetes (Krentz and Bailey, 2005). Although metformin has been in clinical use for decades, its mechanism of action has only been partly solved. The major action of metformin is thought to be the suppression of hepatic glucose output mediated through the down-regulation of the expression of the rate-limiting gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) (Minassian et al., 1998; Yuan et al., 2002)
PPARγ coactivator 1α (PGC-1α) is an inducible transcriptional coactivator involved in number of metabolic functions in high-energy tissues (Lin, 2009). PGC-1α is an important regulator of mitochondrial biogenesis and function (Handschin, 2009). In addition it has many tissue specific functions. In liver, PGC-1α stimulates gluconeogenesis, fatty acid oxidation and haeme biosynthesis (Handschin, 2009). Hepatic PGC-1α is induced by fasting and subsequently up-regulates gluconeogenesis and the key enzymes involved, PEPCK and G6Pase. As PGC-1α has been shown to play an important role in regulation of gluconeogenesis, it has been suggested that it is involved in the hepatic action of metformin (Viollet et al., 2009). Shaw et al. reported that metformin, by affecting LKB1/AMPK activity, phosphorylates and translocates cAMP-responsive element-binding protein (CREB) coactivator (CRTC2; also known as TORC2) out of the nucleus (Shaw et al., 2005). In turn, CRTC2 up-regulates the levels of PGC-1α expression, which may have a role in the antihyperglycaemic mechanism of action of metformin (Viollet et al., 2009). However, according to a recent study, neither AMPK nor LKB1 is needed for the suppression of hepatic glucose output by metformin (Foretz et al., 2010). Interestingly, although PGC-1α is a key regulator of energy metabolism, the effects of metformin on hepatic PGC-1α expression and function have not been specifically studied.
In the current study, we have assessed the effects of metformin on PGC-1α. In contrast to previous assumptions, we showed that metformin induces PGC-1α expression in primary hepatocytes. Moreover, metformin selectively modified PGC-1α-regulated liver functions and impaired the ability of PGC-1α to up-regulate gluconeogenic genes.
Methods
Reagents
DMSO, dibutyryl-cAMP (db-cAMP), 8-bromo-cAMP, metformin, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), compound C, esiGFP, esiAMPKα1, esiAMPKα2, siSIRT, qPCR oligos, FLAG immunoprecipitation kit and mouse anti-β-actin IgG (A1978), were purchased from Sigma-Aldrich (St. Louis, MO, USA). A769662 was from Tocris Bioscience (Bristol, UK). PEPCK, AMPKα1, AMPKα2 gene expression assays and negative control siRNAs used with siSIRT1 were from Life Technologies (Carlsbad, CA, USA). Rabbit anti-PGC-1α IgG (sc-13067), rabbit anti-hepatocyte NF 4α (HNF-4α) IgG (sc-8987) and HRP-conjugated rabbit IgG (sc-2004) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse anti-acetylated lysine (Ac-Lys) IgG (#9681) and rabbit anti-phospho-acetyl-CoA carboxylase (pACC) IgG (#3661) were from Cell Signaling Technology (Danvers, MA, USA). HRP-conjugated anti-mouse IgG was from GE Healthcare (Little Chalfont, UK). Mouse anti-PGC-1α IgG (ST1202), used in the immunodetection of immunoprecipitated PGC-1α was from Calbiochem (Darmstadt, Germany).
Cell cultures
Fresh, human primary hepatocytes from one donor were from Biopredic international (Rennes, France). Cells were incubated with 0.5, 1 and 2 mM metformin for 48 h in standard William's E medium, which was replaced after 24 h. In another experiment with human primary hepatocytes from three donors, cells were prepared as described previously (Krausova et al., 2011) Hepatocytes were treated with 0.5, 1 and 2 mM metformin for 24 h in standard cultivation medium. Mouse primary hepatocytes were isolated from male DBA/2 or C57BL/6 mice (Center for Experimental Animals, University of Oulu, Finland) aged 8–10 weeks, as described previously (Arpiainen et al., 2008), and cultivated in William's E medium containing 2.0 g·L−1 D-glucose and ITS (insulin 5 mg·L−1, transferrin 5 mg·L−1, sodium selenate 5 μg·L−1, Sigma-Aldrich). The cultures were maintained for an additional 12–24 h before being infected with an adenovirus or subjected to various treatments.
HepG2 cells (ATCC, Manassas, VA, USA) were cultured in DMEM medium [Invitrogen (Carlsbad, CA, USA), Life Technologies] containing 4.5 g·L−1 D-glucose, 10% (v v−1) FBS, 100 U·mL−1 of penicillin and 100 U·mL−1 streptomycin (Invitrogen, Life Technologies).
Adenoviruses and treatments of cells
Recombinant adenoviruses expressing green fluorescent protein (GFP-Ad) and PGC-1α were generated as described previously (Arpiainen et al., 2008). Adenovirus expressing human constitutively active AMPKα1 (AMPK-Ad) subunit containing amino acids 1–321 was a gift from Prof. Risto Kerkelä (University of Oulu). PGC-1α phosphorylation mutant (T177A S538A) plasmid was from Addgene (http://www.addgene.org plasmid 18093) and adenovirus was generated as described previously (Arpiainen et al., 2008). Mouse primary hepatocytes were either infected or treated with chemicals in William's E growth medium (Sigma-Aldrich) without serum for the indicated time periods before RNA or protein extractions. A769662, Compound C and db-cAMP were dissolved in DMSO and all other chemicals were dissolved in water. Control samples were either treated with equal amount of vehicle or left untreated.
siRNA experiments
Mouse primary hepatocytes were transfected with X-tremeGENE siRNA transfection reagent (Roche, Basel, Switzerland) using 150 pmol per well (in six-well plates) or 45 pmol per well (in 12-well plates) of siRNAs and 2000 ng per well (in six-well plates) of esiRNAs. Three pooled negative control siRNAs were used with siSIRT1 and esiGFP was used with esiAMPK as a negative control (Table 1). After an initial 4 h incubation period siSIRT1 transfection medium was replaced by fresh medium containing 0.25 mM metformin. esiAMPK samples were treated with 0.25 mM metformin 30 h post-transfection. After 42–48 h, cells were collected for RNA isolation.
Table 1.
Sequences of the PCR primers and siRNA oligos
| Gene | Forward | Reverse |
|---|---|---|
| mPGC-1α | GCAGGTCGAACGAAACTGAC | CTCAGCCTGGGAACACGTTA |
| CTGCTCTGGTTGGTGAGGAa | GCAGGCTCATTGTTGTACTGa | |
| mPEPCK | GGTGTTTACTGGGAAGGCATC | CAATAATGGGGCACTGGCTG |
| mSHP | GTACCTGAAGGGCACGATCC | ACCAGGGCTCCAAGACTTCAC |
| mG6Pase | CATCAATCTCCTCTGGGTGG | TGCTGTAGTAGTCGGTGTCC |
| hPGC-1α | CCCAGATCTTCCTGAACTTG | GCTAGCAAGTTTGCCTCATTC |
| 18S | CGCCGCTAGAGGTGAAATTC | CCAGTCGGCATCGTTTATGG |
| mCycs | CCAAATCTCCACGGTCTGTTC | ATCAGGGTATCCTCTCCCCAG |
| mATP5b | GGTTCATCCTGCCAGAGACTA | AATCCCTCATCGAACTGGACG |
| mCox4i1 | ATTGGCAAGAGAGCCATTTCTAC | CACGCCGATCAGCGTAAGT |
| mCox1 | ATTCCTTCATGTCGGACGAG | ACTGAGAAGCCCCCTCAAAT |
| mCox2 | ACGAAATCAACAACCCCGTA | GGCAGAACGACTCGGTTATC |
| mNRF1 | AGCACGGAGTGACCCAAAC | TGTACGTGGCTACATGGACCT |
| mKLF15 | GAGACCTTCTCGTCACCGAAA | GCTGGAGACATCGCTGTCAT |
| mFOXO1 | CCCAGGCCGGAGTTTAACC | GTTGCTCATAAAGTCGGTGCT |
| mHNF-4α | GTGGCGAGTCCTTATGACACG | GCTGTTGGATGAATTGAGGTTGG |
| hPEPCK | Hs00159918_m1 (Life Technologies) | |
| mAMPKα1(Prkaa1) | Mm01296700_m1 (Life Technologies) | |
| mAMPKα2(Prkaa2) | Mm01264789_m1 (Life Technologies) | |
| siRNA SIRT1 | AAATGTCTCCACGAACAGC | GCTGTTCGTGGAGACATTT |
| siRNA neg control | AM4635, AM4637, AM4390844 (Life Technologies) | |
| esiRNA EGFP | EHUEGFP (Sigma-Aldrich) | |
| esiRNA mouse Prkaa1 | EMU190491 (Sigma-Aldrich) | |
| esiRNA mouse Prkaa2 | EMU052501 (Sigma-Aldrich) |
For adenoviral PGC-1α detection.
Metformin in vivo experiment
The in vivo experiments were approved by the Finnish Animal Experiment Board (Permit ESAVI-2010-06188). C57/BL6NHsd mice (Harlan Laboratories, NL) were housed in animal room at 21 ± 3°C with a 12h:12h dark:light cycle. Mice were on a chow diet (69 kcal% carbohydrates, 22 kcal% protein and 9 kcal% fat; CRM(E), SDS, UK) and food and water were available ad libitum. Female, C57BL/6, pregnant mice were administered metformin 300 mg·kg−1 day−1, i.g. (n = 3), or vehicle being either saline or tap-water (n = 5) for 18 days (E0.5–17.5) as described previously (Salomaki et al., 2013). The animals were killed 24 h after the last metformin dose and the livers were collected. Alternatively, 16–17-week-old male C57BL/6 mice were given metformin (300 mg·kg−1) i.g. for 7 days between 07:00 and 10:00 h. On the seventh day of the treatment, metformin (n = 8) or saline (n = 8) was given 3 h prior to killing. Mixed medetomidine and ketamine anaesthesia combined with decapitation were used to kill the mice in all the experiments (Salomaki et al., 2013). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). According to an allometric body surface area method (Reagan-Shaw et al., 2008), 300 mg·kg−1 for mice corresponds to a dose of 1.7 g metformin day−1 for a 70 kg man.
RNA preparation and quantitative RT-PCR
Total RNA was isolated using TRI-Reagent (Molecular Research Center, Cincinnati, OH, USA) or RNeasy mini kit for in vivo mice (Qiagen, Hilden, Germany) and cDNA generated as described previously (Buler et al., 2012). Quantitative PCR reactions were performed with Applied Biosystems 7300 real time PCR system (Applied Biosystems, Carlsbad, CA, USA) using FastStart Universal SYBRGreen Master Mix or Probe Master Mix (Roche). Sequences of the primers and probes are listed in Table 1. Fluorescence values of the qPCR products were corrected with the fluorescence signals of the passive reference dye (ROX). qPCR results are presented as relative mRNA levels normalized against reference control 18S values (typically between 13 and 17 Ct) using the comparative Ct method (2-ΔΔCt method).
Western blot
Total and nuclear protein fractions were prepared as described previously (Schreiber et al., 1989; Lamsa et al., 2010) and were subjected to SDS–PAGE (10–12% in polyacrylamide) and transferred to a PVDF membrane (Millipore, Billerica, MA, USA). Membranes were incubated with an appropriate primary antibody in 5% skimmed milk in Tris-buffered saline with 0.1% Tween usually overnight followed by incubation with a secondary HRP-conjugated antibody. The immunoreactive bands were visualized by use of a chemiluminescent peroxidase substrate 1 reaction (Sigma-Aldrich).
Plasmids, transient transfection assays and immunoprecipitation
The PGC-1α 5′-Luc and PGC-1α 5′-ΔCRE-Luc reporter constructs were from Addgene (http://www.addgene.org plasmids 8887 and 8888) (Handschin et al., 2003). Of the reporter plasmid, 0.5 μg were transfected per well (12-well plates at a density of 3 × 105 mouse primary hepatocytes) using Tfx-20 reagent (Promega, Madison, WI, USA). Twenty-four hours post transfection, cells were treated with chemicals as indicated earlier. After 24 h, cells were assayed for luciferase activity with the Dual-Luciferase Reporter Assay System (Promega).
HNF-4α response element in front of TATA box was cloned into pGL3-Basic reporter vector. Of the reporter vector, 0.5 μg was transfected per well (24-well plates at 80% confluence of HepG2 cells) using Fugene HD transfection reagent (Roche). Twenty-four hours post transfection cells were treated with metformin and 24 h later assayed for luciferase as mentioned earlier.
Of the PGC-1α-FLAG, 15 μg was transfected per plate (10 cm plates of HepG2 cells) using Fugene HD transfection reagent (Roche). Twenty-four hours post transfection, cells were treated with metformin and collected after another 72 h. PGC-1α-FLAG was immunoprecipitated using a FLAG immunoprecipitation kit and immunoblotted against acetylated lycine (anti-Ac-lys), mouse PGC-1α antibody was used as loading control.
Statistical analysis
Statistical analysis of the data was performed using GraphPad Prism Software (La Jolla, CA, USA). Unless stated otherwise, the comparison of means of two groups was done by Student's t-test, whereas multiple groups or two variables were compared by one-way and two-way ANOVAs followed by Dunnett's post hoc test. Statistically significant values are marked *P < 0.05, **P < 0.01 or ***P < 0.001 compared with untreated control or #P < 0.05, ##P < 0.01, ###P < 0.001 compared with the indicated treatment.
Results
Metformin down-regulates gluconeogenic genes, but induces PGC-1α expression in hepatocytes
The effect of metformin on the expression of PEPCK, G6Pase and PGC-1α was studied in mouse primary hepatocytes. After 48 h of treatment, the expressions of PEPCK and G6Pase mRNAs were significantly down-regulated. In contrast, PGC-1α mRNA was increased almost threefold by 1 mM metformin (Figure 1A). To determine the time course of PGC-1α induction by metformin, 1 mM metformin was used as this concentration induced a maximal increase in PGC-1α after a 48 h. The metformin concentrations used are higher than the observed blood concentrations in clinical use. However, because of hepatic accumulation, these experimental concentrations may reflect the in vivo intrahepatic concentrations as discussed by Foretz et al. (2010). An increase in PGC-1α was detected after 12 h, but the highest level of PGC-1α was observed at the last time point of 72 h. As expected, PEPCK and G6Pase were down-regulated (Figure 1B). The functional significance of the induction of PGC-1α mRNA by metformin was also confirmed at the protein level; PGC-1α protein level was increased fourfold after 48 h (Figure 1C).
Figure 1.
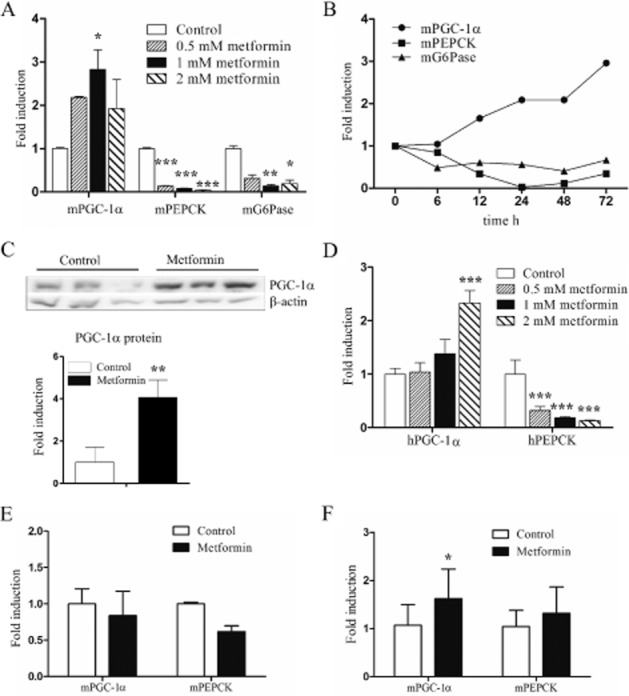
Effect of metformin treatment on PGC-1α and gluconeogenic gene expression. (A) Mouse primary hepatocytes were treated with metformin for 48 h after which mRNAs were measured by RT-qPCR (n = 3). (B) Mouse primary hepatocytes were treated with 1 mM metformin for the indicated time periods after which mRNAs were measured by RT-qPCR (n = 3, but combined before analysis). (C) PGC-1α protein was detected by immunoblotting in total protein fractions of mouse primary hepatocytes treated with metformin for 48 h. β-Actin was measured as a loading control. (n = 3) (D) Human primary hepatocytes were treated with metformin for 48 h and measured by RT-qPCR (n = 3). (E) Mice were administered metformin i.g. 300 mg·kg−1 daily for 18 days. Livers were collected 24 h after the last dose after which mRNA was isolated from the vehicle (n = 5) or metformin (n = 3) treated individuals and measured by RT-qPCR. Statistical significance was not calculated. (F) Mice were administered metformin i.g. 300 mg·kg−1 daily for 7 days. Livers were collected 3 h after the last dose after which mRNA was isolated from the vehicle (n = 8) or metformin (n = 8) treated individuals and measured by RT-qPCR. qPCR and Western blot results represent means of fold change + SD.
We next studied whether metformin also induces PGC-1α expression in human hepatocytes. Human hepatocytes were treated for 48 h with 0.5, 1 and 2 mM metformin. Similar to the mouse, human PGC-1α mRNA was also up-regulated by metformin (Figure 1D). However, compared with mouse hepatocytes, a higher concentration of metformin was required to produce this effect. Furthermore, PEPCK was significantly down-regulated by metformin in the hepatocytes (Figure 1D). Essentially similar results were also observed after 24 h of metformin treatment of a separate batch of human hepatocytes (data not shown).
We also measured the effect of metformin in vivo. Female pregnant mice were treated with 300 mg·kg−1 day−1 metformin, i.g., for 18 days. The livers were collected 24 h after the last drug dose and analysed for PGC-1α and PEPCK. PGC-1α mRNA was not affected by metformin, instead, PEPCK was down-regulated by 40% (Figure 1E). We further studied the in vivo effects by treating male mice for 7 days with 300 mg·kg−1 day−1 metformin, i.g.; however, the samples were collected only 3 h after the last dose. In this experiment, PGC-1α mRNA was increased 1.62-fold by metformin, but PEPCK was not affected (Figure 1F).
Metformin induces PGC-1α through an AMPK and SIRT1 mediated mechanism
Metformin is a known activator of the energy sensor AMPK. We next investigated the involvement of AMPK in the PGC-1α induction of metformin. Treatment of hepatocytes with two pharmacological activators of AMPK; AICAR and metformin resulted in opposite effects on PGC-1α expression. While metformin time-dependently increased PGC-1α, AICAR effectively down-regulated PGC-1α expression (Figure 2A). Nevertheless, both compounds activated AMPK as assessed by phosphorylation of the AMPK target pACC (Figure 2B). The role of AMPK was further studied by using compound C, a pharmacological inhibitor of AMPK. Compound C abolished the induction of PGC-1α by metformin, even reducing the expression level below that of the untreated control. In contrast, compound C potentiated the effect of AICAR (Figure 2C). A769662 is a small-molecular compound that directly activates AMPK. A769662 0.1 mM slightly increased the expression of PGC-1α, but this was not statistically significant (Figure 2D). To study the effect of AMPK directly, hepatocytes were transduced with an adenovirus coding for the constitutively active mutant of AMPKα1 (AMPK-Ad). After 24 h, the expression of PGC-1α was increased dose-dependently up to fivefold (Figure 2E). AMPK-Ad slightly down-regulated the expression of PEPCK. We also performed AMPK knockdown experiments with siRNAs against AMPKα1 and AMPKα2. In particular, AMPKα2 was effectively knocked down by the siRNA used, whereas the effect of the AMPKα1 siRNA was less noticeable (Figure 2F). The basal level of PGC-1α was significantly reduced by the combination of AMPKα1 and AMPKα2 siRNAs. The effect of 0.25 mM metformin on PGC-1α was modest in this particular experiment and did not reach statistical significance. However, the combination of AMPKα1 and AMPKα2 siRNAs also reduced PGC-1α expression in metformin-treated samples. Altogether, these results suggest that AMPK is involved in the up-regulation of PGC-1α induced by metformin, whereas the down-regulation of PGC-1α induced by AICAR is mediated by an AMPK-independent mechanism.
Figure 2.
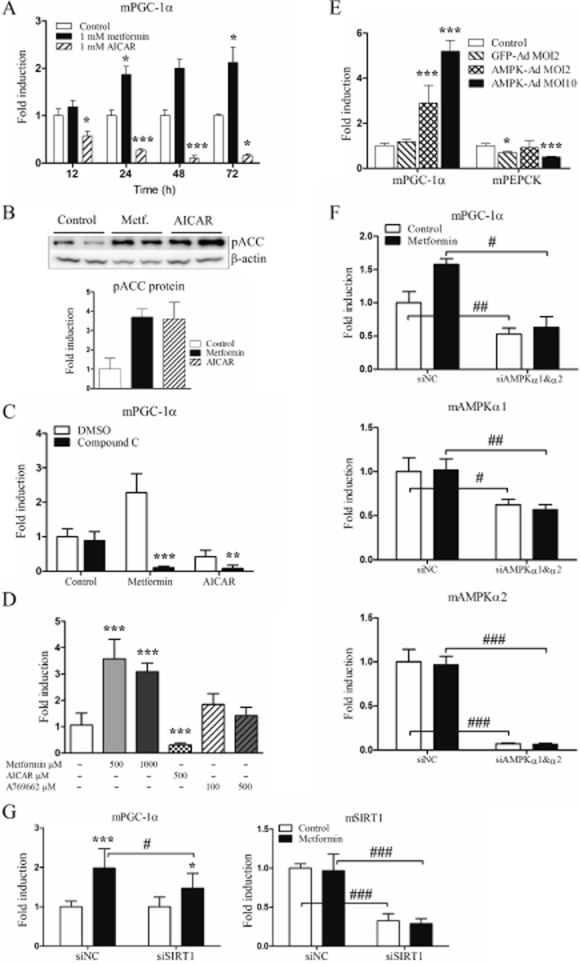
AMPK modulators and AMPK overexpression affect mouse PGC-1α expression level. (A) Mouse primary hepatocytes were treated with 1 mM metformin or 1 mM AICAR for 12, 24, 48 and 72 h after which mRNAs were analysed by RT-qPCR (n = 4). (B) Mouse primary hepatocytes were treated with 1 mM metformin or 1 mM AICAR for 8 h after which cells were sonicated and centrifuged at 13 000× g, supernatant was collected and immunoblotted against pACC antibody. β-Actin was measured as a loading control (n = 2, statistical significance was not calculated). (C) Mouse primary hepatocytes were treated with either 1 mM metformin or 1 mM AICAR alone or combined with 20 μM compound C in DMSO for 24 h after which mRNAs were analysed by RT-qPCR. (n = 4) (D) Mouse primary hepatocytes were treated with metformin, AICAR or A769662 for 48 h after which mRNAs were analysed by RT-qPCR (n = 3). (E) Mouse primary hepatocytes were infected with GFP-Ad (MOI 2) or AMPK-Ad (MOI 2 and 10) for 24 h after which mRNAs were measured using RT-qPCR (n = 3). (F) Mouse primary hepatocytes were transfected with siRNAs; negative control siGFP (siNC) or siAMPKα1 combined with siAMPKα2. Metformin 0.25 mM was added 30 h post-tranfection and incubated for 42 h after which samples were analysed by RT-qPCR (n = 3). (G) Mouse primary hepatocytes were transfected with siRNAs; negative control (siNC) or siSIRT1. Metformin 0.25 mM was added 5 h post-tranfection and incubated for 48 h after which samples were analysed by RT-qPCR (n = 9). qPCR results represent means of fold change + SD and Western blot results means of fold change + range.
AMPK activates SIRT1 by increasing cellular nicotinamide adenine dinucleotide (NAD)+ levels; also, metformin induces SIRT1 expression (Canto et al., 2009; Caton et al., 2010). Furthermore, in skeletal muscle, SIRT1 activation increases PGC-1α mRNA expression (Amat et al., 2009). We, therefore, tested whether SIRT1 is involved in the increase in PGC-1α induced by metformin in hepatocytes. Indeed, SIRT1 siRNA attenuated the increase in PGC-1α induced by metformin (Figure 2G).
Rotenone is a complex I inhibitor that has been reported to induce PGC-1α in hepatic cells through activation of CRTC3 (Than et al., 2011). As metformin also inhibits complex I (El-Mir et al., 2000), we studied the involvement of CRTC3 in the increase in PGC-1α induced by metformin. CRTC3 siRNA had no effect on this increase in PGC-1α, indicating that CRTC3 plays no role in this effect of metformin (data not shown).
Metformin attenuates the increase in PGC-1α induced by cAMP
Fasting is known to induce PGC-1α expression through a mechanism involving CREB activation by cAMP (Herzig et al., 2001). Furthermore, in a diabetic state, the altered balance of glucagon and insulin results in cAMP elevation in liver. Therefore, we studied the effect of metformin on the increase in PGC-1α induced by cAMP. Treatment of primary hepatocytes with db-cAMP alone for 2 h increased PGC-1α and PEPCK mRNAs 22-and 33-fold respectively (Figure 3A). After 24 h, this increase in PGC-1α was no longer observed while PEPCK still remained up-regulated by 15-fold. Metformin treatment alone increased PGC-1α 3.6-fold and down-regulated PEPCK by 0.2-fold compared with untreated controls after 24 h. The combination of db-cAMP with metformin attenuated the increase in PGC-1α induced by cAMP to 37% of that induced by cAMP alone after 2 h. Similarly, although less efficiently, metformin also affected the increase in PEPCK induced by cAMP after 2 h. These results indicate that although metformin per se induces PGC-1α expression, it has an antagonistic effect on cAMP-mediated up-regulation of PGC-1α.
Figure 3.
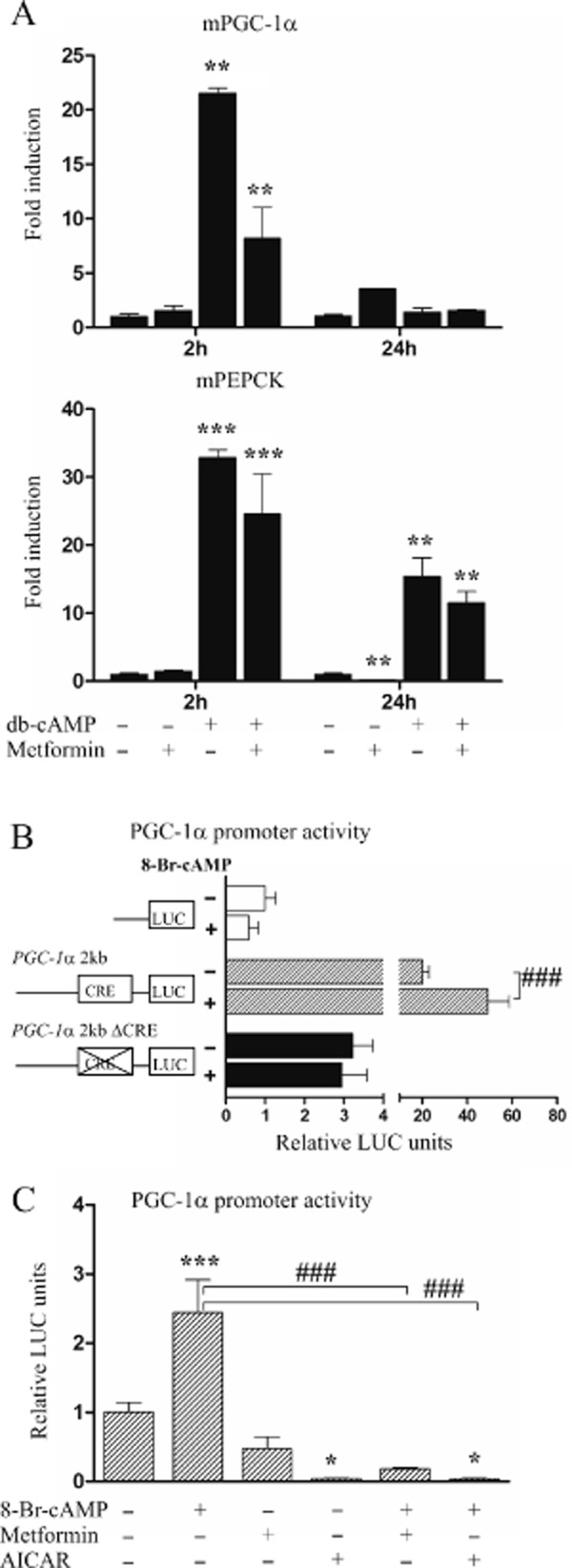
Effect of metformin and cAMP on PGC-1α mRNA expression and promoter activity. (A) Mouse primary hepatocytes were treated with 1 mM metformin or 25 μM db-cAMP alone or combined for 2 and 24 h after which mRNA expressions were analysed by RT-qPCR (n = 2). (B) Mouse primary hepatocytes were transfected with 2 kb PGC-1α-LUC wild-type (pGL3-Basic-PGC-1α 2 kb) construct, CRE site mutant construct (pGL3-Basic-PGC-1α-ΔCRE) or promoterless control plasmid (pGL3-Basic) and after 24 h treated with 25 μM 8-bromo-cAMP (8-Br-cAMP) for 24 h (n = 4). (C) Mouse primary hepatocytes were transfected with 2 kb PGC-1α-LUC wild-type (pGL3-Basic-PGC-1α 2 kb) construct and treated with 25 μM 8-Br-cAMP, 1 mM metformin, 500 μM AICAR or combinations for 24 h (n = 4). qPCR results represent means of fold change + range and luciferase results means of relative LUC units + SD.
Induction of PGC-1α by metformin is not mediated by the proximal PGC-1α promoter
Next, we studied the involvement of the PGC-1α gene proximal promoter in the regulation of PGC-1α by metformin. We first transfected luciferase reporter construct regulated by −2533 to +78 bp of the PGC-1α 5′-flanking region (pGL3-basic-PGC-1α 2 kb) to mouse primary hepatocytes. cAMP has been shown to regulate PGC-1α through a CREB-binding element located at −146 to −129 in the PGC-1α 5′-flanking region. Therefore, we also transfected a PGC-1α construct with mutated cyclic AMP-responsive element (CRE) site (pGL3-Basic-PGC-1α-ΔCRE). As expected, cAMP increased the wild-type construct, but not the CRE mutant construct. Interestingly, the CRE element also appeared to contribute to the constitutive expression of the PGC-1α gene, and the CRE mutant construct was significantly less active than the wild type (Figure 3B).
Treatment of pGL3-Basic-PGC-1α 2 kb transfected hepatocytes with metformin alone had no significant effect, while AICAR strongly down-regulated the luciferase activity. Both metformin and AICAR abolished the cAMP-mediated induction of PGC-1α 2 kb-regulated luciferase activity (Figure 3C). Together, these results indicate that the induction of PGC-1α by metformin is not mediated by the PGC-1α 5′-flanking region studied. However, the metformin-induced suppression of the increase in PGC-1α induced by cAMP is mediated by the previously identified CRE element.
Metformin selectively affects PGC-1α regulated functions
PEPCK is an established target gene of PGC-1α and overexpression of PGC-1α strongly induces PEPCK expression (Yoon et al., 2001). However, we found that metformin up-regulates PGC-1α while simultaneously down-regulating the expression of PEPCK mRNA. This suggests that metformin affects the ability of PGC-1α to induce PEPCK transcription. To test this hypothesis, we transduced mouse primary hepatocytes with PGC-1α-Ad and treated the cells with metformin. Both PEPCK and G6Pase mRNA induction were reduced significantly by metformin indicating that metformin is able to reduce the co-activator function of PGC-1α with regard to these genes (Figure 4A).
Figure 4.
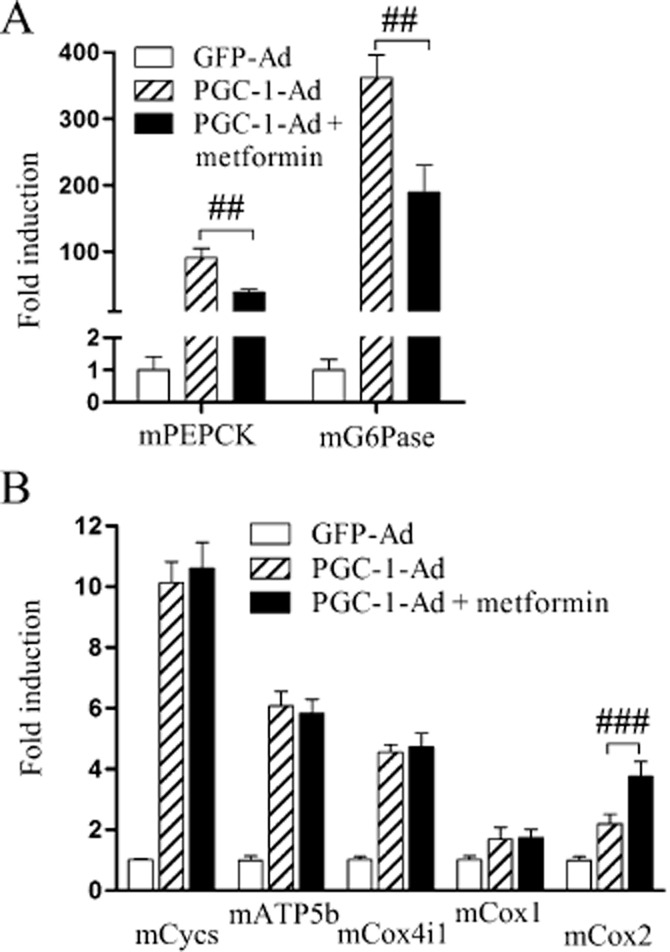
The functional effect of metformin on gluconeogenesis and selected mitochondrial-acting genes. Mouse primary hepatocytes were infected with PGC-1α-Ad at MOI 1 for 24 h after which 1 mM metformin was added. After 48 h cells were collected and mRNAs analysed by RT-qPCR for measuring (A) PEPCK and G6Pase expression and (B) selected genes encoding mitochondrial proteins. Results are compared with GFP-Ad treatment, set as onefold and normalized to equate the levels of PGC-1α expression between the treatments; (n = 3). qPCR results represent means of fold change + SD.
Mitochondrial biogenesis is another key cell function regulated by PGC-1α (Wu et al., 1999). Next, we studied if metformin also diminishes stimulation of mitochondrial biogenesis by PGC-1α. PGC-1α-Ad induced the expression of several genes coding mitochondrial proteins. Treatment of the cells with metformin did not affect the induction of these genes by PGC-1α, indicating that metformin differentially affects the regulation of mitochondrial biogenesis and gluconeogenesis by PGC-1α (Figure 4B).
Characterization of mechanisms by which metformin selectively prevents PGC-1α-mediated up-regulation of gluconeogenesis
Next, we studied potential mechanisms that could prevent the induction of the gluconeogenic genes by PGC-1α. PGC-1α function is regulated by post-transcriptional modifications. Two modifications potentially relevant for metformin's action include phosphorylation by AMPK (Jager et al., 2007) and deacetylation by SIRT1 (Rodgers et al., 2005). Furthermore, a priming signal by AMPK-mediated phosphorylation appears to be necessary for SIRT1-mediated deacetylation of PGC-1α (Canto et al., 2009). To study the potential effect of PGC-1α phosphorylation by AMPK, we prepared an adenovirus with mutated AMPK phosphorylation sites in the PGC-1α expression vector (T177A, S538A). Primary hepatocytes were transduced with the phospho-mutant or the wild type PGC-1α adenoviruses and treated with metformin. After 48 h, the cells were collected and mRNA expression of the PEPCK and G6Pase mRNA was measured. Mutation of the phosphorylation sites did not affect the induction of the genes studied by PGC-1α. Furthermore, metformin similarly down-regulated the induction of some genes despite the mutation (data not shown). This indicates that the attenuation of the PGC-1α-mediated gluconeogenic gene induced by metformin does not involve PGC-1α phosphorylation by AMPK. Furthermore, we studied the effect of metformin on PGC-1α acetylation status in HepG2 cells. Treatment with metformin resulted in 40% deacetylation of PGC-1α compared with the control (Figure 5A) in agreement with SIRT1 activation by metformin.
Figure 5.

Effect of metformin on acetylation of PGC-1α and genes regulating PEPCK and G6Pase. (A) HepG2 cells were transfected with PGC-1α-FLAG plasmid and 24 h post transfection treated with 2 mM metformin for 72 h after which FLAG fusion protein was isolated by immunoprecipitation. The protein fractions were immunoblotted against acetylated lysine antibody (Ac-Lys) and PGC-1α antibody was used as a loading control (n = 2, statistical significance was not calculated). (B) Mouse primary hepatocytes were treated with 1 mM metformin or 1 mM AICAR for 12 and 24 h after which mRNAs were analysed by RT-qPCR (n = 4). (C) Mouse primary hepatocytes were treated with 1 mM metformin for 48 and 72 h after which nuclear protein fractions were immunoblotted against HNF-4α antibody and β-actin was used as a loading control (n = 2 for 48 h/n = 3 for 72 h, two/three parallel experiments combined). Immunoblot is representative of one experiment. (D) HepG2 cells were transfected with HNF-4α-TATA-LUC or promoterless control plasmid pGL3-Basic (LUC), and after 24 h were treated with metformin for 24 h (n = 6). Results are relative luciferase units normalized to control LUC set as onefold. qPCR and Western blot results represent means of fold change + range and luciferase results means of relative LUC units + SD.
We also considered other putative mechanisms for the suppression of gluconeogenic genes. The small heterodimer partner NR0B2 (SHP) is supposedly induced by metformin through AMPK and inhibits HNF-4α-mediated induction of PEPCK (Kim et al., 2008). However, in contrast to the result expected, we were unable to detect any up-regulation of SHP expression in primary hepatocytes treated with metformin. In fact, SHP mRNA levels were down-regulated by both metformin and AICAR (Figure 5B).
Next, we hypothesized that metformin could modulate the expression of the PGC-1α target genes by regulating the DNA binding transcription factors interacting with PGC-1α. Krüppel-like factor 15 (KLF15), forkhead box protein O1 (FOXO1) and HNF-4α have all been shown to regulate PEPCK and G6Pase genes, and are co-activated by PGC-1α (Puigserver et al., 2003; Rhee et al., 2003; Takashima et al., 2010). We, therefore, investigated the effect of metformin on the expression of these transcription factors in primary hepatocytes. Metformin down-regulated the mRNAs of all three transcription factors after 12 and 24 h treatment (Figure 5B). Also AICAR down-regulated KLF15 and HNF-4α, but had no effect on FOXO1 (Figure 5B). Furthermore, treatment with 1 mM metformin down-regulated HNF-4α protein levels after 48 and 72 h (Figure 5C).
PGC-1α induces the expression of many nuclear-and also indirectly mitochondrial-encoded mitochondrial proteins through a mechanism involving nuclear respiratory factor 1 (NRF1) (Wu et al., 1999). We therefore analysed the effect of metformin treatment on NRF1 expression. In contrast to the transcription factors involved in the regulation of the gluconeogenic genes, NRF1 was increased by metformin (and also AICAR) treatment suggesting that the availability of the DNA binding transcription factors co-activated by PGC-1α influences the functional selectivity of PGC-1α.
Furthermore, we investigated the effect of metformin on the co-activation of HNF-4α by PGC-1α. We transfected HepG2 cells with a reporter construct under the control of the HNF-4α site. HepG2 cells express constitutively high levels of HNF-4α, but little PGC-1α. Cotransfection of the cells with a PGC-1α plasmid further activated luciferase activity indicating co-activation of HNF-4α by PGC-1α. Treatment of the cells with metformin down-regulated the luciferase activity 1.6-fold both with and without PGC-1α compared with untreated cells (Figure 5C). These results suggest that metformin does not affect the co-activation function of the PGC-1α or its interaction with HNF-4α. Instead, the diminished gene expression is mediated by a down-regulation of the DNA binding transcription factors, in this case HNF-4α.
Discussion and conclusions
PGC-1α has an important role in regulating hepatic energy balance and has been associated with the pharmacodynamic effects of metformin. Shaw et al. suggested that transcriptional regulation of PGC-1α through co-activator CRTC2 is a key step in the mechanism of action of metformin (Shaw et al., 2005). Our current study, however, shows that transcriptional regulation of the PGC-1α gene is not a major mechanism by which metformin inhibits gluconeogenesis. Indeed, the ability of metformin to down-regulate gluconeogenic genes is not dependent on the expression level of PGC-1α. However, PGC-1α-mediated gene regulation is affected selectively by metformin and activation of the mitochondrial biogenesis by PGC-1α is not disrupted by metformin.
We showed for the first time that the primary effect of metformin is mediated by the induction of both mRNA and protein levels of hepatic PGC-1α. The mRNA induction was confirmed in both mouse and human primary hepatocytes. However, the 18 day treatment of mice with 300 mg·kg−1 metformin in vivo did not result in changes in PGC-1α expression in the liver. The reasons for this lack of PGC-1α induction in liver in vivo are unclear. One possibility is an insufficient hepatic metformin concentration (average blood concentration 174 nmol·L−1 at the time of sample collection). Indeed, the induction of PGC-1α by metformin in skeletal muscle was demonstrated with a much higher dose (631 mg·kg−1) (Suwa et al., 2006). Furthermore, it is possible that PGC-1α is induced only transiently during the intrahepatic peak concentrations of metformin. In support of this hypothesis, when we collected the liver samples 3 h after the last metformin dose (altogether 7 days, 300 mg·kg−1) we detected a modest, but statistically significant, increase in PGC-1α. Similar to skeletal muscle (Suwa et al., 2006; Jager et al., 2007), we demonstrated that the effect of metformin on PGC-1α expression is mediated by AMPK in hepatocytes. This is supported by several lines of evidence. Firstly, compound C completely abolished the induction of PGC-1α by metformin; and secondly, AMPK-Ad induced PGC-1α mRNA expression. In addition, the knocking down of AMPK resulted in diminished PGC-1α levels in both untreated and metformin-treated hepatocytes, further suggesting a role for AMPK in the regulation of PGC-1α levels. Interestingly, another established AMPK modulator, AICAR had an opposite effect on PGC-1α expression and inhibited the transcription of PGC-1α very efficiently. AICAR and metformin have distinct ways to activate AMPK. While metformin targets mitochondrial respiratory complex I (El-Mir et al., 2000) causing inhibition of ATP production (Owen et al., 2000), AICAR, in contrast, is quickly converted to 5-aminoimidazole-4-carboxyamide-1 (ZMP) in cells mimicking 5′-AMP (Sullivan et al., 1994). Indeed, both compounds also activated AMPK in our current study. Nevertheless, AICAR must regulate PGC-1α in an AMPK-independent fashion. Interestingly, several such functions of AICAR have been described (Viollet et al., 2009; Santidrian et al., 2010). In support of the AMPK-independent repression of PGC-1α by AICAR, A769662, a direct, small molecular activator of AMPK (Oakhill et al., 2009) did not suppress PGC-1α expression, but rather had a tendency to increase PGC-1α. AICAR is a popular experimental compound to study AMPK function, but our observations indicate that the results obtained using AICAR should be interpreted with caution.
Furthermore, we showed that the induction of PGC-1α by metformin involves SIRT1. This is in agreement with previous results obtained in skeletal muscle (Amat et al., 2009). Metformin induces SIRT1 activity by increasing the NAD+/NADH ratio and protein level through an AMPK-mediated mechanism in hepatocytes (Caton et al., 2010). Metformin also induces SIRT1 mRNA, but a relatively high concentration is needed (Buler et al., 2012). With the low, 0.25 mM concentration of metformin, we did not detect any increase in SIRT1 mRNA. However, SIRT1 siRNA clearly attenuated the increase in PGC-1α induced by metformin indicating that SIRT1 activation, presumably through an increased NAD+/NADH ratio, plays a role in this effect of metformin. In skeletal muscles, PGC-1α T177/S538 phosphorylation by AMPK results in autoregulation through the proximal promoter (Jager et al., 2007). However, in hepatocytes, the overexpression of PGC-1α by an adenovirus had no effect on the expression of endogenous PGC-1α suggesting that autoregulation plays no significant role (data not shown). Furthermore, metformin did not affect luciferase activity under the control of the 2 kb proximal 5′-flanking region of the PGC-1α gene in primary hepatocytes. Thus the exact regulatory mechanisms mediating the increase in PGC-1α induced by metformin in liver appear to be different from those in skeletal muscle.
While metformin primarily induced PGC-1α expression, it was still able to attenuate the increase in PGC-1α induced by cAMP. This is in agreement with the previous report showing that AMPK-mediated phosphorylation of CRTC2 reduces PGC-1α expression (Shaw et al., 2005). Indeed, this may play some role in the action of metformin in diabetic patients. However, a down-regulation in PGC-1α expression is obviously not needed for the reduction in gluconeogenesis induced by metformin. In fact, metformin attenuated the induction of PEPCK and G6Pase induced by PGC-1α overexpression indicating that metformin controls the ability of PGC-1α to regulate target gene activation. Besides stimulating gluconeogenesis, PGC-1α is a critical regulator of mitochondrial biogenesis (Handschin, 2009). In particular, PGC-1α-Ad induced nuclear-encoded cytochrome c (Cycs), ATP synthase subunit β (ATP5B) and cytochrome c subunit 4 (Cox4i1), and to a lesser extent mitochondrial encoded cytochrome c oxidase I and II (Cox1 and Cox2, respectively). While metformin reduced this PGC-1α-stimulated induction of gluconeogenetic genes, mitochondrial genes were equally induced with and without metformin indicating a selective effect of metformin on PGC-1α-mediated gene regulation.
The selective effect of metformin on PGC-1α function resembles that recently described for PGC-1α phosphorylation by S6 kinase activated upon feeding (Lustig et al., 2011). However, metformin has been reported to inhibit the mammalian target of rapamycin/S6 kinase pathway (Dowling et al., 2007) suggesting that phosphorylation by S6 kinase may not be involved in this effect of metformin on PGC-1α. We, therefore, investigated several mechanisms that could potentially prevent the activation of gluconeogenesis by PGC-1α. It has been suggested that metformin up-regulates SHP, which then acts as a repressor of HNF-4α-and FOXO1-mediated regulation of gluconeogenic gene transcription (Kim et al., 2008). Unexpectedly, we were unable to corroborate these results. In fact, SHP was significantly down-regulated in primary hepatocytes by 12 h after metformin treatment. Also, Krausova et al. did not detect an increase in SHP induced by metformin in primary hepatocytes (Krausova et al., 2011). The reason for these differences is currently unknown. We also investigated the possibility that posttranslational modification of PGC-1α by AMPK or SIRT1 could modulate PGC-1α function after metformin treatment, because both are activated by the drug. However, mutation of the PGC-1α AMPK phosphorylation sites threonine-177/serine-538 did not abolish the effect of metformin on PGC-1α function in the context of gluconeogenic genes (data not shown). However, we observed that metformin treatment resulted in deacetylation of PGC-1α, probably mediated by activation of SIRT1. Nevertheless, PGC-1α deacetylation should lead to increased gluconeogenic gene activation (Rodgers et al., 2005) and, therefore, would not explain the observed down-regulation of the gluconegenic genes.
KLF15, FOXO1 and HNF-4α are the transcription factors that activate the gluconeogenic genes together with PGC-1α (Puigserver et al., 2003; Rhee et al., 2003; Takashima et al., 2010). We observed that metformin down-regulated all of these DNA binding transcription factors. While KLF15 and HNF-4α were also reduced by AICAR, FOXO1 was not, again indicating differences in the response to these two AMPK activators. Recently, KLF15 was shown to contribute to suppression of hepatic glucose production induced by metformin. Administration of metformin to mice has been found to lower gluconeogenic gene levels as well as KLF15 and an upregulation in the expression of KLF15 restored the expression of PEPCK (Takashima et al., 2010). Previously, FOXO1 has not been associated with the inhibition of gluconeogenesis by metformin, but metformin has been reported to reduce the expression of fatty acid-binding protein 4 by inhibiting FOXO1 (Song et al., 2010). The down-regulation of HNF-4α by metformin has not been reported previously. However, several studies have shown that AMPK and AICAR down-regulate and destabilize HNF-4α protein (Leclerc et al., 2001; Hong et al., 2003; Prieur et al., 2005). We also hypothesized that metformin could decrease the expression of HNF-4α. Indeed, we found that both HNF-4α mRNA and protein level as well as HNF-4α-mediated transcriptional activity were reduced by metformin. However, in the context of the reporter gene, PGC-1α was able to activate HNF-4α-mediated transcriptional activity regardless of the metformin treatment suggesting that metformin does not significantly inhibit the PGC-1α–HNF-4α interaction. Altogether, these results suggest that metformin down-regulates multiple key transcription factors and PGC-1α interaction partners, which attenuates the ability of PGC-1α to induce gluconeogenic genes. In contrast, the effect of PGC-1α on mitochondrial biogenesis mediated by NRF1 is not down-regulated by metformin, which would explain its selective influence on different PGC-1α-regulated cell functions. A proposed model of metformin's effects on PGC-1α expression and function is presented in Figure 6.
Figure 6.
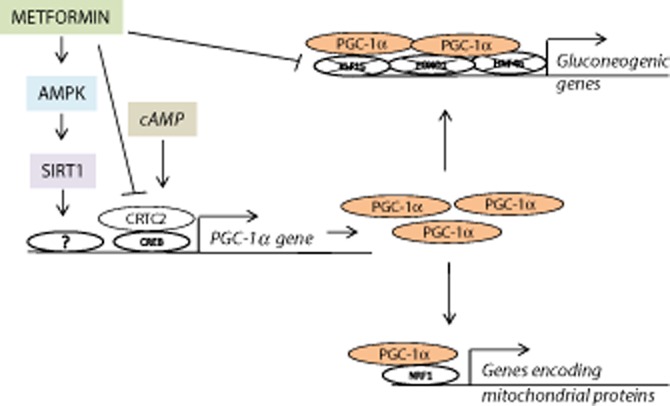
A proposed model for the effect of metformin on hepatic PGC-1α regulation and function. We propose that metformin activates PGC-1α gene transcription through AMPK and SIRT1. However, metformin represses the increase in PGC-1α induced by cAMP. Furthermore, metformin prevents the ability of PGC-1α to induce gluconeogenic genes.
In summary, our results indicate that the down-regulation of PGC-1α is not necessary for suppression of gluconeogenic genes by metformin. However, metformin regulates PGC-1α expression and function at multiple levels and by various mechanisms. Metformin increases PGC-1α expression as such, but attenuates cAMP-mediated up-regulation of PGC-1α. Importantly, metformin selectively affects hepatic PGC-1α-mediated gene regulation, and prevents activation of gluconeogenesis, but does not influence its regulation of mitochondrial genes.
Acknowledgments
The skilful assistance of Ms. Päivi Tyni and Ms. Ritva Tauriainen is greatly acknowledged. This study was funded by the Finnish Diabetes Research Foundation, the Sigrid Jusélius Foundation, the Academy of Finland (contract 110591), the EU (Marie Curie RTN NucSys) and the Czech Scientific Agency GACR303/12/0472.
Glossary
- ACC
acetyl-CoA
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- ATP5β
ATP synthase subunit β
- Cox
cytochrome c oxidase
- Cox4i1
cytochrome c oxidase IV isoform 1
- CRTC
cAMP response element-binding protein coactivator
- Cycs
cytochrome c
- db-cAMP
dibutyryl-cAMP
- FOXO1
forkhead box protein O1
- G6Pase
glucose-6-phosphatase
- HNF-4α
hepatocyte NF 4α
- KLF15
Krüppel-like factor 15
- LKB1
liver kinase B1
- NAD
nicotinamide adenine dinucleotide
- NRF1
nuclear respiratory factor 1
- PEPCK
phosphoenolpyruvate carboxykinase
- PGC-1α
PPARγ coactivator 1α
- SHP
small heterodimer partner NR0B2
- SIRT1
sirtuin 1
Conflict of interest
None.
References
- Amat R, Planavila A, Chen SL, Iglesias R, Giralt M, Villarroya F. SIRT1 controls the transcription of the peroxisome proliferator-activated receptor-gamma co-activator-1alpha (PGC-1alpha) gene in skeletal muscle through the PGC-1alpha autoregulatory loop and interaction with MyoD. J Biol Chem. 2009;284:21872–21880. doi: 10.1074/jbc.M109.022749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpiainen S, Jarvenpaa SM, Manninen A, Viitala P, Lang MA, Pelkonen O, et al. Coactivator PGC-1alpha regulates the fasting inducible xenobiotic-metabolizing enzyme CYP2A5 in mouse primary hepatocytes. Toxicol Appl Pharmacol. 2008;232:135–141. doi: 10.1016/j.taap.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Buler M, Aatsinki SM, Izzi V, Hakkola J. Metformin reduces hepatic expression of SIRT3, the mitochondrial deacetylase controlling energy metabolism. PLoS ONE. 2012;7:e49863. doi: 10.1371/journal.pone.0049863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton PW, Nayuni NK, Kieswich J, Khan NQ, Yaqoob MM, Corder R. Metformin suppresses hepatic gluconeogenesis through induction of SIRT1 and GCN5. J Endocrinol. 2010;205:97–106. doi: 10.1677/JOE-09-0345. [DOI] [PubMed] [Google Scholar]
- Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C. The biology of PGC-1alpha and its therapeutic potential. Trends Pharmacol Sci. 2009;30:322–329. doi: 10.1016/j.tips.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci U S A. 2003;100:7111–7116. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–27501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YD, Park KG, Lee YS, Park YY, Kim DK, Nedumaran B, et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes. 2008;57:306–314. doi: 10.2337/db07-0381. [DOI] [PubMed] [Google Scholar]
- Krausova L, Stejskalova L, Wang H, Vrzal R, Dvorak Z, Mani S, et al. Metformin suppresses pregnane X receptor (PXR)-regulated transactivation of CYP3A4 gene. Biochem Pharmacol. 2011;82:1771–1780. doi: 10.1016/j.bcp.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krentz AJ, Bailey CJ. Oral antidiabetic agents: current role in type 2 diabetes mellitus. Drugs. 2005;65:385–411. doi: 10.2165/00003495-200565030-00005. [DOI] [PubMed] [Google Scholar]
- Lamsa V, Levonen AL, Leinonen H, Yla-Herttuala S, Yamamoto M, Hakkola J. Cytochrome P450 2A5 constitutive expression and induction by heavy metals is dependent on redox-sensitive transcription factor Nrf2 in liver. Chem Res Toxicol. 2010;23:977–985. doi: 10.1021/tx100084c. [DOI] [PubMed] [Google Scholar]
- Leclerc I, Lenzner C, Gourdon L, Vaulont S, Kahn A, Viollet B. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes. 2001;50:1515–1521. doi: 10.2337/diabetes.50.7.1515. [DOI] [PubMed] [Google Scholar]
- Lin JD. Minireview: the PGC-1 coactivator networks: chromatin-remodeling and mitochondrial energy metabolism. Mol Endocrinol. 2009;23:2–10. doi: 10.1210/me.2008-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig Y, Ruas JL, Estall JL, Lo JC, Devarakonda S, Laznik D, et al. Separation of the gluconeogenic and mitochondrial functions of PGC-1{alpha} through S6 kinase. Genes Dev. 2011;25:1232–1244. doi: 10.1101/gad.2054711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian C, Tarpin S, Mithieux G. Role of glucose-6 phosphatase, glucokinase, and glucose-6 phosphate in liver insulin resistance and its correction by metformin. Biochem Pharmacol. 1998;55:1213–1219. doi: 10.1016/s0006-2952(97)00576-5. [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Scott JW, Kemp BE. Structure and function of AMP-activated protein kinase. Acta Physiol (Oxf) 2009;196:3–14. doi: 10.1111/j.1748-1716.2009.01977.x. [DOI] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- Prieur X, Schaap FG, Coste H, Rodriguez JC. Hepatocyte nuclear factor-4alpha regulates the human apolipoprotein AV gene: identification of a novel response element and involvement in the control by peroxisome proliferator-activated receptor-gamma coactivator-1alpha, AMP-activated protein kinase, and mitogen-activated protein kinase pathway. Mol Endocrinol. 2005;19:3107–3125. doi: 10.1210/me.2005-0048. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, et al. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci U S A. 2003;100:4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Salomaki H, Vahatalo LH, Laurila K, Jappinen NT, Penttinen AM, Ailanen L, et al. Prenatal metformin exposure in mice programs the metabolic phenotype of the offspring during a high fat diet at adulthood. PLoS ONE. 2013;8:e56594. doi: 10.1371/journal.pone.0056594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santidrian AF, Gonzalez-Girones DM, Iglesias-Serret D, Coll-Mulet L, Cosialls AM, Frias de M, et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. Blood. 2010;116:3023–3032. doi: 10.1182/blood-2010-05-283960. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Ren P, Zhang L, Wang XL, Chen L, Shen YH. Metformin reduces lipid accumulation in macrophages by inhibiting FOXO1-mediated transcription of fatty acid-binding protein 4. Biochem Biophys Res Commun. 2010;393:89–94. doi: 10.1016/j.bbrc.2010.01.086. [DOI] [PubMed] [Google Scholar]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–36. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- Suwa M, Egashira T, Nakano H, Sasaki H, Kumagai S. Metformin increases the PGC-1{alpha} protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J Appl Physiol. 2006;101:1685–1692. doi: 10.1152/japplphysiol.00255.2006. [DOI] [PubMed] [Google Scholar]
- Takashima M, Ogawa W, Hayashi K, Inoue H, Kinoshita S, Okamoto Y, et al. Role of KLF15 in regulation of hepatic gluconeogenesis and metformin action. Diabetes. 2010;59:1608–1615. doi: 10.2337/db09-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than TA, Lou H, Ji C, Win S, Kaplowitz N. Role of cAMP-responsive element-binding protein (CREB)-regulated transcription coactivator 3 (CRTC3) in the initiation of mitochondrial biogenesis and stress response in liver cells. J Biol Chem. 2011;286:22047–22054. doi: 10.1074/jbc.M111.240481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Guigas B, Leclerc J, Hebrard S, Lantier L, Mounier R, et al. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf) 2009;196:81–98. doi: 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- Yuan L, Ziegler R, Hamann A. Inhibition of phosphoenolpyruvate carboxykinase gene expression by metformin in cultured hepatocytes. Chin Med J (Engl) 2002;115:1843–1848. [PubMed] [Google Scholar]