

Abstract
Background and Purpose
cAMP and pharmacological inhibition of PDE4, which degrades it, are promising therapeutic targets for the treatment of spinal cord injury (SCI). Using our previously described in vitro SCI model, we studied the mechanisms by which cAMP modulators promote neurite outgrowth and myelination using enantiomers of the PDE4-specific inhibitor rolipram and other modulators of downstream signalling effectors.
Experimental Approach
Rat mixed neural cell myelinating cultures were cut with a scalpel and treated with enantiomers of the PDE4-specific inhibitor rolipram, Epac agonists and PKA antagonists. Neurite outgrowth, density and myelination were assessed by immunocytochemistry and cytokine levels analysed by qPCR.
Key Results
Inhibition of the high-affinity rolipram-binding state (HARBS), rather than the low-affinity rolipram binding state (LARBS) PDE4 conformer promoted neurite outgrowth and myelination. These effects were mediated through the activation of Epac and not through PKA. Expression of the chemokine CXCL10, known to inhibit myelination, was markedly elevated in astrocytes after Rho inhibition and this was blocked by inhibition of Rho kinase or PDE4.
Conclusions and Implications
PDE4 inhibitors targeted at the HARBS conformer or Epac agonists may provide promising novel targets for the treatment of SCI. Our study demonstrates the differential mechanisms of action of these compounds, as well as the benefit of a combined pharmacological approach and highlighting potential promising targets for the treatment of SCI. These findings need to be confirmed in vivo.
Keywords: cAMP, Epac, neurite outgrowth, PDE4, spinal cord injury, myelination
Introduction
Spinal cord injury (SCI) generates a wide range of pathophysiological markers, including demyelination, axonal damage/death, astrocyte reactivity, formation of glial scars, and a marked reduction in the level of the ubiquitous second messenger, cAMP (Silver and Miller, 2004). cAMP is essential for the regulation of intracellular functions, including cellular proliferation, differentiation, metabolism and survival. Thus, a reduction in its levels as a result of trauma to the CNS could have widespread detrimental consequences (Pearse et al., 2004).
Elevating neuronal cAMP, through a cAMP analogue or exposure to neurotrophins, overcomes inhibition by myelin-associated glycoprotein (MAG) and myelin in vitro (Cai et al., 1999; Hannila and Filbin, 2008) and in vivo (Cai et al., 2001; Neumann et al., 2002; Qiu et al., 2002; Nikulina et al., 2004). Although the biochemical mechanisms by which cAMP promotes axonal regeneration are poorly understood, two signalling systems activated by cAMP have been identified, namely the kinase PKA, and more recently, Epac, an exchange protein activated by cAMP (nomenclature follows Alexander et al., 2013). Both PKA and Epac can independently mediate cAMP-regulated axon growth and regeneration, and thus represent suitable therapeutic targets for SCI (Murray and Shewan, 2008). Ultimately, cAMP signalling affects the cytoskeletal dynamics in the growth cones. In particular, members of the Rho family of GTPases, including Rho, Rac and cdc42, are known effectors of PKA signalling in both non-neuronal and neuronal cells (Lang et al., 1996; Forgione and Fehlings, 2013).
Cyclic nucleotide PDEs provide the sole means for degrading cAMP in cells and so are poised to play a key role in regulating cAMP-directed processes (Lugnier, 2006; Conti and Beavo, 2007). The availability of specific inhibitors, together with the cell-type-specific expression of different PDE forms has allowed for the development of PDE inhibitors with specific functional properties and therapeutic exploitation. PDE4 family members specifically hydrolyse cAMP, are expressed in a cell-type-specific fashion and, through intracellular targeting, play a major role in underpinning the compartmentalization of cAMP signalling (Houslay, 2010; Keravis and Lugnier, 2010). There has been considerable interest in developing selective PDE4 inhibitors for therapeutic use in inflammatory diseases, particularly chronic obstructive pulmonary disease (COPD) and psoriasis (Houslay et al., 2005; Spina, 2008). However, post-translational modification and interaction with partner proteins allows PDE4 enzymes to adopt distinct conformational states. These can be detected, and exploited pharmacologically, by differences in sensitivity to certain PDE4 selective inhibitors, of which the archetype is rolipram. In this instance, the rolipram S-enantiomer selectively inhibits the low-affinity rolipram-binding site (LARBS) PDE4 conformer, whereas the rolipram R-enantiomer preferentially inhibits the high-affinity rolipram-binding site (HARBS) PDE4 conformer (Souness and Rao, 1997; Houslay and Adams, 2003; Burgin et al., 2010).
We have developed our myelinating co-cultures to closely mimic SCI in vivo (Boomkamp et al., 2012). These co-cultures reproduce many features of the CNS, including nodes of Ranvier and compact myelin (Sørensen et al., 2008; Thomson et al., 2008). After creating a lesion using a scalpel blade, these cultures show significant loss of myelinated fibres, reduced neurite density around the lesion, increased astrocyte reactivity and a persistent lack of spontaneous neurite outgrowth (Boomkamp et al., 2012). Thus, these cultures provide a valuable tool for studying pathogenesis associated with SCI, as well as a throughput screen for potential treatment options before validation in animal models. In this study, we have used this in vitro model of SCI to dissect the mechanisms by which cAMP and its downstream effectors mediate neurite outgrowth and myelination after injury, with comparison with inhibitors of the Rho/Rho kinase (ROCK) pathway. Our results have shown that it is the HARBS PDE4 conformer that provides the functional target.
Methods
Animals
All animal care and experimental procedures complied with the Home Office Regulations and were approved by the Ethics Committee of the University of Glasgow. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Animals were supplied by Charles River, Margate, UK).
Astrocytes derived from neurospheres
Neurospheres were prepared and differentiated into astrocytes as described by Sørensen et al. (2008). Briefly, striata were dissected from the forebrains of 1-day-old Sprague-Dawley rat pups, triturated in Leibovitz L-15 media, centrifuged (136 × g) and resuspended in 20 mL neurosphere medium consisting of DMEM/F12 (1:1, DMEM containing 4.5 g L−1 glucose), supplemented with 0.105% NaHCO3, 2 mM L-glutamine, 5000 IU mL−1 penicillin, 5 μg mL−1 streptomycin, 5.0 mM HEPES, 0.0001% BSA (all from Invitrogen, Life Technologies, Paisley, UK), 25 μg mL−1 insulin, 100 μg mL−1 apotransferrin, 60 μM putrescine, 20 nM progesterone and 30 nM sodium selenite (all from Sigma, Poole, Dorset, UK). The tissue suspension was placed in a 75 cm3 tissue culture flask (Greiner Bio One, Gloucestershire, UK) and supplemented with 20 ng mL−1 mouse submaxillary gland EGF (R&D Systems, Abingdon, UK). The cultures were incubated at 37°C in a humidified atmosphere of 7% CO2/93% air, and 5 mL fresh media and 20 ng mL−1 EGF was added every 2–3 days. After 7–10 days in vitro, the spheres were triturated using a Pasteur pipette to produce smaller spheres/cell clusters. Spheres were differentiated into astrocytes 1–2 days after trituration by plating onto poly-L-lysine (PLL)-coated coverslips in low-glucose DMEM supplemented with 10% FBS and 2 mM L-glutamine (DMEM-FBS). When a confluent layer of astrocytes had formed, dissociated spinal cord cells were plated on top.
Myelinating spinal cord cultures
The method of generating myelinating spinal cord cultures (myelinating cultures) was based on those previously described (Sørensen et al., 2008; Nash et al., 2011) with some minor modifications. The spinal cords from Sprague-Dawley E 15.5 embryos were dissected, stripped of meninges, minced with a scalpel blade, and placed in 1 mL of Hank's balanced salt solution without Ca2+ and Mg2+ (Invitrogen). The tissue was enzymically dissociated by adding trypsin (100 μL of 2.5%, Invitrogen) and collagenase (100 μL 1.33%, Gibco, Life Technologies) for 15 min at 37°C. Enzymic activity was stopped by adding 2 mL of SD solution [L15medium (Life Technologies) containing 0.52 mg mL−1 soybean trypsin inhibitor, 3.0 mg mL−1 BSA, and 0.04 mg mL−1 DNase (Sigma)] to prevent cell clumping. The cells were triturated, centrifuged (136 × g) and the cell pellet resuspended in 2 mL plating medium (PM) that contained 50% DMEM, 25% horse serum, 25% HBSS with Ca2+ and Mg2+, and 2 mM L-glutamine (Invitrogen). The dissociated spinal cord cells were plated onto coverslips supporting a monolayer of neurosphere-derived astrocytes at a density of 150 000 cells per 100 μL. The cells were left to attach for 2 h at 37°C after which 300 μL PM and 500 μL differentiation medium [DMEM containing 4.5 g mL−1 glucose, 10 ng mL−1 biotin, 0.5% hormone mixture (1 mg mL−1 apotransferrin, 20 mM putrescine, 4 μM progesterone, 6 μM selenium, 50 nM hydrocortisone and 0.5 mg mL−1 insulin (all from Sigma)] was added. The cultures were fed three times a week with differentiation media and, after 12 days in culture, insulin was excluded from the differentiation media. Cultures were maintained for 22–37 days in a humidified atmosphere of 7% CO2/93% air at 37°C.
For modelling SCI, mature myelinating cultures were subjected to a cut with a no. 11 scalpel blade (WPI; Herfordshire, UK) at typically day 24 when mature myelin is present as previously described (Boomkamp et al., 2012). If applicable, potentially therapeutic compounds were added to the cut cultures typically 1 day after cutting, and added at every feed two to three times a week.
Purification of microglia from P1 cortex
Cortices from 1-day-old Sprague-Dawley rats were dissected, and triturated in L-15 media prior to digestion with collagenase (500 μL 1.33%, ICN Pharmaceuticals). Enzymic activity was stopped by adding 2 mL of SD solution. The cells were triturated, centrifuged (136 × g), the cell pellet resuspended in low-glucose DMEM supplemented with 10% FBS and 2 mM L-glutamine (DMEM-FBS), and cultured in PLL-coated flasks. Upon confluence, the flasks were shaken on an orbital shaker at 37°C for 4 h after which the supernatant was collected, and placed onto a Petri dish. After a 20 min incubation at 37°C, the supernatant was removed (containing oligodendrocyte precursor cells; OPCs) and the remaining microglia adhering to the dish, transferred to PLL-coated coverslips.
Immunocytochemistry and antibodies
Mature myelin (proteolipid protein, PLP) was detected using the AA3 antibody (1:100, anti-rat; hybridoma supernatant; Yamamura et al., 1991) directed against PLP/DM20. SMI-31 antibody (mouse IgG1, 1:1500) labelled the phosphorylated neurofilament, whereas astrocytes were detected using a polyclonal antibody against glial fibrillary acidic protein (GFAP, rabbit IgG, 1:100, Dako, Cambridgeshire, UK). Antibodies raised against phosphomyosin (phospho-Ser19; rabbit IgG, 1:50, Merck Millipore, Watford, UK) and phalloidin (mouse IgG1, Alexa Fluor 488 conjugated, 1:200, Invitrogen) were used to demonstrate inhibitory activity towards Rho/ROCK. Antibody against CXCL10 was provided by R&D Systems. For intracellular labelling, the cultures were fixed in 4% paraformaldehyde for 20 min at room temperature followed by washes in PBS and permeabilization in 0.2% Triton-X100 for 15 min at room temperature. The cultures were washed in PBS and exposed to blocking buffer for 20 min at room temperature. The primary antibodies were added, diluted in blocking buffer, and the cells incubated for 45 min at room temperature. After washes in PBS, the cultures were incubated with the appropriate secondary antibodies at room temperature for 45 min. The cells were washed in PBS followed by distilled H2O, mounted in Vectashield (Vector Laboratories, Peterborough, UK) and sealed with nail varnish.
Imaging
Cells were imaged using an Olympus BX51 fluorescence microscope (Olympus, Southend-on-Sea, UK) or a Zeiss LSM 510 confocal microscope (Carl Zeiss Ltd., Cambridge, UK). For quantitative analysis of neurite density (SMI-31) and myelination (PLP), 20 images per condition were taken from each coverslip (in duplicate). Neurite density was analysed using Image J software (version 1.41o, NIH Systems, rbsweb.nih.gov) and SMI-31 immunoreactivity, which was calculated by measuring the occupation of neurites immunoreactivity (in pixels) per field of view and dividing that by the total number of pixels. To calculate the percentage of myelinated axons, immunoreactivity associated with PLP-positive myelin sheaths was also measured using Image J. This is explained in detail in Sørensen et al., 2008; briefly, the myelin sheaths were manually drawn over using Adobe Photoshop software, and the percentage of myelin calculated using the area obtained for the highlighted myelin divided by the area obtained for neurite density. To quantify the areas adjacent to the lesion, 20 representative images of the field of view were taken (0–670 μm for ×100 magnification or 0–335 μm for ×200 magnification). Areas that were folded due to the mechanical effect of cutting were not taken into account. All experiments were carried out at least three times (n = 3) in duplicate. The intensities of GFAP, actin and phosphomyosin were calculated using ImageJ software (NIH Systems) by measuring the integrated density of the individual colour per image and normalizing the value to the number of DAPI-positive nuclei. To determine the number of neurites growing into the lesion, 20 images per condition (×100 magnification) were acquired, and the average number of outgrowing neurites per image counted.
RhoA activation assay
For assessment of RhoA activation, cultures required three lesions per coverslip in order to achieve detectable changes in protein expression (data not shown). Cultures were lysed using lysis buffer (Millipore) and the protein concentration determined using the NanoDrop spectrophotometer (Thermo Scientific, Loughborough, UK). A RhoA activation assay kit [Cytoskeleton, Inc., Universal Biologicals (Cambridge), Ltd., Cambridge, UK], which involves the pull-down of GTP-Rho complexed with rhotekin-RBD beads, was used, according to the manufacturer's instructions, to determine the extent of Rho inhibition via Western blotting. The samples were run on a NUPAGE NOVEX Tris-acetate gel (4–12%; Invitrogen) at 200 V for 45 min. The gels were then transferred to a nitrocellulose membrane using the iBlot Western Detection system (Invitrogen). The membranes were incubated in blocking buffer containing 5% skimmed milk powder and 0.2% Triton-X100 in PBS overnight at 4°C. Determination of total Rho of whole cell lysates was assessed for comparison. Quantification of band intensities was performed using Image J, and the RhoA intensity normalized to that of total Rho.
Quantitative real-time PCR
Cells, either control, cut and/or treated with compounds, were lysed and the RNA extracted using the PureLink RNA Mini Kit purification kit according to the manufacturers' instructions (Life Technologies). DNA synthesis was performed using the Quantitect reverse transcription kit (Qiagen) and qRT-PCR carried out using Quantifast SYBR Green PCR kit containing ROX passive reference dye and specific primers (Qiagen). The relative quantities were measured on 7900HT Fast Real-time PCR machine (Applied Biosystems, Warrington, UK), and normalized to that of GAPDH (Qiagen) using the Δ−ΔCT method.
Data analysis
Data are expressed as means ± SEM, from a minimum of three experiments per condition. To compare means between groups of conditions, data were analysed by calculating ratios and analysing these with one-sample Student's t-tests, using 1 as the null hypothesis mean (or 0 when log10means were used). P < 0.05 was considered to show a significant difference between means.
Materials
The compounds used and their suppliers are listed below (Table 1): a cell-permeable version of Clostridium botulinum-derived Rho inhibitor, C3 ADP-ribosyltransferase (C3, Cytoskeleton); the ROCK inhibitor, Y27632 (Calbiochem, Merck Millipore), the PDE4 inhibitors, rolipram (Sigma), R-rolipram and S-rolipram (Tocris Bioscience, R & D Systems), which specifically inhibit the HARBS and LARBS respectively. RS25344 hydrochloride (Tocris) and roflumilast (Sigma), which like rolipram inhibit both HARBS and LARBS; the PKA inhibitors KT-5720 (Sigma) and Rp-cAMPS (Biolog), and the Epac agonist 8-pMeOPT-2′-O-Me-cAMP (Me-cAMP; Biolog, Bremen, Germany).
Table 1.
Summary of the compounds used and their mechanism of action
| Compound | Affects |
|---|---|
| Racemic rolipram | –HARBS + LARBS-PDE4 |
| S-Rolipram | –LARBS-PDE4 |
| R-Rolipram | –HARBS-PDE4 |
| RS25344 | –HARBS-PDE4 |
| Roflumilast | –HARBS + LARBS-PDE4 |
| KT5720 | –PKA |
| RpcAMPS | –PKA |
| Me-cAMP | +Epac |
| C3 | –Rho |
| Y27632 | –ROCK |
Results
Rolipram induces neurite outgrowth and enhances myelination
The myelinating cultures described here have been fully characterized as described elsewhere (Sørensen et al., 2008). The PDE4 isoforms present in the myelinating cultures, as ascertained by qPCR, were predominantly be PDE4A1, A8, A11, B2, B5, D3 and D8 (data not shown). After confirming the abundance of myelinated fibres by immunocytochemistry, the cultures were cut using a scalpel blade, typically at day 24 of culture. It was important that no lateral movement occurred to prevent tearing of the cell sheet. This treatment reliably generated a lesion, which usually was between 200 and 500 μm in width. As demonstrated previously, cutting induces the formation of features typical of the glial scar, including astrogliosis, loss of myelinated fibres, reduced neurite density and neurite damage (Boomkamp et al., 2012). Importantly, this culture system is characterized by a persistent lack of spontaneous neurite outgrowth (Figure 1E). Throughout the study, photomicrographs were taken directly adjacent to the lesion (0–670 μm from the lesion edge away from the lesion (see Boomkamp et al., 2012 for details). In all experiments describing neurite density, the intensity of SMI-31-positive pixels was calculated in areas directly adjacent to the lesion.
Figure 1.
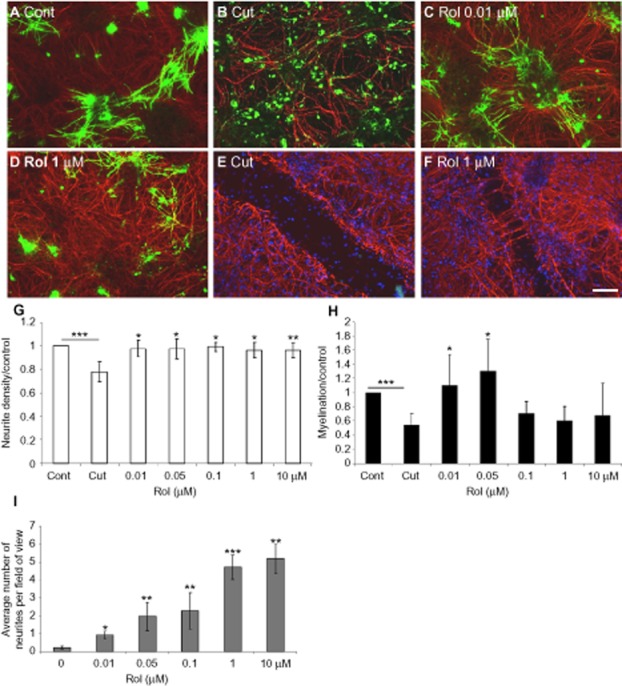
Rolipram induces myelination and neurite outgrowth. Immunolabelling of control, (A), cut (B: adjacent to the lesion, E: lesion site), and cut cultures treated with 10 nM (C) or 1 μM (D: adjacent to the lesion, F: lesion site) rolipram (Rol) with SMI-31 (red) and anti-PLP/DM20 (green) antibody. Cultures were cut at day 24, and treated with varying concentrations of rolipram (μM) 1 day after cutting. The cultures were treated for 6 days prior to immunocytochemistry and quantification of neurite density (G) and myelination (H) surrounding the lesion, as well as neurite outgrowth into the lesion (I). Scale bar 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001, significant differences between control and cut, and treatments and cut.
One day after cutting, the cultures were treated with varying concentrations of rolipram for up to 7 days, and the extent of neurite outgrowth and myelination evaluated via immunocytochemistry using SMI-31 (red) and anti-PLP/DM20 (green) respectively (Figure 1A–F).
We observed that the PDE4 selective inhibitor, rolipram (as the racemic mixture, affecting both HARBS and LARBS), enhanced neurite density in areas closely surrounding the lesion (0–670 μm) and induced significant neurite outgrowth across the lesion over a broad concentration range of 10 nM–10 μM (Figure 1F, G, I), suggesting that PDE4 subpopulations adopting both the HARBS and LARBS conformations were likely to be involved in influencing neurite outgrowth. However, in marked contrast to this, the ability of racemic rolipram to enhance myelination surrounding the lesion was only observed over a more restricted concentration range (10–50 nM) that was indicative of inhibition of the HARBS PDE4 conformer (Figure 1C, D, H), which is highly susceptible to the action of rolipram (Souness and Rao, 1997; Houslay and Adams, 2003; Zhang et al., 2006; Burgin et al., 2010). Intriguingly, this effect of rolipram on myelination was biphasic, with higher concentrations of rolipram actually leading to an inhibition of myelination (Figure 1H) due to inhibition of the LARBS PDE conformer. This process presumably acts to increase cAMP in a locality where it blocks the stimulatory effect on myelination mediated by inhibition of the PDE4 HARBS conformer.
Rolipram did not affect OPC proliferation (data not shown).
High concentrations of rolipram prevents myelination via PKA
In order to determine whether the effects of rolipram on myelination were mediated through PKA we employed the PKA antagonist, Rp-cAMPS, which inhibits cAMP binding to the PKA cAMP binding domain. Cut cultures (Figure 2B) were treated with 10 nM, 50 nM or 1 μM rolipram in either the absence or presence of 10 nM Rp-cAMPS (Figure 2D–F). Inhibition of PKA, at the high concentration of rolipram (1 μM) required to inhibit the LARBS PDE4 conformer, resulted in an increase in myelination that was similar in magnitude to that observed using low concentrations of racemic rolipram (10–50 nM) that inhibit the HARBS PDE4 conformer. The PKA antagonist thus blocked the biphasic response of rolipram action on myelination. In particular, it prevented the inhibition exerted by high-rolipram concentrations, indicating that this action is mediated through the action of a population of PKA that is regulated by the LARBS conformer of PDE4. Similar results were obtained using the PKA antagonist, KT5720, which acts on the PKA catalytic unit (data not shown). In contrast, neurite outgrowth was not significantly altered in the presence of Rp-cAMPS (Figure 2H).
Figure 2.
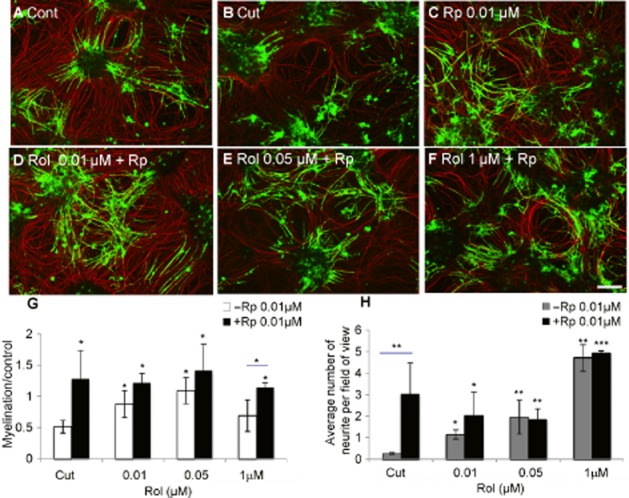
At high concentrations, rolipram inhibits myelination through PKA. Cultures were cut (B) and treated with varying concentrations of rolipram (Rol, 10 nM: D, 50 nM: E, 1 μM: F) in the presence (D–F) or absence of 10 nM Rp-cAMPS (Rp) (C). The cultures were treated for 6 days prior to immunocytochemistry using SMI-31 (red) and anti-PLP (green) and quantification of neurite density (data not shown) and myelination (G) surrounding the lesion, as well as neurite outgrowth into the lesion (H). Scale bar 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001; significant differences between cut with/without Rp-cAMPS and rolipram-treated cut cultures with/without Rp-cAMPS.
Rolipram primarily mediates its effects on myelination via the HARBS PDE4 conformer signalling to Epac
We have inferred that the HARBS PDE4 conformer was involved in myelination because of the sensitivity of this process to stimulation by low nanomolar concentrations of rolipram. To gain further support for this notion, we explored the sensitivity of this process to S-rolipram, which preferentially acts on the LARBS PDE4 conformer; R-rolipram, which acts on the HARBS PDE4 conformer; RS25344 hydrochloride, a potent PDE4 inhibitor acting on the HARBS conformer, and roflumilast, a PDE4 inhibitor used clinically for the treatment of COPD and which interacts equally with the HARBS and LARBS PDE4 conformers (Souness and Rao, 1997; Houslay and Adams, 2003; Zhang et al., 2006; Day et al., 2011). Additionally, we used Rp-cAMPS and KT-5720, which are both selective PKA antagonists, and Me-cAMP, a selective Epac agonist.
The effects of these compounds on neurite outgrowth into the lesion and neurite density and myelination surrounding the lesion was determined by immunocytochemistry. All compounds restored neurite density surrounding the lesion to control levels (data not shown). R-Rolipram, which acts selectively on the HARBS PDE4 conformer, was more effective on myelination than S-rolipram (LARBS PDE4 conformer selective), which was without effect at any of the concentrations tested (Figure 3A, B). RS25344 significantly enhanced myelination at low doses, consistent with its selective action on the HARBS conformer (10 nM; Figure 3C). Roflumilast, which interacts equally with the HARBS and LARBS conformers, induced neurite outgrowth and myelination over the extended low- and high-affinity concentration ranges (Figure 3D).
Figure 3.
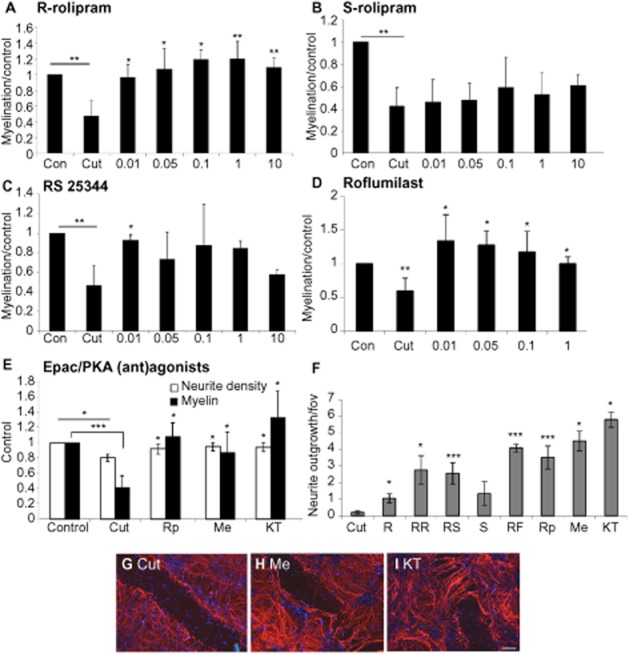
Myelination is primarily mediated via HARBS and Epac. Cut myelinating cultures were treated with varying concentrations (in μM) of R-rolipram (RR; A), S-rolipram (S; B), RS25344 (RS; C),roflumilast (RF; D), Epac/PKA (ant)agonists RpcAMPS (Rp), Me-cAMP (Me), KT5720 (KT; E), and the change in myelination surrounding the lesion, normalized to control, quantified. Neurite density surrounding the lesion was restored to control levels by all drugs tested (data not shown). In addition, the extent of neurite outgrowth into the lesion is shown in F (all at 10 nM), and representative images of the lesion site are shown of untreated cut cultures (G), cultures treated with Me-cAMP (Me; H) and KT5720 (KT; I). Scale bar 100 μm. *P < 0.05, **P < 0.01, ***P < 0.001; significant differences between control and cut, and treatments and cut.
The PKA antagonist, Rp-cAMPS, failed to prevent HARBS conformer-inhibited PDE4 from enhancing myelination, suggesting that such actions may be mediated through the alternative cAMP effector, Epac. Therefore, we treated our culture system with the Epac agonist, Me-cAMP, and found it to increase myelination levels (Figure 3E). As indicated earlier, inhibition of PKA, using either Rp-cAMPS or KT5720, also led to increased myelination levels (Figure 3E). In addition, these agents, which mediated Epac activation or PKA inhibition, all induced major neurite outgrowth across the lesion (Figure 3F–I).
Consistent with a role for the HARBS PDE4 conformer in controlling the pool of cAMP that activates Epac to elicit neurite outgrowth, we noted that both R-rolipram and RS25344 significantly promoted neurite extension across the lesion, while the LARBS selective PDE4 inhibitor, S-rolipram, was ineffective (Figure 3F). With regards to myelination, however, high concentrations of racemic rolipram (10 μM) failed to stimulate neurite outgrowth, indicating an inhibitory effect of cAMP when elevated through inhibition of the LARBS conformer (Figure 3).
These data demonstrate novel, key roles of the pool of cAMP controlled by inhibition of the PDE4 HARBS conformer and activation of Epac in mediating neurite outgrowth and myelination in this in vitro model of SCI.
Rolipram inhibits Rho
The C3 Rho inhibitor induces neurite outgrowth in cut myelinating cultures, whereas the ROCK inhibitor Y27632 enhances myelination (Boomkamp et al., 2012). The stimulatory effect of low concentrations of rolipram on neurite outgrowth suggests that it might mediate its effects on neurite outgrowth via Rho. Indeed, phosphomyosin (pMLC) levels were reduced within the lesion of cultures treated with 50 nM rolipram, to the same extent those after treatment with 1 μg mL−1 C3 or 1 μM Y-27632 (Figure 4A–D). Moreover, pull-down of GTP-Rho using rhotekin-RBD beads and densitometry of Western blot analysis revealed that rolipram (50 nM) reduced levels of activated RhoA, up-regulated after cutting, to the same extent as C3 (1 μg mL−1) and Y27632 (1 μM; Figure 4E). Cutting induced a twofold increase in activated Rho compared with control, whereas treatment with rolipram, C3 or Y27632 reduced this to control levels. Thus, these findings indicate that the neurite outgrowth induced by treatment with rolipram may, at least in part, be mediated by Rho inhibition.
Figure 4.

Rolipram inhibits Rho activation. Cultures were cut and treated with 50 nM rolipram for 6 days prior to immunolabelling with phalloidin (green) and anti-phosphomyosin (red; A–C). Quantification was performed by measuring the intensities of each antibody staining, normalized to the number of DAPI-nuclei, and compared with the effects of Rho/ROCK inhibitors C3 (1 μg mL−1) and Y27632 (1 μM; D). Scale bar 25 μm. *P < 0.05, **P < 0.01, ***P < 0.001; significant differences between control and cut, and treatments and cut. Western blotting for activated RhoA using beads and total Rho was performed on these cell lysates (E). The blots shown are representative of experiments performed three times.
C3 Rho transferase enhances CXCL10 and IL-1β expression
The chemokine CXCL10 is secreted from reactive astrocytes and reduces levels of myelination (Nash et al., 2011). Because of the differential effects of C3, Y27632 and rolipram on myelination and neurite outgrowth, qRT-PCR was performed to determine whether the differential effects of these compounds on myelination and neurite outgrowth were reflected in chemokine levels. Changes in the mRNA for cytokines (CXCL10 and IL-1β) and astrocyte reactivity (nestin and GFAP) in neurosphere-derived astrocytes were measured. Whereas neither Y27632 (1 μM) nor (racemic) rolipram (10 nM) induced any changes in the astrocytic RNA levels after 1 day of treatment, C3 (1 μg mL−1) treatment reduced GFAP and nestin mRNA expression (Figure 5A, B), suggesting reduced astrocyte reactivity. By contrast, levels of CXCL10 and IL1β were up-regulated upon C3 treatment (Figure 5C, D; ∼10- and 30-fold respectively).
Figure 5.
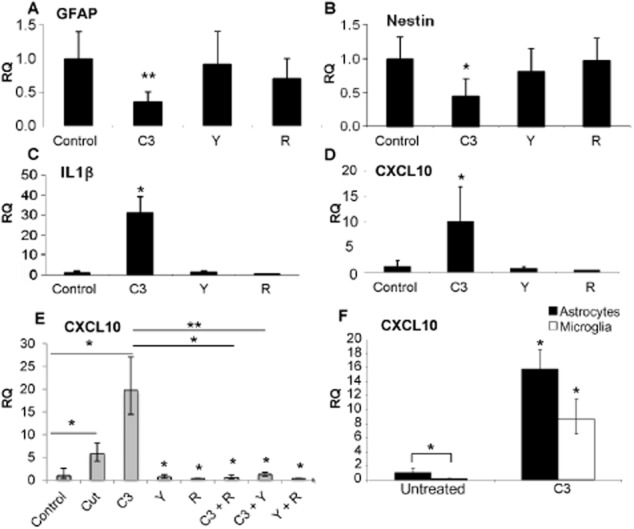
C3 Rho inhibitor enhances expression of mRNA for CXCL10 and IL-1β. qRT-PCR was performed on neurosphere-derived astrocytes (A–D) treated with 1 μg mL−1 C3, 1 μM Y27632 or 10 nM rolipram for 1 day, and levels of GFAP (A), nestin (B), IL-1β (C), CXCL10 (D) assessed. Changes in the RNA levels of CXCL10 were also elucidated in the cut myelinating cultures after treatment with these inhibitors, individually and combined (E) and purified microglia (F). The asterisks indicate changes. *P < 0.05, **P < 0.01; significant differences between control and treatments (A–D, F) and in (E), control and cut, and cut and treatment.
The mRNA levels of CXCL10 were also increased in the cut mixed myelinating cultures (Figure 5E), and further enhanced upon C3 treatment. Interestingly, co-treatment with either Y27632 or (racemic) rolipram with C3 resulted in a reduction of CXCL10 compared with C3 alone (Figure 5E). CXCL10 was mainly produced by astrocytes (Figure 5F), with levels 10-fold higher than in purified microglia; CXCL10 produced by purified OPCs was negligible (data not shown). These data suggest that C3 transferase primarily affects the astrocytes, thereby rendering them unable to support myelination after injury.
Treatment of cut cultures with C3 and antibody to CXCL10 induces neurite outgrowth and myelination
C3 does not promote myelination after cutting of mixed myelinating cultures (Boomkamp et al., 2012). Because of evidence presented earlier, we determined whether the up-regulation of CXCL10 in C3-treated cultures was directly responsible for the absence of enhanced myelination. Cut cultures were treated with C3 (Figure 6C), α-CXCL10 (0.2, 2 and 20 μg mL−1; Figure 6D) or C3 and anti-CXCL10 (Figure 6E, F). Neutralizing CXCL10 antibody alone did not have a significant effect on myelination or neurite density surrounding the lesion, nor neurite outgrowth into the lesion (Figure 6G–I). However, co-treatment with C3 and anti-CXCL10 induced neurite outgrowth to a similar extent as C3 alone, in addition to enhanced myelination surrounding the lesion. These data suggest that the up-regulation of CXCL10 by C3-treated cells affects myelination, which can be overcome by antibody neutralization and illustrates the differing mechanisms of the tested compounds.
Figure 6.
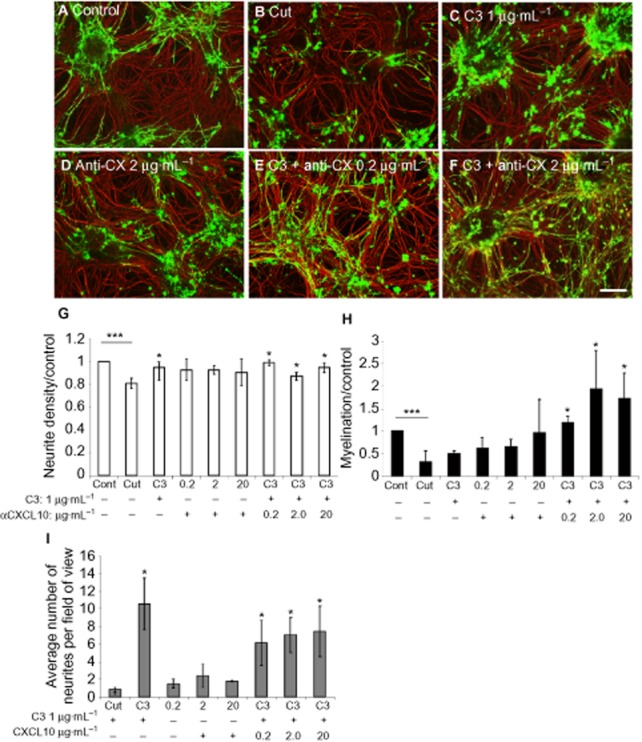
Treatment with C3 and anti-CXCL10 induces neurite outgrowth and myelination. Cut cultures (B) were immunolabelled with SMI-31 (red) and anti-PLP antibody (green) upon treatment with C3 1 μg mL−1 (C), anti-CXCL10 2 μg mL−1 (D), C3 and anti-CXCL10 0.2 μg mL−1 (E) and C3 and anti-CXCL10 2 μg mL−1 (F). The average neurite density (G) and myelination surrounding the lesion (H), normalized to control, and neurite outgrowth across the lesion (I) are shown. Scale bar 100 μm. *P < 0.05, **P < 0.001; significant differences between control and cut, and treatments and cut.
Co-treatments
Because of the ability of (racemic) rolipram to reduce CXCL10 expression by C3-treated cells, we tested whether combined treatment with C3 (1 μg mL−1) and rolipram (50nM) could promote myelination as well as neurite outgrowth (Figure 7). Indeed, enhanced myelination was observed (Figure 7H), and neurite density was restored with all combined treatments (Figure 7G). In addition, whereas Y27632 (1 μM) induced myelination, but not neurite outgrowth (Boomkamp et al., 2012), co-treatment with 50 nM rolipram (racemic) resulted in significant neurite outgrowth (Figure 7D, F, I). Overall, these data suggest that by targeting different, yet converging, pathways, additive effects on the repair of SCI can be achieved (Figure 8).
Figure 7.
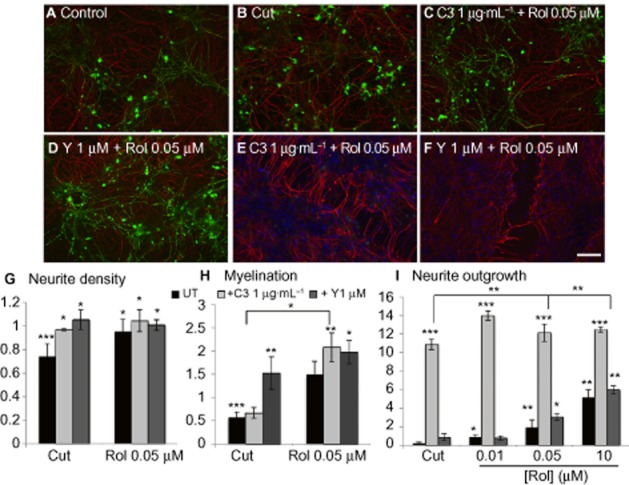
Rho/ROCK and PDE4 inhibition produces additive effects. Cultures were cut (B) and treated with C3 1 μg mL−1 and rolipram (C, E) or Y27632 and rolipram (D, F) for 6 days prior to immunocytochemistry with SMI-31 (red) and anti-PLP (green) labelling. Neurite density (G) and myelination (H) surrounding the lesion, normalized to control and neurite outgrowth (I) were calculated. Scale bar 100 μm. The asterisks indicate changes between control and cut, and treatments and cut. *P < 0.05, **P < 0.01, **P < 0.001; significant differences between control and cut, and treatments and cut. UT, untreated.
Figure 8.
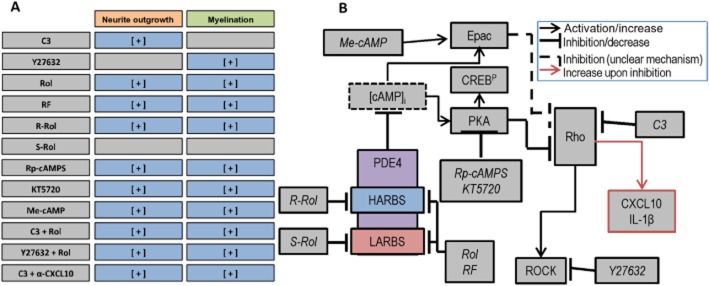
Proposed mechanisms of action of cAMP and Rho/ROCK modulators of neurite outgrowth and myelination. (A) Summary of the effects of the tested compounds (Rol, rolipram; RF, roflumilast; α-CXCL10, antibody to CXCL10) on neurite outgrowth and myelination. (B) Proposed mechanisms and targets of the compounds. cAMP (degraded by PDE4) can activate signalling molecules PKA and Epac. In turn, these effectors can inhibit downstream effector molecules Rho and ROCK. Targets of the inhibitors are indicated.
Discussion
Because of its multifaceted nature, treatment of SCI is one of the most challenging tasks facing regenerative medicine, and it is currently believed that a combination of treatments is required for optimal functional recovery (Lu et al., 2004; Pearse et al., 2004). Extensive research has identified several potential cellular targets that could be modified to facilitate repair. These include neutralizers of inhibitory molecules associated with failure of regeneration, such as Nogo-A and MAG, Rho/ROCK, chondroitin sulphate proteoglycans and also cAMP elevation through inhibition of its degradation through PDE4.
The mechanism by which PDE4 inhibitors promote regeneration is poorly understood. In order to gain insight into this important issue, we set out here to exploit our in vitro mixed myelinating culture system. Cutting mature myelinating cultures induces features typical of the in vivo SCI pathology (Boomkamp et al., 2012). Several cultures of SCI models have been described (Guzmán-Lenis et al., 2009; Zhang et al., 2010; Que et al., 2011), and while they all offer their own advantages, these models are limited in that they do not study myelination. Moreover, some of these cultures lack a complete mix of the cell types that reside in the CNS, which means the cross-talk among various CNS cell types may be lacking, and thus represent an oversimplified system. To overcome this criticism, organotypic cultures have been used, which have merit, but tend to have limited long-term viability, preventing a full characterization of repair strategies over time.
The PDE4 selective inhibitor rolipram promoted neurite outgrowth in a saturable, monophasic manner, suggesting that neurite outgrowth is potentially mediated via a cAMP-signalling pathway. Indeed, reduced levels of activated Rho and phosphomyosin were observed following PDE4 inhibition, which is consistent with studies performed using in vivo models showing Rho involvement (Hannila and Filbin, 2008; Yin et al., 2013). Furthermore, elevated cAMP levels are known to decrease the phosphorylation of cofilin, thus enhancing actin depolymerization and leading to a reduction in stress fibres (Ramachandran et al., 2011). Our data are consistent with the notion that neurite outgrowth induced by PDE4 inhibitors may be mediated via Rho.
In contrast to this, the PDE4 selective inhibitor rolipram promoted myelination surrounding the lesion, with a dose-dependent, bell-shaped curve. This suggests that there may be two processes controlled by cAMP: (i) a stimulatory effect seen when inhibiting PDE4 at low doses of rolipram; and (ii) an inhibitory effect seen when inhibiting PDE4 at high doses of rolipram. There are paradigms for contrasting actions of cAMP, such as in the regulation of DNA-dependent PK intracellular localization and activation, which involve spatially distinct pools of cAMP regulated by different forms of PDE4, where the stimulatory signal is mediated via Epac and the inhibitory signal is mediated via PKA (Huston et al., 2008). It is well-established that PDE4 exists in conformationally distinct states that show different sensitivities to inhibition by rolipram (Souness and Rao, 1997; Houslay and Adams, 2003). These are the so-called HARBS-PDE4 conformer and the LARBS-PDE4 conformer. PDE4 conformers have been traditionally defined pharmacologically by their profound differences in sensitivity to inhibition by rolipram enantiomers, as we do here. Until very recently the structural basis of this profound difference has not been known, as it has proved impossible to crystallize full-length PDE4 enzymes because of their susceptibility to aggregation at high concentration, when purified. Thus, until recently, all structure studies have been undertaken using the truncated catalytic unit. While such studies indicated that rolipram could indeed adopt two binding modes in the catalytic site, this was insufficient to explain the profound difference in binding affinities of the enantiomers. However, a more recent structural study (Houslay and Adams, 2010) used a construct of the N-terminal regulatory region together with the catalytic unit. The basis of the increased binding affinities of rolipram and similar HARBS-related inhibitors is their ability to interact with and stabilize a helix that can ‘cap’ the binding pocket. The HARBS PDE4 conformer is thus a subpopulation of PDE4 enzymes whose capping helix, found N-terminal to the catalytic unit, is not sequestered, for instance through interaction with binding partner proteins, but is freely able to interact with the catalytic pocket, and as such, can be stabilized in the capped configuration by inhibitors such as rolipram. Unfortunately, there are currently no means of identifying the status of the capping helix other than crystallographic analyses or, as used here, by differential inhibition through the action of R- and S-rolipram enantiomers.
Here we exploited the R- and S- enantiomers of rolipram, which selectively inhibit the HARBS and LARBS PDE4 conformers, respectively (Souness and Rao, 1997; Houslay and Adams, 2003), to assess the role of these PDE4 conformers in neurite outgrowth and myelination. R-Rolipram was more potent and effective in enhancing myelination than S-rolipram, which exerted little action. This clearly demonstrated that it was a PDE4 subpopulation that adopts the HARBS conformation, where the regulatory UCR2 domain is able to dock across the catalytic site and enhance the binding of R-rolipram (Burgin et al., 2010; Houslay and Adams, 2010), which was primarily involved in the promotion of myelination after injury. Consistent with this, RS25344, which selectively interacts with the HARBS conformer, potently enhanced myelination, while Roflumilast, which interacts equally with the HARBS and LARBS, enhanced myelination in a bell-shaped manner, comparable to that seen using racemic rolipram.
Rolipram has been reported to be neuroprotective, preserving white matter, promoting axonal regeneration, and reducing oligodendrocyte cell death and central myelinated axon loss after SCI (Nikulina et al., 2004; Whitaker et al., 2008; Schaal et al., 2012), although there have been some controversial findings regarding whether cAMP is always beneficial (Nout et al., 2011). Also, rolipram increased the number of mature oligodendrocytes in purified OPC cultures as well as increasing remyelination in lysolecithin-induced demyelination in cerebellum slice culture and cuprizone-induced demyelination in mice (Sun et al., 2012). Moreover, rolipram enhanced myelination after contusive thoracic SCI (Beaumont et al., 2009). Thus, although rolipram is associated with increased myelination, there have been no studies reported on using rolipram enantiomers in these models.
PDE4 enzymes able to adopt the HARBS conformation have long been thought to mediate the emetic side effects that have compromised the clinical use of PDE4 inhibitors (Burgin et al., 2010). The HARBS conformers may also preferentially promote learning and memory processes (Rose et al., 2005). Nevertheless, over the past decade or more, there has been a drive to generate PDE4 selective inhibitors that do not preferentially target the HARBS conformer for therapeutic use as anti-inflammatory agents (Giembycz and Newton, 2011). Roflumilast, which has now been approved for use as a therapy in COPD (Huang and Mancini, 2006) does not discriminate HARBS from LARBS and, given its higher tolerability in vivo compared with rolipram, may provide a more suitable therapeutic agent for treating SCI. However, as the HARBS PDE4 conformer provides the target for enhanced myelination, administering a HARBS-selective PDE4 inhibitor locally to maximize repair while minimizing systemic availability and access to the emetic centre in the area postrema, may provide a new therapeutic challenge.
In addition to identifying the HARBS conformer of PDE4 as the main inducer of myelination, we found that at high concentrations of rolipram, myelination was not enhanced because of an inhibitory action mediated by PKA. It has been shown by various investigators that inhibition of cAMP breakdown by distinct PDEs, including distinct PDE4 conformers, can induce an increase in cAMP in the intracellular compartment controlled by that particular PDE form (Houslay, 2010). Such spatial actions are then interpreted either by localized PKA or Epac, which will activate distinct processes that can even act antagonistically (Huston et al., 2008). PKA is involved in both Schwann cell myelination (Howe and McCarthy, 2000) and axonal regeneration dependent on developmental age (Cai et al., 2001). However, we show here for the first time that PKA neither promotes myelination nor neurite outgrowth in our CNS culture system. Therefore, a compound that selectively inhibits PKA could provide a therapeutic strategy for the treatment of SCI. By contrast, the Epac agonist Me-cAMP, which selectively activates Epac and does not affect PKA, was found to exert beneficial effects on both neurite outgrowth and myelination. Consistent with this, Me-cAMP promoted neurite outgrowth in neonatal and adult DRG neurons (Murray and Shewan, 2008). Thus, our novel discovery that Epac agonists can promote myelination suggests a therapeutic potential for such compounds in the treatment of SCI and potentially other demyelinating diseases.
In addition to cAMP-elevating drugs, inhibitors of the Rho/ROCK pathway, which act downstream from cAMP, have also gained much interest as a therapeutic strategy for SCI. It has been suggested that the inhibition of neurite outgrowth by the MAG acts by inhibiting cAMP increase and activation of Rho, providing a link between the two pathways (Hannila and Filbin, 2008). The Rho inhibitor C3 transferase (Cethrin) has been tested in a phase I/IIa clinical trial of acute SCI patients (Fehlings et al., 2011). The motor recovery observed suggests that this inhibitor may increase neurological recovery after SCI. In our in vitro model of SCI, the Rho inhibitor C3 induced neurite outgrowth, but failed to induce myelination, whereas the ROCK inhibitor Y27632 promoted myelination, but failed to affect neurite outgrowth into the lesion (Boomkamp et al., 2012). Thus, our findings suggest that treatment of SCI patients with C3 alone may not be the optimum strategy and further investigation is needed into the mechanisms of action of C3 and Y27632 to allow identification of molecules that play a role in the neurite outgrowth and myelination process respectively. Moreover it should be noted, when translating to the clinic, that manipulating cAMP and PKA will affect a wide range of cells and tissues.
In this study, we found that the lack of enhanced myelination in C3-treated cultures was due to elevated CXCL10, a chemokine secreted by activated astrocytes (Ransohoff et al., 1993). Not only does CXCL10 play a role in the recruitment of leukocytes into the CNS, but also the pathogenesis of multiple sclerosis (MS), where it is highly expressed by hypertrophic astrocytes surrounding active demyelinating MS plaques (Sørensen et al., 2002; Omari et al., 2005; Carter et al., 2007; Müller et al., 2007). Monocytes/macrophages have been found to express CXCL10 to a lesser extent (Ransohoff et al., 1993), consistent with our findings. Moreover, studies have shown that CXCL10 can directly affect oligodendrocyte myelination (Nash et al., 2011). In our study, the increased expression of the mRNA for CXCL10 by C3-treated cells was prevented by co-treatment with anti-CXCL10, Y27632 or rolipram, resulting in an increase in myelination, suggesting that these co-treatments are more beneficial than C3 administration alone in promoting features of CNS repair.
In summary, our study shows that the rolipram-induced neurite outgrowth and enhanced myelination in the SCI-simulating cultures occurs primarily via binding to PDE4 isoforms that adopt the HARBS conformation, suggesting the role of distinct PDE4 subpopulations of this nature in the repair of SCI. Intriguingly, both an Epac agonist and PKA antagonists induced similar effects, demonstrating the beneficial role of Epac and the detrimental role of PKA in myelination and neurite outgrowth. Downstream from cAMP, Rho inhibition increased CXCL10 secretion, thought to prevent myelination after injury. This was overcome by neutralizing this chemokine or co-treatment with ROCK or co-inhibition of PDE4. Taken together, using an in vitro model, we can replace and reduce the number of animals necessary to test combinations of therapeutic agents for the potential repair of SCI and have identified more specific and suitable therapeutic targets, including HARBS, Epac and co-inhibition of PDE4 and Rho/ROCK for the treatment of SCI. These findings require validation in animal models of SCI. The interactions between the various inhibitors and their targets are summarized in Figure 8.
Acknowledgments
We thank the National Centre for Replacement, Refinement and Reduction, of Animals in Research for funding (SDB and MAM).
Glossary
- C3
ADP-ribosyltransferase
- COPD
chronic obstructive pulmonary disease
- CSPG
chondroitin sulphate proteoglycan
- GFAP
glial fibrillary acidic protein
- HARBS
high-affinity rolipram-binding site
- LARBS
low-affinity rolipram-binding site
- MAG
myelin-associated glycoprotein
- Me-cAMP
8-pMeOPT-2′-O-Me-cAMP
- MS
multiple sclerosis
- OPCs
oligodendrocyte precursor cells
- PLL
poly-L-lysine
- PLP
proteolipid protein
- PM
plating medium
- pMLC
phosphomyosin
- ROCK
Rho kinase
- SCI
spinal cord injury
Conflict of interest
The authors declare no competing financial interests.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. British Journal of Pharmacology. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont E, Whitaker CM, Burke DA, Hetman M, Onifer SM. Effects of rolipram on adult rat oligodendrocytes and functional recovery after contusive cervical spinal cord injury. Neuroscience. 2009;163:985–990. doi: 10.1016/j.neuroscience.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomkamp SD, Riehle MO, Wood J, Olson M, Barnett SC. The development of a rat in vitro model of spinal cord injury demonstrating the additive effects of Rho and ROCK inhibitors on neurite outgrowth and myelination. Glia. 2012;3:441–456. doi: 10.1002/glia.22278. [DOI] [PubMed] [Google Scholar]
- Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010;28:63–70. doi: 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]
- Cai D, Shen Y, De Bellard M, Tang S, Filbin MT. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron. 1999;22:89–101. doi: 10.1016/s0896-6273(00)80681-9. [DOI] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–4739. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Müller M, Manders PM, Campbell IL. Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFNγ but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia. 2007;55:1728–1739. doi: 10.1002/glia.20587. [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- Day JP, Lindsay B, Riddell T, Jiang Z, Allcock RW, Abraham A, et al. Elucidation of a structural basis for the inhibitor-driven, p62 (SQSTM1)-dependent intracellular redistribution of cAMP phosphodiesterase-4A4 (PDE4A4) Med Chem. 2011;54:3331–3347. doi: 10.1021/jm200070e. [DOI] [PubMed] [Google Scholar]
- Fehlings MG, Theodore N, Harrop J, Maurais G, Kuntz C, Shaffrey CI, et al. A phase I/IIa clinical trial of a recombinant Rho protein antagonist in acute spinal cord injury. J Neurotrauma. 2011;28:787–796. doi: 10.1089/neu.2011.1765. [DOI] [PubMed] [Google Scholar]
- Forgione N, Fehlings MG. Rho-ROCK inhibition in the treatment of spinal cord injury. World Neurosurg. 2013;S1878–8750:16–18. doi: 10.1016/j.wneu.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Giembycz MA, Newton R. Harnessing the clinical efficacy of phosphodiesterase 4 inhibitors in inflammatory lung disease; dual-selective phosphodiesterase inhibitors and novel combination therapies. Handb Exp Pharmacol. 2011;204:415–446. doi: 10.1007/978-3-642-17969-3_18. [DOI] [PubMed] [Google Scholar]
- Guzmán-Lenis MS, Navarro X, Casas C. Drug screening of neuroprotective agents on an organotypic-based model of spinal cord excitoxic damage. Restor Neurol Neurosci. 2009;27:335–349. doi: 10.3233/RNN-2009-0482. [DOI] [PubMed] [Google Scholar]
- Hannila SS, Filbin MT. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp Neurol. 2008;209:321–332. doi: 10.1016/j.expneurol.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD. Underpinning compartmentalised cAMP through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003;370:1–18. doi: 10.1042/BJ20021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD, Adams DR. Putting the lid on phosphodiesterases 4. Nat Biotechnol. 2010;298:38–40. doi: 10.1038/nbt0110-38. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Schafer P, Zhang K. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- Howe DG, McCarthy KD. Retroviral inhibition of cAMP-dependent protein kinase inhibits myelination but not Schwann cell mitosis stimulated by interaction with neurons. J Neurosci. 2000;20:3513–3521. doi: 10.1523/JNEUROSCI.20-10-03513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Mancini JA. Phosphodiesterase 4 inhibitors for the treatment of asthma and COPD. Curr Med Chem. 2006;13:3252–3262. doi: 10.2174/092986706778773040. [DOI] [PubMed] [Google Scholar]
- Huston E, Lynch MJ, Mohamed A, Collins DM, Hill EV, Krause E, et al. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. PNAS. 2008;105:12791–12796. doi: 10.1073/pnas.0805167105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keravis T, Lugnier C. Cyclic nucleotide phosphodiesterases (PDE) and peptide motifs. Curr Pharm Des. 2010;16:1114–1125. doi: 10.2174/138161210790963760. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]
- Lu P, Yang H, Jones LL, Filbin MT, Tuszynski MH. Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J Neurosci. 2004;24:6402–6409. doi: 10.1523/JNEUROSCI.1492-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109:366–398. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Shewan DA. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol Cel Neurosci. 2008;38:578–588. doi: 10.1016/j.mcn.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Müller M, Carter SL, Hofer MJ, Manders P, Getts DR, Getts MT, et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol. 2007;179:2774–2786. doi: 10.4049/jimmunol.179.5.2774. [DOI] [PubMed] [Google Scholar]
- Nash B, Thomson C, Linington C, Athur A, McCLure JD, McBride MW, et al. Modulating astrocyte phenotype affects myelination and is negatively mediated by cxcl10. J Neurosci. 2011;31:13028–13038. doi: 10.1523/JNEUROSCI.1449-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893. doi: 10.1016/s0896-6273(02)00702-x. [DOI] [PubMed] [Google Scholar]
- Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101:8786–8790. doi: 10.1073/pnas.0402595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nout YS, Culp E, Schmidt MH, Tovar CA, Pröschel C, Mayer-Pröschel M, et al. Glial restricted precursor cell transplant with cyclic adenosine monophosphate improved some autonomic functions but resulted in a reduced graft size after spinal cord contusion injury in rats. Exp Neurol. 2011;227:159–171. doi: 10.1016/j.expneurol.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omari KM, John GR, Sealfon SC, Raine CS. CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain. 2005;128:1003–1015. doi: 10.1093/brain/awh479. [DOI] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610–616. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai D, McAtee M, Hoffman PN, Bregman BS, et al. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Que H, Liu Y, Jia Y, Liu S. Establishment and assessment of a simple and easily reproducible incision model of spinal cord neuron cells in vitro. In vitro Cell Dev Biol Anim. 2011;47:558–564. doi: 10.1007/s11626-011-9443-2. [DOI] [PubMed] [Google Scholar]
- Ramachandran C, Partil RV, Sharif NA, Srinivas SP. Effect of elevated intracellular cAMP levels on actomyosin contraction in bovine trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2011;52:1474–1485. doi: 10.1167/iovs.10-6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, et al. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- Rose GM, Hopper A, De Vivo M, Tehim A. Phosphodiesterase Inhibitors for cognitive enhancement. Curr Pharm Des. 2005;11:3329–3334. doi: 10.2174/138161205774370799. [DOI] [PubMed] [Google Scholar]
- Schaal SM, Garg MS, Ghosh M, Lovera L, Lopez M, Patel M, et al. The therapeutic profile of rolipram, PDE target and mechanism of action as a neuroprotectant following spinal cord injury. PLoS ONE. 2012;7:e43634. doi: 10.1371/journal.pone.0043634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Souness JE, Rao S. Proposal for pharmacologically distinct conformers of PDE4 cyclic AMP phosphodiesterases. Cell Signal. 1997;9:227–236. doi: 10.1016/s0898-6568(96)00173-8. [DOI] [PubMed] [Google Scholar]
- Sørensen A, Moffat K, Thomson C, Barnett SC. Astrocytes, but not olfactory ensheathing cells or Schwann cells, promote myelination of CNS axons in vitro. Glia. 2008;56:750–763. doi: 10.1002/glia.20650. [DOI] [PubMed] [Google Scholar]
- Sørensen TL, Trebst C, Kivisäkk P, Klaege KL, Majmudar A, Ravid R, et al. Multiple sclerosis: a study of CXCL0 and CXCR3 co-localisation in the inflamed central nervous system. J Neuroimmunol. 2002;127:59–68. doi: 10.1016/s0165-5728(02)00097-8. [DOI] [PubMed] [Google Scholar]
- Spina D. PDE4 inhibitors: current status. Br J Pharmacol. 2008;155:308–315. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Liu Y, Liu B, Xiao Z, Zhang L. Rolipram promotes remyelination possibly via MEK-ERK signal pathway in cuprizone-induced demyelination mouse. Exp Neurol. 2012;237:304–311. doi: 10.1016/j.expneurol.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Thomson CE, McCulloch M, Sorenson A, Barnett SC, Seed BV, Griffiths IR, et al. Myelinated, synapsing cultures of murine spinal cord – validation as an in vitro model of the central nervous system. Eur J Neurosci. 2008;28:1518–1535. doi: 10.1111/j.1460-9568.2008.06415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker CM, Beaumont E, Wells MJ, Magnuson DS, Hetman M, Onifer SM. Rolipram attenuates acute oligodendrocyte death in the adult rat ventrolateral funiculus following contusive cervical spinal cord injury. Neurosci Lett. 2008;438:200–204. doi: 10.1016/j.neulet.2008.03.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura T, Konola JT, Wekerle H, Lees MB. Monoclonal antibodies against myelin proteolipid protein: Identification and characterization of two major determinants. J Neurochem. 1991;57:1671–1680. doi: 10.1111/j.1471-4159.1991.tb06367.x. [DOI] [PubMed] [Google Scholar]
- Yin Y, Sun W, Li Z, Zhang B, Cui H, Deng L, et al. Effects of combining methylprednisolone with rolipram on functional recovery in adult rats following spinal cord injury. Neurochem Int. 2013;62:903–912. doi: 10.1016/j.neuint.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Zhang HT, Zhao Y, Huang Y, Deng C, Hopper AT, De Vivo M, et al. Antidepressant-like effects of PDE4 inhibitors mediated by the high-affinity rolipram binding state (HARBS) of the phosphodiesterase-4 enzyme (PDE4) in rats. Psychopharmacology (Berl) 2006;186:209–217. doi: 10.1007/s00213-006-0369-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, O'Carroll SJ, Wu A, Nicholson LF, Green CR. A model for ex vivo spinal cord segment culture – a tool for analysis of injury repair strategies. J Neurosci Methods. 2010;192:49–57. doi: 10.1016/j.jneumeth.2010.07.012. [DOI] [PubMed] [Google Scholar]