

Abstract
Background and Purpose
1,4-Benzoquinones are well-known inhibitors of 5-lipoxygenase (5-LOX, the key enzyme in leukotriene biosynthesis), but the molecular mechanisms of 5-LOX inhibition are not completely understood. Here we investigated the molecular mode of action and the pharmacological profile of the novel 1,4-benzoquinone derivative 3-((decahydronaphthalen-6-yl)methyl)-2,5-dihydroxycyclohexa-2,5-diene-1,4-dione (RF-Id) in vitro and its effectiveness in vivo.
Experimental Approach
Mechanistic investigations in cell-free assays using 5-LOX and other enzymes associated with eicosanoid biosynthesis were conducted, along with cell-based studies in human leukocytes and whole blood. Molecular docking of RF-Id into the 5-LOX structure was performed to illustrate molecular interference with 5-LOX. The effectiveness of RF-Id in vivo was also evaluated in two murine models of inflammation.
Key Results
RF-Id consistently suppressed 5-LOX product synthesis in human leukocytes and human whole blood. RF-Id also blocked COX-2 activity but did not significantly inhibit COX-1, microsomal PGE2 synthase-1, cytosolic PLA2 or 12- and 15-LOX. Although RF-Id lacked radical scavenging activity, reducing conditions facilitated its inhibitory effect on 5-LOX whereas cell stress impaired its efficacy. The reduced hydroquinone form of RF-Id (RED-RF-Id) was a more potent inhibitor of 5-LOX as it had more bidirectional hydrogen bonds within the 5-LOX substrate binding site. Finally, RF-Id had marked anti-inflammatory effects in mice in vivo.
Conclusions and Implications
RF-Id represents a novel anti-inflammatory 1,4-benzoquinone that potently suppresses LT biosynthesis by direct inhibition of 5-LOX with effectiveness in vivo. Mechanistically, RF-Id inhibits 5-LOX in a non-redox manner by forming discrete molecular interactions within the active site of 5-LOX.
Keywords: 5-lipoxygenase; leukotriene; inflammation; 1,4-benzoquinone; leukocytes
Introduction
5-Lipoxygenase (5-LOX), a non-haeme iron-containing dioxygenase, initiates the biosynthesis of leukotrienes (LTs) from arachidonic acid (AA). LTs are involved in the pathogenesis of asthma and allergic rhinitis, but may also play a role in atherosclerosis and cancer (Peters-Golden and Henderson, 2007; Radmark et al., 2007). Upon cell stimulation, the cytosolic PLA2 (cPLA2) releases AA that is converted by the enzyme 5-LOX into LTA4. The conversion of AA induced by 5-LOX is facilitated by the nuclear membrane-bound 5-LOX-activating protein (FLAP), which will ultimately determine the biosynthesis of the LTs (Evans et al., 2008). LTA4 is then converted to other LTs (i.e. LTB4 or cysteinyl-LTs) by LTA4 hydrolase or LTC4 synthase, depending on the cell type (Radmark et al., 2007). LTB4 acts as potent pro-inflammatory agent by inducing chemotaxis and activation of leukocytes, whereas the cys-LTs essentially cause vaso- and bronchoconstriction (Back et al., 2011). Because of the significant pathophysiological role of LTs, pharmacological concepts have been developed to either block the action of LTs or to inhibit their biosynthesis (Werz and Steinhilber, 2006).
Inhibition of cPLA2 or of 5-LOX as well as competition with FLAP are effective pharmacological strategies that interfere with LT biosynthesis and there are currently novel 5-LOX and FLAP inhibitors undergoing clinical trials (Tardif et al., 2010; Wasfi et al., 2012; Bain et al., 2013). 5-LOX inhibitors are classified into (i) redox-type inhibitors that interfere with the redox cycle of the active-site iron, (ii) iron ligand-type inhibitors that chelate the active-site iron, (iii) non-redox-type inhibitors that compete with AA and/or fatty acid hydroperoxides and (iv) ‘novel type’ 5-LOX inhibitors with distinct modes of action (Pergola and Werz, 2010). However, only zileuton (Figure 1), an iron ligand-type 5-LOX inhibitor has been approved as a LT synthesis inhibitor for pharmacotherapy (Pergola and Werz, 2010).
Figure 1.
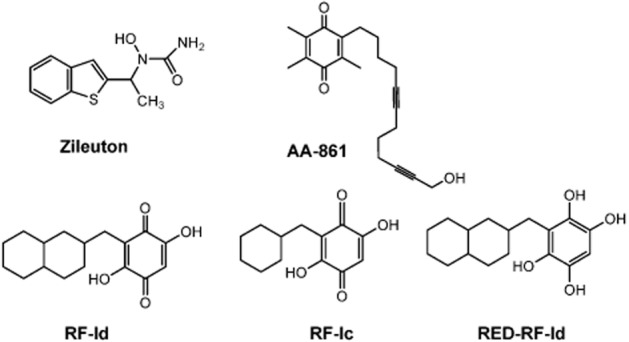
Chemical structures of zileuton, AA-861, RF-Id, RED-RF-Id and RF-Ic.
We have recently discovered a series of 1,4-benzoquinones, exemplified by 3-((decahydronaphthalen-6-yl)methyl)-2,5-dihydroxycyclohexa-2,5-diene-1,4-dione (RF-Id; Figure 1), that act as potent inhibitors of 5-LOX in human neutrophils (IC50 = 0.58 μM; Filosa et al., 2013). Compounds possessing a 1,4-benzoquinone moiety exhibit multiple biological properties, including antioxidant, anti-inflammatory and anti-cancer activities (Dandawate et al., 2010; Petronzi et al., 2011; 2013; Schaible et al., 2013). The anti-inflammatory effects of 1,4-benzoquinones have been found to be associated with suppression of LT formation (Werz, 2007), and the 1,4-benzoquinone AA-861 (Figure 1) is a well-recognized 5-LOX inhibitor (Yoshimoto et al., 1982). Even though AA-861 was shown to be effective in numerous animal models of inflammation, the molecular mode of action of 5-LOX inhibition is still unclear and inhibition of other relevant targets (e.g. 12-LOX) or mechanisms (PGE2 suppression) have been proposed (Ohuchi et al., 1983; Nakadate et al., 1985). Due to the reducing properties of the intracellular milieu (e.g. presence of glutathione in the millimolar range), the 1,4-benzoquinone moiety is thought to be reduced to a 1,4-diphenol structure (=1,4-hydroquinone) that may then either reduce the 5-LOX active-site iron or scavenge lipid hydroperoxides that are needed to activate 5-LOX, suggesting that the reducing character of the substance eventually confers 5-LOX inhibition (Ohkawa et al., 1991; Poeckel et al., 2006). However, the potency of 1,4-benzoquinones does not solely depend on its reducing properties, as its lipophilicity seems to be another specific determinant of potency (Ford-Hutchinson et al., 1994; Werz, 2007). Interestingly, a competitive mode of 5-LOX inhibition has been proposed for AA-861 (Hofmann et al., 2012).
Here we present the results of an analysis of the molecular mode of action and the pharmacological profile of compound RF-Id acting as a 5-LOX inhibitor. We have also demonstrated that it has an anti-inflammatory effect in two mice models. We propose that RF-Id inhibits the activity of the enzyme 5-LOX as a result of its discrete molecular interactions with 5-LOX, but does not affect the redox cycle of its active-site iron.
Methods
Materials
Compounds RF-Id and RF-Ic (used as racemates) were synthesized and characterized as reported previously (Filosa et al., 2013). RED-RF-Id (used as a racemate) was synthesized as follows: tin granulate (4 mg, 0.033 mmol) was added slowly to a suspension of RF-Id (0.028 mmol) in HCl 37% (0.5 mL). The reaction was warmed to 100°C for 1 h. After being cooled to room temperature, the mixture was diluted with water (3 mL) and ethyl acetate (3 mL). The organic layer was washed with water (3 mL) and saline solution (3 mL), separated and dried over Na2SO4. The solvent was evaporated under reduced pressure and RED-RF-Id was produced as a brown solid (yield 86%). 1H NMR (CD3OD, 300 MHz) δ 0.96–1.88 (m, 17H), 2.25–2.40 (m, 1H), 2.51–2.68 (m, 1H), 3.88 (br, 4H), 6.77 (s, 1H). Because docking simulations revealed comparable or even identical GoldScores for the R- and S-enantiomers (see below), we used the racemic mixtures of RF-Id, RF-Ic and RED-RF-Id for the biological evaluations. Hyperforin and MK886 were generous gifts from Schwabe AG (Karlsruhe, Germany) and Merck Frosst (Montreal, Canada) respectively. Zileuton was obtained from Sequoia Research Products (Oxford, UK), PGH2 from Larodan (Malmö, Sweden) and IL-1β, ReproTech (Hamburg, Germany). RSC-3388, ovine isolated COX-1 and human recombinant COX-2 were from Cayman Chemical (Ann Arbor, MI, USA). HPLC solvents were from VWR International GmbH (Darmstadt, Germany). Arachidonic acid, the Ca2+ ionophore A23187, celecoxib, diamide, DTT, N-formyl-methionyl-leucyl-phenylalanine (fMLP), indomethacin, LPS and all other fine chemicals were from Sigma-Aldrich (Taufkirchen, Germany) unless stated otherwise. Nomenclature for the receptors and molecular targets studied here conforms to Alexander et al., 2013.
Docking simulation
Docking was performed with the GOLD software (5.01) (GOLD 5.01, 2012) package under the Suse LINUX operating system. The starting geometry of the ligand was calculated with OMEGA 2.2.1. (OMEGA version 2.2.1, 2007). The X-ray crystal structure data of 5-LOX was obtained from the Protein Data Bank (PDB; Berman et al., 2000). The A chain of the PDB entry 3o8y (Gilbert et al., 2011) was prepared as the binding protein. Water molecules inside the binding pocket were set on toggle and spin. The binding site was defined around the iron (Fe2_1_A, X = 4.05, Y = 21.35, Z = −0.28) in a 10 Å radius. The programme was set to calculate the 10 best ranked poses. As a scoring function, Goldscore fitness was calculated. Otherwise, default settings were used. The resulting poses and potential interactions were visualized in LigandScout 3.02 (Wolber et al., 2009).
Cells
Neutrophils, monocytes and platelets were isolated from human blood from healthy adult volunteers, with consent, obtained from the Institute of Transfusion Medicine, University Hospital Jena, as described previously (Schaible et al., 2013). Washed platelets were finally resuspended in PBS pH 7.4 and 1 mM CaCl2, whereas neutrophils and monocytes were finally resuspended in PBS pH 7.4 containing 1 mg·mL−1 glucose and 1 mM CaCl2 (PGC buffer). A549 cells were grown in DMEM/high glucose (4.5 g·L−1) medium supplemented with heat-inactivated fetal calf serum (10%, v v−1), 100 U·mL−1 penicillin/100 μg·mL−1 streptomycin at 37°C in a 5% CO2 incubator.
For cytotoxicity analysis, neutrophil viability was analysed by trypan blue exclusion with a Vi-cell counter (Beckmann Coulter GmbH, Krefeld, Germany). Cytotoxicity of RF-Id in monocytes incubated for 24 h at 37°C was analysed by use of the MTT assay as described previously (Koeberle et al., 2008).
Determination of 5-LOX product formation in cell-based assays
For assays of intact cells stimulated with Ca2+-ionophore A23187 or 0.3 M NaCl, 5 × 106 freshly isolated neutrophils were resuspended in 1 mL PGC buffer. After pre-incubation with the compounds (15 min, 37°C), 5-LOX product formation was started by addition of 1 mM CaCl2 and 2.5 μM A23187 or 0.3 M NaCl plus AA at the indicated concentrations respectively. After 10 min at 37°C, the reaction was stopped by addition of 1 mL of methanol. For stimulation with fMLP, neutrophils were first primed with 1 μg·mL−1 LPS for 15 min at 37°C before the addition of the compounds. After another 5 min, 0.3 U·mL−1 adenosine deaminase and 10 min later, 1 μM fMLP was added. After 5 min at 37°C, the reaction was stopped. For monocytes (5 × 106 cells·mL−1), cells were primed with 1 μg·mL−1 LPS at 37°C. After 15 min, compounds were added and incubated for another 15 min at 37°C followed by stimulation with 1 μM fMLP for 10 min at 37°C. Cells were placed on ice, centrifuged at 800× g for 5 min at 4°C and the supernatant was added to methanol. The 5-LOX metabolites formed were extracted and analysed by HPLC as described previously (Pergola et al., 2012). 5-LOX product formation is expressed as ng of 5-LOX products per 106 cells, which includes LTB4 and all of its trans isomers, 5(S),12(S)-di-hydroxy-6,10-trans-8,14-cis-eicosatetraenoic acid (5(S),12(S)-DiHETE), and 5(S)-hydro(pero)xy-6-trans-8,11,14-cis-eicosatetraenoic acid (5-H(p)ETE). Cysteinyl LTs C4, D4 and E4 were not detected, and oxidation products of LTB4 were not determined. For the determination of cysLTs in supernatants of monocytes, a cys-LT elisa kit from Enzo Life Sciences International Inc., Lörrach (Germany) was used; this detects LTC4, LTD4 and LTE4.
For whole blood assays, freshly withdrawn blood was obtained by venipuncture and collected in monovettes containing 16 I.E. heparin mL−1. Aliquots of 2 mL were pre-incubated with the test compounds or with vehicle (0.1% DMSO) for 15 min at 37°C, as indicated, and formation of 5-LOX products was started by addition of 30 μM A23187 for 10 min at 37°C or first primed with 1 μg·mL−1 LPS for 30 min at 37°C and then stimulated with 1 μM fMLP for 15 min at 37°C. The reaction was stopped on ice and the samples were centrifuged (600× g, 10 min, 4°C). Aliquots of the resulting plasma (500 μL) were then mixed with 2 mL of methanol. The samples were placed at −20°C for 2 h and centrifuged again (600× g, 15 min, 4°C). The supernatants were collected and diluted with 2.5 mL PBS and 75 μL of 1 N HCl. The 5-LOX metabolites formed were extracted and analysed by HPLC as described for intact neutrophils.
Expression and purification of human recombinant 5-LOX from Escherichia coli, and determination of 5-LOX activity in cell-free systems
E.coli BL21 was transformed with pT3-5-LOX plasmid and the human recombinant 5-LOX protein expressed at 37°C was purified as described previously (Pergola et al., 2012) and immediately used for 5-LOX activity assays. The 5-LOX (0.5 μg) was diluted with PBS/EDTA and pre-incubated with the test compounds. After 15 min at 4°C, samples were pre-warmed for 30 s at 37°C, and 2 mM CaCl2 plus the indicated concentrations of AA were added to start 5-LOX product formation. For the determination of products in cell homogenates, neutrophils (5 × 106) were resuspended in 1 mL PBS containing 1 mM EDTA for 5 min at 4°C and sonicated (4 × 10 s, 4°C). After addition of 1 mM ATP, the homogenates were incubated with the indicated compounds for 15 min at 4°C, pre-warmed for 30 s at 37°C and the reaction was started by the addition of 2 mM CaCl2 and 20 μM AA. After 10 min at 37°C, the metabolites formed were analysed by HPLC as described for intact cells.
Analysis of subcellular redistribution of 5-LOX
The subcellular fractionation of neutrophils by mild detergent lysis was performed as described previously (Pergola et al., 2008). Briefly, neutrophils were pre-incubated for 15 min at 37°C with test compounds, stimulated with 2.5 μM A23187 for 5 min, and chilled on ice. Cells were then lysed by additions of 0.1% Nonidet P-40, and subsequent centrifugation yielded a nuclear and a non-nuclear fraction. 5-LOX in these fractions was analysed by SDS-PAGE and Western Blotting using mouse anti-5-LOX primary monoclonal antibody (generous gift from Dr Dieter Steinhilber, Goethe University, Frankfurt am Main, Germany), and infrared labelled secondary antibody IRDye 800CW, goat anti-mouse (LI-COR Biosciences, Lincoln, NE). For detection of 5-LOX the Odyssey Infrared Imaging System (LI-COR Bioscience) was used and for its analysis, the Odyssey application software (version 3.0.25) was used (Pergola et al., 2012).
Diphenylpicrylhydrazyl radical (DPPH) assay
The antioxidant activity of test compounds was assessed by the method of Blois (1958), with slight modifications. Briefly, 100 μL of 25, 50 or 100 μM compound in DMSO (corresponding to 2.5, 5 or 10 nmol) were added to 100 μL of a solution of the stable free radical DPPH in ethanol (50 μM, corresponding to 5 nmol), buffered with acetate to pH 5.5, in a 96-well plate. After 30 min incubation under gentle shaking in the dark, the absorbance was recorded at 520 nm.
Activity assays of isolated COX-1 and COX-2
Purified COX-1 (ovine, 50 U) or COX-2 (human recombinant, 20 U) was diluted in 1 mL reaction mixture containing 100 mM Tris buffer pH 8, 5 mM glutathione, 5 μM haemoglobin, and 100 μM EDTA at 4°C and pre-incubated with the test compound for 5 min. Samples were pre-warmed for 60 s at 37°C, and AA (5 μM for COX-1, 2 μM for COX-2) was added to start the reaction (Koeberle et al., 2008). After 5 min at 37°C, the reaction was stopped, PGB1, as standard, was added and the COX product 12-hydroxy-5,8,10-heptadecatrienoic acid (12-HHT) was extracted and then analysed by HPLC.
Determination of the COX-1-derived product 12-HHT in human platelets
Freshly isolated human platelets (108 mL−1 PGC buffer) were pre-incubated with test compounds for 15 min at 37°C and stimulated for 10 min at 37°C with 5 μM AA. The COX reaction was stopped after 10 min at 37°C and the 12-HHT formed was analysed by HPLC as described previously (Albert et al., 2002).
Determination of the COX-2-derived product 6-keto PGF1α in intact A549 cells
A549 cells were stimulated with 2 ng·mL−1 IL-1β for 72 h to induce the expression of COX-2 expression and then pre-incubated (2 × 106 cells mL−1 PGC buffer) with the indicated compounds for 15 min at 37°C. After stimulation for 15 min at 37°C with 3 μM AA, the reaction was stopped on ice, the supernatants were recovered after centrifugation and 6-keto PGF1α formation was measured with elisa [6-keto PGF1α was from Sapphire Bioscience (Waterloo, Australia)].
Determination of the activity of isolated human recombinant cPLA2α in a cell-free assay
Expression of His-tagged human recombinant cPLA2α in baculovirus-infected Sf9 cells and its isolation using Ni-NTA agarose beads was performed as described previously (Hoffmann et al., 2010). The release of AA from large unilamellar vesicles, consisting of 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphocholine and 1-palmitoyl-2-oleoyl-sn-glycerol (ratio of 2:1), induced by his-tagged cPLA2 was determined and subsequent analysis of AA by RP-HPLC was performed as described previously (Hoffmann et al., 2010).
Determination of microsomal PGE2 synthase-1 (mPGES-1) activity
Preparation of A549 cells and determination of the activity of mPGES-1 was performed as described previously (Koeberle et al., 2008). In brief, IL-1β-treated A549 cells overexpressing mPGES-1 were sonicated and the microsomal fraction was prepared by differential centrifugation at 10 000× g for 10 min and at 174 000× g. The resuspended microsomal membranes were pre-incubated with the test compounds or vehicle (DMSO). After 15 min, PGE2 formation was initiated by addition of PGH2 (final concentration, 20 μM). After 1 min at 4°C, the reaction was terminated, and PGE2 was separated by solid-phase extraction (RP-18 material) and analysed by RP-HPLC as described previously (Koeberle et al., 2008).
Animals
Male adult CD1 mice (25–35 g, Harlan, Milan, Italy) were housed in a controlled environment (23°C, humidity range of 40 to 70% and 12 h light/dark cycles) and provided with standard rodent chow and water. The experimental protocols were approved by the Animal Care Committee of the University of Naples. Animal care complied with Italian regulations on protection of animals used for experimental and other scientific purpose (Ministerial Decree 116192) as well as with the European Economic Community regulations (Official Journal of E.C. L 358/1 12/18/1986). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Mouse paw oedema
Mice were lightly anaesthetized by inhalation of enflurane and depth of anaesthesia was assessed by checking both abdominal and pedal withdrawal reflex. They were then given a subplantar injection of 50 μL of carrageenan 1% (w v−1). Paw volume was measured using a hydroplethismometer specially modified for small volumes (Ugo Basile, Milan, Italy) immediately before the subplantar injection (basal value) and 2, 4, 6, 24, 48 and 72 h thereafter. Mice were divided into seven groups (n = 6) and received an i.p. injection of compound RF-Id (0.1, 1 and 10 mg·kg−1) or vehicle (DMSO), 30 min before the carrageenan injection.
Mouse air pouch
Mice (28–30 g, two groups, n = 6, each) received an i.p. injection of compound RF-Id (0.1 or 1 mg kg−1), indomethacin (2.5 mg·kg−1) or vehicle (DMSO), 30 min before induction of inflammation. Mice were then lightly anaesthetized with enflurane. Air pouches were developed by s.c. injection of 2.5 mL sterile air into the back of the mice. Three days later, 2.5 mL of sterile air was re-injected into the same cavity. After another 3 days, 1 mL of zymosan 1% (w v−1) or vehicle (saline) was injected into the air pouch, and after another 4 h, mice were killed by CO2 exposure and exudate in the pouch was collected with 1 mL of saline, placed in graduated tubes and centrifuged at 125× g for 10 min. The supernatant was analysed for LTB4 and PGE2 by elisa (Enzo Life Sciences, Lörrach, Germany, and Biotrend, Cologne, Germany, respectively) according to the manufacturer's instructions. The pellet was suspended in 500 μL of saline and leukocytes were evaluated by optical microscopy of the cell suspension diluted with Turk's solution.
Statistics
Data are expressed as mean ± SEM. IC50 values were calculated by non-linear regression using GraphPad Prism software (San Diego, CA, USA) one-site binding competition. Statistical evaluation of the data was performed by one-way anova followed by a Bonferroni or Tukey-Kramer post hoc test for multiple comparisons respectively. A P-value <0.05 (*) was considered significant.
Results
Inhibition of 5-LOX product synthesis by compound RF-Id in isolated leukocytes
In agreement with our previous findings (Filosa et al., 2013), RF-Id concentration-dependently suppressed the synthesis of 5-LOX products in A23187-stimulated human neutrophils (Figure 2A, IC50 = 0.9 ± 0.1 μM). Supplementation with exogenous AA (20 or 40 μM) did not alter the potency of RF-Id, suggesting that RF-Id does not primarily act at the level of substrate supply by inhibiting cPLA2 or at the level of AA transfer (via FLAP) to 5-LOX. Interestingly, the structural derivative RF-Ic, which carries a cyclohexyl moiety instead of the decahydronaphthalene (Figure 1) was inactive in this respect (Figure 2B).
Figure 2.
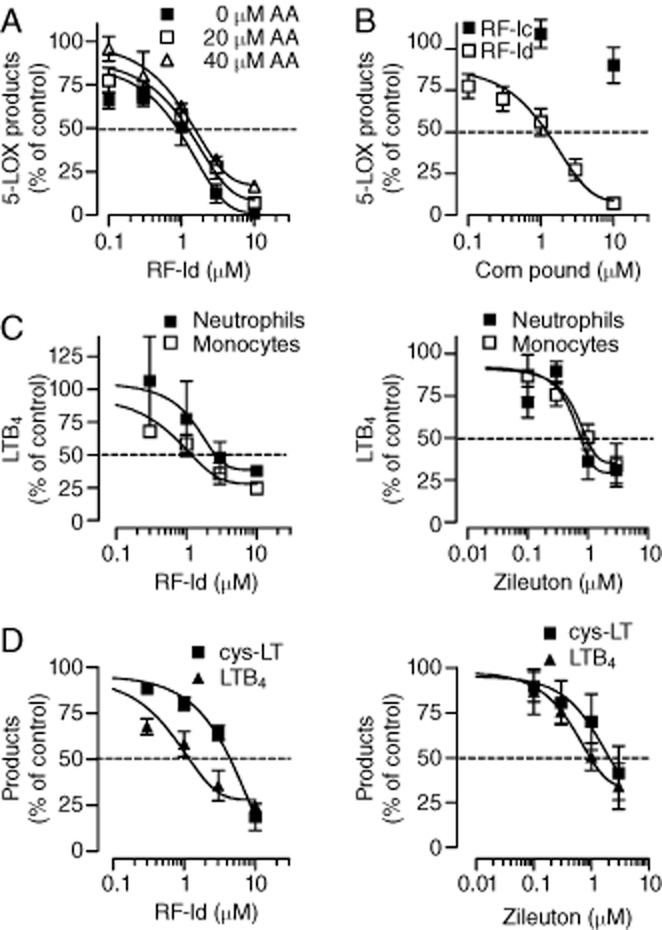
Inhibition of 5-LOX product formation by RF-Id in intact cells. (A) Neutrophils were pre-incubated with RF-Id for 15 min at 37°C and stimulated with 2.5 μM A23187 plus AA as indicated for 10 min at 37°C. Data are means ± SEM; n = 3. (B) Intact neutrophils were pre-incubated with RF-Ic or RF-Id for 15 min, 37°C and stimulated with 2.5 μM A23187 plus 20 μM AA for 10 min at 37°C. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3. (C) Neutrophils were primed for 15 min with 1 μg·mL−1 LPS at 37°C, after 5 min 0.3 U·mL−1 Ada was added for 10 min, and cells were then pre-incubated with the compounds (RF-Id, left panel; zileuton, right panel) for 15 min at 37°C before stimulation with 1 μM fMLP for 5 min at 37°C. Mononcytes were primed with 1 μg mL−1 LPS at 37°C for 5 min, then compounds were added for 15 min, and cells were stimulated for 10 min at 37°C with 1 μM fMLP. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3. (D) Effects of RF-Id on LTB4 and cysLT formation in monocytes. Monocytes were primed with 1 μg mL−1 LPS at 37°C for 5 min, then compounds (RF-Id, left panel; zileuton, right panel) were added for 15 min, and the cells stimulated for 10 min at 37°C with 1 μM fMLP. LTB4 was quantified by HPLC and cysLT levels were analysed by elisa in supernatants respectively. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3.
In order to apply more biologically relevant assay conditions, neutrophils were stimulated with LPS and the chemotactic peptide fMLP. Furthermore, human isolated monocytes, a major source for biosynthesis of LTB4 and of cysLTs (Pergola et al., 2011), were used as an additional cell-based test system. As shown in Figure 2C, RF-Id concentration-dependently and equally well inhibited LTB4 formation in both neutrophils and monocytes (IC50 = 2.6 and 1.6 μM, respectively) that had been primed with LPS and challenged with fMLP, in a similar manner to zileuton (IC50 = 0.8 and 1.5 μM, respectively), a well-recognized and clinically used 5-LOX inhibitor (Carter et al., 1991), which was applied as a reference drug. RF-Id and zileuton blocked the formation of LTB4 and of cysLTs in monocytes with comparable potencies, with RF-Id being slightly more potent at suppressing the formation of LTB4 than the cys-LTs (IC50 = 1.6 and 4.2 μM respectively; Figure 2D). Taken together, these results indicate that RF-Id inhibits the formation of 5-LOX products in intact cells with consistent potency that is not markedly affected by the cell type, the type of stimulus or the quantity of the substrate supplied.
Investigations into the mechanism of the inhibitory effect of RF-Id on 5-LOX
As reported previously, RF-Id also inhibited 5-LOX activity under cell-free conditions when isolated 5-LOX was used as the enzyme source (Filosa et al., 2013). RF-Id was less active in neutrophil homogenates (IC50 = 10 ± 0.6 μM) than in intact neutrophils (IC50 = 0.9 ± 0.1 μM; Figure 3A) implying that for efficient suppression of 5-LOX activity, the intact cellular environment is needed. The possibility that the cytotoxic effects of RF-Id lead to impaired 5-LOX product formation in intact cells can be excluded, as RF-Id up to 10 μM had no significant detrimental effects on the viability of neutrophils or monocytes after 1 and 24 h of incubation respectively (not shown). Some inhibitors of LT synthesis act by preventing the nuclear translocation of 5-LOX and its interaction with FLAP at the nuclear membrane (Werz, 2002; Evans et al., 2008). However, A23187-induced 5-LOX translocation from the cytosol to the nucleus in neutrophils was unaffected by RF-Id (Figure 3B), whereas the novel-type 5-LOX inhibitor hyperforin blocked this process (Figure 3B), as expected (Feisst et al., 2009).
Figure 3.
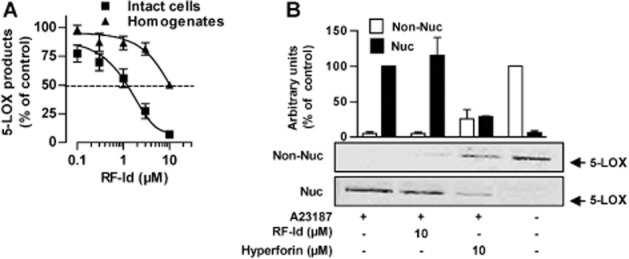
Effects of RF-Id on 5-LOX activity in intact cells and homogenates and on 5-LOX translocation. (A) Intact neutrophils were pre-incubated with RF-Id for 15 min, 37°C and stimulated with 2.5 μM A23187 plus 20 μM AA for 10 min at 37°C. Neutrophil homogenates were incubated with RF-Id for 15 min at 4°C. After addition of 1 mM ATP, samples were warmed up for 30 s at 37°C and stimulated with 2 mM CaCl2 and 20 μM AA for 10 min at 37°C. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3. (B) Effects of RF-Id and hyperforin on 5-LOX subcellular localization in neutrophils following stimulation with 2.5 μM A23187 for 10 min at 37°C. The distribution of 5-LOX was analysed by Western blot in the nuclear and non-nuclear fraction of mild detergent (0.1% NP-40)-lysed cells. One representative experiment of three independent experiments is shown. Values are given as arbitrary densitometric units. Data are expressed as percentage of control (the 5-LOX band from unstimulated neutrophils was set to 100% in Non-Nuc, the 5-LOX band from A23187-activated neutrophils was set 100% in Nuc), means + SEM., n = 3.
In contrast to the antioxidants ascorbic acid and L-cysteine, RF-Id (and also RF-Ic) showed fairly weak radical scavenging activity in the DPPH assay (Figure 4A). Hence, it is possible that RF-Id requires the reducing milieu of the cell (abundance of thiols like glutathione, approx. 5 mM) in order to be transformed into a radical scavenging form of hydroquinone, which has been proposed as a requirement for 5-LOX inhibition for other 1,4-benzoquinones (Ohkawa et al., 1991). Of interest, restoring the reducing milieu in neutrophil homogenates (where intracellular thiols are diluted) by addition of DTT (1 mM; Werz et al., 1998) increased the potency of RF-Id and lowered the IC50 value from 10 to 1.7 μM (Figure 4B, left panel). Similarly, for isolated human recombinant 5-LOX, the potency of RF-Id was enhanced when 1 mM DTT was included in the assay (Figure 4B, right panel). Furthermore, the reduced form of RED-RF-Id was more potent against isolated 5-LOX (IC50 = 0.4 μM), and DTT did not enhance the potency of this compound further (Figure 4C). Moreover, a competitive inhibition of 5-LOX by RF-Id appeared possible under non-reducing conditions. However, there was no significant difference in the potency of RF-Id at AA concentrations between 1 and 30 μM; the IC50 values were in the range of 7.2 to 10.1 μM (not shown) for isolated 5-LOX.
Figure 4.
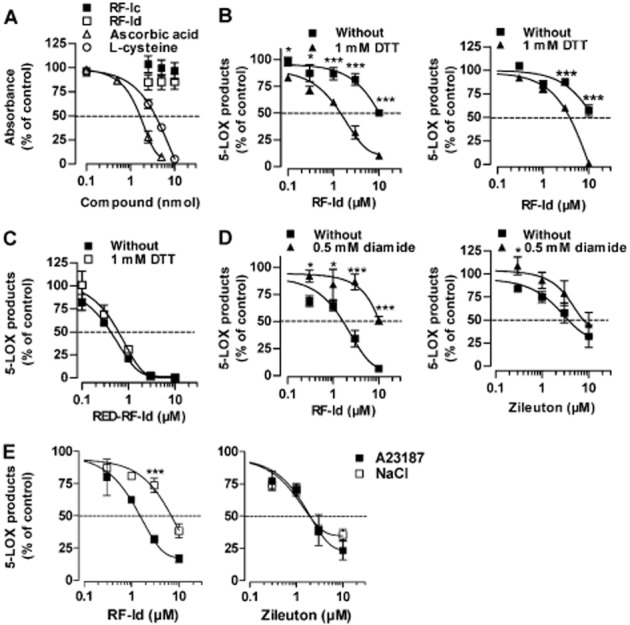
Influence of the redox state on the inhibition of 5-LOX induced by RF-Id. (A) Reduction of the DPPH radical (5 nmol) by RF-Id and RF-Ic, L-cysteine and ascorbic acid. Data are expressed as percentage of DPPH radical (100%); means ± SEM; n = 3. (B) Neutrophil homogenates (left panel) or partially purified human recombinant 5-LOX (right panel) were incubated with RF-Id (or DMSO as vehicle) for 15 min at 4°C. 5 min before stimulation, 1 mM DTT was added as indicated. After addition of 1 mM ATP, samples were warmed up for 30 s at 37°C and stimulated with 2 mM CaCl2 and 20 μM AA for 10 min at 37°C. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3. *P < 0.05, ***P <0.001 DTT treatment versus without, at indicated concentrations. (C) Partially purified 5-LOX was incubated with RED-RF-Id (or DMSO as vehicle) for 15 min at 4°C. 5 min before stimulation, 1 mM DTT was added as indicated. After addition of 1 mM ATP, samples were warmed up for 30 s at 37°C and stimulated with 2 mM CaCl2 and 20 μM AA for 10 min at 37°C. Data are expressed as percentage of vehicle control; means ± SEM; n = 3. (D) To intact neutrophils, 500 μM diamide or vehicle (DMSO) was added 7.5 min before addition of RF-Id (left panel) or zileuton (right panel) at 37°C. After addition of compounds for another 7.5 min at 37°C, cells were stimulated with 2.5 μM A23187 plus 20 μM AA for 10 min at 37°C. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3. *P <0.05, ***P <0.001 diamide treatment versus without, at indicated concentrations. (E) Neutrophils were pre-incubated with RF-Id (left panel) or zileuton (right panel) at 37°C. After 15 min, either 2.5 μM A23187 plus 40 μM AA or 0.3 M NaCl plus 40 μM AA was added, as indicated. After 10 min at 37°C, 5-LOX products were determined. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3. ***P <0.001 A23187 versus NaCl at indicated concentrations.
To confirm that the high efficacy of RF-Id in the cell is due to the reducing intracellular milieu, we pretreated neutrophils with the thiol-oxidizing agent diamide in order to elevate the cellular oxidative tone. As expected, the inhibitory effect of compound RF-Id (but not of zileuton) on 5-LOX was impaired by diamide, and the IC50 value shifted from 1.5 to 10 μM (Figure 4D). Finally, the inhibitory effects of RF-Id and zileuton on 5-LOX were determined in neutrophils subjected to stress using hyperosmotic shock (0.3 M NaCl), in comparison to cells stimulated with A23187. While zileuton was equally effective, RF-Id lost potency in cells subjected to stress (Figure 4E).
Docking simulation
To obtain further insights into the molecular mode of action of RF-Id, docking of the oxidized quinone as well as the reduced hydroquinone form of RF-Id in 5-LOX was performed. With each form, both enantiomers were docked into the X-ray crystal structure of 5-LOX (PDB entry 3o8y; Gilbert et al., 2011). The inactive RF-Ic was docked for comparison. Ten docking poses were calculated for each molecule. Since the docking algorithm tries different starting conformations within the binding pocket and minimizes them, it points towards a clear energetically preferred pose if only very similar binding poses are found (Jones et al., 1997). In this case, the docking poses of all structures were very similar, suggesting a reliable pose prediction. The hydrophobic rings filled the hydrophobic channel of 5-LOX running by the catalytic iron, where the oxidation of AA would take place. The hydroxylated quinone (or hydroquinone) ring was coordinated between several amino acids and water molecules mediating several hydrogen bonds with the protein (Figure 5). Note that the inactive cyclohexyl-substituated quinone RF-Ic did not fill the hydrophobic channel to the same extent as the decahydronaphthalene-group of RF-Id. Of interest, the redox state of RF-Id had a considerable effect on the predicted interaction patterns. For the quinone state (RF-Id) several interactions were calculated, depending on the exact orientation of the ring: hydrogen bonds with Gln557, His367, Tyr181 and Asn425 were observed, as well as interactions with the two water molecules H2O855 and H2O724. In the hydroquinone state (RED-RF-Id), even more interactions were observed because the additional hydroxyl groups can act both as donors and acceptors of hydrogen bonds. In terms of scoring of the R- and S-enantiomers, comparable or identical GoldScores (26 vs. 30 for RF-Id and 28 vs. 28 for RED-RF-Id) were obtained.
Figure 5.
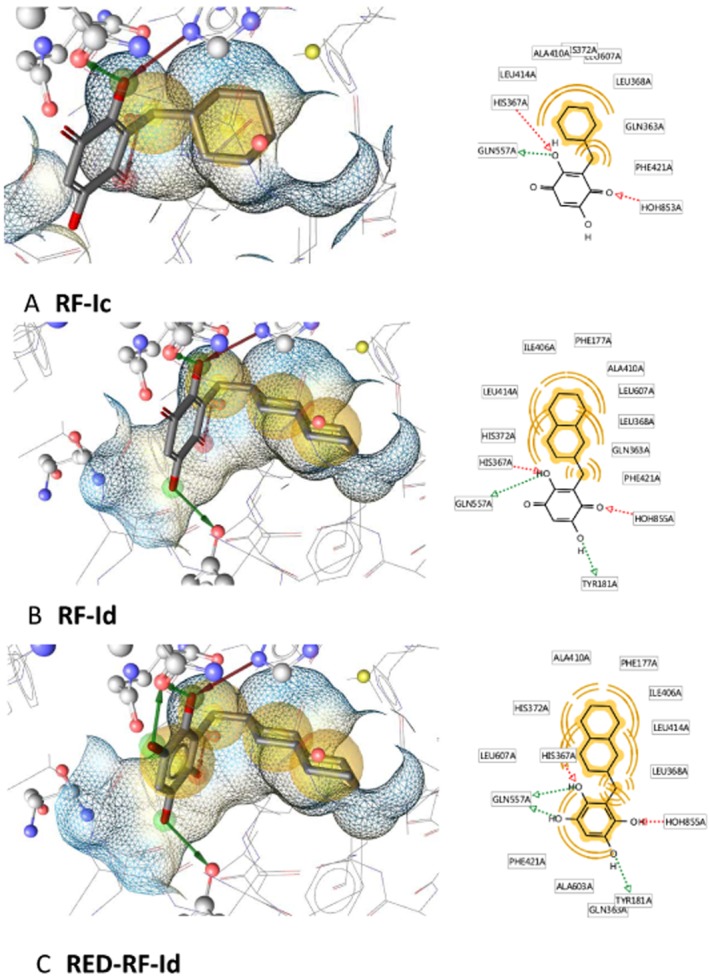
Docking poses for RF-Ic, RF-Id and RED-RF-Id. The wireframe represents the surface of the binding pocket. The yellow spheres indicate hydrophobic parts of the molecule and the arrows mark hydrogen bond donors (green) and acceptors (red). (A) Docking pose of inactive RF-Ic. The quinone part of the molecule forms hydrogen bonds with His367, Gln557 and H2O853. However, the hydrophobic channel of 5-LOX is only incompletely filled by the ligand. (B) Docking pose of active RF-Id within the binding pocket. Interactions with Gln557, Tyr181, His367 and H2O855 are shown. The hydrophobic part of the molecule effectively fills the substrate channel. (C) RED-RF-Id within the binding pocket forms more bidirectional hydrogen bonds than in the oxidized quinone form (RF-Id, see B). Interactions with Gln557, Tyr181, His367 and H2O855 are shown.
Analysis of RF-Id for inhibition of 5-LOX product synthesis in human whole blood and anti-inflammatory effectiveness in vivo
The human whole blood assay includes important parameters (e.g. plasma protein binding, interaction with other cells and fatty acids) that may reduce the bioactivity of test compounds (e.g. for the FLAP inhibitor MK886) in vivo, and data from such experiments might be appropriate to predict the in vivo activity of a test compound (Pergola and Werz, 2010). In human whole blood stimulated with A23187, RF-Id blocked LTB4 and 5-HETE synthesis by approximately 60% at a concentration of 10 μM (IC50 = 9.1 μM, Figure 6A). When fMLP was utilized as stimulus, after priming with LPS, a slightly more efficient suppression of 5-LOX product synthesis by RF-Id was evident, with an IC50 = 4.1 ± 0.6 μM (Figure 6B).
Figure 6.
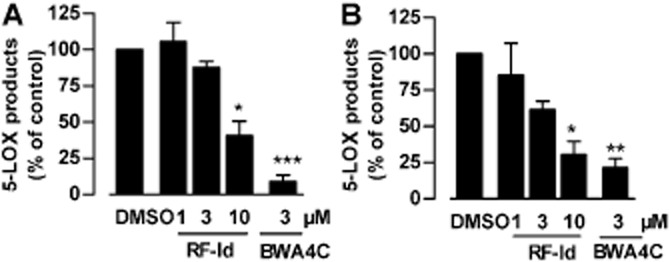
Inhibition of 5-LOX product formation by RF-Id in human whole blood. (A) Human whole blood was pre-incubated with the test compounds (or DMSO as vehicle) for 15 min at 37°C, followed by stimulation with 30 μM A23187 for 10 min at 37°C. (B) Human whole blood was primed for 15 min at 37°C with 1 μg·mL−1 LPS, treated with the test compounds (or DMSO as vehicle) for 15 min at 37°C, and then stimulated with 1 μM fMLP. After 15 min at 37°C, the formation of 5-LOX products was determined (right panel). *P <0.05, **P <0.01, ***P <0.001 versus vehicle control. Data are expressed as percentage of vehicle (0.1% DMSO); means + SEM; n = 3.
In order to evaluate the anti-inflammatory effectiveness of RF-Id in vivo, two well-established animal models of inflammation, mouse carrageenan-induced paw oedema and the mouse air pouch model were used. Intraplantar injection of carrageenan to mice caused a pronounced inflammatory reaction visualized as a massive swelling of the paw within 2 h. Pretreatment (30 min before carrageenan, i.p.) with RF-Id (0.1 – 10 mg·kg−1) significantly reduced paw swelling with maximal effects at a dose of 1 mg·kg−1, and the inhibitory action was essentially observed at all time points investigated (Figure 7A). Because LTB4 is a well-recognized chemotactic and chemokinetic agent that causes migration of various leukocytes, we determined whether RF-Id impairs cell migration in vivo in an air pouch model. RF-Id, given i.p. at 1 mg·kg−1, blocked zymosan-induced cell migration in this model (Figure 7B) and this was accompanied by reduced LTB4 levels (Figure 7C). Note that PGE2 was not lowered by RF-Id (Figure 7D), whereas the COX inhibitor indomethacin reduced cell migration by 54.5 ± 4.7% and PGE2 levels to 20.8 ± 5% but failed to repress LTB4 synthesis (83.0 ± 21.8% remaining).
Figure 7.
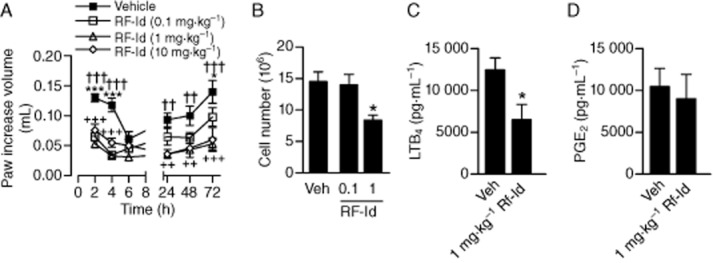
Effect of RF-Id on carrageenan-induced oedema and on zymosan-induced cell migration and LTB4 and PGE2 levels in vivo. (A) Carrageenan-induced oedema. Mice were divided into four groups (n = 6) and received i.p. administration of RF-Id (0.1, 1 or 10 mg·kg−1 30 min before subplantar injection of 50 μL of carrageenan (1%, w v−1). Paw volume was measured using a hydroplethismometer, specially modified for small volumes, immediately before the subplantar injection (basal value) and 2, 4, 6, 24, 48 and 72 h thereafter. Data are means ± SEM., n = 6. *P <0.05; ***P <0.001; 1 mg·kg−1: ††P <0.01; †††P <0.001; ++P <0.01; +++P <0.001. (B–D) Cell migration in air pouches of mice. Mice (n = 5–6 per group) received i.p. administration of RF-Id (0.1 and 1 mg·kg−1), indomethacin (2.5 mg·kg−1) or vehicle (veh, DMSO). After 30 min, air pouches were developed by subcutaneous injection of 2.5 mL sterile air into the backs of the mice. Zymosan 1% (w v−1) was used as an inflammatory agent to induce cell migration. After 4 h of zymosan injection, mice were killed by CO2 exposure and exudate in the pouch was collected and the total leukocyte count was evaluated by optical microscopy of the cell suspension diluted with Turk's solution (B). The supernatants of the exudate were analysed for LTB4 (C) or PGE2 (D) by elisa. Data are means + SEM., n = 5–6. *P <0.05, anova + Bonferroni (B) and Student's t-test (C).
Effects of RF-Id on the activity of other LOXs, COX-1/2, mPGES-1 and cPLA2
Based on the potent anti-inflammatory efficacy of RF-Id in vivo, the compound may also interfere with other enzymes involved in the generation of pro-inflammatory eicosanoids. RF-Id up to 10 μM had no significant inhibitory effect on 12-H(P)ETE or 15-H(P)ETE formation in neutrophil homogenates; IC50 values were >30 μM, regardless of the absence or presence of DTT (Figure 8A), implying that 12/15-LOXs are not markedly affected by RF-Id. Neither the formation of the COX-derived product 12-HHT in intact human platelets nor 12-HHT synthesis by isolated ovine COX-1 enzyme was suppressed by RF-Id (10 μM), while the reference drug indomethacin significantly inhibited 12-HHT synthesis (Table 1). This indicates that RF-Id does not have an effect on COX-1. Also, RF-Id (10 μM) failed to markedly inhibit the enzymatic activity of human mPGES-1 in a microsomal assay and of isolated human recombinant cPLA2 (Table 1); as expected, the reference compounds MK886 (10 μM) and RSC-3388 (5 μM), respectively, inhibited the enzymes. Interestingly, RF-Id blocked COX-2 activity in a cell-based assay (6-keto-PGF1α is a COX-2-derived product) using IL-1β-stimulated A549 cells that were incubated with 3 μM exogenous AA (Figure 8B), although the potency (IC50 = 7.3 μM) was approximately 10-fold lower as compared with suppression of 5-LOX activity in neutrophils (IC50 = 0.58 μM). Along these lines, RF-Id (10 μM) inhibited the activity of isolated human recombinant COX-2 enzyme in a cell-free assay even though by only 25%, while the reference COX-2 inhibitor celecoxib effectively suppressed COX-2 activity (Table 1). Again, as observed for inhibition of 5-LOX, the improved potency of COX-2 in the cell-based assay may be due to the reducing milieu in the cell that facilitates the bioactivity of RF-Id.
Figure 8.
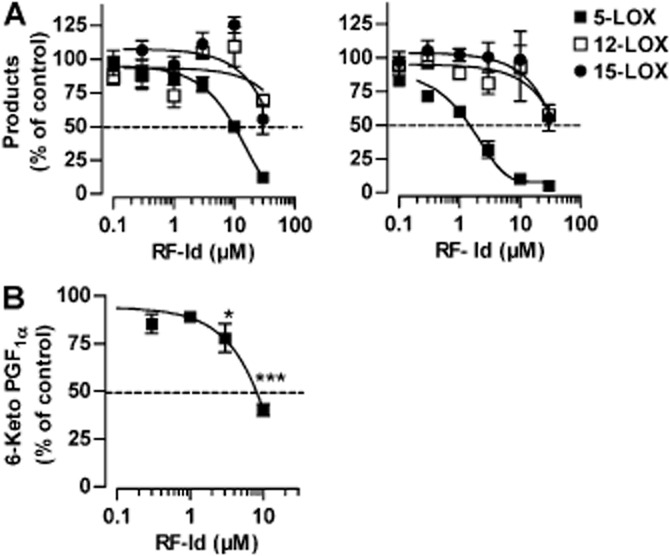
Effects of RF-Id on various lipoxygenases and COX-2 activity. (A) Neutrophil homogenates were incubated with RF-Id for 15 min at 4°C. Left panel: without DTT; right panel with 1 mM DTT, added 5 min before stimulation. After addition of 1 mM ATP, samples were warmed up for 30 s at 37°C and stimulated with 2 mM CaCl2 and 20 μM AA for 10 min at 37°C. 5-LOX products, 12-H(P)ETE and 15-H(P)ETE were extracted and analysed by HPLC. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3–4. (B) Effect of RF-Id on the formation of the COX-2-derived 6-keto PGF1α. IL-1β-stimulated A549 cells were pre-incubated with RF-Id for 15 min at 37°C and stimulated with 3 μM AA for 15 min at 37°C. Then 6-keto PGF1α was analysed by elisa. Data are expressed as percentage of vehicle control (0.1% DMSO); means ± SEM; n = 3. *P <0.05, ***P <0.001 versus vehicle control.
Table 1.
Effects of RF-Id on the activity of COX-1/2, mPGES-1 and cPLA2
| Enzyme/assay | Remaining activity at 10 μM (%) or IC50 of compound (μM) | Remaining activity at given concentration of reference compound (%) |
|---|---|---|
| Isolated COX-1 | 89.6 ± 13.6% | (Indomethacin, 10 μM) 22.2 ± 2.6% |
| COX-1 in platelets | 93.8 ± 3.3% | (Indomethacin, 10 μM) 5.9 ± 3.1% |
| Isolated COX-2 | 76.4 ± 7.6% | (Indomethacin, 10 μM) 43.0 ± 2.7% |
| COX-2 in A549 cells | 7.3 ± 0.5 μM | (Celecoxib, 5 μM) 25.2 ± 4.9% |
| Isolated cPLA2 | 79.9 ± 8.2% | (RSC-3388, 5 μM) 11.6 ± 5.9% |
| mPGES-1, cell-free | 71.6 ± 6.9% | (MK886, 10 μM) 26.7 ± 8.0% |
Data are given as mean ± SEM, n = 3–4.
Discussion and conclusions
Here we provide a detailed analysis of the molecular mode of action and the pharmacological profile of the novel 1,4-benzoquinone RF-Id that inhibits 5-LOX and possesses anti-inflammatory efficacy in vivo. RF-Id consistently suppressed the biosynthesis of various 5-LOX products in human neutrophils and monocytes with similar potencies independent of the stimulus, and also blocked 5-LOX product synthesis in human whole blood. Except for COX-2, no relevant enzyme involved in eicosanoid synthesis other than 5-LOX was markedly suppressed by RF-Id, and cellular determinants of 5-LOX product synthesis, such as FLAP, 5-LOX translocation, cell viability and substrate supply, were also not affected. Mechanistically, RF-Id directly inhibits 5-LOX, which is governed by a reducing environment that may allow the hydroquinone form to perform more bidirectional hydrogen bonds with 5-LOX, and the potency strongly depends on the structural nature of the lipophilic residue (i.e. the decahydronaphthalene moiety). Finally, the pharmacological potential of RF-Id is further substantiated by the observed marked anti-inflammatory efficacy of the compound in vivo in two LT-related animal models. Because of the continuing search for effective inhibitors of LT biosynthesis and the current intensive evaluation of respective candidates in clinical trials (Pergola and Werz, 2010), our data provide significant advances and insights into the development of such drugs.
Several synthetic compounds possessing a quinone structure, exemplified by AA-861 (Yoshimoto et al., 1982) or by 2-[4′-(iso-propylphenyl)-amino]-5,6-dimethyl-1,4-benzoquinone (Poeckel et al., 2006), as well as naturally occurring 1,4-benzoquinones like ardisiaquinone A, thymoquinone, aethiopinone and maesanin have also been reported to be potent 5-LOX inhibitors (reviewed in Werz, 2007). However, most of these 1,4-benzoquinones were not selective for 5-LOX and interfered with other pro-inflammatory targets (e.g. synovial PLA2, LTC4 synthase, COX-1) within the AA cascade. In contrast, with RF-Id only a moderate suppression of COX-2 in A549 cells and of isolated COX-2 was evident; neither cPLA2, COX-1, mPGES-1 nor 12/15-LOXs were strongly affected by RF-Id implying a certain degree of selectivity for 5-LOX.
The mechanism by which 1,4-benzoquinones inhibit 5-LOX activity is a matter of debate: on the one hand, they may compete with AA as a substrate for binding to 5-LOX (Hofmann et al., 2012) favoured by the fact that the potency is paralleled by the lipophilicity of the compounds. On the other hand, the 1,4-benzoquinones may act as reducing agents thereby uncoupling the redox cycle of the active-site iron of 5-LOX. The latter requires the reduction to the corresponding hydroquinone (e.g. in intact cells) that then may interrupt the redox cycle of the active-site iron of the 5-LOX due to its radical scavenging properties (Ohkawa et al., 1991). However, in cell-free assays, various 1,4-benzoquinones have been shown to inhibit the activity of isolated 5-LOX despite the absence of reducing agents (Filosa et al., 2013). As under these conditions, the quinone form seemingly predominates, the reduction of the 5-LOX active-site iron or interference with the redox cycle is an unlikely occurrence. Our data and those of others (Czapski et al., 2012) showed that 1,4-benzoquinones, which potently inhibit 5-LOX, possess only weak antioxidant or radical scavenging activity in cell-free assays. Interestingly, no clear correlation between antioxidant activity and inhibition of 5-LOX could be demonstrated for a series of synthetic 1,4-benzoquinone derivatives (Wurm and Schwandt, 2003). Along these lines, the interaction of the hydroquinone-like α-tocopherol (an antioxidant and radical scavenger) with purified 5-LOX was found to be unrelated to its antioxidant function, but instead was shown to be associated with the selective and tight binding of α-tocopherol to 5-LOX (Reddanna et al., 1985). Also, inhibition of 5-LOX by the 1,4-benzoquinone AA-861 is competitive with regard to AA (Hofmann et al., 2012), and AA-861 offered little protection of lipids against lipid peroxidation and had no effect on DPPH scavenging (Czapski et al., 2012). In our study, replacement of the decahydronaphthalene moiety of RF-Id by the physicochemically related cyclohexyl group in RF-Ic caused complete loss of 5-LOX inhibitory activity in intact cells even though the 1,4-benzoquinone core was not altered. It should be noted that neither RF-Ic nor RF-Id up to 50 μM caused significant radical scavenging activity in the DPPH assay. Moreover, RF-Id failed to strongly inhibit the related 12- and 15-LOXs that both recognize AA as a substrate and possess a non-haem iron in the active site with the same catalytic principle and redox cycle as 5-LOX. Hence, interference with the redox cycle of the 5-LOX active-site iron as an underlying molecular mode of action of RF-Id and 1,4-benzoquinones is doubtful and rather unlikely. Furthermore, the potency of RF-Id was unaffected by the AA concentration, which also excludes a competitive mode of action.
Our data show that reducing conditions favour 5-LOX inhibition by RF-Id, as in the cell-free assay RF-Id was significantly less efficient but after addition of DTT its high potency could be restored. Accordingly, the (reduced) hydroquinone form RED-RF-Id was more potent on isolated 5-LOX than RF-Id and, as expected, DTT failed to further improve 5-LOX inhibition. Also, the addition of the thiol-oxidizing agent diamide, which elevates the cellular oxidative tone in neutrophils, was found to impair the potency of RF-Id. Note also that the potency of so-called non-redox-type 5-LOX inhibitors (i.e. ZM230 487, L-739 010, CJ-13 610), which are structurally unrelated to RF-Id, is strongly improved under reducing conditions (Werz et al., 1998; Fischer et al., 2013). These compounds are thought to act by competing with fatty acid hydroperoxides for binding to 5-LOX (Werz et al., 1998) and this reaction appears to be prevented by the phosphorylation of 5-LOX caused by hyperosmotic shock (Fischer et al., 2013). It is conceivable that RF-Id inhibits 5-LOX by a similar mechanism, as its inhibitory effect was also impaired by hyperosmotic shock. However, the results of the docking study support the idea that the inhibitory effect of RF-Id on 5-LOX is not based on an interference with the redox state of the active-site iron. Instead, the redox state of the inhibitor itself may play a role in its potency by stabilizing it within the binding pocket. In the reduced hydroquinone form, more bidirectional hydrogen bonds can be formed with the 5-LOX ligand-binding site and RED-RF-Id inhibited 5-LOX more potently than RF-Id.
A consequence of the requirement of reducing conditions for non-redox-type 5-LOX inhibitors is that they potently inhibit 5-LOX in intact cells but only moderately in cell-free assays (Werz et al., 1998), and this pattern was also found for RF-Id. The higher potency of RF-Id in intact cells could also be related to it having an effect on additional targets/events that are involved in 5-LOX product synthesis, such as cPLA2, FLAP, CLP and 5-LOX phosphorylation. Because exogenous AA (20 or 40 μM) was provided in the cell-based assay, the possibility that RF-Id affects the level of substrate for 5-LOX, by inhibiting cPLA2- or FLAP-mediated AA transfer to 5-LOX, is unlikely. Also, RF-Id failed to inhibit cPLA2 in a cell-free assay or to affect the FLAP-dependent translocation of 5-LOX to the nuclear membrane. Hence, from our results we conclude that inhibition of 5-LOX by RF-Id, and probably also by other 1,4-benzoquinone-type 5-LOX inhibitors, is not mediated by an effect on the redox cycle of the active-site iron, even though these substances have been traditionally categorized as redox-type inhibitors. Instead, the selective inhibition of 5-LOX induced by RF-Id is likely to result from the formation of discrete bidirectional hydrogen bonds of the (hydro-)quinone core with the 5-LOX active site, and hydrophobic contacts of the decahydronaphthalene moiety within the fatty acid substrate channel of 5-LOX.
Many 5-LOX and FLAP inhibitors that potently repressed 5-LOX product synthesis in isolated cells failed to do so in whole blood due to albumin binding, interaction with other blood cells or competition with fatty acids (Pergola and Werz, 2010). Therefore, the efficacy of RF-Id in human whole blood (IC50 = 4.1 μM) is encouraging and supports the potent anti-inflammatory effectiveness observed in the animal models at a dose of 1 mg·kg−1 with an estimated plasma concentration of 3.5 μM. Carrageenan- or zymosan-induced acute inflammation (e.g. in paw oedema) is accompanied by elevated LT levels (Doherty et al., 1985; Xu et al., 2009), and subplantar injections of LTD4 or LTB4 induce increases in paw thickness (Rackham and Ford-Hutchinson, 1983). In fact, in the exudates of the zymosan-elicited air pouch in mice, RF-Id significantly reduced LTB4 levels while PGE2 was not lowered, which excludes the COX enzymes as targets. Indeed, RF-Id may suppress other pro-inflammatory events such as inflammatory cytokine release (Syahida et al., 2013) and NFκB signalling (Israf et al., 2010), as demonstrated for the related 1,4-benzoquinone atrovirinone that had little effect on 5-LOX (Syahida et al., 2013).
In conclusion, we demonstrated that the novel 1,4-benzoquinone derivative RF-Id is a selective and potent inhibitor of 5-LOX, and we shed light on the molecular mode of action underlying its inhibition of 5-LOX activity, with RF-Id acting as a representative of other 1,4-benzoquinone-type inhibitors. RF-Id was also shown to be a highly effective inhibitor in isolated leukocytes and in human whole blood, and have marked anti-inflammatory efficacy in vivo, further indicating its suitability for future preclinical investigations along these lines.
Acknowledgments
We thank Heidi Traber, Katrin Fischer, Bärbel Schmalwasser and Petra Wiecha for expert technical assistance. Dr Johannes Kirchmair is thanked for discussions on the docking algorithm. This work was supported by the Austrian Science Fund (project Drugs from Nature Targeting Inflammation, project part 11, S10711). We thank OpenEye Scientific for providing OMEGA within their academic free licensing programme and Inte:Ligand for providing LigandScout free of charge for this study.
Glossary
- 5-H(P)ETE
5(S)-hydro(pero)xy-6-trans-8,11,14-cis-eicosatetraenoic acid
- 5-LOX
5-lipoxygenase
- AA
arachidonic acid
- cPLA2
cytosolic PLA2
- DPPH
diphenylpicrylhydrazyl radical
- FLAP
5-LOX-activating protein
- fMLP
N-formyl-methionyl-leucyl-phenylalanine
- mPGES-1
microsomal PGE2 synthase-1
- PDB
Protein Data Bank
- PGC
PBS plus 1 mg·mL−1 glucose and 1 mM CaCl2
Conflicts of interest
None.
References
- Albert D, Zundorf I, Dingermann T, Muller WE, Steinhilber D, Werz O. Hyperforin is a dual inhibitor of cyclooxygenase-1 and 5-lipoxygenase. Biochem Pharmacol. 2002;64:1767–1775. doi: 10.1016/s0006-2952(02)01387-4. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back M, Dahlen SE, Drazen JM, Evans JF, Serhan CN, Shimizu T, et al. International union of basic and clinical pharmacology. LXXXIV: leukotriene receptor nomenclature, distribution, and pathophysiological functions. Pharmacol Rev. 2011;63:539–584. doi: 10.1124/pr.110.004184. [DOI] [PubMed] [Google Scholar]
- Bain G, King CD, Schaab K, Rewolinski M, Norris V, Ambery C, et al. Pharmacodynamics, pharmacokinetics and safety of GSK2190915, a novel oral anti-inflammatory 5-lipoxygenase-activating protein inhibitor. Br J Clin Pharmacol. 2013;75:779–790. doi: 10.1111/j.1365-2125.2012.04386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;281:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blois MS. Antioxidant determinations by the use of a stable free radical. Nature. 1958;181:1199–1200. [Google Scholar]
- Carter GW, Young PR, Albert DH, Bouska J, Dyer R, Bell RL, et al. 5-Lipoxygenase inhibitory activity of zileuton. J Pharmacol Exp Ther. 1991;256:929–937. [PubMed] [Google Scholar]
- Czapski GA, Czubowicz K, Strosznajder RP. Evaluation of the antioxidative properties of lipoxygenase inhibitors. Pharmacol Rep. 2012;64:1179–1188. doi: 10.1016/s1734-1140(12)70914-3. [DOI] [PubMed] [Google Scholar]
- Dandawate PR, Vyas AC, Padhye SB, Singh MW, Baruah JB. Perspectives on medicinal properties of benzoquinone compounds. Mini Rev Med Chem. 2010;10:436–454. doi: 10.2174/138955710791330909. [DOI] [PubMed] [Google Scholar]
- Doherty NS, Poubelle P, Borgeat P, Beaver TH, Westrich GL, Schrader NL. Intraperitoneal injection of zymosan in mice induces pain, inflammation and the synthesis of peptidoleukotrienes and prostaglandin E2. Prostaglandins. 1985;30:769–789. doi: 10.1016/0090-6980(85)90006-1. [DOI] [PubMed] [Google Scholar]
- Evans JF, Ferguson AD, Mosley RT, Hutchinson JH. What's all the FLAP about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sci. 2008;29:72–78. doi: 10.1016/j.tips.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Feisst C, Pergola C, Rakonjac M, Rossi A, Koeberle A, Dodt G, et al. Hyperforin is a novel type of 5-lipoxygenase inhibitor with high efficacy in vivo. Cell Mol Life Sci. 2009;66:2759–2771. doi: 10.1007/s00018-009-0078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa R, Peduto A, Aparoy P, Schaible AM, Luderer S, Krauth V, et al. Discovery and biological evaluation of novel 1,4-benzoquinone and related resorcinol derivatives that inhibit 5-lipoxygenase. Eur J Med Chem. 67:269–279. doi: 10.1016/j.ejmech.2013.06.039. [DOI] [PubMed] [Google Scholar]
- Fischer L, Steinhilber D, Werz O. Molecular pharmacological profile of the nonredox-type 5-lipoxygenase inhibitor CJ-13610. Br J Pharmacol. 2013;142:861–868. doi: 10.1038/sj.bjp.0705860. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford-Hutchinson AW, Gresser M, Young RN. 5-Lipoxygenase. Annu Rev Biochem. 1994;63:383–417. doi: 10.1146/annurev.bi.63.070194.002123. [DOI] [PubMed] [Google Scholar]
- Gilbert NC, Bartlett SG, Waight MT, Neau DB, Boeglin WE, Brash AR, et al. The structure of human 5-lipoxygenase. Science. 2011;331:217–219. doi: 10.1126/science.1197203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLD 5.01. 2012. The Cambridge Crystallographic Data CentreCambridge, UK. Available at: http://www.ccdc.cam.ac.uk(accessed 11/3/2014)
- Hoffmann M, Lopez JJ, Pergola C, Feisst C, Pawelczik S, Jakobsson PJ, et al. Hyperforin induces Ca(2+)-independent arachidonic acid release in human platelets by facilitating cytosolic phospholipase A(2) activation through select phospholipid interactions. Biochim Biophys Acta. 2010;1801:462–472. doi: 10.1016/j.bbalip.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Hofmann B, Rodl CB, Kahnt AS, Maier TJ, Michel AA, Hoffmann M, et al. Molecular pharmacological profile of a novel thiazolinone-based direct and selective 5-lipoxygenase inhibitor. Br J Pharmacol. 2012;165:2304–2313. doi: 10.1111/j.1476-5381.2011.01707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israf DA, Tham CL, Syahida A, Lajis NH, Sulaiman MR, Mohamad AS, et al. Atrovirinone inhibits proinflammatory mediator synthesis through disruption of NF-kappaB nuclear translocation and MAPK phosphorylation in the murine monocytic macrophage RAW 264.7. Phytomedicine. 2010;17:732–739. doi: 10.1016/j.phymed.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberle A, Siemoneit U, Buhring U, Northoff H, Laufer S, Albrecht W, et al. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakadate T, Yamamoto S, Aizu E, Kato R. Inhibition of mouse epidermal 12-lipoxygenase by 2,3,4-trimethyl-6-(12-hydroxy-5,10-dodecadiynyl)-1,4-benzoquinon e (AA861) J Pharm Pharmacol. 1985;37:71–73. doi: 10.1111/j.2042-7158.1985.tb04938.x. [DOI] [PubMed] [Google Scholar]
- Ohkawa S, Terao T, Murakami M, Matsumoto T, Goto G. Reduction of 2,3,5-trimethyl-6-(3-pyridylmethyl)-1,4-benzoquinone by PB-3c cells and biological activity of its hydroquinone. Chem Pharm Bull (Tokyo) 1991;39:917–921. doi: 10.1248/cpb.39.917. [DOI] [PubMed] [Google Scholar]
- Ohuchi K, Watanabe M, Taniguchi J, Tsurufuji S, Levine L. Inhibition by AA861 of prostaglandin E2 production by activated peritoneal macrophages of rat. Prostaglandins Leukot Med. 1983;12:175–177. doi: 10.1016/0262-1746(83)90081-1. [DOI] [PubMed] [Google Scholar]
- OMEGA version 2.2.1. 2007. OpenEye scientific softwareI., Santa Fe, NM, USA. Available at: http://www.eyesopen.com(accessed 11/3/2014)
- Pergola C, Werz O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert Opin Ther Pat. 2010;20:355–375. doi: 10.1517/13543771003602012. [DOI] [PubMed] [Google Scholar]
- Pergola C, Dodt G, Rossi A, Neunhoeffer E, Lawrenz B, Northoff H, et al. ERK-mediated regulation of leukotriene biosynthesis by androgens: a molecular basis for gender differences in inflammation and asthma. Proc Natl Acad Sci U S A. 2008;105:19881–19886. doi: 10.1073/pnas.0809120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergola C, Rogge A, Dodt G, Northoff H, Weinigel C, Barz D, et al. Testosterone suppresses phospholipase D, causing sex differences in leukotriene biosynthesis in human monocytes. FASEB J. 2011;25:3377–3387. doi: 10.1096/fj.11-182758. [DOI] [PubMed] [Google Scholar]
- Pergola C, Jazzar B, Rossi A, Northoff H, Hamburger M, Sautebin L, et al. On the inhibition of 5-lipoxygenase product formation by tryptanthrin: mechanistic studies and efficacy in vivo. Br J Pharmacol. 2012;165:765–776. doi: 10.1111/j.1476-5381.2011.01605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- Petronzi C, Filosa R, Peduto A, Monti MC, Margarucci L, Massa A, et al. Structure-based design, synthesis and preliminary anti-inflammatory activity of bolinaquinone analogues. Eur J Med Chem. 2011;46:488–496. doi: 10.1016/j.ejmech.2010.11.028. [DOI] [PubMed] [Google Scholar]
- Petronzi C, Festa M, Peduto A, Castellano M, Marinello J, Massa A, et al. Cyclohexa-2,5-diene-1,4-dione-based antiproliferative agents: design, synthesis, and cytotoxic evaluation. J Exp Clin Cancer Res. 2013;32:24. doi: 10.1186/1756-9966-32-24. . doi: 10.1186/1756-9966-32-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeckel D, Niedermeyer TH, Pham HT, Mikolasch A, Mundt S, Lindequist U, et al. Inhibition of human 5-lipoxygenase and anti-neoplastic effects by 2-amino-1,4-benzoquinones. Med Chem. 2006;2:591–595. doi: 10.2174/1573406410602060591. [DOI] [PubMed] [Google Scholar]
- Rackham A, Ford-Hutchinson AW. Inflammation and pain sensitivity: effects of leukotrienes D4, B4 and prostaglandin E1 in the rat paw. Prostaglandins. 1983;25:193–203. doi: 10.1016/0090-6980(83)90104-1. [DOI] [PubMed] [Google Scholar]
- Radmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci. 2007;32:332–341. doi: 10.1016/j.tibs.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Reddanna P, Rao MK, Reddy CC. Inhibition of 5-lipoxygenase by vitamin E. FEBS Lett. 1985;193:39–43. doi: 10.1016/0014-5793(85)80075-2. [DOI] [PubMed] [Google Scholar]
- Schaible AM, Traber H, Temml V, Noha SM, Filosa R, Peduto A, et al. Potent inhibition of human 5-lipoxygenase and microsomal prostaglandin E synthase-1 by the anti-carcinogenic and anti-inflammatory agent embelin. Biochem Pharmacol. 86:476–486. doi: 10.1016/j.bcp.2013.04.015. [DOI] [PubMed] [Google Scholar]
- Syahida A, Israf DA, Permana D, Lajis NH, Khozirah S, Afiza AW, et al. Atrovirinone inhibits pro-inflammatory mediator release from murine macrophages and human whole blood. Immunol Cell Biol. 2013;84:250–258. doi: 10.1111/j.1440-1711.2006.01426.x. 2006. [DOI] [PubMed] [Google Scholar]
- Tardif JC, L'Allier PL, Ibrahim R, Gregoire JC, Nozza A, Cossette M, et al. Treatment with 5-lipoxygenase inhibitor VIA-2291 (Atreleuton) in patients with recent acute coronary syndrome. Circ Cardiovasc Imaging. 2010;3:298–307. doi: 10.1161/CIRCIMAGING.110.937169. [DOI] [PubMed] [Google Scholar]
- Wasfi YS, Villaran C, de Tilleghem CLB, Smugar SS, Hanley WD, Reiss TF, et al. The efficacy and tolerability of MK-0633, a 5-lipoxygenase inhibitor, in chronic asthma. Respir Med. 2012;106:34–46. doi: 10.1016/j.rmed.2011.08.019. [DOI] [PubMed] [Google Scholar]
- Werz O. 5-Lipoxygenase: cellular biology and molecular pharmacology. Curr Drug Targets Inflamm Allergy. 2002;1:23–44. doi: 10.2174/1568010023344959. [DOI] [PubMed] [Google Scholar]
- Werz O. Inhibition of 5-lipoxygenase product synthesis by natural compounds of plant origin. Planta Med. 2007;73:1331–1357. doi: 10.1055/s-2007-990242. [DOI] [PubMed] [Google Scholar]
- Werz O, Steinhilber D. Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol Ther. 2006;112:701–718. doi: 10.1016/j.pharmthera.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Werz O, Szellas D, Henseler M, Steinhilber D. Nonredox 5-lipoxygenase inhibitors require glutathione peroxidase for efficient inhibition of 5-lipoxygenase activity. Mol Pharmacol. 1998;54:445–451. doi: 10.1124/mol.54.2.445. [DOI] [PubMed] [Google Scholar]
- Wolber G, Bendix F, Ibis G, Kosara R, Seidel T, Dornhofer A. 2009. LigandScout, 3.0. Inte:Ligand GmbH: Vienna, Austria. Available at: http://www.inteligand.com(accessed 11/3/2014)
- Wurm G, Schwandt S. Methylated 2-aryl-1,4-naphtoquinone derivatives with diminished antioxidative activity. Pharmazie. 2003;58:531–538. [PubMed] [Google Scholar]
- Xu GL, Liu F, Ao GZ, He SY, Ju M, Zhao Y, et al. Anti-inflammatory effects and gastrointestinal safety of NNU-hdpa, a novel dual COX/5-LOX inhibitor. Eur J Pharmacol. 2009;611:100–106. doi: 10.1016/j.ejphar.2009.03.062. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Yokoyama C, Ochi K, Yamamoto S, Maki Y, Ashida Y, et al. 2,3,5-Trimethyl-6-(12-hydroxy-5,10-dodecadiynyl)-1,4-benzoquinone (AA861), a selective inhibitor of the 5-lipoxygenase reaction and the biosynthesis of slow-reacting substance of anaphylaxis. Biochim Biophys Acta. 1982;713:470–473. [PubMed] [Google Scholar]