

Abstract
Background and Purpose
Microglial cells are important mediators of the immune response in the CNS. The phytocannabinoid, cannabidiol (CBD), has been shown to have central anti-inflammatory properties, and the purpose of the present study was to investigate the effects of CBD and other phytocannabinoids on microglial phagocytosis.
Experimental Approach
Phagocytosis was assessed by measuring ingestion of fluorescently labelled latex beads by cultured microglial cells. Drug effects were probed using single-cell Ca2+ imaging and expression of mediator proteins by immunoblotting and immunocytochemistry.
Key Results
CBD (10 μM) enhanced bead phagocytosis to 175 ± 7% control. Other phytocannabinoids, synthetic and endogenous cannabinoids were without effect. The enhancement was dependent upon Ca2+ influx and was abolished in the presence of EGTA, the Ca2+ channel inhibitor SKF96365, the transient receptor potential (TRP) channel blocker ruthenium red, and the TRPV1 antagonists capsazepine and AMG9810. CBD produced a sustained increase in intracellular Ca2+ concentration in BV-2 microglia and this was abolished by ruthenium red. CBD rapidly increased the expression of TRPV2 and TRPV1 proteins and caused a translocation of TRPV2 to the cell membrane. Wortmannin blocked CBD enhancement of BV-2 cell phagocytosis, suggesting that it is mediated by PI3K signalling downstream of the Ca2+ influx.
Conclusions and Implications
The TRPV-dependent phagocytosis-enhancing effect of CBD suggests that pharmacological modification of TRPV channel activity could be a rational approach to treating neuroinflammatory disorders involving changes in microglial function and that CBD is a potential starting point for future development of novel therapeutics acting on the TRPV receptor family.
Keywords: microglia, phagocytosis, cannabinoid, cannabidiol, calcium influx
Introduction
Microglial cells function as the immune cells of the CNS. They are present throughout the CNS and the spinal cord, with a suggested population of up to 12% of all cells in the CNS (Lawson et al., 1990). Microglia are activated by pathogens and products from injured neurons, alongside a wide variety of chemical threat signals including viral, fungal and bacterial structures, complement factors, antibodies, chemokines and cytokines. Activation transforms microglia into motile amoeboid macrophages that move to the site of injury or invasion. The cells are believed to be neuroprotective; for example, it has been shown that the Alzheimer's disease-associated accumulation of amyloid- can be phagocytosed by microglia (Bard et al., 2000), and microglial activation and migration is evident in other pathological conditions, including stroke, neurodegenerative disease, tumour invasion and neuropathic pain (Cao and Zhang, 2008; Kettenmann et al., 2011). However, microglial activation also has the potential to be damaging through the release of cytokines and NO (Woodroofe et al., 1991; Minghetti and Levi, 1998).
Cannabinoids have been shown to engage neuroprotective mechanisms against acute brain damage and toxicity both in vivo and in vitro. Their effects include decreases in pro-inflammatory cytokines, NO formation, antioxidant activity and reductions in excitotoxic calcium influx (Mechoulam et al., 2002).
There has been a growing interest in the phytocannnabinoid cannabidiol (CBD), which comprises up to 40% of Cannabis sativa extracts and represents one of the most promising cannabinoid drugs in clinical development due to its lack of psychoactive effects and its high level of tolerability in humans (Mechoulam et al., 2002; 2007). CBD has a surprisingly wide range of reported pharmacological effects such as anticonvulsant, hypnotic, anti-inflammatory, anxiolytic, antipsychotic and neuroprotective actions (Barichello et al., 2012). The latter property is supported by preclinical research, showing that administration of CBD reduces neuroinflammation in mice injected with intraventricular amyloid-β (Martin-Moreno et al., 2011). Low doses of CBD protect oligodendrocyte progenitor cells during immune system insult (Mecha et al., 2012) and it has been found that CBD has anti-inflammatory effects by inhibiting iNOS expression, COX-2, NO generation and by reducing pro-inflammatory cytokine production induced by bacterial lipopolysaccharide (LPS) (Kozela et al., 2010).
The aim of the present study was, therefore, to investigate the effects of CBD and other cannabinoids on microglial cell phagocytosis in vitro. The results demonstrate a robust activation of the microglia by CBD, but not by other cannabinoids, which appears to be mediated, at least in part, by transient receptor potential (TRP) channels (for nomenclature see Alexander et al., 2013).
Methods
Phytocannabinoids were a generous gift from GW Pharmaceuticals (Salisbury, UK). All other chemicals and cannabinoid agents were purchased from Sigma-Aldrich Chemical Company (Poole, UK) or Tocris Bioscience (Bristol, UK).
Cell culture
Mouse primary microglial cultures
Two male adult 8- to 10-week-old C57bl6 mice were deeply anaesthetized with sodium pentobarbital, transcardially perfused with heparin (1 U·mL−1)-treated 0.9% saline and decapitated according to UK Home Office guidelines, and the whole spinal cord (C1-S1) was dissected. The brain and spinal cord were placed in ice-cold modified Hibernate A media (MHibA) (Life Technologies, Paisley, UK) (20 mL of Hibernate A, 0.4 mL of B27 supplement, 50 μL of 200 mM glutamine and 174 μL of 454 mM EGTA). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Meninges were removed, and the brain and spinal cord were cut into 0.5 mm slices using a McIlwain tissue chopper. The tissue was incubated in MHibA containing papain (20 mL of Hibernate A, 0.4 mL of B27 supplement, 50 μL of 200 mM glutamine and 174 μL of 454 mM EGTA plus 12 mg worthington, 15–23 U·mg−1 protein at 30°C for 30 min on shaker). The cell suspension was centrifuged, and the pellet was re-suspended in 1 mL of medium (DMEM, glutamax-1, 15% heat-inactivated FBS and 1% penicillin, streptomycin), and then the cells were plated onto 13 mm glass coverslips at a density of 80 000 cells·mL−1. The cells were incubated at 37°C with 5% CO2 for 4 days.
BV-2 mouse microglial cells (a gift from Dr Nephi Stella, Washington University, Seattle, WA, USA) were grown in DMEM supplemented with 10% FBS, HEPES (10 mM), NaHCO3 (5 mM), penicillin (100 U·mL−1) and streptomycin (100 μg·mL−1).
HAPI cells (highly aggressive proliferating rat microglia; a gift from Prof JR Connor, Department of Neuroscience and Anatomy, M.S. Hershey Medical Center, Pennsylvania State University, State College, PA, USA) were maintained in 5% FBS-supplemented DMEM, 2 mM l-glutamine and antibiotics. All cell lines were cultured at 37°C with 5% CO2.
RAW-264.7, a monocyte/macrophage-like cell line, were expanded in DMEM with 10% heat-inactivated FBS, l-glutamine (2 mM), penicillin (100 U·mL−1) and streptomycin (100 μg·mL−1). The cells were maintained in a humidified incubator with 5% CO2 at 37°C.
Assessment of phagocytosis
Phagocytosis was assessed in BV-2, HAPI and RAW 264.7 cell lines and in primary microglial cells using two sorts of latex (amino-modified polystyrene) beads with or without fluorescent tagging (1 μm diameter; Sigma; excitation/emission 470/540) as phagocytosis targets.
To observe the phagocytic activity of microglia, cells (5 × 105) were cultured on glass coverslips (19 mm) in 12-well plates and treated with drugs (as indicated in individual experiment descriptions) for 24 h, after which the medium was removed and different volumes of fluorescent latex beads from original stocks were re-suspended in 0.1% BSA with PBS, without Ca2+ and Mg2+, for 2 h at 37°C. Different volumes (0.25, 0.5 and 1 μL) of beads were tested to determine the optimum bead concentration that the cells would phagocytose. After 2 h of incubation, the assay was stopped and non-phagocytosed beads were removed via several rinses of 1 mL of cold PBS to each well. The cells were then fixed onto coverslips with 500 μL of warm paraformadehyde (4% paraformadehyde/400 mM sucrose in PBS) at room temperature for 30 min.
The cells were washed several times with warm PBS and permeabilized with 500 μL of 0.1% Triton X-100 in 1% BSA-containing PBS for 15 min at room temperature. To visualize the phagocytosed beads, F-actin was detected by staining with rhodamine phalloidin (a high-affinity F-actin probe-conjugated red fluorescent dye) (Thermo Fisher Scientific, Loughborough, UK), which binds to the polymerized form of actin (F-actin) diluted in PBS for 15 min at room temperature. Coverslips were then incubated with DAPI (1:1000) for staining of cell nuclei. The coverslips were mounted on glass slides with one drop of warm-mounting medium without DAPI. The cells were viewed by confocal microscopy under 20× magnification using an oil immersion lens.
All images of the cells were recorded using the same microscope, objective lens and exposure time to allow comparison of measurements. Fluorescence intensity in each channel was corrected for background noise and quantified using Velocity 6 Demo image analysis software (Velocity Imaging Products Inc, La Mesa, CA, USA). Fluorescent beads were quantified on consecutive images using Velocity 6 Demo software to measure pixel intensity along the line scans at the bead site. The number of ingested beads per cell and total cell number were counted to determine the mean number of ingested beads per cell.
Quantitative real-time PCR (qPCR)
RNA was extracted from BV-2 cells using Trizole reagent and RNA samples were reverse transcribed using the superscript Reverse Transcription (Thermo Fisher Scientific). Expression of TRPV2 mRNA was determined by qPCR (Thermo Fisher Scientific), using 18S ribosomal RNA as a normalizing gene. Normal and mock reverse-transcribed samples (in the absence of reverse transcriptase), in addition to no-template controls (total mix without cDNA), were run in parallel. For TRPV2 expression in BV2: forward primer, ACTAAGGTGGAGGGTGGACGAT-3′; reverse primer, CCAAGCCTAGCGGGACTCT-3′; probe, CCAGCTACGGAGGCTTCCGCG-3′.
Immunoblotting
Protein samples were separated (45 min, 200 V) on a 10% PAG. Gels were electroblotted onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) and then incubated in blocking buffer [1X Tris-buffered saline with 0.1% Tween 20 (TBS-T), and 3% fish skin gelatin] for 1 h at room temperature with shaking.
Immunodetection of the anti-TRPV2 (1:1000, PC421; Calbiochem, San Diego, CA, USA) and anti-TRPV1 (1:1000, sc-28759; Santa Cruz Biotechnology, Dallas, TX, USA) subunits was performed overnight at 4°C. After being washed, membranes were incubated with secondary (IRDye infrared, Li-Cor Biosciences, Lincoln, NE, USA; 1:2000 dilution) goat anti-rabbit IgG or goat anti-mouse IgG for 1 h at 37°C with shaking. Equal loading for whole protein lysates and cytoplasmic protein was determined with a mouse anti-GAPDH primary antibody using an Odyssey imaging system (Li-Cor Biosciences).
Fura-2 AM Ca2+ imaging
Experiments were typically conducted using BV-2 cultures at 5 × 105 cells seeded onto 19 mm glass coverslips coated with poly-l-lysine. Cultures were washed three times in buffer (NaCl 145 mM, KCl 5 mM, CaCl, 2 mM, MgSO4 1 mM, HEPES 10 mM, glucose 10 mM and 2 g·L−1 BSA) and loaded for 30 min in the dark at 37°C, with 10 μM cell-permeable fluorescent Ca2+ indicator, Fura-2 AM (Cambridge Bioscience, Cambridge, UK). Cells were then washed again with buffer and all experiments were performed at room temperature in the dark. Cultures were suprafused at a rate of 2 mL·min−1. Using the laser scanning microscope, emission intensity was measured up to 20 min, every 200–400 ms, using excitation at 430 nm and emission detection range from 480 to 536 nm. Stimulating drugs were added after 10–30 s.
Immunocytochemistry
Cells were seeded on 13 mm glass coverslips and cultured for 24 h in medium, as indicated in the figure legends. Cells were fixed in 4% paraformaldehyde for 30 min, washed with a PBS solution and then permeabilized using 0.1% Triton X-100 for 5 min. Non-specific binding sites were blocked with PBS containing 1% BSA for 60 min. Cells were incubated with the primary TRPV2 and IBA-1 antibodies; 1:100, Alpha Laboratories; 019-19741, Hampshire, UK) overnight at 4°C. After three PBS washes, fluorescent secondary antibody, FIT-C-conjugated goat anti-rabbit IgG (1:800) or FIT-C-conjugated goat anti-mouse IgG (1:500, both IgG from Jackson Immuno Research, West Grove, PA, USA), were added for 60 min at 37°C. To visualize the cells present on coverslips, cell nuclei were stained with DAPI (1:1000; Sigma) for 15 min at room temperature. After extensive washing, coverslips were mounted and viewed with a Leica DMRA2 fluorescent microscope (Leica Microsystems, Milton Keynes, UK). Images were cropped, and brightness and contrast were adjusted equally using the ImageJ software (NIH, Bethesda, MD, USA). Negative controls without primary antibody were included to identify non-specific staining.
Statistical analysis
Student's two-tailed, unpaired t-test and one-way anova for multiple comparisons were employed where applicable. Post hoc comparisons were performed with Bonferonni's multiple comparison or Dunnett's test, as indicated. P < 0.05 was taken to indicate statistical significance.
Results
Preliminary experiments using BV-2 and HAPI (not shown) microglial cell lines and primary mouse microglial cells indicated that fluorescently labelled latex beads were accumulated in cells in a time- and concentration-dependent fashion. All further phagocytosis experiments were conducted after an incubation period of 2 h with 1 μL (of original manufacturer's suspension)·mL−1 of 1-μm-diameter beads (Figure 1).
Figure 1.
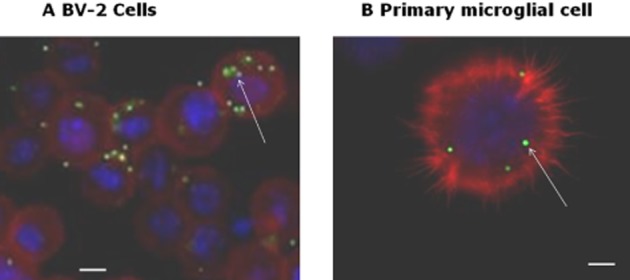
Images of latex bead phagocytosis in (A) BV-2 cells and (B) mouse primary microglia cell. BV-2 (5 × 105 cells per well) were loaded with fluorescent beads in BSA for 2 h and then incubated at 37°C before staining with rhodamine phalloidin (a high-affinity F-actin probe conjugated with red fluorescent dye) and DAPI to label cell nuclei (blue). The arrows indicate examples of the ingested fluorescent beads (40× magnification). Scale bar = 5 μm.
Bead phagocytosis in BV2 cells was examined in response to physical and chemical influences (Figure 2). A reduced incubation temperature of 4°C resulted in a significant reduction in phagocytosis, as did the presence of the actin polymerization inhibitor cytochalasin D (1 μM) (to 10.3% control). However, phagocytosis was not inhibited by 0.1% ethanol (the highest concentration of drug vehicle employed), 1 μM oligomycin (a mitochondrial ATPase inhibitor, 101.0% control) or 1 μM FCCP (a mitochondrial uncoupler that blocks oxidative phosphorylation, 94.9% control). Somewhat surprisingly, minocycline (10 μM), which is commonly used to inhibit microglial activity in vivo, had no effect, at least over the incubation period employed, on BV-2 phagocytosis.
Figure 2.
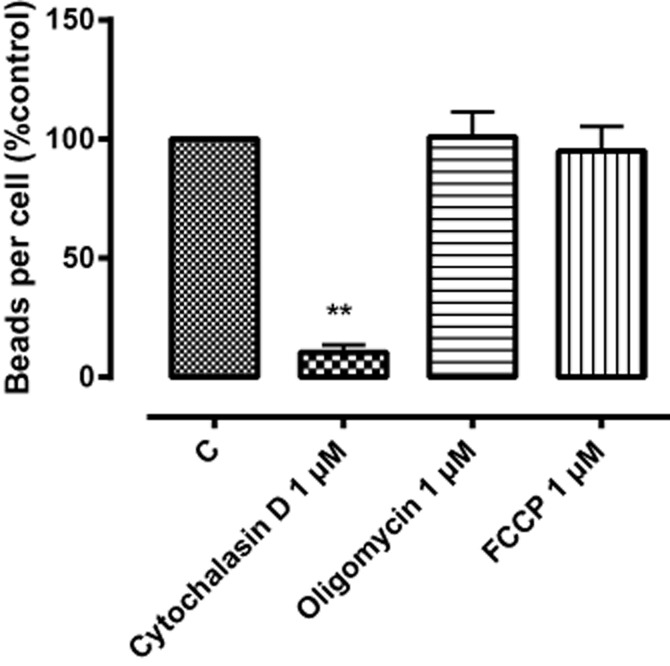
The effect of cytochalasin D (a cytoskeleton inhibitor), oligomycin (a mitochondrial ATPase inhibitor) and FCCP (a mitochondrial uncoupler) on phagocytosis in BV-2 cells. 5 × 105 BV-2 cells per well were pre-incubated with the indicated drugs (1 μM) for 1 h. Control wells (C) were incubated with culture medium alone. BV-2 cells were loaded with fluorescent beads (0.5 μL·mL−1) for 2 h. The figure represents means ± SEM of triplicate determinations in three independent experiments. The data are presented as a percentage of control and were analysed using one-way anova, followed by post hoc Dunnett's test; **P < 0.01 cytochalasin compared with control.
Phagocytosis was able to be facilitated, as demonstrated by the enhanced bead ingestion induced in the presence of UDP (10 μM), but not ATP (100 μM), the pro-inflammatory mediators IFN-γ (50 U·mL−1) and LPS (100 ng·mL−1), and by the anti-inflammatory steroid dexamethasone (1 μM) (Figure 3).
Figure 3.
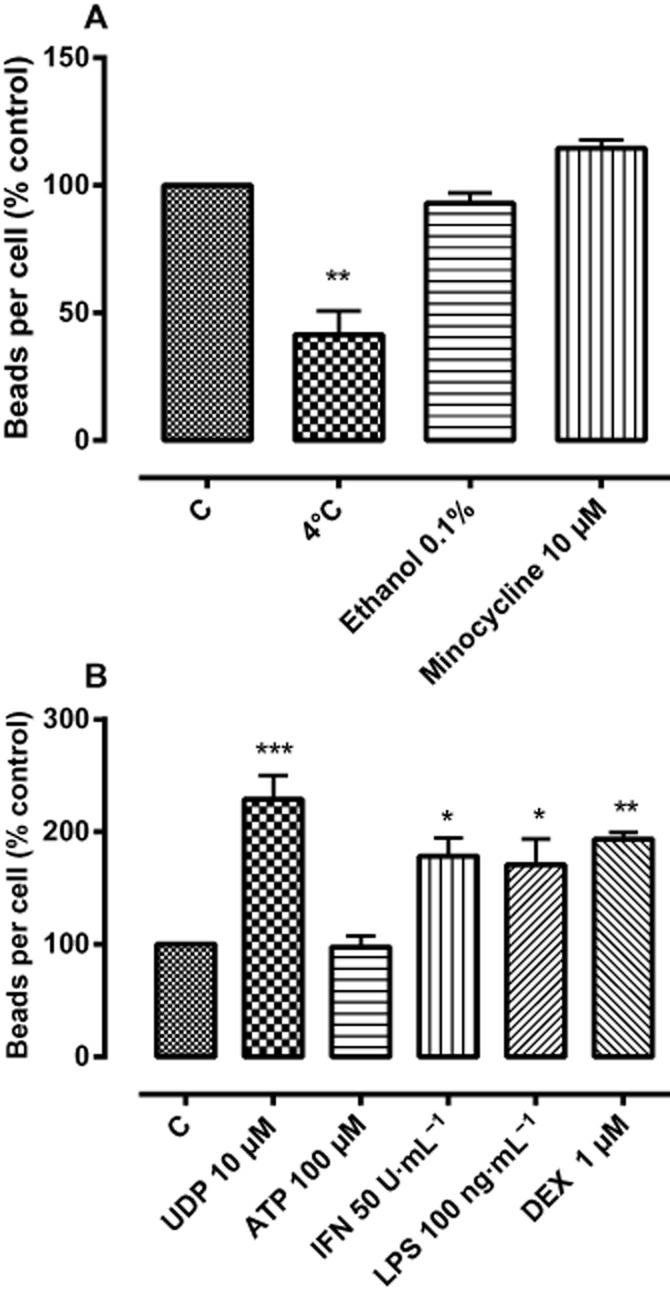
(A) The effects of temperature, ethanol and minocycline on phagocytosis in BV-2 cells. (B) The effect of UDP, ATP, IFN-γ, LPS and dexamethasone (DEX) on phagocytosis. BV-2 cells (5 × 105 cells per well) were loaded with fluorescent latex beads (0.5 μL·mL−1) for 2 h then incubated at 4 or 37°C with or without ethanol. The figure represents means ± SEM of triplicate determinations from three independent experiments, presented as a percentage of control values. Data were analysed using one-way anova, followed by post hoc Dunnett's test; *P < 0.05 ethanol versus control; ***P < 0.001 UDP versus control; **P < 0.01 DEX versus control; *P < 0.05 IFN, LPS versus control.
A variety of endogenous cannabinoids and synthetic cannabinoid receptor agonists failed to have any effect on phagocytosis in BV2 cells (Figure 4). There was a small inhibitory effect of the CB1/CB2 receptor agonist WIN55212-2, although the concentration used (10 μM) was well above that required to activate either CB1 or CB2 cannabinoid receptors.
Figure 4.
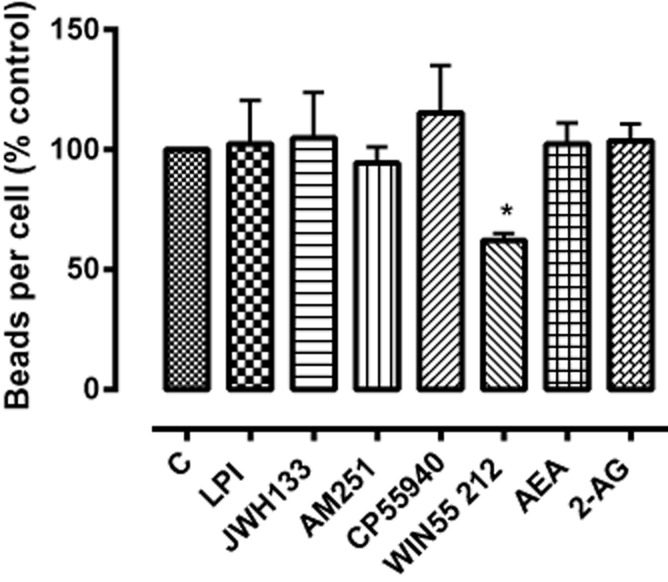
The effect of cannabinoids and endocannabinoids on phagocytosis in BV-2 cells. JWH133 is a CB2 receptor agonist; AM251 is a CB1 receptor antagonist; CP55940 and WIN 55212 (synthetic) and anandamide (AEA) and 2-AG (endogenous) are agonists of CB1 and CB2 receptors; lysophosphatidylinositol (LPI) is a putative endogenous GPR55 agonist. Cells (5 × 105 per well) were pretreated with the indicated drugs (all 10 μM) for 24 h and then incubated with fluorescent latex beads (0.5 μL·mL−1 for 2 h). The figure represents means ± SEM of three independent experiments each conducted in triplicate presented as a percentage of control. Data were analysed using one-way anova, followed by post hoc Dunnett's test; *P < 0.05 versus control (C).
The endogenous CB1/CB2 receptor agonists anandamide and 2-AG (10 μM), as well as lysophosphatidylinositol (10 μM), which is suggested to be an endogenous activator of the cannabinoid receptor-like orphan receptor GPR55 (Oka et al., 2007), were also without effect.
A number of phytocannabinoids were examined for effects on microglial phagocytosis. Of these, only CBD had an effect, significantly increasing phagocytosis in a concentration-related fashion (Figure 5A,B). Similar enhancements in the presence of 10 μM CBD were observed in three independent experiments in HAPI cells (131 ± 9% basal), mouse primary microglia (155 ± 7% basal) and in the RAW-264 murine monocyte/macrophage cell line (179 ± 16% basal).
Figure 5.
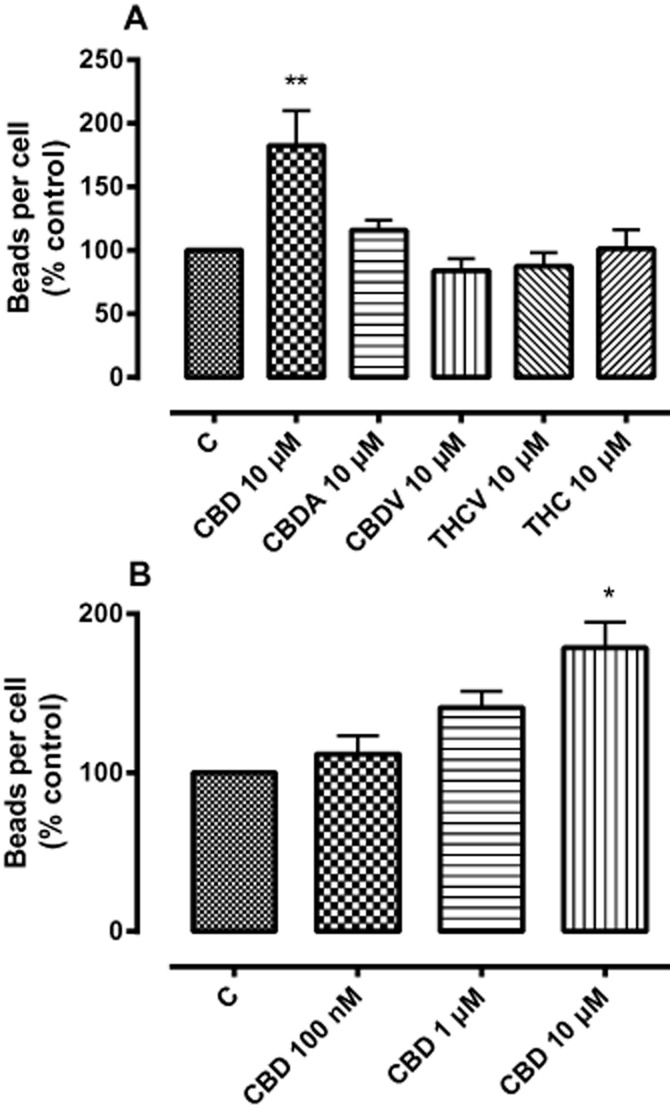
(A) The effects of phytocannabinoids (10 μM) in BV-2 cells. (B) The effect of CBD in BV-2 cells on phagocytosis. BV-2 cells were incubated with the indicated drugs for 24 h and then loaded with fluorescent beads (0.5 μL·mL−1) for 2 h. The figure represents means ± SEM of triplicate determinations of three independent experiments, presented as a percentage of control. The vertical axis represents the number of beads ingested/number of the cells expressed as a percentage of no drug. Data were analysed using one-way anova, followed by post hoc Dunnett's test; *P < 0.05; **P < 0.01 CBD 10 μM versus control (C). CBDA, cannabidiolic acid; CBDV, cannabidivarin; THC, Δ9 tetrahydrocannabinol; THCV, tetrahydrocannabivarin.
Although BV-2 cells have been reported previously to express cannabinoid receptors (Martin-Moreno et al., 2011), RT-PCR showed that the particular cells used in these studies expressed no significant gene products encoding either CB1 or CB2 receptors (data not shown), leading to the conclusion that the effect of CBD was not cannabinoid receptor-mediated. This conclusion was supported by the finding that pertussis toxin (PTX, 50 ng·mL−1, 18 h), which inactivates signalling through Gi-associated GPCRs, had no effect on either basal (116 ± 18% control) or CBD-enhanced bead ingestion [CBD (10 μM) = 161 ± 5% control; CBD + PTX = 153 ± 8% control].
The enhancement of phagocytosis due to CBD was, however, dependent upon extracellular Ca2+. The phagocytic-enhancing effect of CBD was unaltered by pre-incubation with the cell-permeable calcium chelator BAPTA-AM, but it was abolished by pre-incubation with 4 mM EGTA (Figure 6). This reduction was reversed by subsequent addition of 4 mM external CaCl2 (data not shown), indicating that a cytotoxic action of EGTA does not underlie this effect.
Figure 6.
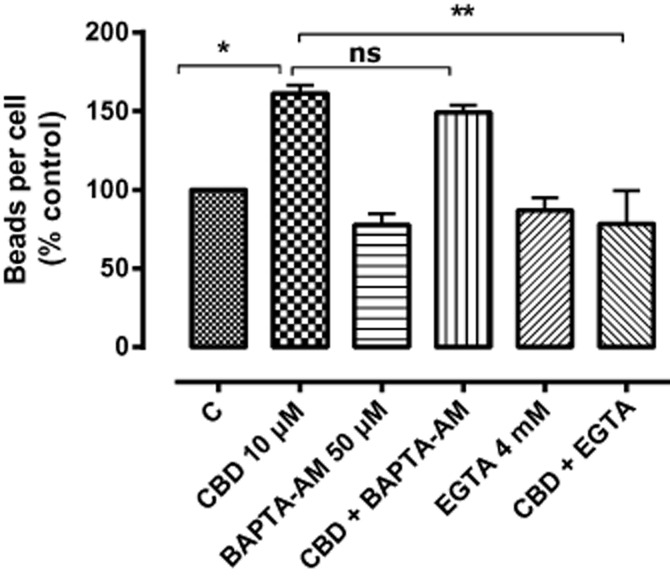
The effects of BAPTA-AM (an intracellular Ca2+ chelator) on CBD-enhanced phagocytosis in BV-2. BV-2 cells were pretreated with BAPTA-AM (50 μM) or with EGTA (4 mM) for 60 min before CBD (10 μM) exposure for 24 h. Control wells were incubated in culture medium alone. Cells were then loaded with fluorescent latex beads (0.5 μL·mL−1) for 2 h. The figure represents means ± SEM of three independent experiments, each conducted in triplicate, presented as a percentage of control. Data were analysed using one-way anova, followed by post hoc Bonferonni's multiple comparison; *P < 0.05; **P < 0.01 CBD versus control; CBD + EGTA versus CBD alone.
CBD was able to induce a significant increase in intracellular calcium concentration ([Ca2+]i) in BV-2 cells (Figure 7). In contrast to the transient effect of ATP (100 μM), CBD produced a sustained increase in [Ca2+]i, which persisted over the course of the incubation period. This increase was abolished in the absence of external Ca2+ and, interestingly, in the presence of the non-selective TRP channel blocker ruthenium red (10 μM).
Figure 7.

The effect of CBD on intracellular calcium ([Ca2+]i) in BV-2 cells using fura-2 ratiometric calcium imaging. In (A), ATP was added to cells to evoke a rapid and transient increase in [Ca2+]i. After a thorough wash with HBSS buffer, a second application of ATP was added to the cells. (B) 10 μM CBD was added after a wash with HBSS buffer. (C) CBD was added with ruthenium red (RR) to cells and changes in [Ca2+]i summarized as the 340/380 ratio of fura-2 fluorescence in (D). Data were obtained from at least three independent experiments (n > 40 cells per experiment).
Accordingly, the effects of ruthenium red on phagocytosis in BV-2 cells were assessed. This non-selective TRP channel blocker had no effect on basal phagocytosis, but, at the highest concentration employed (10 μM), it blocked the enhancement due to CBD.
SKF-96365 [a blocker of TRPC and TRPV subfamily channels and of receptor-mediated and voltage-gated Ca2+ entry (Merritt et al., 1990; Kim et al., 2003; Bomben and Sontheimer, 2008)] also completely prevented the enhancement of phagocytosis due to CBD (Figure 8A), indicating that the effect of CBD is dependent upon the influx of extracellular Ca2+, probably via TRP channels. The inhibition of the CBD effect by the selective TRPV1 antagonist capsazepine supports this contention. The rather more selective competitive TRPV1 antagonist, AMG9810, reported to block all known modes of TRPV1 activation (Gavva et al., 2005), also significantly reduced the effect of CBD but by a lesser amount than the other antagonists (Figure 8A). None of the antagonists alone affected phagocytosis at the concentrations employed, but an attempt to block TRPV2 receptors with tranilast (75 μM) (Hisanaga et al., 2009) showed that this putative antagonist alone increased phagocytosis by a degree equivalent to that of 10 μM CBD (data not shown). To further probe the involvement of TRPV2 in microglial phagocytosis, BV-2 cells were incubated with increasing concentrations of the uricosuric agent probenecid, which is reported to selectively activate TRPV2 but not TRPV1, TRPV3, TRPV4, TRPM8 and TRPA1 (Bang et al., 2007). Probenecid enhanced phagocytosis significantly in a concentration-dependent fashion (Figure 8B).
Figure 8.
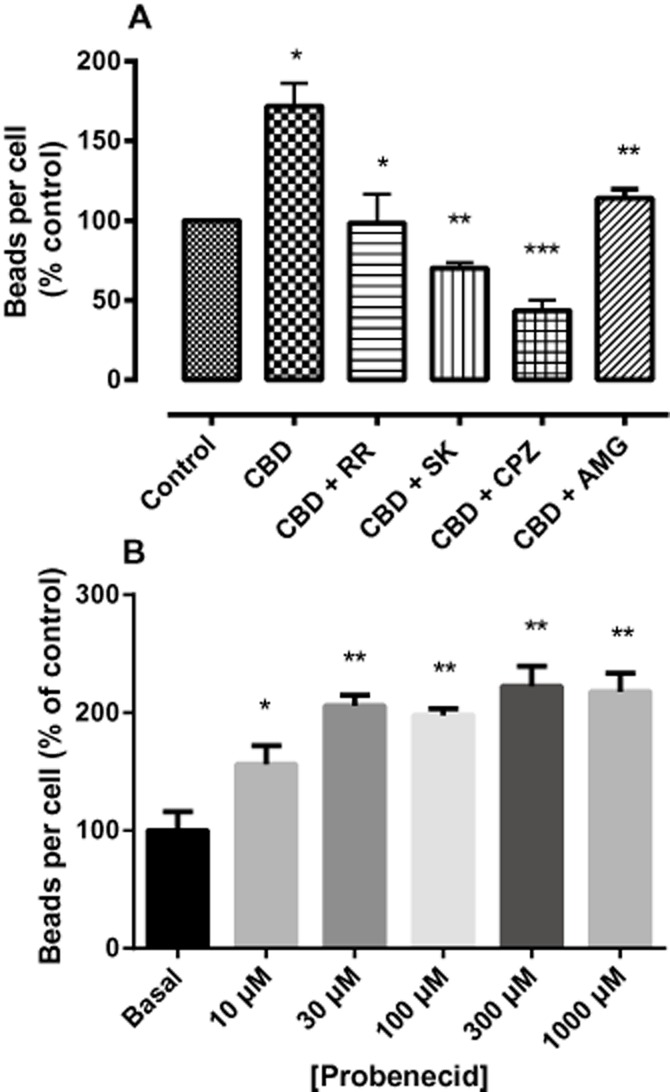
(A) The effect of ruthenium red (RR), SKF96365 (SK), capsazepine (CPZ) and AMG9810 on CBD-enhanced phagocytosis in BV-2 cells. BV-2 (5 × 105 cells per well) were pre-incubated with indicated drugs (10 μM except AMG, 1 μM) for 1 h, and then treated with CBD alone or in combination with CBD for 24 h. Control wells (C) were incubated with culture medium alone. The cells were loaded with 0.5 μL·mL−1 fluorescent latex beads for 2 h. The figure represents means ± SEM of three independent experiments each conducted in triplicate, presented as percentages of control. Data were analysed using one-way anova, followed by post hoc Bonferonni's multiple comparison; *P < 0.05 compared with CBD control; **P < 0.01; ***P < 0.001. (B) Enhancement of BV2 latex bead phagocytosis by the putative TRPV2 agonist probenecid. BV-2 (5 × 105 cells per well) were pre-incubated with probenecid at the indicated concentrations for 24 h prior to loading with latex beads as in (A). *P < 0.05 compared with basal; **P < 0.01 compared with basal.
The effects of CBD on expression and distribution of TRP channels were then investigated. To monitor the expression of TRPV2, which has been shown to play a key role in macrophage phagocytosis (Link et al., 2010) and to evaluate the effect of CBD, BV-2 cells were serum-starved 3 h prior to treatment with CBD alone or in the presence of the protein synthesis inhibitor cycloheximide or a PI3K inhibitor, wortmannin.
TRPV2-like immunoreactivity was evident throughout the cytoplasm in untreated BV-2 cells (Figure 9A). In the cells treated with CBD for 1 h, TRPV2-like staining was clearly enhanced in the region of the cell membrane with apparently reduced staining in the cytoplasm (Figure 9B), suggesting that CBD caused a translocation of TRPV2 to the membrane; this was abolished in the presence of cycloheximide (Figure 9C) and with the PI3K inhibitor wortamannin (10 μM) (Figure 9E). The ingestion of non-fluorescent latex beads also appeared to enhance TRPV2-like staining (Figure 9D), suggesting that the process of phagocytosis itself stimulates TRP channel expression. Intriguingly, CBD-enhanced TRPV2 translocation was also inhibited in the presence of ruthenium red, indicating that it was mediated by TRP channel activation (Figure 9F).
Figure 9.
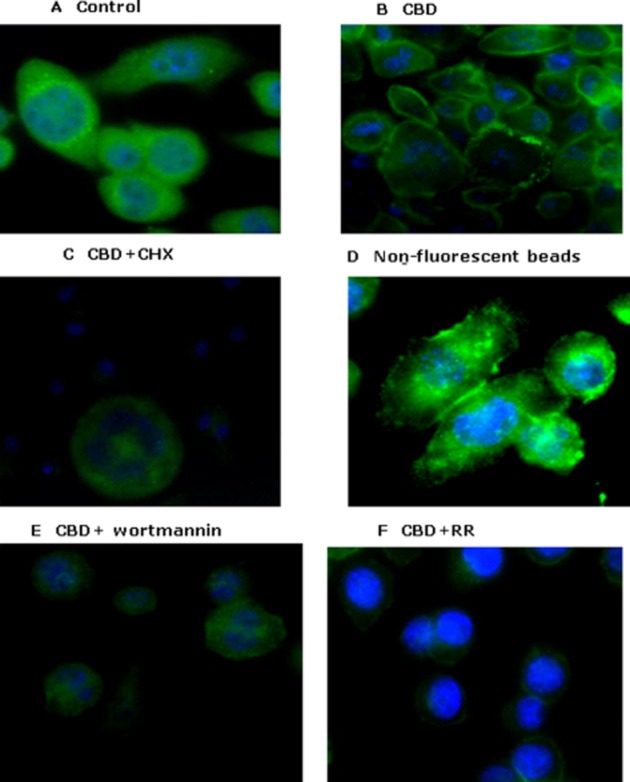
Localization of TRPV2 immunoreactivity in BV-2 cells by immunocytochemical analysis: (A) control; (B) cells incubated with CBD (10 μM) for 1 h; (C) cells incubated with cycloheximide (CHX, 1 μM) 1 h prior to treatment with CBD for 1 h; (D) cells incubated with non-fluorescent latex beads 0.5 μL·mL−1 for 2 h; (E) cells incubated with wortmannin 10 μM 1 h before 10 μM CBD for 1 h; and (F) cells incubated with ruthenium red (RR, 10 μM) 1 h prior to CBD for 1 h. BV-2 cells were fixed with formaldehyde, permeabilized with 0.1% Triton X-100, incubated overnight with the TRPV2 antibody and for 60 min with a fluorescent secondary antibody. The cells were visualized by confocal microscopy under 63× magnification using a glycerine immersion objective lens.
Using immunoblotting, the expression of TRPV2 protein in whole cell and membrane fractions of BV-2 cells was assessed (Figure 10). There was a rapid cycloheximide-sensitive increase in whole cell TRPV2-like protein, which reduced over the course of 24 h, at which point, it was predominantly expressed in the membrane fraction, indicating a translocation to the membrane (Figure 11).
Figure 10.
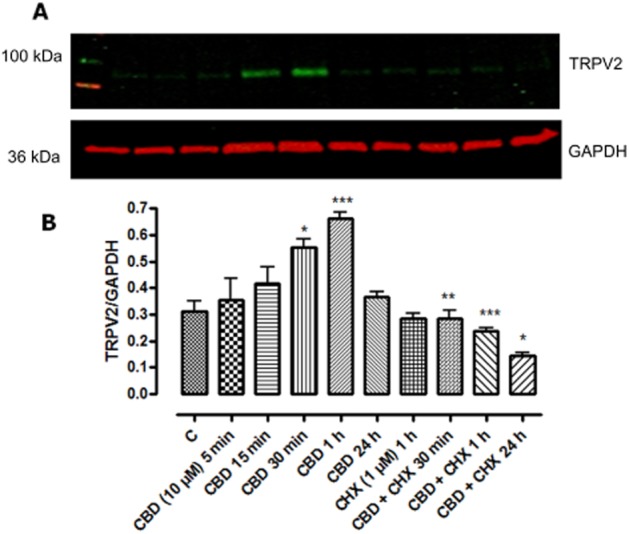
Increase in TRPV2 protein immunoreactivity induced by CBD in whole BV2 cell lysates. Cells were incubated with CBD (10 μM) for 5, 15, 30 and 60 min and 24 h alone. The cycloheximide (CHX 1 μM) was incubated with the cell lysates for 6 h prior to CBD. Data shown are ratios of TRPV2 : GAPDH expression and represent the means ± SEM of three independent experiments. Immunoblot treatments (A) are in the same order as those in the corresponding histograms (B). Data were analysed using one-way anova, followed by post hoc Bonferonni's multiple comparison test; *P < 0.05 CBD 30 min versus control; ***P < 0.001 CBD 1 h versus control; **P < 0.01 CBD + CHX 30 min versus CBD 30 min; ***P < 0.001 CBD + CHX 1 h versus CBD 1 h. *P < 0.05 CBD + CHX 24 h versus CBD 24 h.
Figure 11.
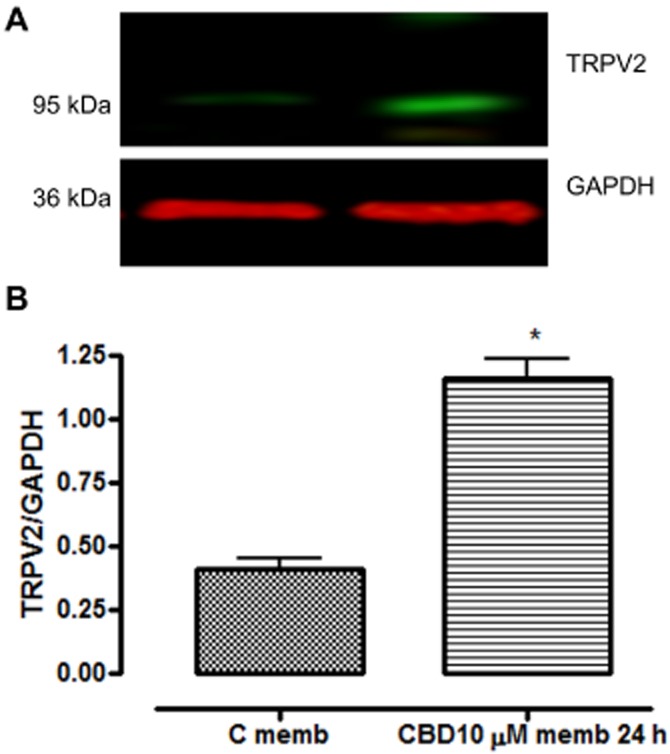
Increased expression of TRPV2 protein in BV2 cell membrane fraction due to CBD. Cells were incubated with no drug (C memb) or CBD (10 μM) for 24 h prior to preparation of membrane fractions. (A) Shows immunoblots and (B) quantification of the expression levels. Data shown are ratios of TRPV2 : GAPDH expression and represent the means ± SEM of three independent experiments. Data were analysed using Student's unpaired t-test (two-tailed). *P < 0.05 CBD 24 (memb) h versus C (control memb).
The CBD enhancement of TRPV2 protein expression was prevented in the presence of the PI3K inhibitor wortmannin (10 μM) and the non-selective TRP channel blocker ruthenium red (Figure 12).
Figure 12.
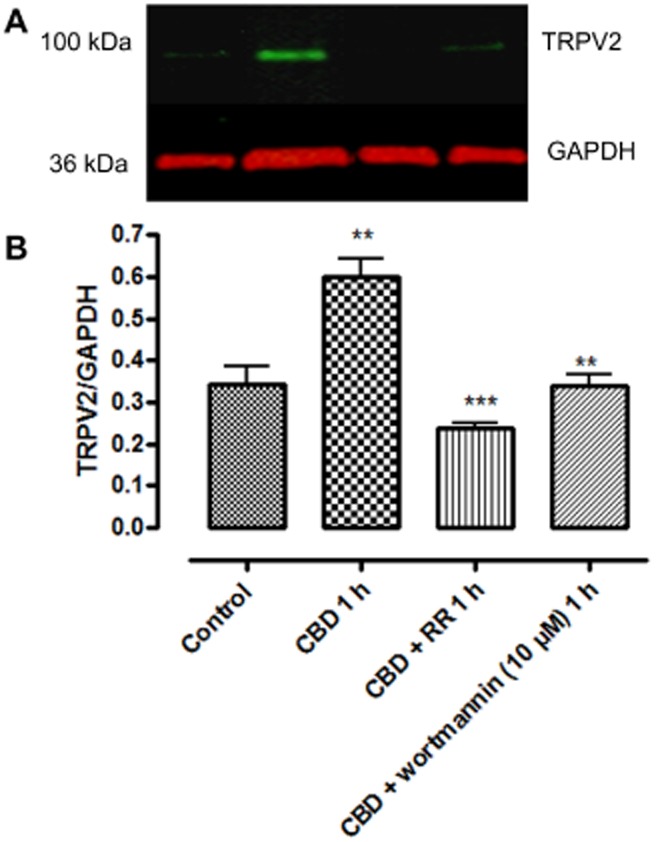
Western immunoblots showing mediation of the enhancing effect of CBD on TRPV2 immunoreactivity in whole cell lysates of BV-2 cells by channel activation and PI3K. Cells were pre-incubated (1 h) with or without ruthenium red (RR) or wortmannin (10 μM) prior to CBD (10 μM). Data shown are ratios of TRPV2: GAPDH expression and represent the means ± SEM of three independent experiments. Data were analysed using one-way ANOVA, followed by post hoc Bonferonni's multiple comparison; **P < 0.01 CBD 1 h versus control; ***P < CBD + RR 1 h versus CBD 1 h alone; **P < 0.01 CBD + wortmannin 1 h versus CBD 1 h alone.
Quantitative RT-PCR analysis indicated that TRPV2 mRNA was expressed at measurable levels in unstimulated BV2 cells. There was a trend towards an increase, albeit non-significant, after 60 min of incubation with CBD (10 μM), and after 24 h, there was a considerable enhancement (Figure 13).
Figure 13.
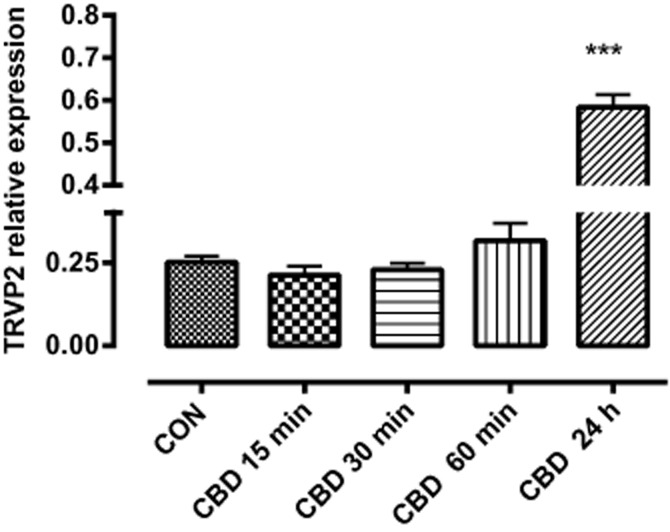
TRPV2 mRNA expression in BV-2 cells and the effect of CBD. RNA was extracted using Trizole reagent and RNA samples were reverse transcribed using the superscript reverse transcription with mouse actin as a normalizing gene. Data are means ± SEM of triplicate determinations of TRPV2/actin mRNA ratios conducted in three separate experiments. Data were analysed using one-way anova, followed by post hoc comparison; ***P < 0.001, compared with control.
Given the sensitivity of CBD enhancement of phagocytosis to the TRPV1 antagonists, the effect of CBD on TRPV1 protein expression was investigated. Similar to its effects on TRPV2, CBD significantly enhanced TRPV1 expression in BV-2 cells at 60 min and this returned to levels not different from control by 24 h. CBD enhancement of TRPV1-like immunoreactivity, as was the case for TRPV2, was blocked by treatment with ruthenium red and wortmannin (10 μM) (Figure 14).
Figure 14.
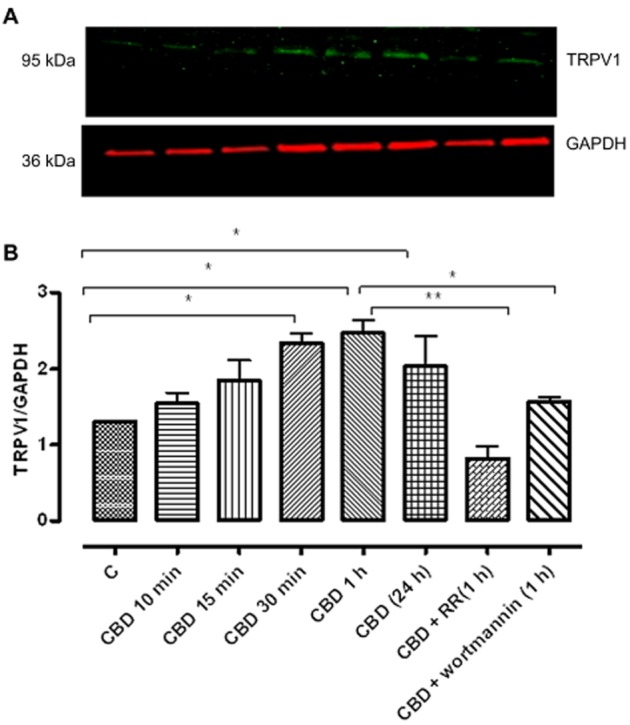
Effect of CBD on TRPV1 protein expression in BV2 whole cell lysates. BV-2 cells were incubated with CBD (10 μM) for 10, 15, 30, 60 min and 24 h. Ruthenium red (RR) (non-selective TRPV channel blocker) and wortmannin (a PI3K inhibitor) (10 μM) significantly inhibited CBD-enhanced TRPV1 expression after 1 h. The data represent the means ± SEM of three experiments, normalized to GAPDH from three independent cultures. Data were analysed using one-way ANOVA, followed by post hoc comparison; *P < 0.05 CBD 30 (min) versus control; *P < 0.05 CBD 1 h versus control; *P < 0.05 CBD 24 h versus control; **P < 0.01 CBD + RR compared with CBD 1 h; *P < CBD + wortmannin 1 h compare with CBD 1 h.
Discussion
CBD has attracted much recent attention as a cannabinoid, albeit with little affinity for G protein-coupled cannabinoid receptors, that has possible medical applications in inflammation, diabetes, cancer, affective and neurodegenerative diseases (Izzo et al., 2009). One of the targets that could mediate some of CBD's central effects is the microglial cell, which plays a crucial role in immune surveillance and response in the CNS. CBD is known to affect some microglial functions; for instance, the control of responses to β-amyloid in Alzheimer's disease models (Martin-Moreno et al., 2011), and to modulate a wide variety of microglial genes involved in the regulation of stress responses and inflammation (Juknat et al., 2012), although its effects on phagocytosis have not been investigated.
In the present study, the effects of CBD and other cannabinoids on phagocytosis were investigated in microglial cell lines and murine microglia in primary culture. All of the microglial cells ingested and accumulated latex beads over time, and the process was able to be positively modulated by the pyrimidine UDP, the pro-inflammatory mediators LPS and IFN-γ, and by the steroid dexamethasone. Of the synthetic cannabinoids tested, only the CB1/CB2 agonist WIN55212-2 significantly reduced phagocytosis, but this was only effective at a concentration well in excess of that required to occupy cannabinoid receptors and, upon investigation by RT-PCR, the BV-2 cells were found not to express either CB1 or CB2 receptors. This is somewhat surprising given the reports that cannabinoid CB2 receptors and abn-CBD receptors mediate the effects of cannabinoids on BV-2 cell migration (Franklin and Stella, 2003), but it is clear that, under the culture conditions employed here, the BV-2 cells failed to express either CB1 or CB2 receptors significantly. It does seem that BV-2 cell expression of CB receptors is not well maintained with increasing passage number (N. Stella, pers. comm.), which might explain this deficit.
However, CBD significantly enhanced latex bead ingestion in BV-2 and HAPI cell lines, in primary mouse microglia and in monocyte/macrophage RAW-264 cells, although the other phytocannabinoids tested were without effect. The Gi-protein inhibitor PTX was unable to affect the enhancement due to CBD, indicating that none of the Gi-linked family of GPCRs was involved. Calcium signalling does seem to play a role in responses to CBD, which has been shown to mobilize intracellular Ca2+ stores in a number of different cell types, including hippocampal neurones and glia (Drysdale et al., 2006). Qin et al. (2008) reported that CBD increased intracellular Ca2+ in dorsal root ganglion neurones via activation of TRPV2 channels and CBD has also been shown to be a partial agonist of TRPV1 and TRPV2 channels, with micromolar potency, and a full agonist at TRPA1 channels with high nanomolar range potency (Bisogno et al., 2001; De Petrocellis et al., 2011). In the present study, CBD's enhancement of BV-2 phagocytosis was not affected by chelation of intracellular Ca2+ via BAPTA-AM treatment but extracellular Ca2+ entry was shown, by reducing extracellular cation levels with EGTA, to be required. Furthermore, the non-selective TRP channel blocker ruthenium red prevented CBD enhancement of phagocytosis as did SKF-96365, a blocker of TRPC and TRPV subfamily channels and of receptor-mediated and voltage-gated Ca2+ entry (Merritt et al., 1990; Kim et al., 2003; Bomben and Sontheimer, 2008). The more selective TRPV1 antagonists capsazepine and AMG9810 were effective blockers of CBD's enhancing effect, although it is notable that the most TRPV1-selective antagonist AMG0910 had a lesser effect than SKF-96365 or capsazepine, indicating that multiple receptors/channels are involved in the control of phagocytosis. The ability of CBD to increase intracellular Ca2+ was confirmed in single-cell imaging studies, which showed it to produce a sustained increase in intracellular Ca2+ in a manner sensitive to blockade by ruthenium red.
These data are consistent with the proposal that CBD-elevated intracellular Ca2+, via activation of one or more TRP channels, mediates the enhanced phagocytosis. A sustained increase in [Ca2+]i seems to be required to enhance phagocytosis since ATP, which produced only a transient [Ca2+]i elevation, was without effect (Figure 3) and TRPV2 channels are known to resist desensitization and to promote calcium entry for a relatively long period (Kanzaki et al., 1999). However, in the absence of more potent and selective antagonists, particularly in the case of TRPV2, it is difficult to fully characterize pharmacologically the receptors involved. Tranilast has been employed as a TRPV2 antagonist (Hisanaga et al., 2009); however, in the present study, it enhanced the basal rate of phagocytosis and produced a somewhat additive effect when combined with CBD. As the receptor/channel pharmacology of tranilast has not been fully characterized, it cannot be concluded whether or not this was an off-target effect of the compound unrelated to TRPV2 blockade. Although it has a variety of other targets (notably in preventing efflux of organic ions from cells), the uricosuric agent probenecid is now known to activate TRPV2 channels with an EC50 of around 30 μM (Robbins et al., 2012), and in the present study, it was found to enhance BV-2 phagocytosis in a concentration-dependent fashion, thereby supporting a modulatory role for TRPV2. Based upon these findings, and given the reports of the key role that TRPV2 channels play in peripheral macrophage phagocytosis (Link et al., 2010), the involvement of this receptor channel was further investigated using a combination of immunoblotting and immunocytochemistry. CBD exposure rapidly increased TRPV2 protein expression and promoted its translocation to the cell surface of BV-2 cells. Blockade by cycloheximide suggests that new protein synthesis was involved, although the corresponding measurements of TRPV2 gene transcription appeared to show that this lagged behind increased protein expression. It is possible that this reflects artifactual differences in the sensitivity of the measurements rather than a real mismatch in gene transcription and translation or that there could be a rapid turnover of the gene transcript, which only accumulates at later time points after early increases in transcription.
Interestingly, TRPV2 expression and its membrane localization appeared to be enhanced simply by the ingestion of latex beads (Figure 9D), suggesting that multiple means of activating phagocytosis might then increase TRPV2 expression, and the observation that blockade of the TRP channels with ruthenium red (Figure 9G) also reduces TRPV2 expression, suggesting that channel activity itself promotes increased expression. This is somewhat counterintuitive as a reactive down-regulation might be predicted upon channel activation to limit Ca2+ entry, and this implies that the TRP channels could be part of a feed-forward amplification of intracellular Ca2+ signalling that enhances microglial activation. However, the changes in channel expression and acute activation might be unrelated phenomena as, for example, the phytocannabinoid cannabichromene modifies the expression of TRPVs1–4 in the gastrointestinal tract but does not activate the channels (De Petrocellis et al., 2012). As TRPV2 channels are mechanoreceptors, it is possible that stressing the cell membrane in the act of phagocytosis (promoted by any appropriate stimulus) would activate the channels, thus engaging the amplification process.
TRPV2 channel activity is reported to be controlled by growth factors acting through a PI3K-dependent signalling pathway (Kanzaki et al., 1999) and, as shown in Figure 10, wortmannin, a PI3K inhibitor, blocked the CBD enhancement of BV-2 phagocytosis. Other examples of PI3K control of TRPV2 channel translocation have been reported; for example, in pancreatic beta cells, insulin, acting via PI3K, increases the insertion of TRPV2 channels into the plasma membrane and accelerates Ca2+-dependent exocytosis, thereby enhancing its own release in an autoregulatory fashion (Aoyagi et al., 2010).
TRPV1 channels seem to have a similar role as TRPV2 in that blockade with capsazepine and AMG9810 prevented CBD-enhanced phagocytosis and, conversely, CBD increased TRPV1 protein expression in a PI3K-dependent fashion. A similar situation is reported regarding dorsal root ganglion cells in which PI3K binds to TRPV1 and mediates nerve growth factor-stimulated TRPV1 trafficking to the plasma membrane, thereby sensitizing the sensory nerves to painful stimuli (Stein et al., 2006). As the endocannabinoid anandamide activates TRPV1 channels (Ross, 2003), it might have been expected to have an effect similar to that of CBD, but anandamide and other agonists appear to bind to an intracellular site on the channel complex (Jung et al., 1999; Ross, 2003) and accessibility through the microglial membrane might be limited or its availability might be reduced by catabolism. Similarly, the other phytocannabinoids, which are TRPV agonists (De Petrocellis et al., 2011), might have been expected to enhance phagocytosis and further studies will be required to clarify which overall properties of the compounds determine efficacy in this regard.
In summary, CBD, but not related phytocannabinoids, enhances microglial phagocytosis, at least in part, via activation of TRPV1 and probably TRPV2 receptor channels with subsequent potential for amplification of the response due to rapidly increased channel expression. The mechanism appears to involve phosphoinositide 3-kinase, although other downstream signalling elements resulting in increased phagocytosis are yet to be identified. Although additional experiments on microglia in primary culture and in situ are required, the results presented herein support the possibility for modulating microglial function using TRP channel ligands, possibly including CBD or a more potent analogue. It is probably simplistic to conclude whether increasing microglial phagocytosis is essentially a positive or a negative strategy for combating neuroinflammatory disorders. For example, enhancing clearance of β-amyloid would probably be of benefit in Alzheimer's disease, but microgliosis is enhanced in other conditions such as neuropathic pain in which inhibiting microglia with minocycline has been shown to be of benefit (Sagar et al., 2011). Clearly, the potential therapeutic outcomes will be dependent on the type, location and chronicity of disease.
Acknowledgments
The authors are grateful to GW Pharma for the gift of phytocannabinoids; to Dr Emma King for the assistance with cell imaging and Mr Liaque Latif for the general technical help. The financial support of the Libyan and Egyptian Cultural Bureaux to S H and K E, respectively, is gratefully acknowledged.
Glossary
- CBD
cannabidiol;
- LPS
bacterial lipopolysaccharide
Conflict of interest
The authors have no conflicts of interest to declare.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi K, Ohara-Imaizumi M, Nishiwaki C, Nakamichi Y, Nagamatsu S. Insulin/phosphoinositide 3-kinase pathway accelerates the glucose-induced first-phase insulin secretion through TrpV2 recruitment in pancreatic beta-cells. Biochem J. 2010;432:375–386. doi: 10.1042/BJ20100864. [DOI] [PubMed] [Google Scholar]
- Bang S, Kim KY, Yoo S, Lee SH, Hwang SW. Transient receptor potential V2 expressed in sensory neurons is activated by probenecid. Neurosci Lett. 2007;425:120–125. doi: 10.1016/j.neulet.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Barichello T, Ceretta RA, Generoso JS, Moreira AP, Simoes LR, Comim CM, et al. Cannabidiol reduces host immune response and prevents cognitive impairments in Wistar rats submitted to pneumococcal meningitis. Eur J Pharmacol. 2012;697:158–164. doi: 10.1016/j.ejphar.2012.09.053. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Hanus L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. 2001;134:845–852. doi: 10.1038/sj.bjp.0704327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomben VC, Sontheimer HW. Inhibition of transient receptor potential canonical channels impairs cytokinesis in human malignant gliomas. Cell Prolif. 2008;41:98–121. doi: 10.1111/j.1365-2184.2007.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Zhang YQ. Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev. 2008;32:972–983. doi: 10.1016/j.neubiorev.2008.03.009. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Ligresti A, Moriello AS, Allara M, Bisogno T, Petrosino S, et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163:1479–1494. doi: 10.1111/j.1476-5381.2010.01166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Orlando P, Moriello AS, Aviello G, Stott C, Izzo AA, et al. Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol (Oxf) 2012;204:255–266. doi: 10.1111/j.1748-1716.2011.02338.x. [DOI] [PubMed] [Google Scholar]
- Drysdale AJ, Ryan D, Pertwee RG, Platt B. Cannabidiol-induced intracellular Ca2+ elevations in hippocampal cells. Neuropharmacology. 2006;50:621–631. doi: 10.1016/j.neuropharm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Franklin A, Stella N. Arachidonylcyclopropylamide increases microglial cell migration through cannabinoid CB2 and abnormal-cannabidiol-sensitive receptors. Eur J Pharmacol. 2003;474:195–198. doi: 10.1016/s0014-2999(03)02074-0. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, et al. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313:474–484. doi: 10.1124/jpet.104.079855. [DOI] [PubMed] [Google Scholar]
- Hisanaga E, Nagasawa M, Ueki K, Kulkarni RN, Mori M, Kojima I. Regulation of calcium-permeable TRPV2 channel by insulin in pancreatic beta-cells. Diabetes. 2009;58:174–184. doi: 10.2337/db08-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Borrelli F, Capasso R, Di M, V, Mechoulam R. Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol Sci. 2009;30:515–527. doi: 10.1016/j.tips.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Juknat A, Rimmerman N, Levy R, Vogel Z, Kozela E. Cannabidiol affects the expression of genes involved in zinc homeostasis in BV-2 microglial cells. Neurochem Int. 2012;61:923–930. doi: 10.1016/j.neuint.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Jung J, Hwang SW, Kwak J, Lee SY, Kang CJ, Kim WB, et al. Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. J Neurosci. 1999;19:529–538. doi: 10.1523/JNEUROSCI.19-02-00529.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki M, Zhang YQ, Mashima H, Li L, Shibata H, Kojima I. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nat Cell Biol. 1999;1:165–170. doi: 10.1038/11086. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature. 2003;426:285–291. doi: 10.1038/nature02162. [DOI] [PubMed] [Google Scholar]
- Kozela E, Pietr M, Juknat A, Rimmerman N, Levy R, Vogel Z. Cannabinoids delta(9)-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2 microglial cells. J Biol Chem. 2010;285:1616–1626. doi: 10.1074/jbc.M109.069294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Link TM, Park U, Vonakis BM, Raben DM, Soloski MJ, Caterina MJ. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat Immunol. 2010;11:232–239. doi: 10.1038/ni.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Moreno AM, Reigada D, Ramirez BG, Mechoulam R, Innamorato N, Cuadrado A, et al. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer's disease. Mol Pharmacol. 2011;79:964–973. doi: 10.1124/mol.111.071290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecha M, Torrao AS, Mestre L, Carrillo-Salinas FJ, Mechoulam R, Guaza C. Cannabidiol protects oligodendrocyte progenitor cells from inflammation-induced apoptosis by attenuating endoplasmic reticulum stress. Cell Death Dis. 2012;3:e331. doi: 10.1038/cddis.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA, Gallily R. Cannabidiol: an overview of some pharmacological aspects. J Clin Pharmacol. 2002;42:11S–19S. doi: 10.1002/j.1552-4604.2002.tb05998.x. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Peters M, Murillo-Rodriguez E, Hanus LO. Cannabidiol – recent advances. Chem Biodivers. 2007;4:1678–1692. doi: 10.1002/cbdv.200790147. [DOI] [PubMed] [Google Scholar]
- Merritt JE, Armstrong WP, Benham CD, Hallam TJ, Jacob R, Jaxa-Chamiec A, et al. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem J. 1990;271:515–522. doi: 10.1042/bj2710515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. 1998;54:99–125. doi: 10.1016/s0301-0082(97)00052-x. [DOI] [PubMed] [Google Scholar]
- Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun. 2007;362:928–934. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci. 2008;28:6231–6238. doi: 10.1523/JNEUROSCI.0504-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins N, Koch SE, Tranter M, Rubinstein J. The history and future of probenecid. Cardiovasc Toxicol. 2012;2:1–9. doi: 10.1007/s12012-011-9145-8. [DOI] [PubMed] [Google Scholar]
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;140:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Burston JJ, Hathway GJ, Woodhams SG, Pearson RG, Bennett AJ, et al. The contribution of spinal glial cells to chronic pain behaviour in the monosodium iodoacetate model of osteoarthritic pain. Mol Pain. 2011;7:88. doi: 10.1186/1744-8069-7-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon SE. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J Gen Physiol. 2006;128:509–522. doi: 10.1085/jgp.200609576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodroofe MN, Sarna GS, Wadhwa M, Hayes GM, Loughlin AJ, Tinker A, et al. Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine production. J Neuroimmunol. 1991;33:227–236. doi: 10.1016/0165-5728(91)90110-s. [DOI] [PubMed] [Google Scholar]