

Abstract
Repair of radiation-induced dna double-strand breaks is a key mechanism in cancer cell radio-resistance. The synthesized compound NU7026 specifically inhibits dna-dependent protein kinase (dna-pk) within the non-homologous end-joining repair mechanism. Earlier studies demonstrated increased radiosensitivity in dna-pk deficient cells compared with wild-type cells. In chronic leukemia cells, NU7026 appears to enhance the cytotoxic effect of chlorambucil. The radio-modifying effects of NU7026 on cell survival, cell cycle, apoptosis, and dna double-strand break repair have yet to be studied in gastric cancer cells.
Methods
The gastric cancer cell line N87 was treated with 0 Gy or 4 Gy in the presence of NU7026 at a dose range of 0–20 μmol/L. Clonogenic assays were used to assess cell survival after treatment. Cell-cycle distribution was analyzed using propidium iodide with fluorescence-activated cell sorting. Apoptosis was detected using annexin-V and propidium iodide with fluorescence-activated cell sorting. The γH2AX assay was used to measure dna double-strand breaks.
Results
Statistically significant increases in G2/M arrest were observed in N87 cells treated with radiation and NU7026 compared with those treated with radiation alone (p = 0.0004). Combined treatment also led to an increase in apoptosis (p = 0.01). At 24 hours, the γH2AX analysis revealed more dna double-strand breaks in N87 cells treated with radiation and NU7026 than in those treated with radiation alone (p = 0.04). Clonogenic assays demonstrated declining cell survival as both the radiation and the NU7026 dose increased. The dose enhancement factor at 0.1 survival fraction was 1.28 when N87 cells were treated with 4 Gy radiation and 5 μmol/L NU7026.
Conclusions
In gastric cancer cells, NU7026 appears to enhance the cytotoxic effect of irradiation as assessed by clonogenic assays. This increased cytotoxicity might be the result of an increase in dna double-strand breaks resulting in G2/M cell arrest and possibly higher levels of apoptosis.
Keywords: Gastric cancer, dna pk inhibitor, NU7026, radiation therapy, radiosensitization
1. INTRODUCTION
The management of gastric cancer patients has evolved since the early 2000s. The combination of primary surgical resection with perioperative chemotherapy or adjuvant chemoradiation is the standard of care for most of these patients. Despite advances in primary and adjuvant treatments, 50%–70% of patients with advanced gastric or gastroesophageal junction tumours will relapse and die of their disease1. Meta-analyses showed modest survival gains with adjuvant chemotherapy2, but no benefit was found for postoperative radiotherapy alone3. The Intergroup 0116 trial demonstrated that postoperative chemoradiation significantly improves survival in patients with gastric and gastroesophageal cancer, and thus established one of the standard approaches for these patients4. Although perioperative chemotherapy and adjuvant chemoradiation have improved overall survival, the results—50% overall survival at 3 years—are still suboptimal and in need of further improvement.
It has been proposed that dna repair enzymes, involved in both single- and double-stand breaks (dsbs) in dna, contribute to the resistance of tumour cells to ionizing radiation and drugs that cause dsbs5. To improve the therapeutic index of proven anticancer drugs, the use of dna repair inhibitors to sensitize cancer cells to chemotherapeutic agents is currently being translated to the clinical setting. The impact of dna repair inhibitors on radiosensitivity in gastric cancer cells remains to be explored.
The homologous recombination and non-homologous end-joining (nehj) mechanisms are both involved in dna dsb repair6. In mammalian cells, nehj is the major repair pathway, operating in the S and G2 phases of the cell cycle. The nehj mechanism is regulated by the dna-dependent protein kinase (dna-pk) complex that consists of dna-pkcs, a catalyst subunit, and its heterodimer Ku regulatory subunit, Ku 70 and Ku 80. The dna-pk complex binds to dna ends and acts as a scaffold for other dna repair; it also phosphorylates a series of dna repair enzymes, including p53, Ku, Artemis, xrcc4, and dna-pkcs itself7–10. Upon auto-phosphorylation, the dna-pkcs is released from the dna ends and processing and ligation of the dna dsbs are initiated by Artemis and ligase iv–xrcc4 complexes.
At least 16 amino acid residues in dna-pkcs are autophosphorylation sites. Mutation in some of these sites results in increased radiosensitivity and impaired dsb repair by inhibition of phosphorylation and decreased processing of dna ends. The specific dna-pk inhibitor NU7026 [2-(morpholin-4-yl)-benzo[h]chomen-4-one] has been demonstrated to impair cell survival and dna dsb repair in dna-pk proficient cells11. The specificity of this compound was confirmed when no effect on cell survival and dna dsb repair was observed in NU7026-treated dna-pk–deficient cell lines11. In vitro testing of NU7026 in combination with chlorambucil demonstrated dna-pk inhibitor–mediated sensitization of primary chronic lymphocytic leukemia cells12. We hypothesized that the use of dna-pk inhibitor in combination with ionizing radiation in gastric cancer cells might increase dna dsb, which might in turn increase cell killing by increased radiosensitivity.
2. METHODS
2.1. Cell Culture
Human N87 metastatic gastric cancer cells (CRL-5822) were obtained from ATCC (Manassas, VA, U.S.A.). Cells were cultured in rpmi-1640 (Gibco Invitrogen, Burlington, ON) supplemented with 10% fetal bovine serum (Wisent, St-Jean-Baptiste, QC) and 1% penicillin–streptomycin. Cells were cultured at 37°C in 5% CO2 and dimethyl sulfoxide dilution. Before irradiation, cells were treated with NU7026 (Sigma–Aldrich, St. Louis, MO, U.S.A.) at various concentrations ranging from 2.5 μmol/L to 20 μmol/L.
2.2. Irradiation
Cells were irradiated at room temperature using a 160 kVp X-ray irradiator (FC-160: Faxitron X-Ray, Wheeling, IL, U.S.A.) at a dose rate of 1.9 Gy/min.
2.3. Cell Proliferation Assay
Growth inhibition was measured using the thiazoyl blue tetrazolium bromide assay. Cells were plated at a density of 10,000 per well in 96-well plates. The cells were treated with NU7026 for 24 hours before irradiation with 0 Gy or 4 Gy. Cells were exposed for an additional 24 hours to NU7026 before being washed with drug-free medium and left to incubate for an additional 72 hours. Thiazoyl blue tetrazolium bromide solution (Sigma–Aldrich, Oakville, ON) was then added for 4 hours, and the reaction was stopped before optical density was measured in a 96-well plate reader at 750 nm.
2.4. Colony-Forming Assay
Cells were plated at specific cell numbers in 6-well plates. They were treated for 24 hours with NU7026 before undergoing irradiation with 0 Gy, 2 Gy, 4 Gy, and 6 Gy. The plated cells were incubated for an additional 24 hours after irradiation before being washed with drug-free medium. Incubation was then continued for a period of 21 days, with the medium being renewed twice weekly. After 21 days, colonies were fixed with 70% ethanol and stained with methylene blue. Only colonies containing more than 50 cells were counted. The plating efficiency was determined by dividing the number of colonies formed in the untreated control plates by the number of cells plated. Survival fraction was determined as colonies formed after the specific radiation dose divided by cells seeded at the same radiation dose, multiplied by the plating efficiency. To plot the survival curve, survival fractions were normalized to the non-irradiated controls. Dose enhancement factor was measured as the ratio of the radiation doses at a survival fraction of 0.1 of non-drug-treated to drug-treated cells.
2.5. Cell Cycle Analysis
Cells were exposed to 20 μmol/L NU7026 for 24 hours before undergoing 0 Gy and 4 Gy irradiation. Twenty-four hours after irradiation, cells were washed, harvested, and fixed with ethanol. Cells were then stained with propidium iodide (Sigma–Aldrich, Oakville, ON) and analyzed by flow cytometry (BD Biosciences, Mississauga, ON). Cell cycle distribution was analyzed using the Mod-Fit LT software package (Verity Software, Topsham, ME, U.S.A.).
2.6. Apoptosis Analysis
Cells were exposed to 0 μmol/L and 20 μmol/L NU7026 24 hours before irradiation with 0 Gy and 4 Gy. Forty eight hours after irradiation, the cells were harvested and washed with phosphate-buffered saline 1× (Gibco Invitrogen). They were then labelled with annexin-V–fluorescein isothiocyanate and propidium iodide according to the manufacturer’s protocol (TACS apoptosis kit: R&D Systems, MI, U.S.A.) and were analyzed by flow cytometry. Data were collected using logarithmic amplification of both the fl1 (fluorescein isothiocyanate) and fl2 (propidium iodide) channels. Cells were characterized as apoptotic when they were positive for annexin-V or annexin-V and propidium iodide. Collected data were analyzed using the CellQuest software (BD Biosciences).
2.7. γH2AX Analysis
Cells were treated with 0 μmol/L and 20 μmol/L NU7026 24 hours before exposure to 0 Gy and 4 Gy irradiation. Cells were then incubated for an additional 24 hours, washed, and harvested. Cells were washed first with methanol-free formaldehyde and then with phosphate-buffered saline and were fixed in 70% ethanol. Cells were then exposed to mouse anti-γH2AX (Ser139) antibody (AbCam, Cambridge, MA, U.S.A.) overnight at 4°C and subsequently to goat anti-mouse immunoglobulin G fluorescein isothiocyanate antibody (Sigma–Aldrich, St. Louis, MO, U.S.A.) in the dark at room temperature. The cells were then stained with propidium iodide for 1 hour in the dark at room temperature. Cells were analyzed by flow cytometry at 1 and 24 hours, and the data collected were analyzed by CellQuest software using a published protocol13. A limit of 95% of untreated cells expressing γH2AX immunofluorescence was set, and that limit was used to quantify relative increases in γH2AX foci in treated cells14.
2.8. Statistical Analysis
Comparisons of the various treatments in all experiments were made using the two-tailed t-test. Differences with a p value less than 0.05 were considered statistically significant. The data are represented as mean and standard error of the mean from 3 independent experiments.
3. RESULTS
3.1. Cell Proliferation Assay
Cell proliferation assays demonstrated a decrease in optical density when cells were treated with radiation or with NU7026. The optical density was 0.754 after treatment with radiation and 0.569 after treatment with NU7026. When the cells were exposed to 4 Gy and 12.5 μmol/L NU7026 in combination, the optical density was 0.389 (p = 0.0004). The half-maximal inhibitory concentration of the drug for N87 cells was 17.5 μmol/L (Figure 1).
FIGURE 1.

Cell viability of N87 when exposed to increasing concentrations of NU7026. The half-maximal inhibitory concentration (IC50) was determined to be 17.5 μmol/L(μM). Data are the mean and standard error of the mean from 3 independent experiments.
3.2. Clonogenic Assays
The clonogenic assays demonstrated a significant decline in clonogenic survival when N87 cells were treated with radiation and NU7026. The dose enhancement factor for 0.1 survival at 5 μmol/L NU7026 was determined to be 1.28 (Figure 2).
FIGURE 2.

Analysis by clonogenic assay of cell survival fraction when N87 cells were treated with NU7026 and varying doses of radiation. Cells were pre-treated with NU7026 24 hours before radiation. Data are the mean and standard error of the mean from at least 3 independent experiments. μM = μmol/L.
3.3. Cell Cycle Analysis
Cell cycle analysis at 48 hours confirmed irradiation-induced G2/M cell cycle arrest when N87 cells were irradiated with 4 Gy compared with 0 Gy for control cells (34.1% vs. 21.2%, p = 0.04). Compared with the control group, cells exposed to 20 μmol/L NU7026 alone showed a non-statistically significant increase in G2/M cell cycle arrest (27.8% vs. 21.2%, p = 0.125). Compared with the treatment group receiving 4 Gy alone, cells receiving pre-irradiation treatment with 20 μmol/L NU7026 showed increased G2/M cell cycle arrest (68.4% vs. 34.1%, p = 0.0004, Figure 3).
FIGURE 3.

Cell cycle analysis by flow cytometry for N87 cells 24 hours after radiation in the presence and absence of NU7026. Representative cell cycle histograms for (A) 0 Gy, (B) 0 Gy and 20 μmol/L NU7026, (C) 4 Gy, and (D) 4 Gy and 20 μmol/L NU7026.
3.4. Apoptosis Analysis
Apoptosis analysis of cells 48 hours after treatment demonstrated a baseline apoptosis level of 5.69% in the control arm. Cells treated with NU7026 alone had an apoptosis level of 7.79%, and those treated with radiation alone had an apoptosis level of 5.29%. When NU7026 and radiation were combined, the apoptosis level was 8.54%, which was statistically significant compared with the control arm (p = 0.0004) and with the radiation-alone arm (p = 0.012, Figure 4).
FIGURE 4.
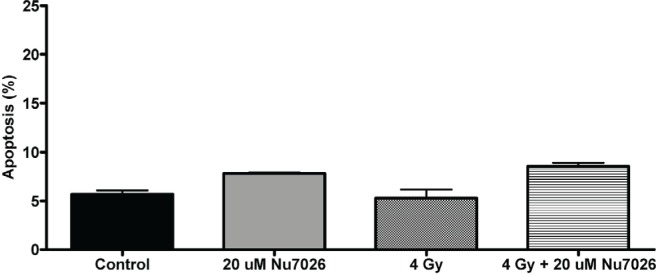
Analysis of early apoptosis in N87 cells, 48 hours after treatment with NU7026, with radiation, or with NU7026 and radiation combined. Data are represented as the mean and standard error of the mean of 3 independent experiments. μM = μmol/L.
3.5. γH2AX Analysis
At 1 and 24 hours post-irradiation, γH2AX levels were determined for the N87 cell lines. At 1 hour, the limit of γH2AX immunofluorescence was set at 5.0%, per protocol. The relative increase in γH2AX foci at 1 hour was measured at 62.1% in the cells treated with 4 Gy (p = 0.0004 compared with control) and 46.4% for cells treated with 4 Gy and 20 μmol/L NU7026 (p = 0.03). At 1 hour, there was no significant difference between cells treated with radiation alone and cells treated with radiation and NU7026. At 24 hours, N87 cells treated with radiation demonstrated 7.2% γH2AX foci compared with the baseline 5.0% in control cells (p = 0.09). In cells treated with NU7026 and radiation, γH2AX foci showed a relative increase at 11.2%, which was significantly different from the foci in control cells (p = 0.008) and in cells treated with radiation alone (p = 0.04, Figure 5).
FIGURE 5.

Gamma-H2AX analysis by flow cytometry of N87 cells treated with NU7026 alone, with radiation alone, or with NU7026 and radiation combined. The flow cytometry analysis was performed at 1 hour and at 24 hours. At 24 hours, relatively higher levels of gamma-H2AX induction was observed in cells treated with radiation and NU7026. Results shown here are representative of 3 independent experiments performed at 24 hours after irradiation. μM = μmol/L.
4. DISCUSSION
Inhibition of dna-pk has previously been shown to sensitize chronic lymphocytic leukemia cells to cytotoxic agents12. The combination of specific NU7026 dna-pk inhibition and ionizing radiation has been demonstrated to increase dna dsb and cell kill in human glioma, multiple myeloma, and ovarian cancer cell lines15–17. To our knowledge, no studies have so far correlated dna-pk inhibition with dna dsbs, apoptosis, and ultimately, cell survival in any human gastric cancer cell line.
We demonstrated that, compared with radiation treatment alone, the combination of NU7026 dna-pk inhibition and radiation increases cell kill in a human gastric cancer cell line. Although the level of γH2AX foci, a sensor of dsb repair, remained elevated in cells treated with radiation and NU7026 (compared with cells receiving radiation alone), the percentage of γH2AX foci was less at 24 hours than at 1 hour after irradiation. That observation might be a result of incomplete dna-pk inhibition or dna dsb repair by an alternative mechanism such as the homologous repair pathway. It might also reflect dna-pk inhibitor delay in dna dsb repair in the combined radiation and NU7206 group at both the 1-hour (hinted at by the increase in γH2AX foci in the radiation-only group compared with the combined-therapy group) and the 24-hour time points (higher level of γH2AX foci in the combined-therapy group compared with the radiation-only group). Future scheduling experiments—for example, drug given before or after radiation—will be conducted.
The current hypothesis proposes that dna-dsb repair is mediated by dna-pk via the nehj mechanism and occurs predominantly in the G2/M phase of the cell cycle12. The resulting G2/M accumulation observed in the combination of radiation and NU7026 is congruent with previous studies combining NU7026 with chlorambucil or etoposide12,18,19. The contribution of apoptosis to cytotoxicity in solid tumours is usually small, unlike that in hematogenous tumour models. Our apoptosis assay demonstrated a slight but statistically significant increase (8.54%) in the apoptosis level for cells receiving radiation and NU7026 than for cells receiving radiation alone. That observation suggests that the method of cell death might only in part be attributable to apoptosis, and that another mechanism, such as mitotic catastrophe, might contribute to the increased cell killing observed in the clonogenic assays.
Our results suggest an increase in cell kill when gastric cancer cells are exposed to both radiation and dna-pk inhibition by NU7026. These findings represent a novel way of increasing the efficacy of irradiation on dna dsb and cell death. However, there is room to further evaluate mechanisms of dna dsb repair and to further characterize the modes of cell death in gastric cancer cells. NU7026 is not a tumour-specific dna-pk inhibitor; any effect on dna dsb and cell kill would also affect normal tissue. To increase the therapeutic index of this drug, it might be necessary to combine dna-pk inhibition with very conformal methods of radiation delivery such as stereotactic radiosurgery or brachytherapy. Another method might be to combine dna-pk inhibitors with targeted drug delivery systems.
5. CONCLUSIONS
Our study showed that a dna-pk inhibitor, NU7026, can be used to enhance radiation-induced cytotoxicity in a sequence-dependent manner by increasing the levels of dna damage and cell-cycle arrest in G2/M. These results set the premise for further investigation into the molecular mechanisms underlying the observed effect and for a demonstration of the efficacy of this novel type of combination in vivo.
6. ACKNOWLEDGMENTS
We thank the Canadian Association for Radiation Oncology for financial support (razcer 2009 grant), and Beatrice Fournier for revision of the manuscript.
7. CONFLICT OF INTEREST DISCLOSURES
The authors do not have a financial relationship with the organization that sponsored the research.
8. REFERENCES
- 1.Sasako M. Principles of surgical treatment for curable gastric cancer. J Clin Oncol. 2003;21(suppl):274s–5s. doi: 10.1200/JCO.2003.09.172. [DOI] [PubMed] [Google Scholar]
- 2.Gianni L, Panzini I, Tassinari D, Mianulli AM, Desiderio F, Ravaioli A. Meta-analyses of randomized trials of adjuvant chemotherapy in gastric cancer. Ann Oncol. 2001;12:1178–80. [PubMed] [Google Scholar]
- 3.Allum WH, Hallissey MT, Ward LC, Hockey MS. A controlled, prospective, randomised trial of adjuvant chemotherapy or radiotherapy in respectable gastric cancer: interim report. British Stomach Cancer Group. Br J Cancer. 1989;60:739–44. doi: 10.1038/bjc.1989.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macdonald JS, Smalley SR, Benedetti J, et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med. 2001;345:725–30. doi: 10.1056/NEJMoa010187. [DOI] [PubMed] [Google Scholar]
- 5.Jackson SP. Sensing and repairing dna double-strand breaks. Carcinogenesis. 2002;23:687–96. doi: 10.1093/carcin/23.5.687. [DOI] [PubMed] [Google Scholar]
- 6.De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of dna interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20:7980–90. doi: 10.1128/MCB.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of dna-pk. Oncogene. 2005;24:949–61. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- 8.Uematsu N, Weterings E, Yano K, et al. Autophosphorylation of dna-pkcs regulates its dynamics at dna double-strand breaks. J Cell Biol. 2007;177:219–29. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114–24. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 10.Salles B, Calsou P, Frit P, Muller C. The dna repair complex dna-pk, a pharmacological target in cancer chemotherapy and radiotherapy. Pathol Biol (Paris) 2006;54:185–93. doi: 10.1016/j.patbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW. Radiosensitization and dna repair inhibition by the combined use of novel inhibitors of dna-dependent protein kinase and poly (adp-ribose) polymerase-1. Cancer Res. 2003;18:6008–15. [PubMed] [Google Scholar]
- 12.Amrein L, Loignon M, Goulet AC, et al. Chlorambucil cytotoxicity in malignant B lymphocytes is synergistically increased by 2-(morpholin-4-yl)-benzo[h]chomen-4-one (NU7026)–mediated inhibition of dna double-strand break repair via inhibition of dna-dependent protein kinase. J Pharmacol Exp Ther. 2007;321:848–55. doi: 10.1124/jpet.106.118356. [DOI] [PubMed] [Google Scholar]
- 13.Muslimovic A, Ismail IH, Gao Y, Hammarsten O. An optimized method for measurement of gamma-H2AX in blood mononuclear and cultured cells. Nat Protoc. 2003;3:1187–93. doi: 10.1038/nprot.2008.93. [DOI] [PubMed] [Google Scholar]
- 14.Huang X, Darzynkiewicz Z. Cytometric assessment of histone H2AX phosphorylation: a reporter of dna damage. Methods Mol Biol. 2006;314:73–80. doi: 10.1385/1-59259-973-7:073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukherjee B, McEllin B, Camacho CV, et al. egfrviii and dna double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009;69:4252–9. doi: 10.1158/0008-5472.CAN-08-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang C, Betti C, Singh S, Toor A, Vaughan A. Impaired nehj function in multiple myeloma. Mutat Res. 2009;660:66–73. doi: 10.1016/j.mrfmmm.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nutley BP, Smith NF, Hayes A, et al. Preclinical pharmacokinetics and metabolism of a novel prototype dna-pk inhibitor NU7026. Br J Cancer. 2005;93:1011–18. doi: 10.1038/sj.bjc.6602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willmore E, de Caux S, Sunter NJ, et al. A novel dna-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase ii poisons used in the treatment of leukemia. Blood. 2004;103:4659–65. doi: 10.1182/blood-2003-07-2527. [DOI] [PubMed] [Google Scholar]
- 19.Zhu H, Gooderham NJ. Mechanisms of induction of cell cycle arrest and cell death by cryptolepine in human lung adenocarcinoma A549 cells. Toxicol Sci. 2006;91:132–9. doi: 10.1093/toxsci/kfj146. [DOI] [PubMed] [Google Scholar]