

Abstract
Background and Aims
Hybridizing species such as oaks may provide a model to study the role of selection in speciation with gene flow. Discrete species' identities and different adaptations are maintained among closely related oak species despite recurrent gene flow. This is probably due to ecologically mediated selection at a few key genes or genomic regions. Neutrality tests can be applied to identify so-called outlier loci, which demonstrate locus-specific signatures of divergent selection and are candidate genes for further study.
Methods
Thirty-six genic microsatellite markers, some with putative functions in flowering time and drought tolerance, and eight non-genic microsatellite markers were screened in two population pairs (n = 160) of the interfertile species Quercus rubra and Q. ellipsoidalis, which are characterized by contrasting adaptations to drought. Putative outliers were then tested in additional population pairs from two different geographic regions (n = 159) to support further their potential role in adaptive divergence.
Key Results
A marker located in the coding sequence of a putative CONSTANS-like (COL) gene was repeatedly identified as under strong divergent selection across all three geographically disjunct population pairs. COL genes are involved in the photoperiodic control of growth and development and are implicated in the regulation of flowering time.
Conclusions
The location of the polymorphism in the Quercus COL gene and given the potential role of COL genes in adaptive divergence and reproductive isolation makes this a promising candidate speciation gene. Further investigation of the phenological characteristics of both species and flowering time pathway genes is suggested in order to elucidate the importance of phenology genes for the maintenance of species integrity. Next-generation sequencing in multiple population pairs in combination with high-density genetic linkage maps could reveal the genome-wide distribution of outlier genes and their potential role in reproductive isolation between these species.
Keywords: Quercus rubra, Q. ellipsoidalis, red oak, CONSTANS-like genes, flowering time, divergent selection, ecological speciation, speciation genes
INTRODUCTION
Natural selection has long been recognized as an important driver of speciation, particularly in the adaptive radiation of allopatric populations (Schluter, 2000). More recently, divergent selection has been suggested to play an important role in the development of intrinsic barriers to gene flow (Barluenga et al., 2006; Savolainen et al., 2006) in a process termed ecological speciation (Schluter, 2009; Via, 2012). In ecological speciation, adaptive divergence and reproductive isolation are both products of genetic adaptation to differing ecological conditions, potentially even in the presence of recurrent gene flow (Rundle and Nosil, 2005; Schluter, 2009). However, both the prevalence and the genetic mechanisms underlying this mode of speciation are still largely unknown. Linkage disequilibrium may facilitate ecological speciation if alleles involved in reproductive isolation are linked to genomic regions subject to strong divergent selection (Via, 2012). Alternatively, a single ‘magic’ trait could be associated with simultaneous adaptive divergence and non-random mating such as flowering phenology in plants (Servedio et al., 2011). By extension, ‘magic genes’ may potentially underlie such traits involved in both ecological adaptation and non-random mating. In either scenario, the genomes of recently diverged ‘ecological species’ are marked by broad regions exhibiting little or no differentiation punctuated by islands of pronounced genetic differentiation. Accordingly, genome screens can be used to identify regions resisting the homogenizing effect of gene flow, which may contain genes subject to divergent selection and/or may be involved in the development of reproductive isolation (Nosil et al., 2009; Via, 2012).
A variety of marker types can be employed in FST-based tests to detect loci putatively affected by divergent selection. In these tests, FST values of individual loci are compared with a simulated null distribution derived from a neutral island model of migration (Beaumont and Nichols, 1996). Loci significantly deviating from neutral expectations of differentiation, or outlier loci, may be closely linked to the target of natural selection or even directly under selection themselves. Such model-based approaches can be used concurrently with other statistics to reduce the rate of false positives due to underlying population structure, varying mutation and/or recombination rates, and population admixture (Storz, 2005). In particular, the LnRH statistic provides a model-independent test of selection and compares the reduction of locus-specific gene diversity between two populations with the approximately normal distribution of genome-wide heterozygosity (Schlötterer and Dieringer, 2005).
Here, we apply complementary selection–detection methods to oaks (Quercus: Fagaceae) to identify genes involved in adaptive divergence and reproductive isolation. Hybridization is common in oaks, yet species identities are maintained despite non-zero levels of interspecific gene flow (Curtu et al., 2009; Lepais and Gerber, 2011). Consistent with ecological speciation, genetic mapping and outlier analyses using 389 genetic markers in two sympatric European oak species with different adaptations to water availability (Q. robur and Q. petraea) revealed largely undifferentiated genomes marked by a few clusters of highly differentiated loci, a pattern probably the result of divergent selection with recurrent interspecific gene flow (Scotti-Saintagne et al., 2004). Furthermore, Goicoechea et al. (2012) studied the same oak species pair and their results indicated lower recombination rates for genomic regions containing outliers as compared with a control region.
We focus on the interfertile species Q. rubra and Q. ellipsoidalis (section Lobatae) which maintain varied adaptations to drought despite recurrent interspecific gene flow (Lind and Gailing, 2013; A. R. Sullivan, in prep.). Both species occur in sympatry in the Great Lakes region of eastern North America but differ in their ecological niches. While Q. ellipsoidalis grows on sandy outwash plains and is considered the most drought-tolerant red oak species (Abrams, 1988; Burns and Honkala, 1990), Q. rubra is more common on north-facing and bottom slopes with fine soils containing more organic matter (Abrams, 1990; Burns and Honkala, 1990). Consistent with their occurrence on sites with differing moisture availability, these two species also differ in morphological and physiological characteristics related to drought response and water-use efficiency (e.g. tissue elasticity, leaf conductance, xylem anatomy and root depth; Abrams, 1990).
Despite the ecological and morphological differences between Q. rubra and Q. ellipsoidalis, they generally show very low genetic differentiation at most genetic markers, and the presence of morphologically and genetically intermediate individuals is consistent with recurrent interspecific gene flow (Hokanson et al., 1993; Lind and Gailing, 2013). In addition, genetic assignment analyses using nuclear [nuclear simple sequence repeat (nSSR)] and genic microsatellite [expressed sequence tag (EST)-SSR] markers have identified putative first-generation hybrids and introgressive forms in adult trees and seedlings. However, adult hybrids were relatively infrequent, suggesting the absence of hybrid swarms and maintenance of species identity by some sort of pre- and/or post-zygotic isolation mechanism (Lind and Gailing, 2013). Differences in flowering time and different adaptations to drought might contribute to the effective reproductive isolation between both species. For example, Q. ellipsoidalis seedlings showed a significantly later, albeit overlapping, bud burst and higher mortality than Q. rubra seedlings from neighbouring populations in a common garden experiment (Gailing, 2013).
Our main objective was to identify genes involved in the adaptive species divergence and reproductive isolation between Q. rubra and Q. ellipsoidalis. For this purpose, we selected genetically mapped genic markers, some of which had annotated functions in drought tolerance and flowering time, to identify loci under divergent selection between these two species. Outlier tests were replicated in three distinct Q. rubra–Q. ellipsoidalis population pairs. Replicated population pairs can provide greater power to outlier screens because divergence in multiple pairs of populations is less likely to be due to non-selective factors such as false positives or genetic drift (Nosil et al., 2009). Specifically, in addition to the eight nSSRs developed for Q. rubra, we adapted 36 gene-linked EST-SSRs to Q. rubra and Q. ellipsoidalis that were originally developed and mapped in Q. robur (Durand et al., 2010). These EST-SSRs provide a focused way to detect selection because they represent gene-associated polymorphic regions (Sullivan et al., 2013). We expect loci involved in adaptive species differences to be under selection in population pairs across different geographic regions. Such replicated outlier loci are candidate genes potentially involved in the evolution and maintenance of species identity between these ecologically divergent oak species.
MATERIALS AND METHODS
Plant material
Two neighbouring population pairs of Quercus rubra and Q. ellipsoidalis within the Ford Center and Baraga Plains area (Baraga County, MI), one population pair in the Chequamegon National Forest (Bayfield County, WI) and one population pair in the Nicolet National Forest (Oconto County, WI) were sampled, resulting in a total number of 319 adult trees (Fig. 1; Table 1). Only adult trees occupying a dominant or co-dominant canopy were sampled. A minimum distance of approx. 30 m was kept between sampled trees to minimize family structure. Species identity in the Baraga Plains was confirmed through both morphological (Gailing et al. 2012) and genetic assignment analyses (Lind and Gailing, 2013). Species identity in the Chequamegon–Nicolet National Forest was determined through whole-tree silvic characters and confirmed by genetic species assignment (Supplementary Data Fig. S1). Total genomic DNA (approx. 20 ng) was isolated from leaves using the DNeasy96 Plant Kit (Qiagen, Valencia, CA, USA) following the manufacturer's instructions.
Fig. 1.

Sampling locations. Soil data are from Soil Survey Staff, Natural Resources Conservation Service, United States Department of Agriculture. Soil Survey Geographic (SSURGO) Database. Available online at http://soildatamart.nrcs.usda.gov. Accessed 6 January 2013. In the case of FC-A, the data were modified slightly to match field soil samples (data not shown).
Table 1.
Sampled Quercus rubra and Q. ellipsoidalis populations
| Abbreviation | Region | Population | Species | Sample size (n) | Soil characteristics* | Latitude | Longitude |
|---|---|---|---|---|---|---|---|
| FC-A | FC/Baraga Plains: MI | Stand A | Q. rubra | 40 | 2 (78C: Keweenaw–Kalkaska Complex; 1–12 % slopes) | 46°39′9″N | 88°30′6″ W |
| FC-B | FC/Baraga Plains: MI | Stand B | Q. rubra | 40 | 3 (25B: Munising–Yalmer loamy sand; 1–6 % slopes) | 46°40′27″N | 88°31′27″W |
| FC-C | FC/Baraga Plains: MI | Stand C | Q. ellipsoidalis | 40 | 1 (10B: Grayling sand; 0–6 % slopes) | 46°39′14″N | 88°35′25″W |
| FC-E | FC/Baraga Plains: MI | Stand E | Q. ellipsoidalis | 40 | 1 (10B: Grayling sand; 0–6 % slopes) | 46°39′55″N | 88°33′19″W |
| N-QR | Nicolet National Forest: WI | Nicolet QR | Q. rubra | 40 | 2 (KaC: Kennan fine sandy loam; 6–15 % slopes) | 45°20′53″N | 88°23′17″W |
| N-QE | Nicolet National Forest: WI | Nicolet QE | Q. ellipsoidalis | 39 | 2 (RsB: Rousseau fine sand; 1–6 % slopes) | 45°19′19″N | 88°19′53″W |
| C-QR | Chequamegon National Forest: WI | Chequamegon QR | Q. rubra | 40 | 3 (480B: Portwing–Herbster complex, 0–6 % slopes | 46°42′54″N | 91°02′8″W |
| C-QE | Chequamegon National Forest: WI | Chequamegon QE | Q. ellipsoidalis | 40 | 1 (74B/C: Vilas loamy sand; 0–15 % slope) | 46°44′43″N | 91°04′20″W |
*Rating developed from drainage classes, with 1 being excessively drained, 2 being well drained, and 3 being moderately well drained; soil type and drainage class identified according to the Soil Survey Staff, Natural Resources Conservation Service, United States Department of Agriculture. Official Soil Series Descriptions available online at http://websoilsurvey.nrcs.usda.gov/app/HomePage.htm (accessed 29 November 2012).
Marker selection and microsatellite genotyping
For all EST-SSRs, the repeat-containing Quercus unigene elements (Durand et al., 2010) were reassembled in CAP3 (Huang and Madan, 1999) and functionally annotated using the BLASTx algorithm as implemented in the Blast2GO software package (Altschul et al., 1997) by comparing the reassembled EST contigs with homologous sequences in the non-redundant NCBI database. Results are reported in Supplementary Data Table S1 for sequences that had an expected value of ≤10−4 and a sequence similarity of >50 %. Fourteen EST-SSRs originally described for Q. robur (Durand et al., 2010) that had putative functions in drought tolerance, flowering time and other functions were chosen and adapted for use in Q. rubra. Previously, 30 markers including seven putatively neutral nSSRs were developed for Q. rubra (Aldrich et al., 2002; Sullivan et al., 2013), as well as one nSSR (Steinkellner et al., 1997) and 22 EST-SSRs adapted for use in Q. rubra (Lind and Gailing, 2013; Sullivan et al., 2013).
All 44 microsatellite markers (eight nSSRs and 36 EST-SSRs), covering ten of the 12 linkage groups in Q. robur (Durand et al., 2010), were amplified in the Ford Center and Baraga Plains populations (FC). Identified outliers in the FC population pairs, along with eight EST-SSRs with pairwise FST values close to the mean and the eight nSSRs were amplified in the Chequamegon National Forest (CNF) and Nicolet National Forest (NNF) populations (Supplementary Data Table S1).
Polymerase chain reaction amplification and electrophoretic separation were performed according to Lind and Gailing (2013) with one modification: the 10 µL PCR mix was scaled to a 15 µL reaction mix. Marker VIT081 amplified two loci: VIT081L1 had a range of 108–112 bp and VIT081L2 had a range of 115–136 bp. Both loci, VIT081L1 and VIT081L2, were in Hardy–Weinberg (HW) equilibrium in all populations.
Genetic structure
MICROCHECKER was used to assess all markers in each population for null alleles (α = 0·05) since they can lead to overestimation of measures of differentiation such as FST (Van Oosterhout et al., 2004). MICROCHECKER identified 13 loci as being in HW disequilibrium in at least three of the FC populations. Only two loci, FIR013 and GOT004, were identified as being in HW disequilibrium in at least three of the CNF and NNF populations, respectively. Deviations from HW equilibrium may be due to the presence of null alleles, but the high number of markers in HW disequilibrium in one Q. rubra and both Q. ellipsoidalis populations in the FC indicates potential inbreeding (Supplementary Data Tables S2 and S3). All outlier screens were conducted with and without corrections using the van Oosterhout correction algorithm, for potential null alleles, resulting in only minor differences in the outliers detected (Supplementary Data Tables S4–S6).
STRUCTURE 2·3 (Pritchard et al. 2000) was used to determine the population structure under the admixture model with correlated allele frequencies without a priori information regarding species identity. Five independent runs of 106 iterations with a burn-in period of 30 000 were performed for each K value (K = 1–4 for FC populations with 44 markers and K = 1–8 for FC, CNF and NNF populations with 20 markers). We carried out both analyses to see if the lower number of markers was sufficient to delineate the species. The most likely number of groups (K) was chosen by using the ad hoc statistic presented in Evanno et al. (2005).
Genetic variation parameters were calculated in GeneAlEx 6·41 (Peakall and Smouse, 2006) including the average number of alleles (Na), Nei's unbiased gene diversity (He) and Wright's inbreeding coefficient (F). FST and pairwise FST with corresponding significances were calculated in GenePop 4·1 (Raymond and Rousset, 1995). To correct for multiple comparisons, sequential Bonferroni adjustments of the significance level (α) were made. An FST-based UPGMA dendrogram was created in Populations 2·0 (Langella, 1999).
Outlier screens
FST (Wright's fixation index) can be used to identify outlier loci between populations based on the assumption that gene loci under selection will show higher differentiation between species than expected under neutral evolutionary conditions (Beaumont and Nichols, 1996). Using coalesecent simulations, the program LOSITAN (Antao et al., 2008) creates a null distribution of FST values from the multilocus data that can be used to identify outlier loci under the finite island model. The run parameters in LOSITAN included 100 000 simulations at a 95 % confidence interval under the stepwise mutation model. In order to account for multiple testing and resultant potential false positives, the default false discovery rate (FDR) of 10 % was used. Both the ‘neutral’ mean FST option to exclude loci potentially under selection for the computation of the initial mean FST and the ‘force’ mean FST option to increase the precision of the simulated mean FST were applied.
The LnRH test statistic estimates variability between populations at individual loci instead of population divergence to identify selection, and is a powerful test statistic for the detection of selective sweeps. LnRH is generally normally distributed as determined by simulation studies, and when plotted against FST values outliers can be found in the tails of the distribution (Schlötterer and Dieringer, 2005). LnRH measures relative gene diversity between two populations to identify putative loci under selection through the calculation of the natural logarithm of the ratio of gene diversity (H) for each locus between a pair of populations as follows:
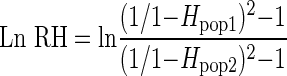 |
The Kolmogorov–Smirnov test for normal distribution was performed on the observed data for each comparison. Significance was determined by standardizing the observed values and applying a 95 % confidence interval, where outliers were identified as being more than ±1·96 standard deviations away from the mean.
Outlier tests were performed for all between- and within-species pairwise comparisons as replicated population pairs provide increased power and reduce the chance of observed divergence being due to non-selective factors (Nosil et al., 2009). They were also performed for all populations within a region pooled by species.
Isolation by distance vs. isolation by adaptation
To confirm further potential loci under divergent selection, identified outliers were amplified in two additional interspecific population pairs in two different regions, CNF and NNF. Each identified outlier was paired with another marker with an FST value close to the mean identified in the FC populations and amplified in the same populations to prevent upward biasing of FST. The outlier methods described above were used to screen this marker set in these additional populations. Outliers that are due to isolation by adaptation (IBA) between species should be identified as outliers in both allopatric and sympatric geographic scenarios. In contrast, loci identified as potentially under divergent selection due to isolation by distance (IBD) should only be identified as distance increases between populations, so that geographically proximal populations do not show divergence but distant populations do (Nosil et al., 2009). Outliers identified in only one geographic region could be involved in local adaptation of populations or, more conservatively, may comprise false positives.
RESULTS
Genetic structure and variation
Two clusters were shown to best fit the data in STRUCTURE using the ad hoc statistic ΔK (Evanno et al., 2005), which correspond to the two species, Q. rubra and Q. ellipsoidalis (Supplementary Data Fig. S1). The Q. ellipsoidalis populations in the CNF and NNF showed higher levels of potential introgression as compared with the FC populations. However, most individuals could be assigned a species designation with high certainty (posterior probability >0·90). The species assignment for the FC populations displayed similar numbers of hybrids and introgressive forms for both the runs using 44 (data not shown) and 20 markers (Supplementary Data Fig. S1). This indicates that the higher number of hybrids and introgressive forms in the CNF and NNF populations are not due to the lower number of markers used for species assignment. Consequently, outlier screens were conducted with all samples and after excluding potential hybrids and introgressive forms. Results were consistent and differed mainly in the number and identity of loci potentially subject to balancing selection which were not considered (Supplementary Data Tables S7–S9). An FST-based UPGMA unrooted dendrogram showed populations separated by species (Fig. 2). All nodes had high bootstrap values whether all individuals or only ‘pure’ species (data not shown) were included in the analysis.
Fig. 2.
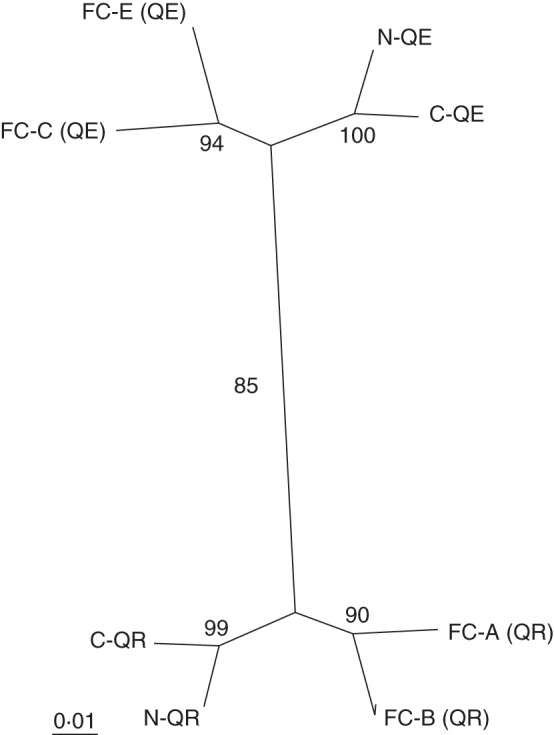
Unrooted tree (UPGMA method) based on genetic distance (FST) at 20 markers; numbers at nodes are percentages over 1000 bootstrap replicates using Populations 2·0 (Langella, 1999).
At the sub-set of 20 markers common to all eight populations, all populations exhibited similar levels of genetic variation (Supplementary Data Table S10). The mean heterozygosity (He) was 0·715 and 0·711 for Q. rubra and Q. ellipsoidalis, respectively, while the mean number of alleles per locus was 15 for both species. Additionally, 19 out of 20 markers showed statistically significant differentiation (FST) between species, with values ranging from 0·007 to 0·668 (Supplementary Data Table S11). Mean genetic diversity was slightly lower at the 44 marker set only used in the four FC populations, with means of 0·660 and 0·642 for Q. rubra and Q. ellipsoidalis, respectively. The average number of alleles at the 44 markers was also slightly lower than at the 20 markers, averaging between nine and ten for both species in the FC populations. This is probably due to the higher number of EST-SSRs used, which showed lower diversity than nSSRs. Thirty-five out of 44 markers showed significant differentiation (FST) between species, with a range from 0·0046 to 0·801 (Supplementary Data Table S12).
Outlier screens
LOSITAN consistently identified FIR013 as an outlier between species in almost all possible comparisons (Fig. 3; Supplementary Data Fig. S2 and S3). This locus showed two major alleles and was nearly fixed on alternative alleles in all populations of both species (Q. ellipsoidalis, 138 bp; Q. rubra 141 bp; Fig. 4). Consistent with this pattern of allelic variation, FIR013 was identified as an outlier between species within and among regions, which strongly implicates isolation by adaptation (Table 2). Locus FIR013 is putatively located in a CONSTANS-like (COL) gene. Together with CONSTANS (CO), COL genes comprise a family encoding transcription factors involved in the photoperiodic regulation of growth and development. Specifically, this family is broken up into three broad groups based on structural differences. In Arabidopsis, CO and homologous COL (1–5) genes belong to Group I and are characterized by the presence of two conserved zinc finger B-boxes near the N-terminus and a CCT domain near the C-terminus (Griffiths et al., 2003). COL genes can be distinguished from CO by the presence of poly(Q) regions following the B-box zinc finger domain (Almada et al., 2009).
Fig. 3.

Outlier loci analysis of 44 markers between species among all four FC/Baraga Plains populations as calculated by LOSITAN (Antao et al., 2008). Outliers are indicated by a star while all others are circles, and black lines represent the 5 % quantiles, with markers outside the lower 5 % quantile under balancing selection and the markers outside the upper 5 % quantile under divergent selection, while the grey line represents the median (50 %).
Fig. 4.
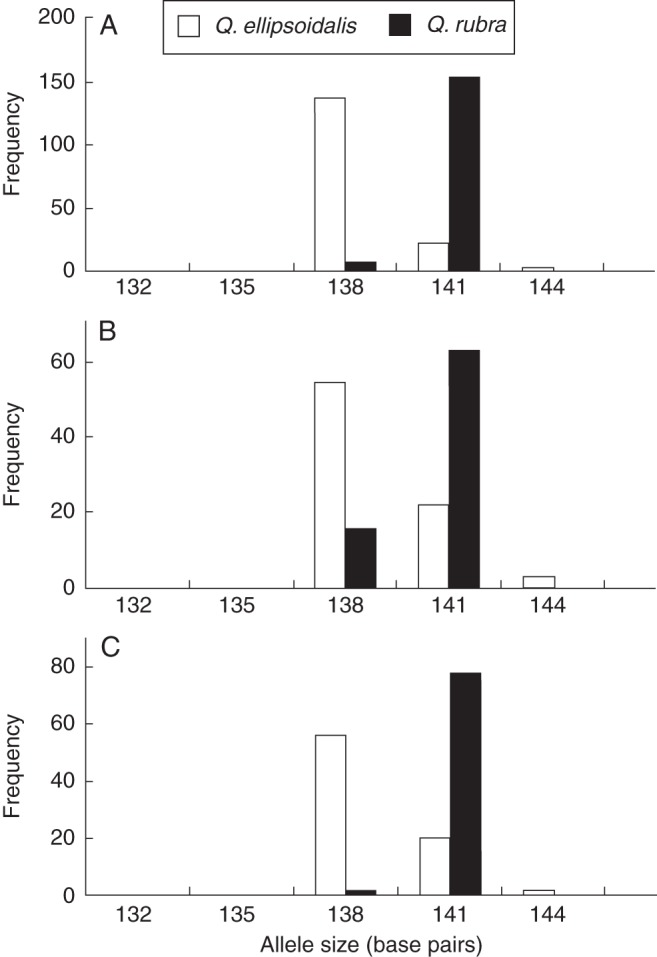
Allele frequency distribution of each species in the (A) FC/Baraga Plains populations, (B) CNF populations and (C) NNF populations for EST-SSR FIR013 (CONSTANS-like 1).
Table 2.
Summary of between-species identified outliers under divergent selection
| Pairwise comparison | LOSITAN | LnRH |
|---|---|---|
| FC (QR vs. QE) | FIR013 | FIR039 |
| POR016 | GOT040 | |
| Chequamegon National Forest (QR vs. QE) | FIR013 | GOT021 |
| Nicolet National Forest QR vs. QE) | FIR013 | FIR013 |
| All (QR vs. QE) | FIR013 | NO |
FC, Ford Center and Baraga Plains; NO, no outliers detected.
Alignment of the Quercus EST unigene element to Populus COL-1 and COL-2 genes shows 81 and 84 % similarity, respectively, and indicates that the FIR013 locus encodes a poly(Q) repeat and is located between a conserved double B-box zinc finger domain and a CCT motif (Yuceer et al., 2002). Additionally, a gene tree created using protein sequences from closely related species and the Quercus unigene indicated strong similarity to the COL genes in Populus spp. with high bootstrap support (Supplementary Data Fig. S4). Together, the presence of characteristic protein domains and the high similarly to putatively homologous genes supports the identity of this Quercus locus as a Group I COL gene.
Even though FIR013 shows high divergence between species, the LnRH statistic only detected it between species in the NNF population (Table 2; Supplementary Data Fig. S5). The LnRH statistic describes significant reductions of heterozygosity, but both Q. rubra and Q. ellipsoidalis exhibit very low heterozygosity at this locus overall, with different alleles being nearly fixed in each species (Q. rubra, He = 0·08; Q. ellipsoidalis, He = 0·25). Additional outliers were identified by either LOSITAN or LnRH, but did not show up consistently across all comparisons, which could indicate a role in local adaptations of specific populations (Table 2; Supplementary Data Tables S13 and S14). However, given the potentially high rate of false positives associated with outlier methods, we used caution in interpreting the results. Thus, we considered the FIR013 locus which was identified as an outlier by the FST-based method in multiple replicate species comparisons to be the best candidate under divergent selection.
DISCUSSION
By employing replicated population pairs, we identified a marker located within the coding sequence of a COL gene as under divergent selection in almost all interspecific comparisons within and among regions, but not for within-species comparisons. Such a pattern of spatially replicated divergent selection in contrasting environments is consistent with a role for this COL gene in ecological speciation, whereby natural selection drives species divergence and reproductive isolation (Nosil et al., 2009; Schluter, 2009). Moreover, COL genes have been implicated in the regulation of both flowering time and growth, which presents an avenue for the development of ecologically mediated reproductive barriers. Putative outlier loci are only rarely confirmed in multiple population pairs (e.g. Nielsen et al., 2009) and have never before been confirmed in forest tree species. Interestingly, single nucleotide polymorphism (SNP) variation in the same COL gene was significantly associated with the timing of vegetative bud burst in a Q. petraea provenance trial (Alberto et al., 2013). Other studies have also shown associations between nucleotide variation in COL genes and phenotypic traits such as flowering time and height in other plant species (e.g. Medicago sativa, Herrmann et al., 2010; Populus nigra, Fabbrini et al., 2012; Populus tremula, Ma et al., 2010). For example, in the association mapping study of M. sativa, a COL gene was shown to be involved in flowering time and plant height (Herrmann et al., 2010).
Together with CO, COL genes form a family of transcription factors involved in the photoperiod pathway of floral transition (Amasino, 2005). Group I COL genes in Arabidopsis are regulated by a circadian clock and, in turn, may help regulate the pace of the circadian oscillator in Arabidopsis as it has been shown to accelerate the circadian clock (Ledger et al., 2001; Griffiths et al., 2003). However, altered expression in transgenic plants of either COL-1 or COL-2 had little effect on flowering time, suggesting that the functions of CO, COL-1 and COL-2 may not completely overlap and may have diverged in function in Arabidopsis (Ledger et al., 2001).
Functional diversification of COL genes is also evident in angiosperm trees. In Populus trichocarpa, PtCOL-2 was shown through RNAi (RNA interference) experiments to play a central role in the CO/Flowering Time (FT) regulon that controls seasonal growth patterns in trees (Böhlenius et al., 2006). While overexpression of PtCOL-1 and PtCOL-2 did not alter normal reproductive onset, bud break or bud set in Populus, overexpression of PtCOL-1 in arabidopsis rescued the late flowering phenotype of the co-1 mutant, suggesting that it may function similarly to CO (Hsu et al., 2012). In addition, overexpression of PtCOL-1 and PtCOL-2 in Populus affected plant height.
Both Q. rubra and Q. ellipsoidalis are nearly fixed on different alleles at the COL microsatellite marker (Fig. 4), which corresponds to the addition or deletion of a glutamine residue as the microsatellite encodes a poly(Q) repeat. While the role of poly(Q) repeats in human genetic disorders is well studied, their role in normal protein function is largely unknown. However, it has been posited that they may be trans-activation sequences involved in stabilization of protein interactions or in regulation of gene transcription activation (Yuceer et al., 2002). More interestingly, it has been proposed that selection drives the evolution of low-complexity sequences such as poly(Q) repeats. This has been demonstrated in the Chinook salmon circadian-regulating CLOCK gene, where repeat lengths are positively correlated with latitude and thus potentially involved in adaptation of these fish to different latitudes (Haerty and Golding, 2010). Notably, one allele of a poly(Q) repeat in the Populus tremula COL2B gene was found to be associated with growth cessation, albeit this effect was not independent of other polymorphisms in the photoperiodic pathway (Ma et al., 2010). While the exact function of COL genes and the poly(Q) length polymorphism in Quercus are currently unknown, they provide excellent candidates for underlying functional polymorphisms in flowering time and growth-related traits, and warrant further investigation.
Differences in flowering time are a clear mechanism of pre-zygotic isolation. Selection on the photoperiod pathway might have been essential in divergence and maintenance of species differences. While oak flowers are difficult to observe in field studies, the timing of vegetative bud burst is strongly associated with flowering time and is used to infer phenological patterns in flowering time (Chesnoiu et al., 2009). Latitudinal and altitudinal gradients in sessile oak (Ducousso et al., 1996) and local environmental conditions have been shown to impact timing of bud burst in two interfertile live oak species (Cavender-Bares and Pahlich, 2009). Our previous study indicated low levels of introgression between adult and seedling populations of the two species, which may be a result of pre-zygotic isolation via flowering time (Lind and Gailing, 2013). Furthermore, significant differences in bud burst were observed in a common garden of Q. ellipsoidalis and Q. rubra seedlings. Quercus ellipsoidalis seedlings exhibited significantly later bud burst and leaf fall (a proxy for bud set) than Q. rubra seedlings, suggesting an underlying genetic mechanism (Gailing, 2013). Also Q. ellipsoidalis seedlings were significantly smaller and showed a lower survival rate than Q. rubra seedlings from neighbouring populations (Gailing, 2013). Interspecific differences in growth and flowering time could possibly be linked to the divergence between the two species at the putative COL gene since similar genes in Populus and other plant species have been shown to be involved in growth and development (Herrmann et al., 2010; Hsu et al., 2012). In natural populations, Q. ellipsoidalis trees are also smaller in habit than Q. rubra trees (personal field observation). Given the xeric nature of the environment in which Q. ellipsoidalis resides, a slower growth strategy, including later flowering, may confer a selective advantage by avoiding late frost damage and conserving energy. In fact, frost pockets and slow forest canopy establishment are characteristic in level xeric areas such as the outwash plains on which Q. ellipsoidalis grows (Motzkin et al., 2002).
Further assessment of flowering time in the natural populations of these two species will be of value in assessing the importance of this trait in maintenance of species identity in interfertile oak species. The genetic assignment analysis suggested recurrent gene flow between species and higher rates of introgression in the CNF and NNF populations than in the FC populations. Gene flow estimates via parentage analysis could confirm recurrent gene flow between the species and its dependence on environmental conditions providing additional support for adaptive species divergence with gene flow. Alternatively, post-zygotic selection on COL could be inferred from reciprocal transplant experiments between parental environments of Q. rubra, Q. ellipsoidalis and hybrid seedlings with different genotypes for COL (138/138, 138/141 and 141/141 bp). Since adult populations are nearly fixed on the alternative alleles, 138 bp in Q. ellipsoidalis and 141 bp in Q. rubra, an increase of species-specific alleles from seedling to adult generation and higher growth and survival of Q. ellipsoidalis and Q. rubra seedlings with species-specific alleles in parental environments would implicate post-zygotic selection on the Quercus COL gene. Finally, expression studies of this gene through quantitative PCR could be carried out to assess differential expression patterns for both species throughout the photoperiod.
Conclusions
We employed outlier screens with three replicated population pairs and identified a putative COL gene potentially involved in adaptive divergence and reproductive isolation of Q. rubra and Q. ellipsoidalis. We show that oaks provide a good model to identify potential genomic regions involved in ecological speciation. In particular, the gene associated with the EST-SSR FIR013, COL-1, could be a ‘magic gene’ involved in ecological speciation because of its potential simultaneous involvement in reproductive isolation and local adaptation (Servedio et al., 2011). Variation in putative ‘speciation genes’ can be associated with traits related to adaptive species differences (e.g. in drought tolerance and bud burst) in quantitative trait locus and association mapping approaches. Alternatively, COL may not be subject to divergent selection per se but could be linked to an ecologically significant gene (Via, 2012). The availability of high-density linkage maps or a whole-genome sequence would allow for the identification of clusters of linked genes, which could help elucidate the relative importance of linkage disequilibrium and single-locus effects. Full-sib families in Q. rubra and high-density genetic linkage maps are currently being constructed. Next-generation sequencing technologies such as restriction site-associated DNA (RAD) sequencing and integration of these and other markers in genetic linkage maps will also help to identify genome-wide patterns of adaptive species divergence.
SUPPLEMENTARY DATA
ACKNOWLEDGEMENTS
We are grateful to Linda Parker, Daniel Hinson and John Lampereur for their assistance in locating appropriate populations in the Chequamegon–Nicolet National Forest. We would also like to thank Jonathan Riehl for his invaluable help with constructing many of the figures, and two anonymous reviewers for suggestions to further improve this paper. This work was supported by the Research Excellent fund of Michigan Technological University, the Hanes Trust, the United States Department of Agriculture's McIntire Stennis fund, the National Science Foundation's Plant Genome Research program (NSF 1025974), the Huron Mountain Wildlife Foundation, the Northern Institute of Applied Climate Science, the United States Department of Education and the European Commission's Directorate General for Education and Culture States' Atlantis Program, and the Michigan Technological University Ecosystem Science and Biotechnology Research Centers.
LITERATURE CITED
- Abrams MD. Comparative water relations of three successional hardwood species in central Wisconsin. Tree Physiology. 1988;4:263–273. doi: 10.1093/treephys/4.3.263. [DOI] [PubMed] [Google Scholar]
- Abrams MD. Adaptations and responses to drought in Quercus species of North America. Tree Physiology. 1990;7:227–238. doi: 10.1093/treephys/7.1-2-3-4.227. [DOI] [PubMed] [Google Scholar]
- Alberto FJ, Derory J, Boury C, Frigerio J-M, Zimmermann NE, Kremer A. Imprints of natural selection along environmental gradients in phenology-related genes of Quercus petraea. Genetics. 2013;195:495–512. doi: 10.1534/genetics.113.153783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich PR, Michler CH, Sun WL, Romero-Severson J. Microsatellite markers for northern red oak (Fagaceae: Quercus rubra) Molecular Ecology Notes. 2002;2:472–474. [Google Scholar]
- Almada R, Cabrera N, Casaretto J, Ruiz-Lara S, González Villanueva E. VvCO and VvCOL1, two CONSTANS homologous genes, are regulated during flower induction and dormancy in grapevine buds. Plant Cell Reporter. 2009;28:1193–1203. doi: 10.1007/s00299-009-0720-4. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amasino RM. Vernalization and flowering time. Current Opinion in Biotechnology. 2005;16:154–158. doi: 10.1016/j.copbio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Antao T, Lopes A, Lopes R, Beja-Pereira A, Luikart G. LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics. 2008;9:323. doi: 10.1186/1471-2105-9-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont M, Nichols R. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society B: Biological Sciences. 1996;263:1619–1626. [Google Scholar]
- Barluenga M, Stölting KN, Salzburger W, Muschick M, Meyer A. Sympatric speciation in Nicaraguan crater lake cichlid fish. Nature. 2006;439:719–723. doi: 10.1038/nature04325. [DOI] [PubMed] [Google Scholar]
- Böhlenius H, Huang T, Charbonnel-Campaa L, et al. CO/FT regulatory module controls timing of flowering and seasonal growth cessation in trees. Science. 2006;312:1040–1043. doi: 10.1126/science.1126038. [DOI] [PubMed] [Google Scholar]
- Burns RM, Honkala BH. Silvics of North America Volume 2: hardwoods. Washington, DC: United States Forest Service; 1990. [Google Scholar]
- Cavender-Bares J, Pahlich A. Molecular, morphological, and ecological niche differentiation of sympatric sister oak species, Quercus virginiana and Q. geminata (Fagaceae) American Journal of Botany. 2009;96:1690–1702. doi: 10.3732/ajb.0800315. [DOI] [PubMed] [Google Scholar]
- Chesnoiu E, Sofletea N, Curtu AL, Toader A, Radu R, Enescu M. Bud burst and flowering phenology in a mixed oak forest from Eastern Romania. Annals of Forest Research. 2009;52:199–206. [Google Scholar]
- Curtu A, Gailing O, Finkeldey R. Patterns of contemporary hybridization inferred from paternity analysis in a four-oak-species forest. BMC Evolutionary Biology. 2009;9:284. doi: 10.1186/1471-2148-9-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducousso A, Guyon J, Kremer A. Latitudinal and altitudinal variation of bud burst in western populations of sessile oak (Quercus petraea (Matt) Liebl) Annals of Forest Science. 1996;53:775–782. [Google Scholar]
- Durand J, Bodenes C, Chancerel E, et al. A fast and cost-effective approach to develop and map EST-SSR markers: oak as a case study. BMC Genomics. 2010;11:570. doi: 10.1186/1471-2164-11-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Fabbrini F, Gaudet M, Bastien C, et al. Phenotypic plasticity, QTL mapping and genomic characterization of bud set in black poplar. BMC Plant Biology. 2012;12:47. doi: 10.1186/1471-2229-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailing O. Difference in growth, survival and phenology in Q. rubra and Q. ellipsoidalis seedlings. Dendrobiology. 2013;70:71–79. [Google Scholar]
- Gailing O, Lind J, Lilleskov E. Leaf morphological and genetic differentiation between Quercus rubra L. and Q. ellipsoidalis E.J. Hill populations in contrasting environments. Plant Systematics and Evolution. 2012;298:1533–1545. [Google Scholar]
- Goicoechea PG, Petit RJ, Kremer A. Detecting the footprints of divergent selection in oaks with linked markers. Heredity. 2012;109:361–371. doi: 10.1038/hdy.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths S, Dunford RP, Coupland G, Laurie DA. The evolution of CONSTANS-like gene families in barley, rice, and Arabidopsis. Plant Physiology. 2003;131:1855–1867. doi: 10.1104/pp.102.016188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerty W, Golding GB. Low-complexity sequences and single amino acid repeats: not just ‘junk’ peptide sequences. Genome. 2010;53:753–762. doi: 10.1139/g10-063. [DOI] [PubMed] [Google Scholar]
- Herrmann D, Barre P, Santoni S, Julier B. Association of a CONSTANS-like gene to flowering and height in autotetraploid alfalfa. Theoretical and Applied Genetics. 2010;121:865–876. doi: 10.1007/s00122-010-1356-z. [DOI] [PubMed] [Google Scholar]
- Hokanson SC, Isebrands JG, Jensen RJ, Hancock JF. Isozyme variation in oaks of the Apostle Islands in Wisconsin: genetic structure and levels of inbreeding in Quercus rubra and Q. ellipsoidalis (Fagaceae) Amercian Journal of Botany. 1993;80:1349–1357. [Google Scholar]
- Hsu C-Y, Adams JP, No K, et al. Overexpression of CONSTANS homologs CO1 and CO2 fails to alter normal reproductive onset and fall bud set in woody perennial poplar. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045448. e45448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Research. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langella O. Populations 1·2·30: a population genetic software. 1999 CNRS UPR9034. [Google Scholar]
- Ledger S, Strayer C, Ashton F, Kay SA, Putterill J. Analysis of the function of two circadian-regulated CONSTANS-like genes. The Plant Journal. 2001;26:15–22. doi: 10.1046/j.1365-313x.2001.01003.x. [DOI] [PubMed] [Google Scholar]
- Lepais O, Gerber S. Reproductive patterns shape introgression dynamics and species succession within the European white oak species complex. Evolution. 2011;65:156–170. doi: 10.1111/j.1558-5646.2010.01101.x. [DOI] [PubMed] [Google Scholar]
- Lind JF, Gailing O. Genetic structure of Quercus rubra L and Quercus ellipsoidalis E. J. Hill populations at gene-based EST-SSR and nuclear SSR markers. Tree Genetics and Genomes. 2013;9:707–722. [Google Scholar]
- Ma X-F, Hall D, St Onge KR, Jansson S, Invgarsson PK. Genetic differentiation, clinal variation, and phenotypic associations with growth cessation across the Populus tremula photoperiodic pathway. Genetics. 2010;186:1033–1044. doi: 10.1534/genetics.110.120873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzkin G, Ciccarello SC, Foster DR. Frost pockets on a level sand plain: does variation in microclimate help maintain persistent vegetation patterns? Journal of the Torrey Botanical Society. 2002;129:154–163. [Google Scholar]
- Nielsen E, Hemmer-Hansen J, Poulsen N, et al. Genomic signatures of local directional selection in a high gene flow marine organism; the Atlantic cod (Gadus morhua) BMC Evolutionary Biology. 2009;9:276. doi: 10.1186/1471-2148-9-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosil P, Funk DJ, Ortiz-Barrientos D. Divergent selection and heterogeneous genomic divergence. Molecular Ecology. 2009;18:375–402. doi: 10.1111/j.1365-294X.2008.03946.x. [DOI] [PubMed] [Google Scholar]
- Peakall ROD, Smouse PE. GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes. 2006;6:288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M, Rousset F. GenePop (Version-1·2) – population genetics software for exact tests and ecumenicism. Journal of Heredity. 1995;86:248–249. [Google Scholar]
- Rundle HD, Nosil P. Ecological speciation. Ecology Letters. 2005;8:336–352. [Google Scholar]
- Savolainen V, Anstett M-C, Lexer C, et al. Sympatric speciation in palms on an oceanic island. Nature. 2006;441:210–213. doi: 10.1038/nature04566. [DOI] [PubMed] [Google Scholar]
- Schlötterer C, Dieringer D. A novel test statistic for the identification of local selective sweeps based on microsatellite gene diversity. In: Nurminsky D, editor. Selective sweep. Georgetown, TX: Landes Bioscience; 2005. pp. 55–64. [Google Scholar]
- Schluter D. The ecology of adaptive radiation. Oxford: Oxford University Press; 2000. [Google Scholar]
- Schluter D. Evidence for ecological speciation and its alternative. Science. 2009;323:737–741. doi: 10.1126/science.1160006. [DOI] [PubMed] [Google Scholar]
- Scotti-Saintagne C, Mariette S, Porth I, et al. Genome scanning for interspecific differentiation between two closely related oak species [Quercus robur L. and Q. petraea (Matt.) Liebl.] Genetics. 2004;168:1615–1626. doi: 10.1534/genetics.104.026849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servedio MR, Doorn GSV, Kopp M, Frame AM, Nosil P. Magic traits in speciation: magic but not rare? Trends in Ecology and Evolution. 2011;26:389–397. doi: 10.1016/j.tree.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Steinkellner H, Lexer C, Turetschek E, Glössl J. Conservation of (GA)n microsatellite loci between Quercus species. Molecular Ecology. 1997;6:1189–1194. doi: 10.1023/a:1005736722794. [DOI] [PubMed] [Google Scholar]
- Storz J. Using genome scans of DNA polymorphism to infer adaptive population divergence. Molecular Ecology. 2005;14:671–688. doi: 10.1111/j.1365-294X.2005.02437.x. [DOI] [PubMed] [Google Scholar]
- Sullivan A, Lind JF, McCleary T, Romero-Severson J, Gailing O. Development and characterization of genomic and gene-based microsatellite markers in North American red oak species. Plant Molecular Biology Reporter. 2013;31:231–239. [Google Scholar]
- Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes. 2004;4:535–538. [Google Scholar]
- Via S. Divergence hitchhiking and the spread of genomic isolation during ecological speciation-with-gene-flow. Philosophical Transactions of the Royal Society B: Biological Sciences. 2012;367:451–460. doi: 10.1098/rstb.2011.0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuceer C, Harkess RL, Land SB, Jr, Luthe DS. Structure and developmental regulation of CONSTANS-LIKE genes isolated from Populus deltoides. Plant Science. 2002;163:615–625. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.