

Anodic oxidation reactions can trigger a variety of interesting, new cyclization reactions.[1] For example, the anodic coupling of an electron-rich olefin and a toluenesulfonamide leads to cyclic amino acid derivatives that contain tetrasubstituted carbons (Scheme 1).[2,3] Such materials can potentially serve as building blocks for constructing a variety of alkaloids and peptidomimetics.[4-8]
Scheme 1.

Synthesis of cyclic amino acid derivatives containing tetrasubstituted α-carbons.
The enthusiasm for using this reaction in a synthesis is enhanced by both the substrates being available in an enantiomeric fashion[9] and a strategy being in place for reversing the stereochemistry of the tetrasubstituted carbon generated.[10] It is dampened by the need for the sulfonyl protecting group and its subsequent removal.[11]
A potentially more useful approach would take advantage of an unprotected amine (4) as the nucleophile (Scheme 2). Yet while this reaction is easy to propose, the oxidation potentials highlighted in Scheme 2 provide reason for concern.[12] As illustrated, the reaction would employ a dithioketene acetal group as the electron-rich olefin for the oxidation. This group was selected because of its low oxidation potential. Even with this group, the oxidation potential of the secondary amine in the product is lower than that of either functional group in the starting material. This is a situation that typically foreshadows over-oxidation of the product. Hence, we protected the amine with a sulfonyl group in order to channel the oxidation toward the dithioketal and away from the amine in the product.
Scheme 2.

Proposed intramolecular anodic coupling of an amine and a dithioketene acetal.
However, this analysis neglects the cyclization reaction and its possible influence on the oxidation potential of the substrate. Rapid cyclization reactions can lower the oxidation potential of a substrate.[13] For example, when the amine in 4 is replaced with an alcohol (6, Figure 1) the substrate has an Ep/2 vs. Ag/AgCl of 0.95 V,[12] a drop in potential from the isolated dithioketene acetal of 110 mV.
This drop in potential can be explained using steady-state kinetics and the Nernst equation (Figure 1). In this treatment, steady state kinetics is used to write an expression for the concentration of the radical cation 7. This expression is then substituted into the Nernst equation to provide equation (1). In this equation, Eo is positive and everything following the minus sign is positive. Hence, as k2 gets larger, and the cyclization faster, the observed potential decreases. One way to make k2 larger would be to increase the nucleophilicity of the XH. Such would be the case with an amine-based substrate like 4. But how much does the potential drop in this case, and is the potential drop large enough to make the potential of 4 lower than that of 5 so that the cyclization proposed in Scheme 2 might work? We report herein that this is the case, and that the sulfonyl protecting group in 1 was never needed.
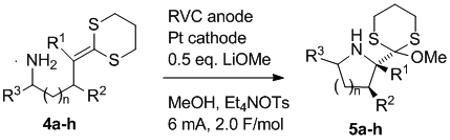
Work to investigate the compatibility of amine trapping groups with the anodic cyclizations began by examining a series of dithioketene acetal substrates shown in Table 1.[14] The first indication of how well the electrolyses would work came when the oxidation potential for substrate 4a (Table 1, entry 1) was measured to be Ep/2 = + 0.60 V vs. Ag/AgCl,[12] a 460 mV drop in potential relative to an isolated dithioketene acetal. This was the largest drop in potential that we have measure to date. Clearly, the cyclization reaction with the more nucleophilic amine is very rapid.
Table 1.
Anodic coupling of amines and dithioketene acetals.
| Entry | Subst. | n | R1,R2,R3 |
Ep/2[a] [V] |
Yield[b] [%] |
|---|---|---|---|---|---|
| 1 | 4a | 1 | R1 = Me R2 = R3 = H |
0.60 | 84 |
| 2 | 4b | 1 | R1 = R2 = Me R3 = H |
0.62 | 81 (>20:1) |
| 3 | 4c | 1 | R1 = R3 = Me R2 = H |
0.58 | 92[c] (3:2) |
| 4 | 4d | 1 | R1 = CH=CHMe R2 = Me, R3 = H |
-- | 90 (10:1) |
| 5 | 4e | 2 | R1 = Me R2 = R3 = H |
0.65 | 72 |
| 6 | 4f | 2 | R1 = R2 = Me R3 = H |
0.68 | 83[d] (10:1) |
| 7 | 4g | 2 | R1 = R3 = Me R2 = H |
0.67 | 64[e] (2:1) |
| 8 | 4h | 3 | R1 = Me R2 = R3 = H |
0.70 | -- |
Measured vs. Ag/AgCl on a carbon anode in acetonitrile.
Ratio in parenthesis is diastereomeric ratio.
A 40% yield (measured by 1H NMR using an internal standard) of product was obtained when 6 eq. of 2,6-lutidine was used as the base instead of LiOMe.
2.3 F/mol.
2.4 F/mol.
The large drop in oxidation potential for 4a led to a substrate potential that was approximately 300 mV lower than that of the product. Electrochemical oxidations can easily differentiate between such molecules. Accordingly, the preparative cyclization reaction led to a high yield of the cyclic product. The oxidation was carried out using an undivided cell, a reticulated vitreous carbon (RVC) anode, a Pt wire cathode, a Et4NOTs in MeOH electrolyte solution, 0.5 eq of LiOMe as a base, and a constant current of 6 mA until 2.0 F/mol of charge was passed. An 84% isolated yield of product 5a was obtained.
For the reaction illustrated in entry 2 of Table 1, a second substituent was added to the allylic carbon of the dithioketene acetal 4b in order to probe the stereoselectivity of the reaction. The cyclization led to an 81% isolated yield of product as a single diastereomer. The stereochemistry was assigned as having the large orthoester trans to the methyl group in position R2 in analogy to earlier cyclizations using both sulfonamide[2] and oxygen[15] nucleophiles.
The orthoester in product 5b was converted to a methyl ester in order to demonstrate how the cyclic products can be used to generate more traditional amino acid derivatives (Scheme 3). The reaction was accomplished using N-chlorosuccinimide in an acetone/water mixture.[14]
Scheme 3.
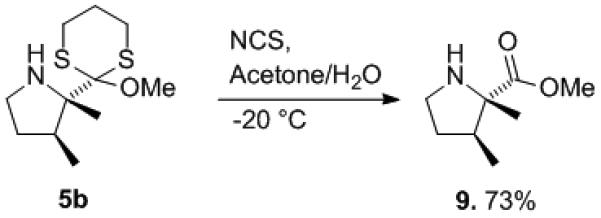
Hydrolysis of the electrolysis product 5b.
The origin of the observed stereoselectivity in 5b was attributed to a steric interaction between the methyl group in position R2 and the larger ketene dithioacetal derived substituent. Evidence to support this suggestion was gathered by the oxidation of 4c (entry 3). In this case, the steric interaction was removed by moving the methyl group to position R3. The cyclization proceeded in a very nice 92% yield, but led to a 3:2 ratio of diastereomers.
The need for LiOMe in the reactions was also probed with substrate 4c. When the LiOMe was replaced with 2,6-lutidine as a proton scavanger, the cyclization afforded the cyclic product in only a 40% yield. The reaction led to a mixture of products. It was difficult to isolate 5c in a pure form, so the yield was obtained by integration of the 1H NMR spectrum for the product relative to an internal standard. There are two possible explanations for the fall-off in yield and reaction cleanliness when LiOMe is not used as the base. First, it is possible that the use of the stronger LiOMe base leads to rapid deprotonation of the initial cyclic intermediate 8 (Figure 1). This would facilitate the cyclization by slowing down the reverse reaction. Second, it is possible that the use of methoxide accelerates trapping of the final cation generated by oxidation of the radical in 8 thereby reducing the chance for secondary reactions. The importance of efficiently trapping this cation has been demonstrated in previous cyclizations.[2, 16]
Figure 1.

Relationship of observed potentials and rate of cyclizations.
The reactions are also compatible with the use of an unsaturated dithioketene acetal (entry 4, Table 1). The oxidation of 4d led to a 90% yield of the cyclic product in a 10:1 ratio of diastereomers. The major product was assigned as having the orthoester group trans to the methyl in position R2 in direct analogy to the corresponding alcohol example.[10] The vinyl group was used because it offers an opportunity to manipulate the stereochemistry of the tetrasubstituted carbon in the product. For example, a similar substitution pattern has been used to reverse the stereochemistry of tetrasubstituted carbons in tetrahydrofuran products. [10]
With the success of the five-membered ring cyclizations, we wondered if the amine was a good enough nucleophile to overcome the twin barriers of six-membered ring and tetrasubstituted carbon formation. If so, then the methodology would also enable the synthesis of pipercolic acid derivatives. Previous reactions using a sulfonamide nucleophile for this purpose were only moderately successful leading to optimal yield of around 50% for the six-membered ring product.
The use of the amine nucleophile for synthesizing pipercolic acid derivatives proved to be quite efficient (Table 1, entries 5-7). The potentials measured for the substrates (4e-g) were lower than that of the product (the six-membered ring product has an Ep/2 = + 0.95 V vs. Ag/AgCl), but not quite as low as those measured for the faster five-membered ring cyclizations. When substrate 4e was oxidized, a 72% isolated yield of the six-membered ring product was generated. This cyclization proceeded much better than the corresponding cyclization using the toluene sulfonamide. When a substitutent was placed on the allylic carbon of the dithioketene acetal (4f), the cyclic product was formed in an 83% isolated yield as a 10:1 ratio of diastereomers. Once again, the stereochemistry in this case was the result of steric interactions between the neighboring groups. When the methyl group was placed on carbon bearing the amine nucleophile (4g), the reaction led to the diasteromeric cyclic products in only a 2:1 ratio. The stereochemical mixture observed in this example suggests that the cyclizations are under kinetic control with an early transition state (no clearly defined equatorial and axial positions). This suggestion is consistent with the very fast cyclization rate.
Finally, an attempt to synthesize a seven-membered ring analog from substrate 4h was made (entry 8, Table 1). Cyclic voltammetry for the substrate indicated a potential drop that was consistent with a rapid cyclization. However, none of the desired cyclic product was obtained from the preparative electrolysis of 4h. Instead, a complex mixture of products was obtained. A proton NMR spectrum of this mixture indicated that substrate 4h was consumed in the reaction. The spectrum showed no evidence of the uncyclized elimination or methanol trapping products typical of failed cyclization reactions. Hence, at this point we feel that the oxidation led to seven-membered ring intermediates that were unstable to the reaction conditions.
With the success of the dithioketene acetal derived cyclizations, we wondered if the cyclizations were fast enough to allow reactions using substrates with even higher oxidation potentials. To this end, the oxidation chemistry of vinylsulfide substrate 10 was examined (Scheme 4).
Scheme 4.

Anodic coupling of an amine and a vinylsulfide.
The oxidation potential for the vinylsulfide employed in 10 is Ep/2 = +1.22 V vs. Ag/AgCl.[12] This value is significantly higher than the potential for the dithioketene acetal and in fact slightly higher than that of the primary amine. The cyclic voltammogram for 10 again showed a significant drop in potential relative to the isolated functional groups (370 mV from the amine to an Ep/2 = + 0.78 V vs. Ag/AgCl).[17] The cyclization was again very fast, but the final potential reached suggested that the reaction might lead to competitive oxidation of the product (Ep/2 = + 0.89 V vs. Ag/AgCl). In practice, this turned out to be the case. After the theoretical amount of current (2.0 F/mole) was passed through the cell, the oxidation of 9 led to only a 20% yield of the cyclized product following its conversion to a toluene sulfonamide derivative 11.[2] In addition, a 17% yield of recovered starting material was obtained indicating a drop-off in the current efficiency of the reaction (an observation consistent with over-oxidation of the product). The reaction led to a number of products and isolation of the product before protection of the secondary amine was difficult. An attempt to completely convert the starting material to product by passing 2.7 F/mol of current through the cell led to no starting material or product being isolated from the reaction. In the end, we concluded that in this case the drop in substrate potential was not enough to avoid complications with product oxidation.
In conclusion, the trapping of a dithioketene acetal derived radical cation can be accomplished using an unprotected amine. The reactions generate amino acid derivatives having a tetrasubstituted α-carbon. Initially, the reactions look like they will fail due to the product having a lower oxidation potential than either of the functional groups in the starting material. However, such an analysis ignores the fact that the cyclization itself can lower the oxidation potential of the substrate. In the examples reported, the cyclizations are fast enough to drop the substrate potential to a point that is significantly lower than that of the product. Clearly, the simple analysis of oxidation potentials for isolated functional groups can be very misleading in terms of predicting the success of an oxidative cyclization reaction. Currently, work to capitalize on the synthetic potential of the cyclization is underway.
Supplementary Material
Footnotes
We thank the National Science Foundation (CHE-0809142) for their generous support of our work. We also gratefully acknowledge the Washington University High Resolution NMR facility, partially supported by NIH grants RR02004, RR05018, and RR07155, and the Washington University Mass Spectrometry Resource Center, partially supported by NIHRR00954, for their assistance.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1].For a review see Moeller KD. Synlett. 2009;8:1208–1218.
- [2].a) Xu H–C, Moeller KD. J. Am. Chem. Soc. 2008;130:13542–13543. doi: 10.1021/ja806259z. [DOI] [PubMed] [Google Scholar]; b) Xu H–C, Moeller KD. J. Am. Chem. Soc. 2010;132:2839–2844. doi: 10.1021/ja910586v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Xu H–C, Moeller KD. Org. Lett. 2010;12:1720–1723. doi: 10.1021/ol100317t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].For reviews of cyclic amino acid derivatives see: Park K–H, Kurth MJ. Tetrahedron. 2002;58:8629–8659. Cativiela C, Diaz-de-Villegas MD. Tetrahedron: Asymmetry. 2000;11:645.
- [5].For recent references see: Mitsunaga S, Ohbayashi T, Sugiyama S, Saitou T, Tadokoro M, Satoh T. Tetrahedron: Asymmetry. 2009;20:1697–1708. Wang Y-G, Mii H, Kano T, Maruoka K. Bioorg. Med. Chem. Lett. 2009;19:3795–3797. doi: 10.1016/j.bmcl.2009.04.025. Kaname M, Yamada M, Yoshifuji S, Sashida H. Chem. Pharm. Bull. 2009;57:49–54. doi: 10.1248/cpb.57.49. Dickstein JS, Fennie MW, Norman AL, Paulose BJ, Kozlowski MC. J. Am. Chem. Soc. 2008;130:15794–15795. doi: 10.1021/ja8073006. Prazeres VFV, Castedo L, Gonzalez-Bello C. Eur. J. Org. Chem. 2008;23:3991–4003. Simila STM, Martin SF. Tetrahedron Lett. 2008;49:4501–4504. doi: 10.1016/j.tetlet.2008.05.073. Undheim K. Amino Acids. 2008;34:357–402. doi: 10.1007/s00726-007-0512-5. and references therein.
- [6].For leading references concerning the use of lactam based peptidomimetics containing cyclic amino acid derivatives see: Cluzeau J, Lubell WD. Biopolymers. 2005;80:98–150. doi: 10.1002/bip.20213. I-lalab L, Gosselin F, Lubell WD. Biopolymers. 2000;55:101–122. doi: 10.1002/1097-0282(2000)55:2<101::AID-BIP20>3.0.CO;2-O. Hanessian S, McNaughton-Smith G, Lombart H-G, Lubell WD. Tetrahedron. 1997;53:12789–12854. For additional lead references see: Polyak F, Lubell WD. J. Org. Chem. 1998;63:5937–5949. doi: 10.1021/jo980596x. Curran TP, Marcaurell LA, O’Sullivan KM. Org. Lett. 1999;1:1225–1228. doi: 10.1021/ol9902069. Gosselin F, Lubell WD. J. Org. Chem. 2000;65:2163–2171. doi: 10.1021/jo991766o. Polyak F, Lubell WD. J. Org. Chem. 2001;66:1171–1180. doi: 10.1021/jo001251t. Feng Z, Lubell WD. J.Org. Chem. 2001;66:1181–1185. doi: 10.1021/jo001252l.
- [7].For synthetic routes to peptidomimetics containing cyclic amino acid derivatives: Scott WL, Alsina J, Kennedy JH, O’Donnell MJ. Org. Lett. 2004;6:1629–1632. doi: 10.1021/ol049547z. Palomo C, Aizpurua JM, Benito A, Miranda JI, Fratila RM, Matute C, Domercq M, Gago F, Martin-Santamaria S, Linden A. J. Am. Chem. Soc. 2003;125:16243–16260. doi: 10.1021/ja038180a. Colombo L, Di Giacomo M, Vinci V, Colombo M, Manzoni L, Scolastico C. Tetrahedron. 2003;59:4501–4513. Dolbeare K, Pontoriero GF, Gupta SK, Mishra RK, Johnson RL. J. Med. Chem. 2003;46:727–733. doi: 10.1021/jm020441o. Khalil EM, Pradhan A, Ojala WH, Gleason WB, Mishra RK, Johnson RL. J. Med. Chem. 1999;42:2977–2987. doi: 10.1021/jm990140n. Aube J. Advances in Amino Acid Mimetics and Peptidomimetics. 1997;1:193–232. Tong Y, Olczak J, Zabrocki J, Gershengorn MC, Marshall GR, Moeller KD. Tetrahedron. 2000;56:9791–9800. Duan S, Moeller KD. Tetrahedron. 2001;57:6407–6415. Liu B, Brandt JD, Moeller KD. Tetrahedron. 2003;59:8515–8523.
- [8].For examples using electrochemistry to functionalize cyclic amino acids see: Beal LM, Liu B, Chu W, Moeller KD. Tetrahedron. 2000;56:10113–10125. Simpson JC, Ho C, Shands EFB, Gershengorn MC, Marshall GR, Moeller KD. Bioorg. Med. Chem. 2002;10:291–302. doi: 10.1016/s0968-0896(01)00287-5. and citations therein.
- [9].Brandt JD, Moeller KD. Heterocycles. 2006;67:621–628. [Google Scholar]
- [10].Xu H-C, Brandt JD, Moeller KD. Tetrahedron Lett. 2008;49:3868–3871. [Google Scholar]
- [11].Wuts PGM, Greene TW. Protective Groups in Organic Synthesis. 4th Ed. John Wiley & Sons; New York: 2007. pp. 855–859. [Google Scholar]
- [12].All potentials were measured by cyclic voltammetry using a BAS 100B Electrochemical Analyzer, a C working electrode, a Pt auxiliary electrode, a Ag/AgCl reference electrode, a 0.1 M Et4NOTs in acetonitrile electrolyte solution, a substrate concentration of 0.025 M, and a sweep rate of 25 mV/sec.
- [13].For other examples see: Moeller KD, Tinao LV. J. Am. Chem. Soc. 1992;114:1033–1041. Reddy SHK, Chiba K, Sun Y, Moeller KD. Tetrahedron. 2001;57:5183–5197. Huang Y, Moeller KD. Tetrahedron. 2006;62:6536–6550.
- [14].For experimental details please see the Supporting Information.
- [15].Liu B, Duan S, Sutterer AC, Moeller KD. J. Am. Chem. Soc. 2002;124:10101–10111. doi: 10.1021/ja026739l. [DOI] [PubMed] [Google Scholar]
- [16].Brandt JD, Moeller KD. Org. Lett. 2005;7:3553–3556. doi: 10.1021/ol051296m. [DOI] [PubMed] [Google Scholar]
- [17].The radical cation generated from the oxidation can undergo a very rapid intramolecular electron-transfer reaction. Hence, it does not matter which of the two functional groups oxidizes first. For example see: Duan S, Moeller KD. J. Am. Chem. Soc. 2002;124:9368–9369. doi: 10.1021/ja027227+.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.