

Abstract
Mammalian somatic cells can be directly reprogrammed into induced pluripotent stem cells (iPSCs) by introducing defined sets of transcription factors. Somatic cell reprogramming involves epigenomic reconfiguration, conferring iPSCs with characteristics similar to embryonic stem cells (ESCs). Human ES cells contain 5-hydroxymethylcytosine (5hmC), which is generated through the oxidation of 5-methylcytosine by the TET enzyme family. Here we show that 5hmC levels increase significantly during reprogramming to human iPSCs mainly due to TET1 activation, and this hydroxymethylation change is critical for optimal epigenetic reprogramming, but does not compromise primed pluripotency. Compared with hES cells, we find iPS cells tend to form large-scale (100 kb-1.3 Mb) aberrant reprogramming hotspots in subtelomeric regions, most of which display incomplete hydroxymethylation on CG sites. Strikingly, these 5hmC aberrant hotspots largely coincide (~80%) with aberrant iPS-ES non-CG methylation regions. Our results suggest that TET1-mediated 5hmC modification could contribute the epigenetic variation of iPSCs and iPSC-hESC differences.
Pluripotency is defined as a stem cell state with the potential to differentiate into any of the three germ layers. Somatic cells can be reprogrammed to a pluripotent state by defined factors such as OCT4, SOX2, KLF4, c-MYC, NANOG and LIN281-3. These iPSCs are extremely similar to ESCs. During the reprogramming process, the global epigenetic landscape in somatic cells has to be reset to reach a pluripotent state via DNA methylation/demethylation and chromatin remodelling processes.
Besides 5-methylcytosine (5mC), which is known to display dynamic changes during early embryonic and germ cell development as well as the reprogramming process, the mammalian genome also contains 5hmC, which is generated by oxidation of 5mC by the TET family of enzymes4, 5. The Tet proteins function in ESCs regulation, myelopoiesis and zygote development6-10. 5hmC was found to be widespread in many tissues and cell types at different levels11, 12. Particularly, 5hmC is abundant in the central nervous system and ESCs. Several reports have explored the genome-wide distribution of 5hmC modification in mES cells and hES cells, and suggest that it is enriched in gene bodies and enhancers13, 14.
Reprogramming toward pluripotency involves a dynamic epigenetic modification process. 5hmC has been implicated in the DNA demethylation process15, pointing to a potential role for 5hmC modification during reprogramming toward pluripotency. Thus, understanding the dynamic 5hmC changes during reprogramming will provide additional insight into somatic cell reprogramming mechanisms.
Multiple studies suggest there are subtle yet substantial genetic and epigenetic differences between iPS cells and hES cells16, 17. The current consensus is that iPS cells and ES cells are two overlapping classes of heterogeneous cells, with iPS cells being more variable than hES cells18. Although iPS cells and hES cells are functionally equivalent in general, the subtle genetic and epigenetic differences could lead to functional consequences among individual lines. Previous study of the base-resolution methylomes of iPSCs and ESCs identified differentially methylated regions (DMRs) between iPSCs and ESCs, consisting of CG-DMRs and non-CG-DMRs16, 17. However, the traditional bisulfite sequencing technique they used could not distinguish 5mC from 5hmC19, which means how these DMRs are caused by hydroxymethylation differences remains unknown.
Here we show that 5hmC levels increase significantly during reprogramming to human iPSCs mainly due to TET1 activation, and this hydroxymethylation change is critical for optimal epigenetic reprogramming. We found that during reprogramming extensive genome-wide 5hmC modification occurs. Importantly, we identified specific aberrant reprogramming hotspots in iPS cells, which cluster on a large-scale (100kb-1.3Mb) at subtelomeric regions bearing incomplete CG hydroxymethylation. These hotspots largely overlap with aberrant non-CG methylation hotspots, suggesting hydroxymethylation contributes to the epigenetic difference between iPS cells and hES cells.
RESULTS
TET1-mediated hydroxymethylation plays a critical role during reprogramming to pluripotency in human cells
DNA methylation is a major barrier to iPS cell reprogramming. Several lines of evidence suggest that 5hmC is involved in the process of DNA demethylation20, 21. We found a significant increase of 5hmC level in human iPS cells compared to their original fibroblasts, with the amount in iPSCs being similar to hES cells (Fig. 1a).
Figure 1. TET1 is associated with increased hydroxymethylation during human iPSC reprogramming.
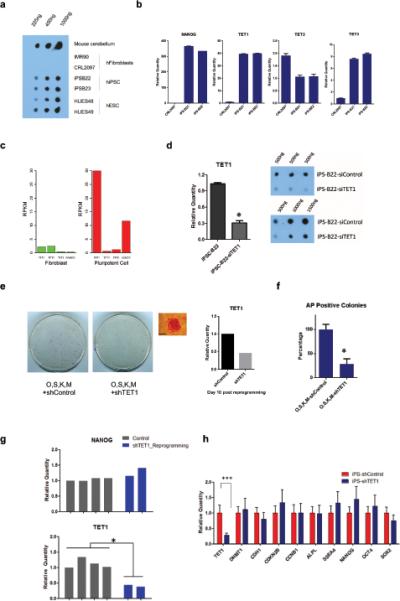
(a) Measurement of 5hmC levels in genomic DNAs from fibroblasts, hiPSCs and hESCs by dot blot using anti-5hmC antibody. Mouse cerebellum genomic DNA was used as a control. 225 ng, 450 ng and 1000 ng DNA were used for each sample. (b) Quantitative RT-PCR to detect mRNA levels of TET1, TET2, TET3 and NANOG in fibroblasts (CRL2097) and hiPSCs (iPSC-B21, iPSC-B22). Error bars represent the standard error of the mean (S.E.M.) collected from three independent experiments. (c) Boxplot of transcript copy numbers of TET1, TET2, TET3, and NANOG in IMR90 (fibroblasts) and H1 (hESCs) represented by RPKM in RNA-seq. (d) Knocking down TET1 by siRNA significantly decreases 5hmC levels in hiPSCs. Left panel represents siTET1 knock down efficiency by quantitative RT-PCR (* t-test, p<0.05). Right panel depicts the effect of total 5hmC levels 48hours post siTET1 transfection. Error bars represent S.E.M. collected from three independent experiments. (e) Alkaline phosphatase (AP) staining of reprogrammed cells treated either with shTET1 lentivirus or an equal titer shControl lentivirus after O,S,K,M retroviral transduction of 100,000 CRL2097 cells on day 20. Cells used for staining were grown in 10 cm dishes. The image on the right shows a representative AP positive colony and TET1 transcript level in shTET1- or shControl-treated cells 10 days post transduction in one representative experiment of three independent experiments. Scale bars: 300 μm. (f) Summary of quantitative analysis of AP-positive colonies in three different experiments (* t-test, p<0.05). Controls were normalized to 100%. Error bars represent the standard deviation (SD). (g) Real time PCR analysis of TET1 and pluripotency marker NANOG. shTET1-treated reprogrammed colonies maintained normal levels of NANOG, but shows decreased TET1 expression (* t-test, p<0.05). Colonies were picked and maintained in puromycin medium (0.5 μg/ml) on puromycin resistant MEFs. (h) Real time PCR analysis of normalized gene expression levels of TET1 and selected pluripotency related factors in stable shTET1 or shControl iPS-B22 cells under the puromycin selection (0.5 μg/ml) (*** t-test, p<0.05). Error bars represent the S.E.M. of three independent experiments. The raw values of related statistical test in this figure are listed in Supplementary Table S1.
TET family proteins (TET1, TET2 and TET3) could convert 5mC to 5hmC6. We found a statistically significant increase of TET1 and TET3; with a more dramatically increase of TET1, and a slight decrease of TET2 expression (Fig. 1b). RNA-seq reveals that TET1 is at a comparable level to NANOG in pluripotent cells, but the expression of TET2 and TET3 are significantly lower (Fig. 1c). Depletion of TET1 but not TET2 and TET3 by siRNA could significantly decrease total 5hmC levels in human iPS cells (Fig. 1d and Supplementary Fig. S1a,b). Therefore, we conclude that TET1 is the main TET protein regulating hydroxymethylation during human iPS cells reprogramming.
Because cellular reprogramming is an epigenetic state reconfiguring process, we next asked whether TET1-mediated hydroxymethylation changes are critical in human iPSC reprogramming. Introducing shTET1 lentivirus with “Yamanka factors” infection could decrease alkaline phosphatase positive colonies when compared with equal titer shControl lentivirus transduction (Fig. 1e,f and Supplementary Fig. S1c,d). shTET1 treated colonies during reprogramming can be further stably maintained, showing decreased TET1 levels, but similar pluripotent gene expression levels compared with iPSCs (Fig. 1g). Furthermore, iPS cells depleted with TET1 maintained a normal undifferentiated stem cell morphology, are positive for alkaline phosphatase, expressed same level pluripotent related factors and stained positive for the pluripotency markers such as NANOG, SOX2, TRA-1-81 (Fig. 1h and Supplementary Fig. S1e-g). Therefore, TET1-mediated hydroxymethylation modification is required for optimal induction of iPSCs, but does not compromise the essential pluripotency of human stem cells.
5hmC epigenomic landscape during reprogramming
We employed 5hmC Capture-Seq to assess genome-wide 5-hmC distributions during reprogramming11. The cell lines and sequencing statistics are summarized on Supplementary Table S2 and S3. Pearson correlation and cluster analysis of the global 5hmC modification pattern suggests a significant difference between iPS cells and fibroblasts (Fig. 2a and Supplementary Table S4).
Figure 2. Reprogramming confers a 5hmC epigenome in a pattern with a bias towards telomere proximal regions in autosomes.
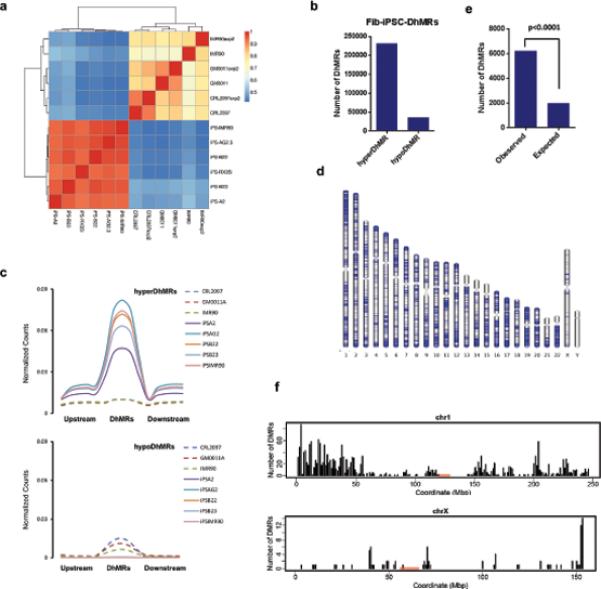
(a) Pearson correlation analysis and cluster among fibroblasts and fibroblast derived iPSCs. The values close to 1 indicate greater similarity. (b) Summary of the numbers of 5hmC differentially modified between fibroblasts and iPSCs, indicated by hyperDhMR (iPSCs>Fibroblast) and hypoDhMR (iPSCs<Fibroblast). The regions enriched either in fibroblasts or in iPSCs were subjected to DhMR calling. 5hmC-enriched regions in 3 fibroblast lines and 5 fibroblast-derived iPSC lines were coalesced into a union window. Then the reads in these windows were recounted and normalized to the total read count from the respective cell line. 267,664 DhMRs were called with a FDR of 0.01 by the Bioconductor Deseq package, which uses a negative binomial model for testing differential expression of sequencing data. Among them, 231,866 are hyperDhMRs, and 35,798 are hypoDhMRs. (c) Composite 5hmC enrichment profile for fibroblasts and iPSCs in the upstream regions of DhMRs, DhMRs, and downstream regions of DhMRs. The length for upstream and downstream of DhMRs is 5kb. (d) Chromosome ideograms showing the genome-wide distribution of the top 20,000 Fib-iPSC-DhMRs ranked by lowest adjusted p-value. Blue lines indicate location of DhMRs. (e) Observed and expected numbers of hyperDhMRs occurring at telomere-proximal regions (chi-square test, p value<0.00001). Telomere-proximal regions were defined as regions at either end of a chromosome with a length equal to 1/20th of the total length of that chromosome. The observed number occurring at telomere-proximal regions is called by overlapping with top 20,000 hyperDhMRs. The expected number is calculated based on the proportion of total telomere-proximal region length compared to the whole length of all chromosomes. The top 20,000 hyperDhMRs were based on the 5hmC profiles of 3 fibroblast lines and 5 fibroblast-derived iPSC lines. (f) The distribution of the top 20,000 Fib-iPSC-hyperDhMRs in Chr1 and ChrX.
Based on a negative binomial model for testing differential expression of sequencing data22, we found 267,664 regions in the genome showing differential 5-hydroxymethylation modification between iPS cells and fibroblast (false discovery rate (FDR): 0.01), which denoted as differential 5-hydroxymethylated regions (DhMRs). Among them, 231,866 are hyperDhMRs (5hmC level is higher in iPS cells), and 35,798 are hypoDhMRs (5hmC level is lower in iPS cells) (Fig. 2b). The hyperDhMRs show higher gain of 5hmC than the loss of 5hmC observed at hypoDhMRs (Fig. 2c). The hyperDhMRs are distributed across all autosomes, but largely missing in sex chromosomes (Fig. 2d). Particularly, of the top 20000 hyperDhMRs (ranked by adjusted p-values), they have a higher probability (p<0.0001) of being located in the telomere proximal regions (Fig. 2e), as shown by example of Chromosome 1 and Chromosome X (Fig. 2f).
5hmC is bi-directionally correlated with DNA methylation changes and associated with pluripotency related gene networks
The analysis described above suggests a global hydroxymethylation change during reprogramming. 5hmC has been suggested linked with gene expression in ES cells and neurons13, 14, 23-26. To assess the correlation between 5hmC modifications and gene expression changes during reprogramming, we stratified genes into 9 categories based on gene expression changes between iPS cells and fibroblasts (category 1: high expression in iPS cells, low expression in fibroblast; category 2: medium expression in iPS cells, low expression in fibroblast, etc). We then quantified the amount of 5hmC around transcription start site (TSS). As a result, those 9 categories can be clustered into 3 distinct patterns (Fig. 3a). Of note, most expressed genes during reprogramming show a bimodal distribution with a depletion of 5hmC in TSS sites, whereas genes remain silenced after reprogramming show a peak in TSS sites. Among 3 clusters, cluster1 has the lowest 5hmC levels in TSS; cluster 3 has the highest levels of 5hmC in TSS, but has lowest 5hmC levels in gene bodies (Fig. 3b).
Figure 3. 5hmC is associated with gene activity and pluripotency regulatory networks in stem cells.

(a) 3 distinct clusters of 5hmC-density pattern at TSS regions (+/− 3kb) in iPSCs and fibroblasts among 9 categories. The 9 categories were classified based on the gene expression changes between iPS cells and fibroblasts: Category 1: high expression in iPS cells, low expression in fibroblast; Category 2: medium expression in iPS cells, low expression in fibroblast, etc. (b) Box plots of hydroxymethylation levels in TSS regions and Gene bodies among the three clusters. *** indicates significantly more 5hmC levels compared with all others (P < 0.001, Wilcoxon rank test). Similarly, * indicates lowest 5hmC levels, ** indicated intermediate 5hmC level. (c) 5hmC enrichment density heatmap. Genes were ordered by expression level from high to low as determined by H1 RPKM27. The TSS and direction of transcription of genes are indicated by the genomic region from –3kb to +3kb and an arrow. The TES is indicated by the genomic region from –3kb to +3kb and vertical lines. The left part of the panel shows genes in fibroblasts, the right part shows the genes in iPSCs. (d) The correlation between PMD (methylation level is higher in stem cells) and DhMRs, and the correlation between hypoDMRs (methylation level is lower in stem cells) and DhMRs. (e) 5hmC density at the NANOG locus in input, iPSCs, and fibroblast cell lines. The position of the loci within the chromosome and the scale are shown above the gene tracks. Black lines indicate the DhMRs. (f) The overlap between NANOG, OCT4, KLF4, SOX2 binding sites in ES cells and 5hmC significant change regions, shown are observed-to-expected ratios. Lower panel shows the overlapping percentage of each binding sites. (g) Gene ontology analysis for genes overlapped with most significant DhMRs. (h) Plot of hyperDhMR and hypoDhMR densities in the context of C+G percent, CG percent, CH percent and CHG percent.
We then examined the correlation between absolute amount of transcripts and 5hmC enrichment. We noticed that hyperDhMRs tend to form bimodal distribution associated with gene activity in iPS cells, with the lowest level similar to the level in fibroblast in TSS regions (Fig. 3c and Supplementary Fig. S2). TES regions also show a bimodal distribution, the depletion is more dramatic in a narrower region centred on TES (Supplementary Fig. S2). However, compared with hypoDhMRs, hyperDhMRs are more enriched in TSS, exons and TES (Supplementary Fig. S3a). We observed a significant negative correlation between 5hmC level of TSS surrounding regions (±200bp) and gene expression levels in iPS cells (Supplementary Fig. S3b).
We also observe bidirectional correlation between 5hmC level and DNA methylation during reprogramming process. 80% of the partially methylated domains (PMD), which displays lower levels of CG methylation in somatic cells than stem cells27, have increased 5hmC levels, with the rest have no 5hmC level change (Fig. 3d). Interestingly, we also found around 60% stem cells hypoDMRs (lower CG methylation in stem cells) shows increased 5hmC modification (Fig. 3b). Collectively, our results suggest that increased hydroxymethylation not only occur in loci with increased methylation but also loci with decreased methylation during reprogramming.
Based on the results of bimodal distribution of 5hmC in TSS and TES, we then determined whether this distribution is associated with core pluripotency regulatory networks. We found that pluripotent master regulators, such as OCT3/4 and NANOG, bear this typical modification in iPSCs but not in fibroblasts (Fig. 3e). We further tested the relation of 5hmC and key pluripotency factors binding sites27. We found a more than 8-fold higher than expected overlap between 5hmC-enriched regions and OCT4, KLF4 binding sties, with a weak association with NANOG and SOX2 binding sites (Fig. 3f). Our results suggest that OCT4 and KLF4 regulatory networks may require 5hmC to regulate pluripotency during reprogramming. Furthermore, gene ontology analysis shows that genes acquiring most 5hmC are involved in stem cell differentiation and patterning process (Fig. 3g), suggesting 5hmC in stem cells are highly correlated with pluripotency.
Sequence preferences of 5hmC modification during reprogramming
We compared the CG, CH (CA, CT, CC), CHG preference of hyperDhMRs and hypoDhMRs. HyperDhMRs tend to be located at higher C and G enriched regions, as well as CHG and CH enriched regions, whereas hypoDhMRs have the same level as the genome background (Fig. 3h). Previous observations suggest that 5hmC modification is related to CpG-density24, 28. We find that in iPSCs, the low CpG content group of CpG islands tend to have more 5hmC modifications (Supplementary Fig. S3c), which is consistent with the observation that DNA methylation occurs more frequently in CpG islands with low CpG content29. Furthermore, 5hmC modifications acquired during reprogramming tend to occur within the unique sequence in which the methylation is evolutionarily less conserved30(Supplementary Fig. S3d-f).
Aberrant 5hmC reprogramming hotspots cluster in telomere-proximal regions
Reprogramming of somatic cells to a pluripotent state requires complete reversion of the somatic epigenome into the pluripotent epigenome, which is an ES-like-state. iPSCs retain some type of somatic memory from their previous identity31-33. We further determined the genome-wide 5hmC modification differences between iPS and ES cells, aiming to understand whether 5hmC modifications underlie the differences between hES cells and iPS cells. To reduce the biases of tissue origins, we used 9 iPS cells derived from different origins, 6 of which are from fibroblasts as mentioned earlier, 2 are derived from peripheral blood cells, and 1 is derived from human exfoliated deciduous teeth cells (SHED).
In general, global DNA hydroxymethylation patterns are very similar between iPS and ES cells (Fig. 4a). A comprehensive analysis of 372,423 5hmC-enriched regions between 4 hES cell and 9 iPS cell lines led to the identification of 113 iPS-ES-DhMRs that were differentially hydroxymethylated in at least one iPS cell or ES cell line (FDR<0.01), as shown for the SIGLEC6 and SIGLEC 12 locus in Fig. 5a. Surprisingly, these regions are not randomly located across the genome; instead, they tend to cluster at the telomere-proximal regions, in particular, at chromosome 3, 7, 8, 12, and 20 (Fig. 4b).
Figure 4. Aberrant 5hmC reprogramming hotspots cluster at subtelomeric regions.
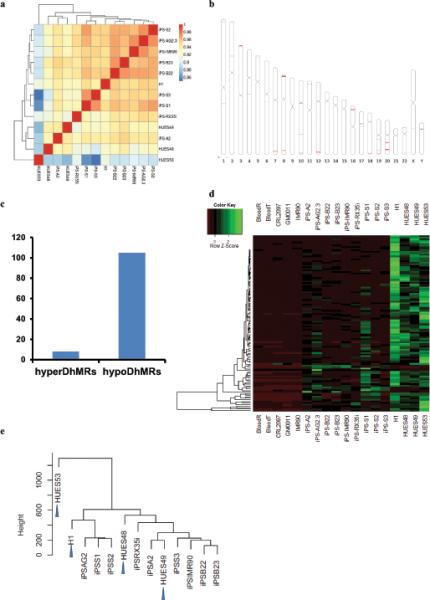
(a) Pearson correlation analysis and clustering among 9 iPSCs and hESCs. Values close to 1 indicate greater similarity. (b) Chromosome ideograms showing the genome-wide distribution of 113 iPSC-ES DhMRs. Red lines indicate locations of DhMRs. (c) The number of iPS-ES-hyperDhMRs and iPS-ES-hypoDhMRs. The 372,423 5hmC-enriched regions either in 9 iPSC lines or 4 hESC lines were subjected to DhMR calling by Bioconductor Deseq package. This analysis led to the identification of 113 iPS-ES-DhMRs that were differentially hydroxymethylated in at least one iPS cell or ES cell line (FDR<0.01). 105 of the 113 iPS-ES DhMRs are hypo-hydroxymethylated, with 5hmC levels similar to their respective progenitors. (d) Complete linkage hierarchical clustering of 5hmC density within the iPS-ES-DhMRs. The raw count values are scaled by rows during clustering. (e) Hierarchical cluster analysis using the top 1,000 most variable 5hmC enriched regions across all iPSC and hESC samples. Arrow indicates hESCs.
Figure 5. 5hmC DhMRs largely overlap with non-CG-DMRs in a large-scale pattern.
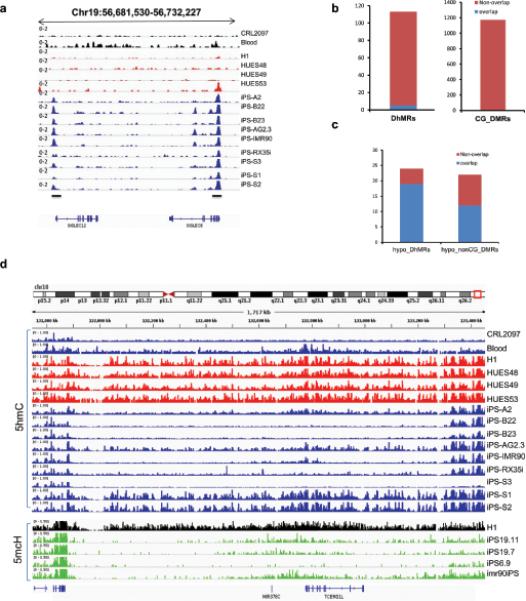
(a) 5hmC density at the iPS-ES-DhMR SIGLEC6, SIGLEC12 locus, in fibroblast (CRL2097), blood, iPS, and ES cell lines. The position of the loci within the chromosome and the scale are shown above the gene tracks. Black bars indicate DhMRs. (b) The number of 5hmC DhMRs that overlaps with CG-DMRs. CG-DMRs were categorized by methylation state relative to the ES cells. (c) The number of 5hmC large-scale hypoDhMRs that overlap with nonCG-DMRs. NonCG-DMRs were categorized by methylation state relative to the ES cells reported previously17. The overlap was called if overlapping length is larger than 1 kb. First bar summarizes the overlap for large-scale hypoDhMRs with hypo-nonCG-DMRs. The second bar summarizes the overlap for hypo-nonCG-DMRs with large-scale hypoDhMRs. The blue colour represents overlap between nonCG-DMR and hypoDhMRs. The red colour represents no overlap. (d) 5hmC density at of iPS-ES-DhMR TCERG1L locus in fibroblast (CRL2097), blood, iPS, and ES cell lines. The position of the loci within the chromosome and the scale are shown above the gene tracks. Lower parts shows the 5mC levels in CH studied by Lister et al, black colour indicates H1 stem cells, green depicts iPSCs.
In contrast to the symmetric pattern of DMRs between iPS and ES cells17, 105 of the 113 iPS-ES DhMRs are hypo-hydroxymethylated, with 5hmC levels similar to their respective progenitors blood cells or fibroblast (Fig. 4c,d). Of these DhMRs, the 5hmC patterns are more variable compared with hES cells (Fig. 4d). Unsupervised hierarchical clustering using the top 1,000 most variable 5hmC modified regions among all samples could not distinguish hESCs from hiPSCs, suggesting that the variability among iPSCs is not due to different levels of pluripotency, and the 5hmC deviation of iPSCs is not a key determinant to distinguish hESCs from iPSCs (Fig. 4e).
Copy number variation (CNV) has been reported to contribute to the variations of iPSCs34,35. Since DhMRs cluster at subtelomeric regions and shows depletion of hydroxymethylation, we further examined whether the DhMRs were simply due to genetic variation, such as CNV, instead of real aberrant 5hmC epigenetic modification. To this end we used high-density comparative genomic hybridization (aCGH) array to examine 3 iPSCs and 2 human ESCs. Array CGH yields an average of 70 CNVs on autosomes, none of which is overlapping with the iPS-ES-DhMRs we identified (Supplementary Fig. S4). Therefore, iPS-ES-DhMRs are caused by aberrant epigenetic modification.
Concordance of large-scale 5hmC hotspots and iPS-ES non-CG DMRs
Our results suggest that iPS-ES-DhMRs tend to cluster at telomere proximal regions, forming aberrant reprogramming hotspots. To better define these large-scale regions, we developed a statistical method to identify potential large-scale aberrant reprogramming hotspots. An aberrant reprogramming hotspot is defined as a genomic region satisfying the following conditions: (1) large variability of 5hmC levels among iPS cells, (2) the average 5hmC difference between iPSCs and ESCs is statistically significant, and (3) longer than100kb. 20 large scale regions were identified. Among them, 19 are hypoDhMRs, all of which have the same epigenetic status as their parent cells, pointing to a “somatic memory” during reprogramming, and 1 is hyperDhMRs (Table 1).
Table 1.
Summary of large-scale hotspots between iPSCs and hESCs
| hypoDhMR(19 regions) | ||||||
|---|---|---|---|---|---|---|
| Chr | Range(bp) | Length (bp) | NonCG-DMR | Aberrant Lines No. | Somatic Memory | Genes |
| Chr1 | 4533001-5059001 | 526,001 | Y | 5 | Y | AJAP1 |
| Chr3 | 474001-592001 | 118,001 | N | 9 | Y | Intergenic |
| Chr3 | 2515001-2907001 | 392,001 | N | 7 | Y | CNTN4 |
| Chr7 | 152805001-153016001 | 211,001 | Y | 8 | Y | Intergenic |
| Chr7 | 153184001-153312001 | 128,001 | Y | 8 | Y | DPP6 |
| Chr7 | 153461001-153856001 | 395,001 | Y | 6 | Y | DPP6 |
| Chr7 | 154010001-154317001 | 307,001 | Y | 6 | Y | DPP6 |
| Chr8 | 2681001-3289001 | 608,001 | Y | 7 | Y | CSMD1 |
| Chr8 | 138881001-139209001 | 328,001 | Y | 7 | Y | CSMD1 |
| Chr8 | 139536001-139818001 | 282,001 | Y | 5 | Y | FAM135B,COL22A1 |
| Chr10 | 132010001-133270001 | 1,260,001 | Y | 7 | Y | TCERG1L,MIR378c |
| Chr12 | 125969001-126071001 | 102,001 | Y | 5 | Y | Intergenic |
| Chr12 | 127355001-127814001 | 459,001 | Y | 5 | Y | TMEM132C |
| Chr16 | 6803001-7330001 | 527,001 | Y | 5 | Y | RBFOX1 |
| Chr18 | 73780001-74420001 | 640,001 | N | 4 | Y | Intergenic |
| Chr20 | 40395001-40593001 | 198,001 | Y | 7 | Y | PTRPT |
| Chr20 | 41004001-41305001 | 301,001 | Y | 7 | Y | PTRPT |
| Chr20 | 53591001-53742001 | 151,001 | Y | 7 | Y | Intergenic |
| Chr22 | 46433001-46536001 | 103,001 | Y | 4 | Y | Intergenic |
| hyperDhMR(1 region) | ||||||
|---|---|---|---|---|---|---|
| Chr | Range | Length | NonCG-DMR | Aberrant Lines No. | Somatic Memory | Genes |
| Chr22 | 46005001-46204000 | 199000 | N | 6 | Y | LOC339685 |
We then compared DhMRs with the DMRs identified previously using whole-genome single base bisulfite sequencing, which would not be able to distinguish 5mC from 5hmC17. Of the total 113 DhMRs, only 5 overlap with 1,175 CG-DMRs (Fig. 5b). Surprisingly, out of the 19 hypo large-scale hotspots, 84.2% overlap with the 24 mega-scale hypo-non-CG-DMRs, whereas the expected percentage is 1.6% based on permutation (Fig. 5c). Fig. 5d shows one of these regions, chr10: 132010002-133270002, 5-mCH are depleted in iPS cells but not hESC lines; similarly, of the 9 total iPS cells, only iPS-S1 and iPS-S2 derived from blood bear similar levels of 5hmC compared with hESC counterparts. Of note, the variances from iPS cells are significantly larger than ES cells (Fig. 6a and Supplementary Fig. S5a, b). None of the iPS cell lines has all of the 19 hypo large-scale DhMRs restored the same level as the 4 human ES cell lines (Fig. 6b). This indicates that these large-scale regions tend to form aberrant reprogramming hotspots that were resistant to reprogramming. We did not observe a statistically significant (p=0.54) correlation between passage number of iPSCs and the number of aberrant hotspots (Supplementary Fig. S5c), implying that passage number may not be a key determinant of hotspots number in each iPSC line.
Figure 6. Large-scale incomplete hydroxymethylation hotspots are characteristics of human iPS cells.
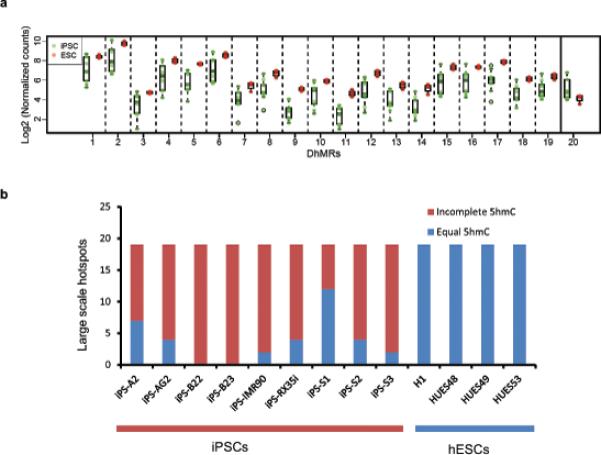
(a) Distribution of 20 5hmC large-scale DhMRs in iPSCs and ESs respectively. Green colour: 9 iPS cell relative enrichment counts, Red colour: 4 hESC cell relative enrichment counts. Solid vertical line separates hyperDhMRs and hypoDhMRs. (b) Summary of 19 hypo large-scale DhMRs in each iPSC line. Blue colour indicates regions have similar 5hmC level compared with ES cells, red colour indicates a lower 5hmC level than ES cells. 5hmC levels were determined by counting 5hmC Capture-Seq reads within each hypo large-scale DhMRs for each cell line. A lower 5hmC level in iPS cells was determined by the criteria that 5hmC levels were less than three standard deviations from the mean among ES cells; if levels were within three standard deviations, the region was considered having similar 5hmC levels.
The aberrant 5hmC reprogramming hotspots we identified may also explain the transcription level variability in iPSCs. Notably, some of the genes such as TCERG1L and FAM19A (Table 1), have been reported to be expressed at a significantly lower level in many but not all iPSCs as compared to ES cells36, 37.
Base-resolution 5hmC analyses reveal large-scale hotspots are mainly caused by aberrant CG hydroxymethylation
The observed extremely high concordance between hypo large-scale DhMRs and non-CG-DMRs is surprising, and might indicate that of the previously identified aberrant 5mCH hotspot regions, a significant portion of CH consists of 5hmC; alternatively, these regions could contain both non-CG (mC) and CG (hmC) aberrant modification. The majority of 5hmC in ESCs is found at CG sites38. In addition, 5hmC quantification by Tet-Asisted-Bisulfite sequencing (TAB-Seq) and the chemical capture approach is well correlated both genome-widely and within the 20 large-scale hotspots regions (Supplementary Fig. S6a,b). Therefore, it is very likely that the aberrant 5hmC is caused by CG modification.
To test this possibility experimentally, we applied TAB-Seq, which can detect hydroxymethylation status at base resolution, to 2 hESCs and 4 iPS cell lines. We performed base-resolution analysis of 5hmC in 3 randomly chosen large-scale regions, chr10, chr18, chr22, and amplified 5hmC enriched regions by PCR (Fig. 7a and Supplementary Table S6,7). We then subjected them to deep sequencing. Deep sequencing of PCR amplicons after traditional bisulfite conversion confirmed that there is epigenetic variation in non-CG sites but not CG sites (Fig. 7b,d). Consistent with the results obtained by capture method, we saw the similar 5hmC variations in iPS cells (Fig. 7c and Supplementary Fig. S6c,d). Importantly, this incomplete hydroxymethylation is caused by CG modification, but not CH modification (Fig. 7c and Supplementary Fig. S6c,d). For example, in the Chr10 hotspot, iPS-B22 and B23 show incomplete 5hmC in CG dinucleotides, but not in CH dinucleotides (Fig. 7e). Therefore, our results suggest the coexistence of aberrant non-CG methylation and CG aberrant hydroxymethylation in subtelomeric hotspots (Fig. 7f). The concordance of aberrant CG hydroxymethylation with those aberrant CH large-scale regions suggests there might be crosstalk between epigenetic pathway regulates hydroxymethylation and pathway regulates CH methylation; this crosstalk may behave more stochastically in those subtelomeric regions.
Figure 7. Large-scale hotspots are caused predominantly by aberrant CpG hydroxymethylation.

(a) Summary of PCR based TAB-Seq. (b) 5hmC+5mC single base density in one of the amplicons by traditional bisulfite sequencing in 2 hESC and 2 iPSC lines. Bisulfite sequencing shows the CH methylation (or methylation plus hydroxymethylation) variation in iPS cells. The position of the loci within the chromosome and the scale are shown above the gene tracks. (c) 5hmC single base density on CG sites in 15 amplicons by TAB-Seq in 2 human ES cells 4 iPS cell lines. iPS-B22 and B23 shows incomplete CG hydroxymethylation. Green colour indicates iPSCs bearing same hydroxymethylation detected by 5hmC Capture-Seq. Blue colour indicates iPSCs bearing incomplete hydroxymethylation detected by 5hmC Capture-Seq in this region. (d) 5hmC+5mC single base density in 15 amplicons by traditional bisulfite sequencing in 2 hESC and 2 iPSC lines. (e) 5hmC single base density on CG dinucleotides and CH dinucleotides in one of the amplicons that are marked by blackdot in (c) by TAB-Seq in 2 human ES and 4 iPS cell lines. Green colour indicates iPSCs bearing the same hydroxymethylation detected by 5hmC Capture-Seq. Blue colour indicates iPSCs bearing incomplete hydroxymethylation detected by 5hmC Capture-Seq in this region. (f) Schematic summary of large scale incomplete hydroxymethylation on CG dinucleotides in iPS cells.
DISCUSSION
Our study suggests that the significant increase of 5hmC during reprogramming is mainly due to the activation of TET1 protein in human iPS cells, which is in contrast to the previous observations that both Tet1 and Tet2 are upregulated in mouse iPS cells. Mouse ESCs are different from human ESCs in many aspects, such as X-chromosome inactivation status in female lines39. From a cell signaling perspective, human pluripotency (primed pluripotency) depends mainly on FGF and Activin-Nodal signaling pathways, whereas mouse pluripotency (naïve/ground state pluripotency) is maintained by LIF-STAT pathways. The difference between human and mouse TET family proteins involved in reprogramming may be caused by FGF signaling selection of a subpopulation of hiPSCs. Several studies of generating naïve human iPSCs under LIF signaling have been reported40, 41. So it is possible that TET1 and TET2 have distinct roles in regulating pluripotency, with TET2 being involved in naïve pluripotency and TET1 functioning in primed pluripotency. On the other hand, it is possible that TET1-mediated 5hmC modification is unique in human regardless of different pluripotent stages. Since TET1/2 is dispensable for maintaining stem cells pluripotency, and their loss are compatible with embryonic and postnatal development42, it is likely that TET2 expression is not under positive section for stem cell functions during evolution, thus eventually silenced in human pluripotent stages.
Reprogramming induces a remarkable epigenomic reconfiguration throughout the somatic cell genome. Recently, it was shown that TET1 and TET2, in synergy with NANOG, enhance the efficiency of mouse iPS cells reprogramming43. Here we show TET1-mediated hydroxymethylation change is critical for optimal human iPS cells reprogramming. We further show that TET1-mediated-5hmC modification only affects reprogramming efficiency, but does not alter the essential pluripotency in human stem cells. The pathways involving TET1 regulation largely remain unknown. It would be interesting to know whether the known epigenetic factors such as DOT1L, Kdm2b, etc 44, 45 which are negative and positive modulators for reprogramming are linked to TET1-regulated hydroxymethylation modification.
Human iPS cells hold great promise for regenerative medicine and for establishing models of specific diseases. iPS and ES cells are known to share key features of pluripotency, including the expression of pluripotency markers, teratoma formation, cell morphology, the ability to differentiate into germ layers, and tetraploid complementation46. Two models depict the equivalence, or lack thereof, between iPSCs and ESCs. One model posits there may be small but consistent differences between ESCs and iPSCs, as suggested before36, 47; the other model states that iPSCs and ESCs should be treated as two partially overlapping groups that share unique features. In this second model, single iPS cell lines cannot be distinguished from ES cell lines, though iPSCs shows more epigenetic variance. Mounting evidence supports the latter model16, 17, 32. Therefore, each iPSC may represent a unique epigenetic status with variable differentiation potential. The cause and degree of variation remain to be determined. Our study integrates the 5hmC epigenomic mark into the investigation of ES-iPS equivalence. We find that 5hmC occurs extensively in iPS cells at levels similar to ES cells, and there are no consistent 5hmC markers that can distinguish iPSCs from hESCs; however, we identified 20 regions in iPSCs that tend to form large scale (100kb-1.3Mb) aberrant reprogramming hotspots, supporting the current consensus that iPSCs are more epigenetically variable than ESCs. Remarkably, these regions with 5hmC variations tend to cluster in telomere-proximal regions. The close proximity of the hotspots to telomeres indicates there may be a distinct cellular process that could impede the reprogramming process.
Almost none of the DhMRs overlap with CG-DMRs, suggesting CG-DMRs identified previously are primarily caused by DNA methylation. DNA methylation in non-CG contexts is abundant in pluripotent stem cells (mCHG and mCHH, where H = A, C or T), comprising almost 25% of all cytosines at which DNA methylation is identified. Strikingly, ~80% of large-scale iPS-ES DhMR regions coincide with previously reported non-CG DNA methylation aberrant hotspots17. Reciprocally, ~50% of non-CG DMRs overlaps with our identified DhMRs. It was reported that non-CG DMRs also occur in the peri-centromeric zones. Notably, these peri-centromeric regions contain low level of 5hmC (stem cells have similar levels of 5hmC as fibroblasts), suggesting cells do not need to establish 5hmC in these regions during reprogramming (Supplementary Fig. S7). Thus, the concordance occurs mainly at telomere proximal regions. By applying TAB-Seq, we show that incomplete hydroxymethylation occur predominantly at CG sites, but not CH sites, suggesting the co-existence of aberrant non-CG methylation and aberrant CG hydroxymethylation in these regions. During reprogramming, both CH methylation and hydroxymethylation need to be established de novo from the somatic epigenome. It is known that non-CG cytosine methylation is exclusively catalysed by Dnmt3a and Dnmt3b48. The concordance suggests there might be crosstalk between epigenetics pathways that regulate the activities of TET and DNMT3, which may behave more stochastically in those subtelomeric regions.
In summary, our results indicate that TET1-mediated 5hmC modification contributes to both the human iPS cell reprogramming process and differences between iPSCs and hESCs. In particular, we identified 20 large-scale aberrant hotspots, suggesting iPSCs are more epigenetically variable than ESCs in terms of 5hmC modification. Our data suggest that, when studying aberrant epigenetic reprogramming events, as well as their functional consequences, at the DNA level, 5hmC modification merits particular consideration, in addition to 5mC.
METHODS
Methods and any associated references are available in the online version of this paper.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Joshua Suhl, Michael Santoro, Steve Bray and Cheryl Strauss for critical reading of the manuscript. We thank Xinping Huang from the Viral Vector Core of the Emory Neuroscience NINDS Core Facilities for preparing the retrovirus/lentivirus used in this study. We are grateful to Julie Mowrey, Viren Patel, Craig Street and Sandeep Namburi for support on Illumina Hiseq2000/Miseq sequencing. This study was supported in part by the National Institutes of Health (NS079625 and HD073162 to P.J.; MH089606 and HD24064 to S.T.W.), the Emory Genetics Discovery Fund, and the Autism Speaks grant (#7660 to X.L.).
Footnotes
AUTHOR CONTRIBUTIONS
T.W., S.T.W. and P.J. designed the study and interpreted the results. T.W. and H.W. analyzed the data. T.W. performed the majority of experiments; Y.L., L.L., X.L. performed 5hmC capture and parts of library preparation. M.Y. C.X.S, H.G. and C.H. assisted with the TAB-Seq experiment and 5hmC capture experiment. A.D. and K.E.S. contributed to the Illumina sequencing, I.G. and K.R. contributed array CGH experiments. I.C., S.C., J.H., M.K., Y.Y., and Q.C. provide some of the hESC and hiPSC lines. T.W., S.T.W. and P.J. wrote the paper with assistance from H.W.
ACCESSION NUMBER
Sequencing data have been deposited to GEO with accession number GSE37050.
Supplementary Information is available in the online version of this paper.
COMPETING FINANCIAL INTEREST
The authors declare no competing financial interests.
REFERENCES
- 1.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 4.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ko M, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koh KP, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell stem cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wossidlo M, et al. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nature communications. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 10.Dawlaty MM, et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell stem cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song CX, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Globisch D, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PloS one. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pastor WA, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szulwach KE, et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS genetics. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes & development. 2011;25:2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bock C, et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y, et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PloS one. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortellino S, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szulwach KE, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nature neuroscience. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams K, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ficz G, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 26.Wu H, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes & development. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Y, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Molecular cell. 2011;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen NM, Kenigsberg E, Tanay A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell. 2011;145:773–786. doi: 10.1016/j.cell.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 31.Kim K, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim K, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nature biotechnology. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polo JM, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nature biotechnology. 2010;28:848–855. doi: 10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hussein SM, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- 35.Laurent LC, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell stem cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin MH, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell stem cell. 2009;5:111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guenther MG, et al. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell stem cell. 2010;7:249–257. doi: 10.1016/j.stem.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nichols J, Smith A. Naive and primed pluripotent states. Cell stem cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, et al. Rapid and efficient reprogramming of somatic cells to induced pluripotent stem cells by retinoic acid receptor gamma and liver receptor homolog 1. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18283–18288. doi: 10.1073/pnas.1100893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–525. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dawlaty MM, et al. Combined deficiency of tet1 and tet2 causes epigenetic abnormalities but is compatible with postnatal development. Developmental cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costa Y, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495:370–374. doi: 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Onder TT, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang G, He J, Zhang Y. Kdm2b promotes induced pluripotent stem cell generation by facilitating gene activation early in reprogramming. Nature cell biology. 2012;14:457–466. doi: 10.1038/ncb2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daley GQ, et al. Broader implications of defining standards for the pluripotency of iPSCs. Cell stem cell. 2009;4:200–201. doi: 10.1016/j.stem.2009.02.009. author reply 202. [DOI] [PubMed] [Google Scholar]
- 47.Stadtfeld M, et al. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dyachenko OV, Schevchuk TV, Kretzner L, Buryanov YI, Smith SS. Human non-CG methylation: are human stem cells plant-like? Epigenetics : official journal of the DNA Methylation Society. 2010;5:569–572. doi: 10.4161/epi.5.7.12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.