

Abstract
Extracellular adenosine 3′,5′-cyclic monophosphate (3′,5′-cAMP) is an endogenous source of localized adenosine production in many organs. Recent studies suggest that extracellular 2′,3′-cAMP (positional isomer of 3′,5′-cAMP) is also a source of adenosine, particularly in the brain in vivo post-injury. Moreover, in vitro studies show that both microglia and astrocytes can convert extracellular 2′,3′-cAMP to adenosine. Here we examined the ability of primary mouse oligodendrocytes and neurons to metabolize extracellular 2′,3′-cAMP and their respective adenosine monophosphates (2′-AMP and 3′-AMP). Cells were also isolated from mice deficient in 2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNPase). Oligodendrocytes metabolized 2′,3′-cAMP to 2′-AMP with 10-fold greater efficiency than did neurons (and also more than previously examined microglia and astrocytes); whereas, the production of 3′-AMP was minimal in both oligodendrocytes and neurons. The production of 2′-AMP from 2′,3′-cAMP was reduced by 65% in CNPase -/- versus CNPase +/+ oligodendrocytes. Oligodendrocytes also converted 2′-AMP to adenosine, and this was also attenuated in CNPase -/- oligodendrocytes. Inhibition of classic 3′,5′-cAMP-3′-phosphodiesterases with 3-isobutyl-1-methylxanthine did not block metabolism of 2′,3′-cAMP to 2′-AMP and inhibition of classic ecto-5′-nucleotidase (CD73) with α,β-methylene-adenosine-5′-diphosphate did not attenuate the conversion of 2′-AMP to adenosine. These studies demonstrate that oligodendrocytes express the extracellular 2′,3′-cAMP-adenosine pathway (2′,3′-cAMP → 2′-AMP → adenosine). This pathway is more robustly expressed in oligodendrocytes than in all other CNS cell types because CNPase is the predominant enzyme that metabolizes 2′,3′-cAMP to 2-AMP in CNS cells. By reducing levels of 2′,3′-cAMP (a mitochondrial toxin) and increasing levels of adenosine (a neuroprotectant), oligodendrocytes may protect axons from injury.
Introduction
Adenosine is a purine nucleoside that exerts a wide range of effects in many organ systems of the body, including the central nervous system (CNS). In the CNS, adenosine is both a neuromodulator and an immunomodulator (Stone et al. 2009). In response to injury in the brain, the extracellular levels of adenosine increase dramatically (Bell et al. 1998; Bell et al. 2001), and extracellular adenosine is neuroprotective primarily via activation of A1 adenosine receptors (Stone et al. 2009). For example, mice lacking the A1 receptor have increased incidence of lethal status epilepticus following experimental traumatic brain injury demonstrating an essential role for adenosine signaling post-injury (Kochanek et al. 2006). Although elevated adenosine levels can lead to increased inflammatory signaling by activation of A2A receptors (Dai and Zhou 2011), adenosine produced by microglia is neuroprotective against excitotoxic death in hippocampal neurons (Lauro et al. 2010) and adenosine is thought to be mainly protective post-CNS injury.
Because adenosine is likely neuroprotective, it is important to elucidate the mechanisms regulating adenosine production in the brain and how these pathways respond to brain injury. Studies show that extracellular adenosine can be formed via multiple pathways. For example, the stepwise metabolism of extracellular 5′-ATP to adenosine (5′-ATP → 5′-ADP → 5′-AMP → adenosine) is a particularly important pathway of extracellular adenosine generation in many organ systems and is mediated by the sequential actions of CD39 and CD73 (Eltzschig 2009; Eltzschig and Carmeliet 2011; Eltzschig et al. 2006). However, evidence is slowly emerging that another source of extracellular adenosine might be via metabolism of extracellular adenosine cyclic monophosphates (cAMPs) to corresponding adenosine monophosphates (AMPs) which are in turn converted to adenosine (Jackson 2011).
There are two potential “cAMP-adenosine pathways,” one that utilizes 3′,5′-cAMP (3,5′-cAMP → 5′-AMP → adenosine) and the other that utilizes 2′,3′-cAMP (2′,3′-cAMP → 2′-AMP + 3′-AMP → adenosine) (Jackson 2011). Although early studies elucidated the existence of the 3′,5′-cAMP-adenosine pathway (Jackson and Raghvendra 2004), more recent studies have focused on the 2′,3′-cAMP-adenosine pathway. 2′,3′-cAMP is a positional isomer of 3′,5′-cAMP that was first detected as being released from the kidney due to metabolic stress and/or injury and was shown to be a source for adenosine formation in the kidney via metabolism of 2′,3′-cAMP to 2′-AMP and 3′-AMP which are in turn converted to adenosine (Jackson et al. 2009).
The extracellular 2′,3′-cAMP-adenosine pathway differs from the extracellular 3′,5′-cAMP-adenosine not only by the starting cAMP substrate and intermediate AMPs formed, but also by the enzymes involved. cAMPs are metabolized to AMPs by phosphodiesterases (PDEs) expressed within the cell or on the cell surface (Lugnier 2006). The extracellular 3′,5′-cAMP-adenosine pathway entails the metabolism of 3′,5′-cAMP to its sole 5′-AMP metabolite by ecto-3′,5′-cAMP-3′-PDEs that are inhibited by 3-isobutyl-1-methylxanthine (IBMX) and 1,3-dipropyl-8-p-sulfophenylxanthine (DPSPX), and 5′-AMP is then converted in the extracellular space to adenosine primarily by ecto-5′-nucleotidase (CD73) (Jackson and Raghvendra 2004).
The 2′,3′-cAMP-adenosine pathway is currently far less well defined than the 3′,5′-cAMP pathway. Extracellular 2′,3′-cAMP is metabolized to 3′-AMP by enzymes resistant to both IBMX and DPSPX and to 2′-AMP by enzymes resistant to IBMX and only partially sensitive to DPSPX, and 2′-AMP and 3′-AMP are converted to adenosine independently of CD73 (Jackson 2011; Jackson and Gillespie 2012; Jackson et al. 2011a; Jackson et al. 2011b; Jackson et al. 2011c; Jackson et al. 2010a; Jackson et al. 2009).
Very recent studies suggest that the extracellular 2′,3′-cAMP-adenosine pathway is functional in vivo in the CNS. The evidence supporting this notion is that: 1) 2′,3′-cAMP, 2′-AMP, 3′-AMP and adenosine are present in the cerebral spinal fluid of patients with traumatic brain injury; 2) the mouse brain in vivo metabolizes 2′,3′-cAMP to 2′-AMP plus 3′-AMP; 3) the mouse brain in vivo further metabolizes 2′-AMP and 3′-AMP to adenosine; and 4) in mice cortical impact elevates brain interstitial levels of 2′,3′-cAMP, 2′-AMP, 3′-AMP and adenosine (Verrier et al. 2012). Moreover, in vitro studies using primary mouse microglia and astrocytes demonstrate that both of these cell types express components of the extracellular 2′,3′-cAMP-adenosine pathway, but appear to lack a significant 3′,5′-cAMP-adenosine pathway (Verrier et al. 2011).
Recent studies also suggest that a long enigmatic enzyme, namely 2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNPase; also known as Cnp), does indeed metabolize 2′,3′-cAMP to 2′-AMP in the mouse brain in vivo and that this mechanism provides an important source of localized adenosine production post-controlled cortical impact (Verrier et al. 2012). CNPase, which comprises about 4% of the total myelin protein in the CNS, is presently thought to be a structural protein primarily expressed in the myelinating glia. It's enzymatic potential to metabolize 2′,3′-cyclic nucleotides to 2′-nucleotides in vitro was puzzling because until the recent detection of 2′,3′-cAMP in vivo, its endogenous substrate was unknown. Interestingly, the CNPase knockout mouse displays a progressive neuropathy with only slight, ultrastructural changes to the myelin. It has been proposed that the lack of CNPase in the oligodendrocytes inhibits an essential glial trophic support mechanism of axons (Edgar et al. 2009), although no such factor has been identified yet. As of this publication the only enzymatic role demonstrated for CNPase in vivo is the metabolism of 2′,3′-cAMP to 2′-AMP, although the samples analyzed were whole brain and the individual contribution of oligodendrocytes and neurons to this pathway have yet to be examined.
In the data presented here, we show that oligodendrocytes are likely the major cell type responsible for the extracellular 2′,3′-cAMP-adenosine pathway in the CNS. Oligodendrocytes are very efficient at metabolizing 2′,3′-cAMP to 2′-AMP and this reaction is predominately performed by the enzyme CNPase. Oligodendrocytes are also able to convert 2′,3′-cAMP to 3′-AMP, but to a much lesser extent than 2′-AMP. Neurons convert 2′,3′-cAMP to its AMPs (and to adenosine) at a much reduced rate when compared to oligodendrocytes suggesting this pathway is primarily non-neuronal. These data establish another role for glial mediated CNS protection via a myelin enriched protein dependent mechanism of localized adenosine formation.
Methods
Primary Oligodendrocyte and Neuron Cultures
Primary oligodendrocytes were isolated from embryonic day 16 mouse pups of either sex bred from a previously characterized CNPase knockout colony (Lappe-Siefke et al. 2003) as described previously (Chen et al. ; Pedraza et al. 2008). Pure CNPase +/+ or CNPase -/- litters were obtained from homozygous breeding pairs. The animals were sacrificed, and for each experiment three brains were obtained and placed in ice-cold Hank's buffered salt solution (HBSS). To obtain enough cells for the experiments, at least 3 litters per genotype were used. After the removal of the meninges, the brains were washed in cold HBSS before the cortices were isolated, triturated and passed through a 50 μm nylon filter to achieve a single cell suspension. The cells were plated onto plastic culture flasks and grown in neurosphere growth media [Dulbecco's Modified Eagle Medium (DMEM) F-12, 25 μg/ml insulin, 100 μg/ml apo-transferrin, 20 nM progesterone, 60 μM putrescine, 30 nM sodium selenite, 20 ng/ml basic fibroblast growth factor, 20 ng/ml epidermal growth factor] and fed every other day for 4 days. At this point, neurospheres were clearly visible and the media was then supplemented with 20 ng/ml bFGF and 40 ng/ml platelet-derived growth factor to induce oligosphere formation. After 2 weeks of oligosphere media (changed every other day), the cells were dissociated and filtered through a 50 μm nylon filter and plated onto poly-d-lysine coated 24 well plates at 1.5 × 105 cells per well in oligodendrocyte precursor media [DMEM/F12, Hepes, penicillin/streptomycin, 4 mM L-glutamine, 1 mM sodium pyruvate, 0.1% bovine serum albumin (BSA), 50 μg/ml apo-transferrin, 5 μg/ml insulin, 30 nM sodium selenite, 10 nM D-biotin, 10 nM hydrocortisone]. The oligodendrocyte precursor cells were allowed to proliferate for 3 days and then oligodendrocyte differentiation media (DMEM/F12, Hepes, penicillin/streptomycin, 100 μg/ml apo-transferrin, 100 μg/ml BSA, 16 μg/ml putrescine, 40 ng/ml sodium selenite, 5 μg/ml N-acetyl-L-cystine, 10 ng/ml biotin, 4.2 μg/ml forskolin, 5 μg/ml insulin, 2 mM glutamine, 1 mM sodium pyruvate, 40 ng/ml 3,3′,5-Triiodo-L-thyronine, 10 ng/ml cilliary neurotrophic factor) was added and cells were re-fed with half fresh media every other day. After 5 days in differentiation media, approximately 85% of the cells were estimated by light microscopy to be mature oligodendrocytes and were then used for the metabolism and immunofluorescence studies.
Primary mouse neurons were isolated and cultured as previously described (Jackson et al. 2010b). In brief, mouse embryos were collected. To obtain enough cells for the experiments, at least four brains were isolated, chopped, trypsinized and triturated from at least 3 separate litters per genotype. Neurobasal media (Neurobasal Media, B27 supplement, L-glutamine, penicillin/streptomycin) was used to culture the cells on poly-d-lysine coated 24-well plates at a density of 2 × 105 cells/well. Cells were placed in a 37°C incubator (5% CO2/95% O2). At day in vitro 3, neurobasal media containing 8 μM arabinofuranosyl cytidine was added to stop glial proliferation in the cultures. Cells were used for metabolism studies at day in vitro 10. All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and under the approval of the University of Pittsburgh Animal Care and Use Committee.
Immunofluorescence and Western Blotting
Primary oligodendroctyes and neurons were isolated as described above and grown on 8-chamber poly-d-lysine coated glass slides. At the indicated time point (day in vitro 10 for neurons, 5 days post-differentiation for oligodendrocytes) the cells were washed twice with phosphate buffered saline (PBS) prior to fixation with 4% paraformaldehyde (in PBS, pH 7.4) for 10 minutes at room temperature. The cells were permeabilized with 0.1% Trition X-100 for 15 minutes, blocked with 10% normal goat serum/PBS for 45 minutes and incubated with primary antibodies at 4°C for 16 hours. The cells were then washed 3 times with PBS and fluorochrome conjugated secondary antibodies (1:500 dilution, Invitrogen, Carlsbad, CA) were added for 1.5 hours. The cells were again washed 3 times with PBS and mounted using a medium containing DAPI (Santa Cruz Biotechnology; Santa Cruz, CA). To demonstrate the presence of neurons, cultures were probed with a mouse anti-neuron specific beta tubulin III antibody (1:1000 dilution; Abcam; Cambridge, MA). To show the presence of mature oligodendrocytes, we employed a mouse anti-CNPase antibody (1:500 dilution; Abcam, Cambridge, MA). The images were captured using a Nikon fluorescent microscope and images were processed using Adobe Photoshop software.
Western blot analysis was performed as described (Jackson et al. 2010b). In brief, CNPase +/+ and CNPase -/- mouse cortical tissue was homogenized in RIPA buffer (Thermo Fisher Scientific, Rockford, IL) with both protease and phosphatase inhibitors. Protein sample concentration was determined using a BCA protein assay kit (Thermo Fisher Scientific). Twenty mg of sample was separated by SDS-PAGE and transferred to a PVDF membrane. Primary antibody (either mouse anti-CNPase, 1:500, Abcam; or rabbit anti-GAPDH, Abcam) was diluted in 5% milk/TBS and incubated on a shaker at 4°C for 16 hours. The blots then washed and probed with HRP-conjugated secondary antibody for 2 hours, washed, and exposed to chemiluminescent substrate prior to development. The resulting films were scanned and processed with Adobe Photoshop software.
Metabolism Studies
To examine purine metabolism experiments were performed as previously described (Verrier et al. 2011). In brief, either 150,000 primary mouse oligodendrocytes or 200,000 primary neurons per well of a 24-well plate were washed twice with HEPES-buffered HBSS and treated with 0.5 ml of PBS with HEPES (25 mmol/L) and NaHCO3 (13 mmol/L) in the presence and absence of substrates (2′,3′-cAMP, 3′-AMP, or 2′-AMP; Sigma, St. Louis, MO). Where indicated enzyme inhibitors were included in the treatment [3-isobutyl-1-methylxanthine {IBMX, broad spectrum phosphodiesterase inhibitor (Beavo and Reifsnyder 1990)}; 1,3-dipropyl-8-p-sulfophenylxanthine {DPSPX, ecto-phosphodiesterase inhibitor (Tofovic et al. 1991)}; α,β-methylene-adenosine-5′-diphosphate {AMPCP, ecto-5′-nucleotidase (CD73) inhibitor (Zimmermann 1992)}]. After one-hour incubation at 37°C, the medium was collected and immediately incubated at 100°C for 3 minutes to denature enzymes. Samples were then stored at −80°C until assayed by mass spectrometry. Total protein content of a least three wells per 24-well plate was measured using the Thermo Scientific Pierce BCA Protein Assay Kit (ThermoFisher Scientific, Waltham, MA).
Analytical Methods
The internal standard (13C10-adenosine) for the following analytical methods was obtained from Medical Isotopes Inc. (Pelham, NH). The purines in our collected samples were resolved by reversed-phase liquid chromatography (Agilent Zorbax eclipse XDB-C-18 column) and further quantified using a triple quadrupole mass spectrometer (LC-MS/MS; TSQ Quantum-Ultra, ThermoFisher Scientific). The methods have been described in detail previously (Jackson et al. 2009). The determined limit of detection for purines in this assay system is estimated to be 0.2 nmol/L.
Statistical Analysis
Data were analyzed by 1-factor analysis of variance (ANOVA), 2-factor ANOVA or Student's t-test as appropriate. If and only if main effects were significant in the ANOVAs, post hoc comparisons were performed with a Fisher's Least Significant Difference test. The criterion of significance was p<0.05. All values in text and figures are means ± SEM. Data were converted to nmol/L/μg protein to normalize between the cell types used in these experiments.
Results
CNPase +/+ and -/- Mouse Colony Primary Cell Cultures
A previously characterized CNPase knockout mouse colony (Lappe-Siefke et al. 2003) was utilized for these studies. We confirmed the complete absence of both type 1 and 2 isoforms of CNPase from these animals using western blot analysis for the CNPase protein. Samples of mouse cortical brain tissue isolated from both wild-type and knockout mice show that neither CNPase isoform is detectable in the knockout mice (Figure 1A). Since we sought to define the cellular metabolism of 2′,3′-cAMP, 2′-AMP and 3′-AMP of both neurons and oligodendrocytes, we isolated these primary cell types from both CNPase +/+ and CNPase -/- embryonic mice. The cell type identification of the cultures was determined by immunofluorescence staining using antibodies for cell-type-specific proteins. For neuron cultures, neuron specific beta tubulin III shows the cultures to be over 95% neurons (Figure 1B). CNPase was used as a label to detect mature, differentiated oligodendrocytes (Figure 1C). The cultures were determined to be approximately 85% oligodendrocytes with few contaminating other glial cell types.
Figure 1.
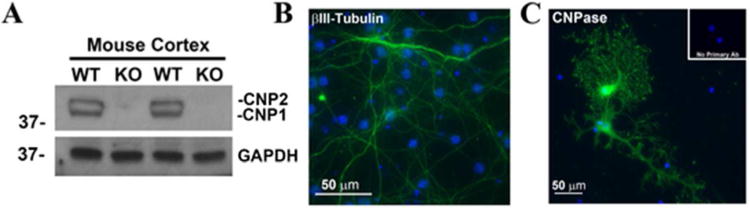
CNPase knockout confirmation and cell type identification. The complete lack of both type 1 (CNP1) and type 2 (CNP2) isoforms of CNPase was confirmed by Western blot on protein extracts from CNPase +/+ and -/- mouse cortical tissue using an anti-CNPase antibody. GAPDH is shown as a loading control (A). A neuron specific beta III-tubulin antibody and a FITC-conjugated secondary antibody was used to confirm pure cultures of primary mouse neurons (Green, DAPI, blue; Scale bar = 50 microns) (B). An anti-CNPase antibody was used to detect mature mouse oligodendrocyte cultures (Green, DAPI, blue; Scale bar = 50 microns) (C).
Metabolism of 2′,3′-cAMP to 2′-AMP and 3′-AMP by Oligodendrocytes and Neurons
We first sought to characterize the ability of primary mouse oligodendrocytes and neurons from both CNPase +/+ and CNPase -/- mice to metabolize extracellular 2′,3′-cAMP. Oligodendrocytes isolated from CNPase +/+ mice were able to convert extracellular 2′,3′-cAMP to 2′-AMP in an efficient manner (Figure 2A), reaching a maximum detection of 203 nmol/L/μg protein when 30 μM of 2′,3′-cAMP was added to the cultures. The oligodendrocytes from the CNPase -/- mice were approximately 65% less effective at forming 2′-AMP from extracellular 2′,3′-cAMP (Figure 2A). The reduced ability of oligodendrocytes from CNPase -/- mice to metabolize 2′,3′-cAMP to 2′-AMP was confirmed by the highly significant statistical interaction between genotype and 2′,3′-cAMP level (P<0.0001) that was revealed by 2-factor ANOVA. In contrast, neurons from CNPase -/- mice were able to convert 2′,3′-cAMP to 2′-AMP approximately 20% more efficiently than the neurons isolated from the CNPase +/+ mice (Figure 2B) (P<0.0001 for genotype × 2′,3′-cAMP; 2-factor ANOVA). Comparing between the two cell types, wild-type oligodendrocytes were able to form approximately 10-fold more 2′-AMP from 2′,3′-cAMP than wild-type neurons.
Figure 2.
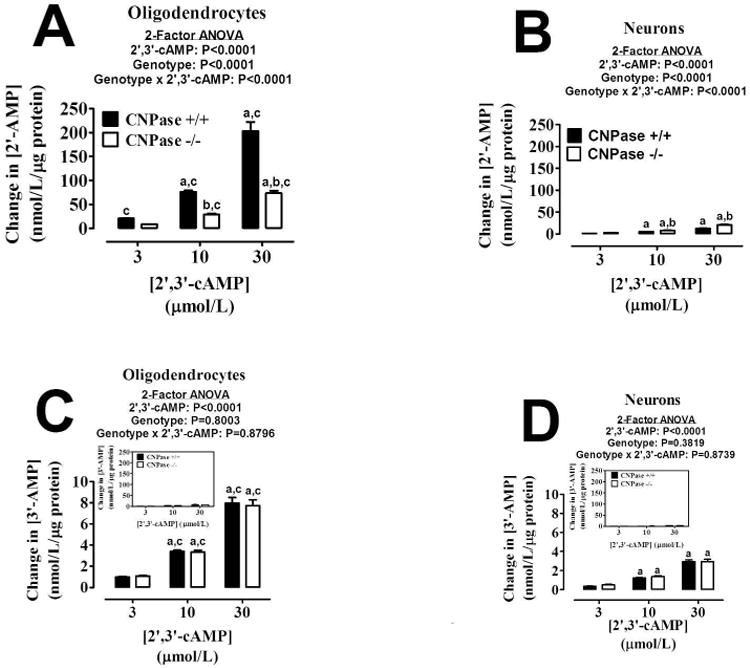
Bar graphs depict the production of extracellular 2′-AMP (A and B) and 3′-AMP (C and D) from extracellular 2′,3′-cAMP in oligodendrocytes (A and C) and neurons (B and D) from CNPase +/+ and CNPase -/- mice. “a” indicates significance difference from corresponding 3 μmol/L concentration of 2′,3′-cAMP; “b” indicates significant difference between genotypes at the same concentration of 2′,3′-cAMP; and “c” indicates significant difference versus neurons with same concentration of 2′,3′-cAMP and same genotype. Significance is defined as P<0.05. Insert graphs show same data plotted on same scale as A and B. Values represent means ± SEM for 6 experiments.
With regard to 3′-AMP formation from extracellular 2′,3′-cAMP, oligodendrocytes (Figure 2C) were over 2-fold more efficient at this process than neurons (Figure 2D). There was no genotype effect observed in either cell type (for oligodendrocytes P=0.8003 and P=0.8796 for genotype and genotype × 2′,3′-cAMP, respectively, by 2-factor ANOVA; for neurons P=0.3819 and P=0.8739 for genotype and genotype × 2′,3′-cAMP, respectively, by 2-factor ANOVA). Also, the amounts of 3′-AMP formed from 2′,3′-cAMP compared to 2′-AMP were over 25-fold less in the oligodendrocytes and over 6-fold less in the neurons.
Metabolism of 2′,3′-cAMP to Adenosine
We also sought to determine if either oligodendrocytes (Figure 3A) or neurons (Figure 3B) possess the capacity to convert extracellular 2′,3′-cAMP to adenosine, and we investigated if lack of CNPase would significantly affect this reaction. CNPase +/+ oligodendrocytes (Figure 3A) were able to convert extracellular 2′,3′-cAMP to adenosine more readily than CNPase -/- oligodendrocytes (P=0.0017 for genotype × 2′,3′-cAMP; 2-factor ANOVA). Neurons (Figure 3B), were also able to convert 2′,3′-cAMP to adenosine but much less efficiently than oligodendrocytes. When comparing the two cell types, at the highest concentration of 2′,3′-cAMP used, wild-type oligodendrocytes were approximately 4-fold more efficient at converting 2′,3′-cAMP to adenosine than wild-type neurons. The ability of CNPase -/- neurons to convert 2′,3′-cAMP to adenosine was only slightly reduced compared with CNPase +/+ neurons.
Figure 3.
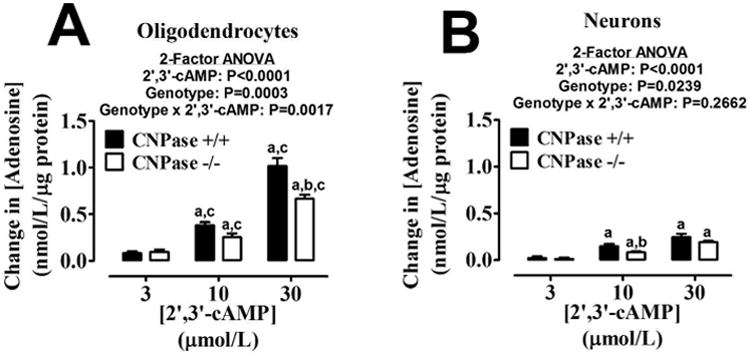
Bar graphs depict the production of extracellular adenosine from extracellular 2′,3′-cAMP in oligodendrocytes (A) and neurons (B) from CNPase +/+ and CNPase -/- mice. “a” indicates significance difference from corresponding 3 μmol/L concentration of 2′,3′-cAMP; “b” indicates significant difference between genotypes at the same concentration of 2′,3′-cAMP; and “c” indicates significant difference versus neurons with same concentration of 2′,3′-cAMP and same genotype. Significance is defined as P<0.05. Values represent means ± SEM for 6 experiments.
Metabolism of 2′-AMP and 3′-AMP to Adenosine
We next sought to evaluate both cell types and genotypes for their ability to convert 2′-AMP and 3′-AMP to adenosine. Oligodendrocytes (Figure 4A) and neurons (Figure 4B) were able to convert 2′-AMP to adenosine in both genotypes. However the CNPase -/- oligodendrocytes were slightly less efficient at the reaction than the CNPase +/+ cells (Figure 4A) (P=0.0015 for genotype; 2-factor ANOVA). 3′-AMP was converted to adenosine by both cell types in similar amounts and at levels slightly higher than 2′-AMP (Figure 4C and D). There was also a small genotype effect (reduced activity) observed in oligodendrocytes (P=0.0032 for genotype; 2-factor ANOVA) and neurons (P=0.0014 for genotype; 2-factor ANOVA) in the conversion of 3′-AMP to adenosine.
Figure 4.
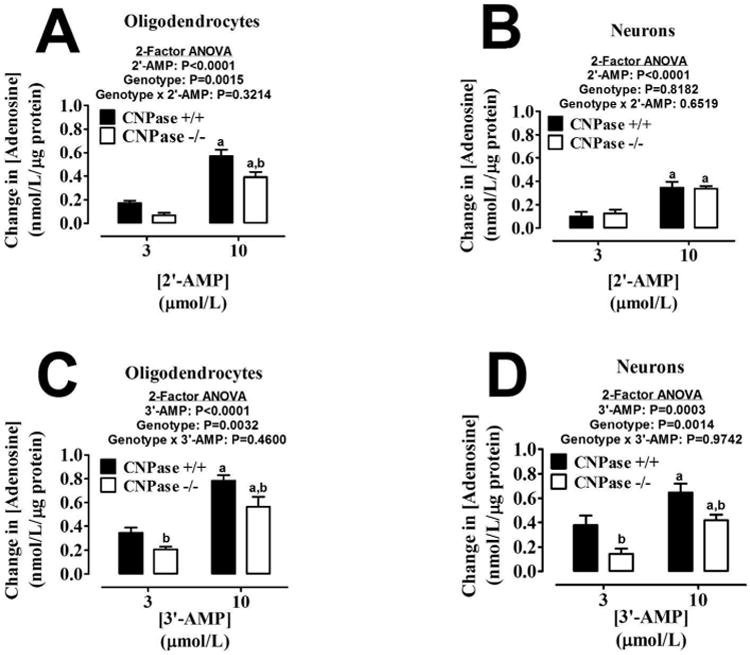
Bar graphs depict the production of extracellular adenosine from extracellular 2′-AMP (A and B) and extracellular 3′-AMP (C and D) in oligodendrocytes (A and C) and neurons (B and D) from CNPase +/+ and CNPase -/- mice. “a” indicates significance difference from corresponding 3 μmol/L concentration of 2′,3′-cAMP; and “b” indicates significant difference between genotypes at the same concentration of 2′,3′-cAMP. Significance is defined as P<0.05. Values represent means ± SEM for 6 experiments.
PDE Inhibitors on 2′,3′-cAMP to 2′-AMP and 3′-AMP Formation
The conversion of extracellular 3′,5′-cAMP to 5′-AMP is blocked by IBMX (1 mmol/liter; a broad spectrum PDE inhibitor) and DPSPX (1 mmol/liter; an ecto-PDE inhibitor) (Jackson 2011; Jackson and Raghvendra 2004). To determine whether the metabolism of extracellular 2′,3′-cAMP is via the same enzymes that metabolize 3′,5′-cAMP to 5′-AMP, we examined in oligodendrocytes and neurons the ability of IBMX and DPSPX to inhibit the metabolism of 2′,3′-cAMP to 2′-AMP and 3′-AMP. IBMX did not affect the metabolism of 2′,3′-cAMP to either 2′-AMP or 3′-AMP regardless of cell type or genotype (Figure 5). However in both oligodendrocytes and neurons, irrespective of genotype, DPSPX did slightly reduce the amount of 2′-AMP formed from the addition of extracellular 2′,3′-cAMP (Figure 5) (P<0.0001; 1-factor ANOVA).
Figure 5.
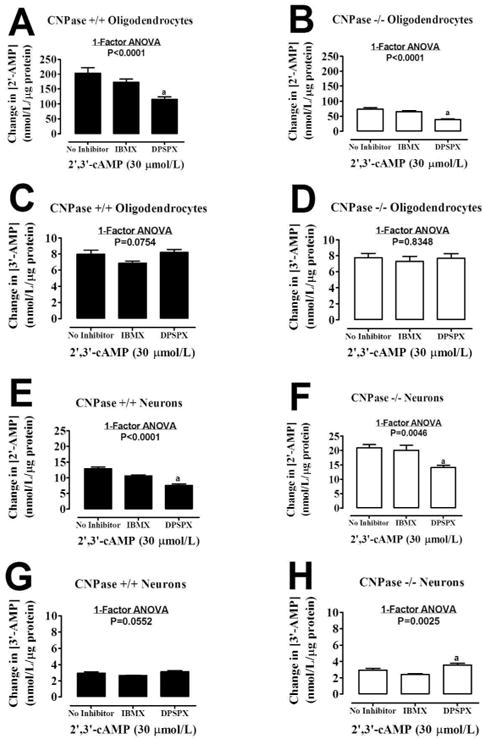
Bar graphs demonstrate the effect of 3-isobutyl-1-methylxanthine (IBMX, 1 mmol/L; broad spectrum phosphodiesterase inhibitor) and 1,3-dipropyl-8-p-sulfophenylxanthine (DPSPX, 1 mmol/L; ecto-phosphodiesterase inhibitor) on the conversion of extracellular 2′,3′-cAMP to 2′-AMP and 3′-AMP by CNPase +/+ or CNPase -/- oligodendrocytes (A - D) and neurons (E -H). “a” indicates significance difference versus No Inhibitor group (same group as in Figure 2). Significance is defined as P<0.05. Values represent means ± SEM for 6 experiments.
CD73 Inhibition on 2′-AMP and 3′-AMP Metabolism to Adenosine
The conversion of extracellular 5′-AMP to adenosine is blocked by AMPCP (Jackson 2011; Jackson and Raghvendra 2004), an inhibitor of ecto-5′-nucleotidase (CD73). To determine whether the metabolism of extracellular 2′-AMP and 3′-AMP to adenosine is via the same enzyme (CD73) that metabolizes 5′-AMP to adenosine, we examined in oligodendrocytes and neurons the ability of AMPCP to inhibit the metabolism of 2′-AMP and 3′-AMP to adenosine. AMPCP did not affect the metabolism of either 2′-AMP or 3′-AMP to adenosine regardless of cell type or genotype (Figure 6).
Figure 6.
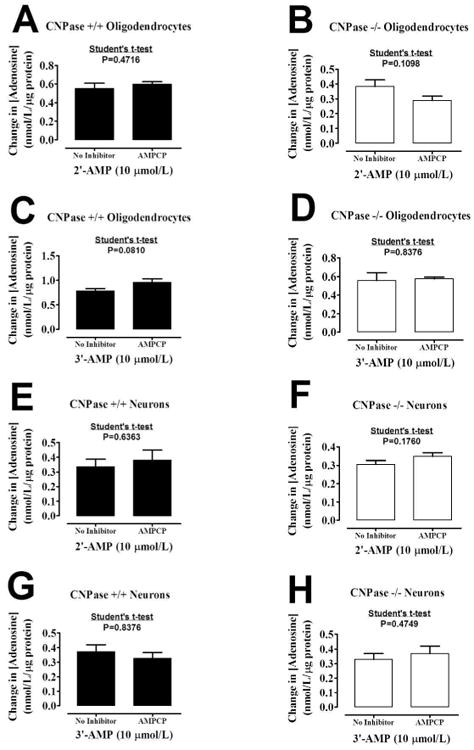
Bar graphs demonstrate the effect of α,β-methylene-adenosine-5′-diphosphate [AMPCP, 0.1 mmol/L; ecto-5′-nucleotidase (CD73) inhibitor] on the conversion of extracellular 2′-AMP and 3′-AMP to adenosine by CNPase +/+ or CNPase -/- oligodendrocytes (A - D) and neurons (E - H). Values represent means ± SEM for 6 experiments.
Discussion
The rapid and robust increase of extracellular adenosine in the brain post-injury is an essential process for endogenously mediated neuroprotection, yet mechanistically it remains poorly defined. The experiments presented here further elucidate the cell types and enzymes responsible for the extracellular 2′,3′-cAMP-adenosine pathway in the CNS. We show that oligodendrocytes metabolize 2′,3′-cAMP to 2′-AMP much more robustly than do neurons. Also, compared to our previous results in astrocytes and microglia (Verrier et al. 2011), oligodendrocytes are much better at generating 2′-AMP from 2′,3′-cAMP. Thus although neurons, astrocytes and microglia can metabolize 2′,3′-cAMP to 2′-AMP, oligodendrocytes are clearly the primary cell type producing extracellular 2′-AMP in the CNS.
We also reveal here that oligodendrocytes process 2′,3′-cAMP mainly to 2′-AMP with only trace amounts of 3′-AMP. In agreement with previous in vivo studies (Verrier et al. 2012), the conversion of 2′,3′-cAMP to 2′-AMP in oligodendrocytes is highly dependent on the expression of CNPase. The observation that in oligodendrocytes lacking CNPase the metabolism of 2′,3′-cAMP to 2′-AMP is reduced by approximately 65% clearly demonstrates that CNPase is the major enzyme responsible for this process.
However, the metabolism of 2′,3′-cAMP to 2′-AMP is not exclusively mediated by CNPase since some 2′-AMP is formed even in CNPase -/- oligodendrocytes, an observation supported by previous in vivo experiments in CNPase -/- mice (Verrier et al. 2012). Also, some of the capacity of CNPase +/+ and -/- oligodendrocytes and neurons to convert 2′,3′-cAMP to 2′-AMP is inhibited by DPSPX, but not by IBMX. This is consistent with our previous observations that a component of the capacity of vascular smooth muscle cells and glomerular mesangial cells to metabolize 2′,3′-cAMP to 2′-AMP is sensitive to DPSPX (Jackson et al. 2010a). Together, these findings imply that 2′,3′-cAMP is metabolized to 2′-AMP by at least three enzymes: 1) CNPase; 2) DPSPX-insensitive, non-CNPase; and 3) DPSPX-sensitive, non-CNPase. Nonetheless, at least in the CNS CNPase is likely the primary enzyme mediating 2′-AMP formation. Interestingly, neurons lacking CNPase have a slightly enhanced ability to produce 2′-AMP from 2′,3′-cAMP, implying an upregulation of expression or activity of other PDEs when CNPase is absent.
Although there is a 65% reduction of 2′-AMP production from 2′,3′-cAMP in the CNPase -/- oligodendrocytes, the conversion of 2′,3′-cAMP into adenosine is reduced by approximately 30%. Thus, CNPase makes only a partial contribution to adenosine production from 2′,3′-cAMP. It could be that the major benefit of CNPase is to prevent the cytotoxic effects of accumulated 2′,3′-cAMP and that the production of adenosine via this pathway is of secondary importance. However, our hypothesis is that both mechanisms participate (Figure 7).
Figure 7.
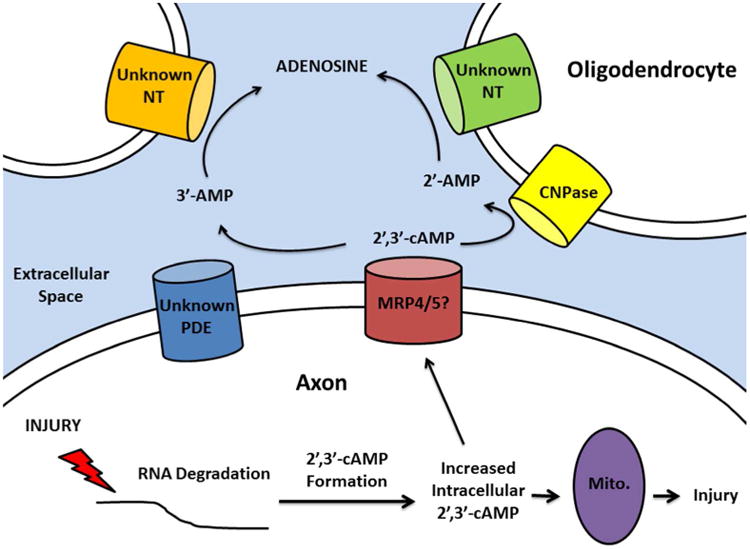
Our working hypothesis is that axonal injury leads to RNA degradation with subsequent formation of intracellular 2′,3′-cAMP. Because 2′,3′-cAMP activates permeability transition pores in mitochondria (Mito.), elevated intracellular 2′,3′-cAMP further promotes axonal injury. However, transport of 2′,3′-cAMP into the extracellular space (perhaps mediated via multidrug resistance proteins 4 and 5; MRP4/5) leads to removal of 2′,3′-cAMP from axons. Clearance of extracellular 2′,3′-cAMP via metabolism to 2′-AMP by oligodendrocytic CNPase would maintain a favorable gradient for efflux of intracellular 2′,3′-cAMP from axons, thus providing protection by facilitating the removal of an intracellular toxin. Moreover, the production of adenosine from 2′-AMP would further protect axons from injury. The precise location of CNPase in the oligodendrocyte responsible for metabolizing extracellular 2′,3′-cAMP is unknown, but could involve intracellular CNPase (via transport of 2′,3′-cAMP into the oligodendrocyte), metabolism by membrane-bound CNPase or metabolism by CNPase shed by oligodendrocytes. The close proximity of oligodendrocytes and axons would maximize the efficiency of this proposed mechanism. NT, nucleotidase; PDE, phosphodiesterase.
The PDEs responsible for the metabolism of 2′,3′-cAMP to 3′-AMP are even more elusive than the 2′-AMP producing PDEs since they are insensitive to both IBMX and DPSPX, findings consistent with what is observed in astrocytes, microglia, vascular smooth muscle cells and glomerular mesangial cells (Jackson et al. 2010a; Verrier et al. 2011). These data do help to limit the possible PDEs that are producing 3′-AMP (and 2′-AMP) in the CNS. Although IBMX can inhibit most PDEs, PDE8 and PDE9 are known to be IBMX insensitive (Lugnier 2006). Future experiments can use specific inhibitors of IBMX-insensitive PDEs to further characterize the 2′,3′-cAMP-adenosine pathway. Our previous studies in astrocytes and microglia show that IBMX and DPSPX significantly inhibit conversion of 3′,5′-cAMP to 5′-AMP (Verrier et al. 2011). These data indicate that this process is performed by several PDEs targeted by these inhibitors, an observation supported by previous studies (Lugnier 2006). Thus the 2′,3′-cAMP-adenosine pathway is distinct in many respects from the 3′,5′-cAMP-adenosine pathway in the major cell types of the CNS.
The present findings also show that both oligodendrocytes and neurons are able to metabolize both 2′-AMP and 3′-AMP to adenosine and that there are only minor differences between the cell types and genotypes in this regard. However, oligodendrocytes do form adenosine from 2′-AMP about 50 percent more efficiently than neurons. Of note, the ability of oligodendrocytes to metabolize 2′-AMP and 3′-AMP to adenosine is modestly reduced in CNPase -/- oligodendrocytes. It is possible that life-long knockout of CNPase may lead to down-regulation or altered activity of other enzymes in these metabolic pathways. Alternatively, it is conceivable that CNPase per se may be involved in the direct metabolism of 2′-AMP and 3′-AMP to adenosine. In this regard, in both cell types the conversion of 2′-AMP and 3′-AMP to adenosine is not affected by inhibition of CD73 with AMPCP, which is known to reliably abolish 5′-AMP to adenosine metabolism. These data suggest that oligodendrocytes and neurons convert both 2′-AMP and 3′-AMP to adenosine via a non-CD73 enzyme and highlight the fact there are still yet to be identified 2′ and 3′ specific nucleotidases.
There are implications from the findings that both 2′-AMP and 3′-AMP are converted to adenosine independent of CD73. First, adenosine derived from 2′-AMP and 3′-AMP cannot be discounted in experiments using CD73 knockout animals or CD73 inhibitors. Another implication is that it is no longer sufficient to refer to all cyclic adenosine monophosphates as just “cAMP” because we show 2′-AMP and 3′-AMP are processed differently by distinct enzymes, likely arise from different sources than 5′-AMP, and yet are precursors for adenosine similar to 5′-AMP. Again these observation reinforce the concept that the extracellular 2′,3′- cAMP-adenosine pathway is a similar yet still distinct mechanism from the 3′,5′-cAMP-adenosine pathway.
CNPase is highly enriched in the myelinating cells of the nervous system, Schwann cells in the PNS and oligodendrocytes in the CNS (Sprinkle 1989). There are reports of this enzyme being detected in other cell types, specifically in the mitochondrial fraction, and CNPase activity is observed in both the spleen and thymus (Weissbarth et al. 1981). However, as indicated by immunological and other protein analytical techniques, and here by the robust levels of the CNPase-dependent reaction (2′,3′-cAMP to 2′-AMP), oligodendrocytes have both the highest levels of expression and activity of the four major cell types of the CNS. Studies show that CNPase is down-regulated in Down syndrome, Alzheimer's disease (Vlkolinsky et al. 2001) and multiple sclerosis, where CNPase may serve as an auto-antigen for the disease (Walsh and Murray 1998). Indeed, CNPase -/- mice develop severe neurodegeneration with age with seemingly normal myelin (Lappe-Siefke et al. 2003), a yet to be explained phenotype. Moreover, even minor brain injury in very young (postnatal day 28) CNPase -/- mice triggers a vicious cycle of neurodegeneration and low-grade inflammation (astrogliosis and microgliosis) accompanied by axonal degeneration and deterioration of working memory 3 months after the initial injury (Wieser et al. 2013). Of great importance is that even CNPase +/- (heterozygous) mice that have only reduced CNPase develop a psychiatric disease with advanced age characterized as a catatonia-depression syndrome (Hagemeyer et al. 2012). Finally, detailed studies in myelin from aged rhesus monkeys demonstrate a striking age-related dysfunction of CNPase in lipid rafts, a finding hypothesized to lead to myelin and axonal pathology (Hinman et al.). These studies, when taken in combination, strongly suggest that decreased CNPase contributes to neurological and psychiatric diseases. It is possibly that a compromised 2′,3′- cAMP-adenosine pathway contributes to this phenomenon but it is premature to state such a link without further studies as CNPase may have other functions. For example, recent studies show that CNPase interacts with calmodulin in addition to CNPase's more well-known interaction with microtubules (Myllykoski et al. 2012). Moreover, CNPase's role in the mitochondria may have significant effects on cell death pathways because intracellular 2′,3′-cAMP may promote mitochondrial permeability transition pore opening (Azarashvili et al. 2009). Intracellular CNPase is primarily localized to the mitochondria implying an endogenous mechanism to prevent 2′,3′-cAMP induced apoptosis under normal physiological conditions. In times of stress with increased formation of 2′,3′-cAMP, it may be essential to export 2′,3′-cAMP into the extracellular space to prevent toxic intracellular accumulation. It is our current hypothesis that the 2′,3′-cAMP-adenosine pathway is a mechanism to rid the cell of a toxic product (2′,3′-cAMP) and in turn transform it to a protective metabolite (adenosine) (Figure 7).
The data presented here provides additional information on the identity of the enzymes used by the extracellular 2′,3′-cAMP-adenosine pathway. The determination of the remaining enzymes is no trivial task. Although CNPase is the only known mammalian PDE known to possess the ability to form 2′-nucleotides there must be other, less cell-specific PDEs that can perform this reaction in a less efficient manner. Recent studies have identified several bacterial PDEs with the capacity to create 3′ and 2′- products but their mammalian homologs have yet to be examined (Rao et al. 2010). The further elucidation of these PDEs and nucleotides may lead to new therapeutic targets as PDE inhibitors are emerging as neuroprotective agents in brain injury. The current studies focus solely on the extracellular role for 2′,3′-cAMP and its AMP metabolites. Since 2′,3′-cAMP is believed to be formed from RNA degradation (Thompson et al. 1994), which occurs within the cell, examining the actions, if any, of intracellular 2′,3′-cAMP and its unique metabolites might reveal new protective or deleterious cell signaling pathways regulated by CNPase. 3′,5′-cAMP acts as a second messenger and has a wide array of actions intracellularly, none of which have been examined for potential interaction with the enzymes that regulate the 2′,3′-cAMP-adenosine pathway. It will also be of use to determine which cell type in the CNS is the predominate source of 2′,3′-cAMP in times of stress and injury and how precisely it is exported into the extracellular space.
In summary, data presented here reveal that CNPase is a critically important enzyme in the 2′,3′-cAMP-adenosine pathway and participates in the production of adenosine, a protective neuromodulator. Future work will seek to identify the remaining PDEs and nucleotidases of this previously uncharacterized pathway. Our findings also demonstrate that the extracellular 2′,3′-cAMP-adenosine pathway is highly enriched in oligodendrocytes. Since oligodendrocytes wrap myelinated neurons, CNPase in the myelin sheath is positioned next to axons, thus likely serving to remove toxic 2′,3′-cAMP and to replace it with adenosine, which is an axonal protectant (Fern et al. 1994). Thus, the 2′,3′-cAMP-adenosine pathway may be critical to the long-term health of axons.
Acknowledgments
This work was supported by NIH grants NS070003 (PMK/EKJ), DK068575 (EKJ), DK079307 (EKJ), HL1090002 (EKJ), DK091190 (EKJ), NS30318 (PMK), and NS38878 (RB).
References
- Azarashvili T, Krestinina O, Galvita A, Grachev D, Baburina Y, Stricker R, Evtodienko Y, Reiser G. Ca2+-dependent permeability transition regulation in rat brain mitochondria by 2′,3′-cyclic nucleotides and 2′,3′-cyclic nucleotide 3′-phosphodiesterase. Am J Physiol Cell Physiol. 2009;296:1428–1439. doi: 10.1152/ajpcell.00006.2009. [DOI] [PubMed] [Google Scholar]
- Beavo JA, Reifsnyder DH. Primary sequence of cyclic nucleotide phosphodiesterase isozymes and the design of selective inhibitors. Trends Pharmacol Sci. 1990;11:150–155. doi: 10.1016/0165-6147(90)90066-H. [DOI] [PubMed] [Google Scholar]
- Bell MJ, Kochanek PM, Carcillo JA, Mi Z, Schiding JK, Wisniewski SR, Clark RS, Dixon CE, Marion DW, Jackson E. Interstitial adenosine, inosine, and hypoxanthine are increased after experimental traumatic brain injury in the rat. J Neurotraum. 1998;15:163–70. doi: 10.1089/neu.1998.15.163. [DOI] [PubMed] [Google Scholar]
- Bell MJ, Robertson CS, Kochanek PM, Goodman JC, Gopinath SP, Carcillo JA, Clark RS, Marion DW, Mi Z, Jackson EK. Interstitial brain adenosine and xanthine increase during jugular venous oxygen desaturations in humans after traumatic brain injury. Crit Care Med. 2001;29:399–404. doi: 10.1097/00003246-200102000-00033. see comment. [DOI] [PubMed] [Google Scholar]
- Chen Y, Balasubramaniyan V, Peng J, Hurlock EC, Tallquist M, Li J, Lu QR. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nature Protocols. 2:1044–51. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
- Dai SS, Zhou YG. Adenosine 2A receptor: a crucial neuromodulator with bidirectional effect in neuroinflammation and brain injury. Rev Neurosci. 2011;22:231–9. doi: 10.1515/RNS.2011.020. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Werner HB, McCulloch MC, Barrie JA, Brown A, Faichney AB, Snaidero N, Nave KA, Griffiths IR. Early ultrastructural defects of axons and axon–glia junctions in mice lacking expression of Cnp1. GLIA. 2009;57:1815–1824. doi: 10.1002/glia.20893. [DOI] [PubMed] [Google Scholar]
- Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009;111:904–15. doi: 10.1097/ALN.0b013e3181b060f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–65. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Weissmuller T, Mager A, Eckle T. Nucleotide metabolism and cell-cell interactions. Methods Mol Biol. 2006;341:73–87. doi: 10.1385/1-59745-113-4:73. [DOI] [PubMed] [Google Scholar]
- Fern R, Waxman SG, Ransom BR. Modulation of anoxic injury in CNS white matter by adenosine and interaction between adenosine and GABA. J Neurophysiol. 1994;72:2609–2616. doi: 10.1152/jn.1994.72.6.2609. [DOI] [PubMed] [Google Scholar]
- Hagemeyer N, Goebbels S, Papiol S, Kästner A, Hofer S, Begemann M, Gerwig UC, Boretius S, Wieser GL, Ronnenberg A, et al. A myelin gene causative of a catatonia-depression syndrome upon aging. EMBO Mol Med. 2012;4:528–539. doi: 10.1002/emmm.201200230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman JD, Chen CD, Oh SY, Hollander W, Abraham CR. Age-dependent accumulation of ubiquitinated 2′,3′-cyclic nucleotide 3′-phosphodiesterase in myelin lipid rafts. GLIA. 56:118–33. doi: 10.1002/glia.20595. [DOI] [PubMed] [Google Scholar]
- Jackson EK. The 2′,3′-cAMP-adenosine pathway. Am J Physiol Renal. 2011;301:F1160–7. doi: 10.1152/ajprenal.00450.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EK, Gillespie DG. Extracellular 2′,3′-cAMP and 3′,5′-cAMP stimulate proliferation of preglomerular vascular endothelial cells and renal epithelial cells. Am J Physiol Renal Physiol. 2012;303:F954–F962. doi: 10.1152/ajprenal.00335.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EK, Gillespie DG, Dubey RK. 2′-AMP and 3′-AMP inhibit proliferation of preglomerular vascular smooth muscle cells and glomerular mesangial cells via A2B receptors. J Pharmacol Exp Ther. 2011a;337:444–450. doi: 10.1124/jpet.110.178137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EK, Raghvendra DK. The extracellular cyclic AMP-adenosine pathway in renal physiology. Annu Rev Physiol. 2004;66:571–99. doi: 10.1146/annurev.physiol.66.032102.111604. [DOI] [PubMed] [Google Scholar]
- Jackson EK, Ren J, Cheng D, Mi Z. Extracellular cAMP-adenosine pathways in the mouse kidney. Am J Physiol Renal Physiol. 2011b;301:F565–F573. doi: 10.1152/ajprenal.00094.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EK, Ren J, Gillespie DG. 2′,3′-cAMP, 3′-AMP and 2′-AMP inhibit human aortic and coronary vascular smooth muscle cell proliferation via A2B receptors. Am J Physiol Heart Circ Physiol. 2011c;301:H391–H401. doi: 10.1152/ajpheart.00336.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EK, Ren J, Gillespie DG, Dubey RK. Extracellular 2′,3′-cyclic adenosine 5′-monophosphate is a potent inhibitor of preglomerular vascular smooth muscle cell and mesangial cell growth. Hypertension. 2010a;56:151–158. doi: 10.1161/HYPERTENSIONAHA.110.152454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EK, Ren J, Mi Z. Extracellular 2′,3′-cAMP is a source of adenosine. J Biol Chem. 2009;284:33097–33106. doi: 10.1074/jbc.M109.053876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TC, Verrier JD, Semple-Rowland S, Kumar A, Foster TC. PHLPP1 splice variants differentially regulate AKT and PKC signaling in hippocampal neurons: characterization of PHLPP proteins in the adult hippocampus. J Neurochem. 2010b;115:941–55. doi: 10.1111/j.1471-4159.2010.06984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, Jenkins LW, Clark RSB, Homanics GE, Dixon CE, et al. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2006;26:565–75. doi: 10.1038/sj.jcbfm.9600218. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–74. doi: 10.1038/ng1095. see comment. [DOI] [PubMed] [Google Scholar]
- Lauro C, Cipriani R, Catalano M, Trettel F, Chece G, Brusadin V, Antonilli L, van Rooijen N, Eusebi F, Fredholm BB, et al. Adenosine A1 receptors and microglial cells mediate CX3CL1-induced protection of hippocampal neurons against Glu-induced death. Neuropsychopharmacology. 2010;35:1550–9. doi: 10.1038/npp.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109:366–98. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Myllykoski M, Itoh K, Kangas SM, Heape AM, Kang SU, Lubec G, Kursula I, Kursula P. The N-terminal domain of the myelin enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase: direct molecular interaction with the calcium sensor calmodulin. J Neurochem. 2012;123:515–24. doi: 10.1111/jnc.12000. [DOI] [PubMed] [Google Scholar]
- Pedraza CE, Monk R, Lei J, Hao Q, Macklin WB. Production, characterization, and efficient transfection of highly pure oligodendrocyte precursor cultures from mouse embryonic neural progenitors. GLIA. 2008;56:1339–52. doi: 10.1002/glia.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao F, Qi Y, Murugan E, Pasunooti S, Ji Q. 2′,3′-cAMP hydrolysis by metal-dependent phosphodiesterases containing DHH, EAL, and HD domains is non-specific: Implications for PDE screening. Biochem Biophys Res Commun. 2010;398:500–5. doi: 10.1016/j.bbrc.2010.06.107. [DOI] [PubMed] [Google Scholar]
- Sprinkle TJ. 2′,3′-cyclic nucleotide 3′-phosphodiesterase, an oligodendrocyte-Schwann cell and myelin-associated enzyme of the nervous system. Crit Rev Neurobiol. 1989;4:235–301. [PubMed] [Google Scholar]
- Stone TW, Ceruti S, Abbracchio MP. Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handb Exp Pharm. 2009;193:535–587. doi: 10.1007/978-3-540-89615-9_17. [DOI] [PubMed] [Google Scholar]
- Thompson JE, Venegas FD, Raines RT. Energetics of catalysis by ribonucleases: fate of the 2′,3′-cyclic phosphodiester intermediate. Biochemistry. 1994;33:7408–14. doi: 10.1021/bi00189a047. [DOI] [PubMed] [Google Scholar]
- Tofovic SP, Branch KR, Oliver RD, Magee WD, Jackson EK. Caffeine potentiates vasodilator-induced renin release. J Pharmacol Exp Ther. 1991;256:850–860. [PubMed] [Google Scholar]
- Verrier JD, Exo JL, Jackson TC, Ren J, Gillespie DG, Dubey RK, Kochanek PM, Jackson EK. Expression of the 2′,3′-cAMP-adenosine pathway in astrocytes and microglia. J Neurochem. 2011;118:979–987. doi: 10.1111/j.1471-4159.2011.07392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier JD, Jackson TC, Bansal R, Kochanek PM, Puccio AM, Okonkwo DO, Jackson EK. The brain in vivo expresses the 2′,3′-cAMP-adenosine pathway. J Neurochem. 2012;122:115–25. doi: 10.1111/j.1471-4159.2012.07705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G. Decreased brain levels of 2′,3′-cyclic nucleotide-3′-phosphodiesterase in Down syndrome and Alzheimer's disease. Neurobiol Aging. 2001;22:547–53. doi: 10.1016/s0197-4580(01)00218-4. [DOI] [PubMed] [Google Scholar]
- Walsh MJ, Murray JM. Dual implication of 2′,3′-cyclic nucleotide 3′ phosphodiesterase as major autoantigen and C3 complement-binding protein in the pathogenesis of multiple sclerosis. J Clin Invest. 1998;101:1923–31. doi: 10.1172/JCI1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbarth S, Maker HS, Raes I, Brannan TS, Lapin EP, Lehrer GM. The activity of 2′,3′-cyclic nucleotide 3′-phosphodiesterase in rat tissues. J Neurochem. 1981;37:677–80. doi: 10.1111/j.1471-4159.1982.tb12540.x. [DOI] [PubMed] [Google Scholar]
- Wieser GL, Gerwig UC, Adamcio B, Barrette B, Nave KA, Ehrenreich H, Goebbels S. Neuroinflammation in white matter tracts of Cnp1 mutant mice amplied by a minor brain injury. GLIA. 2013 doi: 10.1002/glia.22480. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. 5′-Nucleotidase: molecular structure and functional aspects. Biochem J. 1992;285:345–65. doi: 10.1042/bj2850345. [DOI] [PMC free article] [PubMed] [Google Scholar]