

Abstract
Acute hypoxia depolarizes carotid body chemoreceptor (glomus) cells and elevates intracellular Ca2+ concentration ([Ca2+]i). Recent studies suggest that AMP-activated protein kinase (AMPK) mediates these effects of hypoxia by inhibiting the background K+ channels such as TASK. Here we studied the effects of modulators of AMPK on TASK activity in cell-attached patches. Activators of AMPK (1 mM AICAR and 0.1–0.5 mM A769662) did not inhibit TASK activity or cause depolarization during acute (10 min) or prolonged (2–3 hr) exposure. Hypoxia inhibited TASK activity by ~70% in cells pretreated with AICAR or A769662. Both AICAR and A769662 (15–40 min) failed to increase [Ca2+]i in glomus cells. Compound C (40 µM), an inhibitor of AMPK, showed no effect on hypoxia-induced inhibition of TASK. AICAR and A769662 phosphorylated AMPKα in PC12 cells, and Compound C blocked the phosphorylation. Our results suggest that AMPK does not affect TASK activity and is not involved in hypoxia-induced elevation of intracellular [Ca2+] in isolated rat carotid body glomus cells.
Keywords: Hypoxia, Carotid body, Chemoreceptors, AMP kinase, Background K+ channels
1. Introduction
In carotid body glomus cells, hypoxia inhibits the outward K+ current, and thereby causes cell depolarization, Ca2+ influx via voltage-dependent Ca2+ channels and secretion of transmitters (Ortega-Saenz et al., 2007; Peers et al., 2010; Prabhakar, 2006). The hypoxia-sensitive K+ current in glomus cells is believed to consist mainly of Kv, BK and TASK (TASK-1, TASK-3 and TASK-1/3) channels, but the signaling pathways by which hypoxia inhibits each of these K+ channels are not well defined. Several mechanisms for hypoxia-induced inhibition of K+ current have been proposed, including inhibition of heme-oxygenase-2 (Williams et al., 2004), inhibition of mitochondrial oxidative phosphorylation (Buckler and Vaughan-Jones, 1998; Wyatt and Buckler, 2004), and an undefined rotenone-sensitive pathway (Ortega-Saenz et al., 2003). It may be that different O2 sensors and signals are involved in the inhibition of specific K+ channels, but this remains to be determined. The inhibition of mitochondrial oxidative phosphorylation is probably responsible for the hypoxia-induced reduction of TASK, as mitochondrial inhibitors and uncouplers reversibly inhibit these two-pore domain background K+ channels (Buckler, 2007, 2012; Kim, 2013).
Inhibition of mitochondrial oxidative phosphorylation results in the reduction of ATP production and rise in [ADP]/[ATP] ratio. Adenylate kinase converts ADP to AMP and ATP, which causes an increase in cell [AMP]/[ATP] ratio (Oakhill et al., 2011). Increases in both [ADP]/[ATP] and [AMP]/[ATP] ratios have been shown to stimulate AMP-activated protein kinase (AMPK) to regulate cell energy consumption (Hardie and Carling, 1997; Steinberg and Kemp, 2009). In glomus cells, AICAR, a well-known activator of AMPK, was found to inhibit the outward whole-cell K+ current sensitive to iberiotoxin, suggesting that BK was a target of AMPK (Wyatt et al., 2007). In the same study, AICAR caused cell membrane depolarization, elevated intracellular calcium concentration ([Ca2+]i) in glomus cells and increased the carotid sinus nerve discharge in carotid body-sinus nerve preparations. AICAR also inhibited a Ba2+-sensitive, voltage-independent K+ current, suggesting that a background K+ current was also targeted by AICAR (Wyatt et al., 2007). In support of this finding, AICAR inhibited TASK-3 expressed in HEK293 cells by ~50%, and this inhibition was blocked by Compound C, an inhibitor of AMPK (Dallas et al., 2009). These findings have led to the hypothesis that AMPK mediates the hypoxia-induced excitation of glomus cells by inhibition of K+ channels such as BK and TASK that are both well expressed in glomus cells (Peers et al., 2010; Wyatt et al., 2007).
In the course of our studies to identify the effect of phosphorylation by AMPK on TASK single channel behavior and potential amino acid residues involved, we tested the effect of AICAR on TASK single channel kinetics to confirm its inhibitory action. Our preliminary tests using cell-attached patches showed no effect of AICAR on TASK function in glomus cells or in COS-7 cells expressing TASK-3. As our findings are in direct contradiction to the proposal that AMPK inhibits TASK and mediates the hypoxia-induced excitation of glomus cells, we further investigated the effects of AMPK on hypoxia-induced inhibition of TASK and intracellular [Ca2+]i in glomus cells. TASK channel activity in cell-attached patches and intracellular [Ca2+]i were recorded in response to modulators of AMPK. Consistent with our preliminary observation, AMPK activators failed to inhibit TASK, and hypoxia still inhibited TASK and produced strong depolarization even after blockade of AMPK with Compound C. Furthermore, AMPK activators failed to produce depolarization or an increase in [Ca2+]i in glomus cells. Our results show that AMPK is unlikely to be a signal for hypoxia-induced inhibition of TASK and depolarization in isolated rat glomus cells.
2. Methods
2.1. Cell isolation
Rats (postnatal 14–18 day; Sprague-Dawley) were anesthetized with isoflurane and used according to the animal protocols approved by the Animal Care and Use Committees of Rosalind Franklin University and University of Arkansas for Medical Sciences. The carotid bodies were removed and placed in icecold low-Ca2+, low-Mg2+ phosphate buffered saline solution (low Ca2+/Mg2+-PBS: 137 mM NaCl, 2.8 mM KCl, 2 mM KH2PO4, 0.07 mM CaCl2, 0.05 mM MgCl2, pH 7.4). Each carotid body was cut into 3–4 pieces and placed in a solution containing trypsin (400 µg /mL) and collagenase (400 µg /mL) in low Ca2+/Mg2+-PBS and incubated at 37°C for 20–25 min. Carotid bodies were gently triturated using a fire polished Pasteur pipette to mechanically dissociate the cells. Growth medium (Ham’s F-12, 10% fetal bovine serum, 23 mM glucose, 2 mM L-glutamine, 10K units penicillin/streptomycin, and 300 µg/ml insulin) was added to stop enzyme activity. After brief trituration, the solution containing the digested carotid bodies was centrifuged for 4 min at ~6000 rpm (~2000×g) using a microcentrifuge. Supernatant was removed and warm growth medium added to gently resuspend the pellet. Suspended cells were placed on glass coverslips coated with poly-L-lysine, and incubated at 37°C in a humidified atmosphere of 95% air-5% CO2. Cells were used within 6 hr.
2.2. Electrophysiological studies
Electrophysiological recording was performed using a patch clamp amplifier (Axopatch 200B, Molecular Devices, Sunnyvale, CA). Patches were formed using borosilicate glass pipettes with 3–5 megaohm tip resistance. The pipette solution contained (mM) 150 KCl, 1 MgCl2, 5 EGTA, 10 glucose and 10 HEPES (pH 7.3), and the bath perfusion solution contained (mM) 117 NaCl, 5 KCl, 23 NaHCO3, 1 CaCl2, 1 MgCl2 and 11 glucose (pH 7.3). Channel current was filtered at 3 kHz using 8-pole Bessel filter (-3 dB; Frequency Devices, Haverhill, MA) and transferred to a computer using the Digidata 1320 interface at a sampling rate of 20 kHz. Single-channel currents were analyzed with the pCLAMP program (Version 10). Channel openings were analyzed to obtain channel activity (NPo, where N is the number of channels in the patch, and Po is the open probability of a channel). NPo was determined from 15–30 s of current recordings. Because glomus cells express both ~16-pS (TASK-1) and ~35-pS (TASK-3 and TASK-1/3) channels, analysis was done to detect all three isoforms by setting the open levels as multiples of ~16-pS channel. Single-channel current tracings shown in figures were filtered at 1 kHz. All electrophysiological experiments were performed at ~35°C.
2.3. [Ca2+]i measurement
[Ca2+]i was measured by quantitative fluorescence imaging using the calcium-sensitive dye fura-2. Cells plated on the coverslip were incubated with 4 µM fura-2 acetoxymethyl ester (fura-2 AM; Molecular Probes) for 30 min at 37 °C. Fura-2 fluorescence emission was measured at 510 nm in response to alternating excitation at 340 and 380 nm. Images were acquired and stored using a NIKON Eclipse TE300 microscope (with 40x oil immersion objective) and CCD (CoolSNAP HQ2) camera under computer control (MetaFluor: Molecular Devices). For each coverslip, the background light levels were determined and subtracted from each image before measurement of the fluorescence intensity ratio. [Ca2+]i was determined using the 340 nm/380 nm fluorescence ratio as described previously (Wasicko et al., 1999). Calibration was performed using cell-free solutions (Grynkiewicz et al., 1985). The perfusion solution used for [Ca2+] measurements contained (mM): 118 NaCl, 23 NaHCO3, 3 KCl, 1 MgCl2, 1.2 CaCl2, 11 glucose.
2.4. Hypoxia studies
Cell-attached patches were formed on glomus cells and perfused with a bicarbonate-buffered solution containing 117 mM NaCl, 5 mM KCl, 23 mM NaHCO3, 1 mM MgCl2 and 11 mM glucose, and bubbled with 5% CO2/95% air mixture (normoxia) for ~60 min. After steady state channel activity was obtained, the perfusion solution was switched to solution bubbled vigorously (for at least 60 min at 37°C) with 5% CO2/95% N2 mixture (hypoxia) for desired period of time. The pipette solution contained (mM) 150 KCl, 1 MgCl2, 5 EGTA, 10 glucose and 10 HEPES (pH 7.3). The temperature of the perfusion solutions was kept at ~35°C, and the cells were perfused at a rate of ~2.2 ml/min. O2 pressure of the solutions was determined using an oxygen meter (ISO2, WPI, Sarasota, USA) that was calibrated to 0% with solution gassed with pure nitrogen for 60 min and to 21% with solution gassed with air for 60 min at 35°C. The O2 partial pressure as judged by the reading on the meter for the hypoxic solution inside the recording chamber used in this study was ~2% (range: 15–18 mmHg O2).
2.5. Western blot analysis
For isolation of total protein, PC12 cells derived from a pheochromocytoma of the rat adrenal medulla were homogenized in a protein extraction solution (PRO-PREP™; iNtRON Biotechnology, Korea) containing 50 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1 mM dithiothreitol (DTT), 0.5% NP-40, 1% Triton X-100, 1% deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 0.1% EDTA, 1 µM leupeptin, 1 µM pepstatin A, 1 mM phenylmethylsulfonyl fluoride, and 0.1 µM aprotinin. The homogenates were incubated for 60 min on ice with intermittent vortexing, and then centrifuged at 13,000 rpm for 20 min at 4°C. The resulting supernatant was separated on an 8% SDS-polyacrylamide gel and then transferred to a polyvinylidene fluoride (PVDF) membrane for 40 min using a semi-dry transfer (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 3% fat-free dry milk and then incubated with anti-AMPK, anti-phospho (p)-AMPK (1:1000 dilution), and anti-α-tubulin (1:1000 dilution) antibodies. These were followed by incubation with a horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody at a 1:1000 dilution (Assay Designs, Ann Arbor, MI, USA). Immuno-positive bands were visualized by enhanced chemiluminescence (ECL Western Blotting Substrate; Promega, Madison, Wl, USA), following the manufacturer’s instructions. Anti-AMPK and anti-p-AMPK antibodies were purchased from Cell Signaling Technology® (Danvers, MA, USA).
2.6. Statistical analysis
Student’s t-test (for comparison of two sets of data) and one-way analysis of variance (for comparison of three data sets) were used. Data were analyzed using PRISM software and represented as mean±SD, unless indicated otherwise. Post hoc testing was based on unpaired t-test with Bonferroni correction. Significance level was set at p < 0.05.
2.7. Materials
AICAR (5-aminoimidazole-4-carboxamide riboside), A769662 and Compound C were purchased from Tocris. All other chemicals were purchased from Sigma-Aldrich Co.
3. Results
3.1. AICAR and A769662 do not affect TASK activity in cell-attached patches
In our recent study, biophysical and pharmacological studies showed that TASK was the primary K+ channels that was open at rest in cell-attached patches of glomus cells (Kim et al., 2009). In this study, TASK was clearly identified by the single channel conductance level (~34-pS) and short mean open time (~1 ms), and was recorded without applying any potential to the pipette (i.e., a pipette potential of 0 mV). Under this condition, single channel openings of TASK current can be recorded, because of the negative resting Em (approximately −60 mV) of glomus cells in the physiological bath perfusion solution (Fig. 1A).
Figure 1. No effect of AICAR and A769662 on TASK activity in glomus cells.
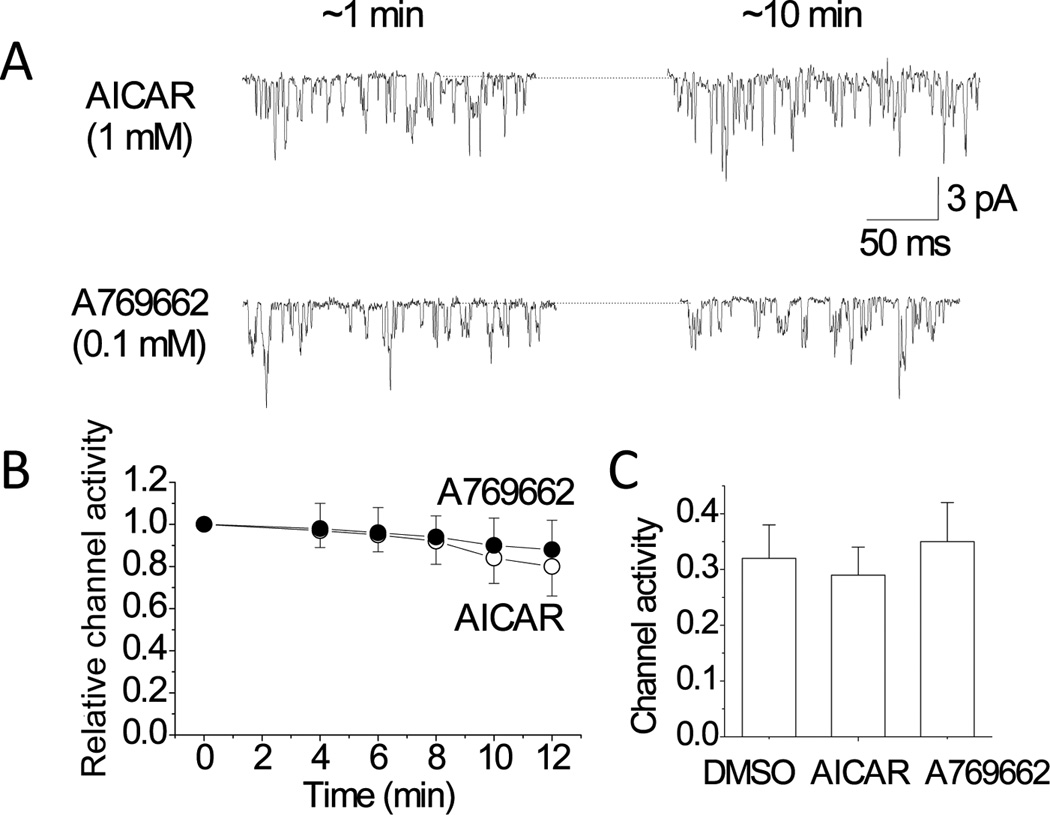
A. Cell-attached patches were perfused with solution 1 mM AICAR or 0.1 mM A769662 until patches broke or up to 12 min. Pipette potential was set at 0 mV. Representative single-channel currents at ~1 min and ~10 min following application of the drugs are shown.
B. TASK activity (NPo) was determined at various time points and plotted as a function of perfusion time. TASK activity was calculated and normalized to that obtained at time zero (1.0). Each point is the mean±SD of 12 determinations from 6 separate cell preparations.
C. TASK activity in cell-attached patches was determined from cells incubated with 0.1% DMSO, 1 mM AICAR or 0.1 mM A769662 for 2–3 h. Each bar is the mean±SD of 12 patches.
If the inhibition of TASK activity by hypoxia is mediated by AMPK, activators of AMPK are expected to inhibit TASK activity. Therefore, the effect of AMPK activation on TASK activity was tested using AICAR and A769662, which are membrane permeable activators of AMPK (Corton et al., 1995; Goransson et al., 2007). Switching of the perfusion solution to that containing 0.1% DMSO showed no significant effect on TASK activity during ~10 min of perfusion in all 7 cells tested (data not shown). Perfusion of cell-attached patches with solution containing 1 mM AICAR or 0.1 mM A769662 also did not produce significant changes in channel activity for the duration of ~10 min (Fig. 1).
When glomus cells depolarize, the single channel amplitude decreases simultaneously, due to the reduced membrane potential difference across the cell-attached patch membrane (Kim et al., 2011). The mean amplitude levels of TASK single channels were not significantly changed (2.0±0.2 pA for AICAR and 1.9±0.2 pA for A769662) from control (2.1±0.2-pA), indicating that the resting Em was unaffected by these drugs. This is also consistent with the lack of effect of AICAR and A769662 on TASK activity. Thus, neither of the two AMPK activators caused depolarization of glomus cells.
It was possible that a ~10 min exposure to AICAR or A769662 was not sufficient to cause activation of AMPK. To test the effect of a longer exposure to AMPK activators, glomus cells were incubated with AICAR, A769662 or 0.1% DMSO (control) at 37°C for 2–3 hr before forming cell-attached patches in perfusion solution containing the same activator. Comparison of TASK activity in control, AICAR-treated and A769662-treated glomus cells showed no significant difference among the three groups (Fig. 1C). These results show that activators of AMPK do not cause inhibition of TASK activity recorded from cell-attached patches of isolated glomus cells even after a long period of exposure to the drugs.
3.2. AMPK modulators do not affect hypoxia-induced inhibition of TASK activity
The finding that AMPK activators do not affect TASK activity suggests that hypoxia should still able to inhibit TASK in glomus cells treated with AMPK activators. We tested the effect of hypoxia on TASK activity after a ~2–3 hr incubation of glomus cells with AICAR and A769662. Hypoxia produced a reversible inhibition of TASK activity in glomus cells incubated with 0.1% DMSO for ~3 hr (Fig. 2A). In glomus cells treated with AICAR and A769662 for 2–3 hr, hypoxia also produced a reversible inhibition of TASK activity that was similar in magnitude to those observed in glomus cells incubated with 0.1% DMSO (Fig. 2B and 2C). In glomus cells incubated with Compound C (40 µM), a well-known inhibitor of AMPK activity, hypoxia was still able to produce a strong inhibition of TASK activity (Fig. 2D). These findings show that AMPK is unlikely to be involved in hypoxia-induced inhibition of TASK activity in isolated glomus cells.
Figure 2. AMPK modulators do not affect the hypoxia-induced inhibition of TASK activity.
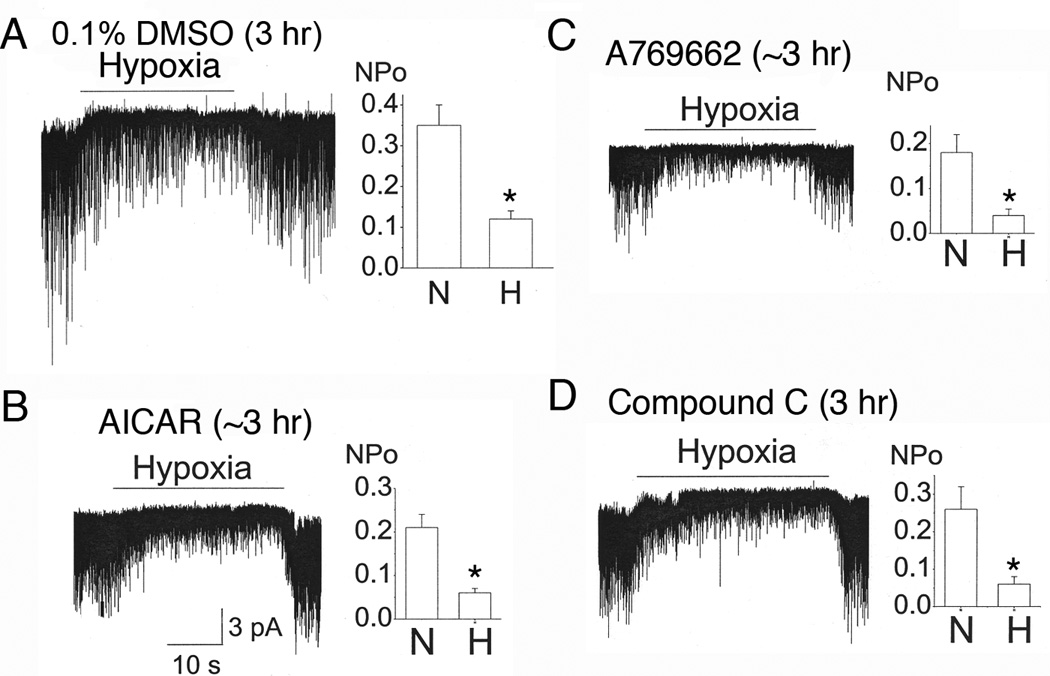
A. Cell-attached patches were perfused with normoxic solution, then with hypoxia solution for ~30 sec, and then back to normoxic solution. Glomus cells were incubated for 2–3 h with 0.1% DMSO.
B-C. Same experiment as in A except that glomus cells were incubated with 1 mM AICAR (B) or A760662 (C) for 2–3 h before recording TASK in cell-attached patches. Perfusion solutions contained either AICAR or A769662.
D. Glomus cells were incubated with 40 µM Compound C for 2–3 hr. Cell-attached patches were then formed and perfused with normoxic and hypoxic solution containing Compound C. For all bar graphs (A-D), each bar is the mean±SD of 6–8 patches. Asterisk (*) indicates a significant reduction from control (normoxia) at p < 0.05. For all experiments in this figure, data were gathered from 4 separate cell preparations.
3.3. Phosphorylation of AMPKα by AICAR in PC12 cells
Activation of AMPK involves its phosphorylation at Thr172 due to binding of AMP to the γ subunit of AMPK. To confirm that AICAR and A769662 used in our experiments indeed caused phosphorylation of AMPK, we incubated PC12 cells with 1 mM AICAR or 100 µM A769662 for 1 hr, and then assessed the level of phosphorylation by Western blot analysis. As shown in Fig. 3, AMPKα was basally phosphorylated, and AICAR and A769662 increased the level of phosphorylation by 3.3±1.5-fold and 2.1±0.5-fold, respectively. Compound C (40 µM) blocked AICAR- and A769662- induced increases in phosphorylation. Although Western blot analyses could not be performed in glomus cells due to technical difficulties (i.e., glomus cell number is very low and cell preparations contain other types of cells), the results suggest that AICAR and A769662 used in our experiments are pharmacologically active, and able to phosphorylate AMPK in isolated cells.
Figure 3. Phosphorylation of AMPKα by AICAR and A769662.
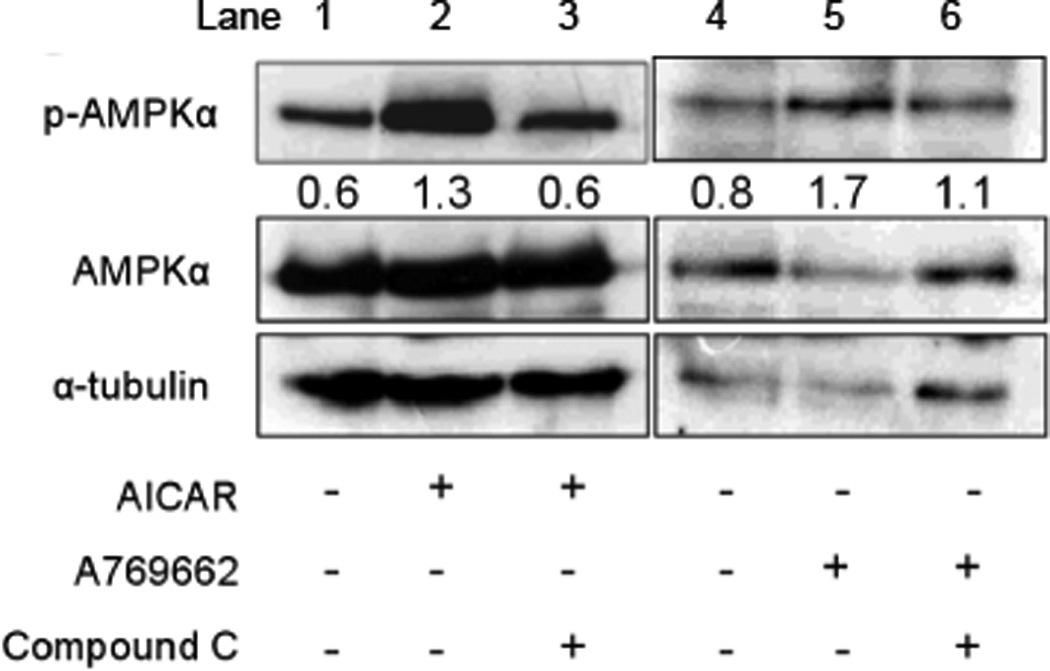
PC12 cells were treated with 1 mM AICAR or 100 µM A769662 for 60 min. Compound C (40 µM) was pretreated for 30 min to block AMPK activation. Activated AMPK (p-AMPKα) was detected in PC12 cells using a specific antibody for the phosphorylated form of AMPKα. Whole-cell lysate was used as total protein. Equal amounts (30 µg) of total protein were loaded in each lane. The plus and minus signs (+ and −) represent treatment conditions with and without chemical, respectively. The bands obtained from Western blot were quantified by Quantity One software (version 4.6.3) attached to GS-800 Calibrated densitometer (Bio-Rad). The numbers below the pAMPKα band represent the ratio of the expression levels of p-AMPKα to those of AMPKα for each lane. For example, in lanes 1and 4 (control), the level of phosphorylated p-AMPKα compared to that of AMPKα is 0.6 and 0.8, respectively. AICAR or A769662 increased the fractional levels of AMPKα phosphorylation (lanes 2 and 5) and Compound C blocked this effect (lanes 3 and 6). The α-tubulin was used as a loading control. The size of molecules: p-AMPKα (62 kDa), AMPKα (62 kDa), and α-tubulin (50 kDa). The experiment was done three times and an image from one experiment is shown.
3.4. AICAR and A769662 do not elevate intracellular [Ca2+] in glomus cells
Earlier studies have reported that AICAR depolarizes glomus cells and elevates [Ca2+]i (Wyatt et al., 2007). Because AICAR did not inhibit TASK activity in our glomus cell preparations, we re-assessed the effects of AICAR and A769662 on [Ca2+]i. Cells were perfused with bicarbonate-buffered solution containing 5 mM KCl and bubbled with 95% air/5% CO2 (control normoxic solution). Perfusion of glomus cells with solution containing 20 mM KCl or solution bubbled with 95% N2/5% CO2 (hypoxic solution) elicited a rapid rise in [Ca2+]i that quickly returned to the basal level when cells were reperfused with the control solution (Fig. 4A and 4B). Cells perfused with control normoxic solution containing either AICAR (1 mM) (Fig. 4A) or A769662 (0.1 mM) (Fig. 4B) showed no significant increase in [Ca2+]i for 15 min, changing only by 9.88 ± 4.04 nM and −3.98 ± 3.85 nM, respectively (p > 0.05). The same cells responded well to 20 mM KCl, hypoxia with 0.1 mM A769662, and hypoxia alone (after washout of 0.1 mM A769662) showing [Ca2+]i increases of 238.0 ± 15.5 nM, 243.9 ± 70.3 and 221.7 ± 21.7 nM, respectively (p > 0.05) (Fig. 4C). Compound C (40 µM) in presence of AICAR showed no significant effect on [Ca2+]i (p > 0.05) (Fig. 4C). These results suggest that AMPK activation does not produce an increase in [Ca2+]i.
Figure 4. AICAR and A769662 do not elevate intracellular [Ca2+].

A. Representative averaged trace of changes in [Ca2+]i in glomus cells in response to 20 mM KCl, hypoxia, AICAR (1 mM), and AICAR (1 mM) + Compound C (40 µM) are shown.
B. Representative averaged trace of changes in [Ca2+]i in glomus cells in response to 20 mM KCl, hypoxia, A769662 (0.1 mM), and A769662 (0.1 mM) + 0% O2 are shown.
C. Averaged [Ca2+]i increase above the basal level in response to 20 mM KCl (n=15), 0% O2 (n=15), 1 mM AICAR (n=15), AICAR (1 mM) + Compound C (40 µM) (n=15), A769662 (0.1 mM) (n=25), and A769662 (0.1 mM) + 0% O2 (n=8). Each bar represents the mean±SE. Asterisk(*) indicates significant difference at p < 0.001. NS= not significant.
D-F. Glomus cells were incubated in medium containing 1 mM AICAR (D), 0.5 mM A769662 (E) or 0.5% DMSO (F) for 30 min at 37°C before recording [Ca2+]. Representative averaged traces of changes in [Ca2+]i in response to hypoxia and 20 mM KCl are shown. For these experiments, cells from 3 separate preparations were used.
Again, there was a possibility that the duration of incubation of cells with AICAR and A769662 was too short to cause sufficient activation of AMPK in glomus cells. Therefore, glass coverslips with attached cells were incubated in the cell culture medium that contains 1 mM AICAR and 0.5 mM A769662 for 30 min at 37°C, and then placed into the recording chamber that already contains the same concentration of the drug. After 30 min of exposure to AICAR in 0.1% DMSO, the levels of [Ca2+]i in glomus cells were low (< 100 nM) and remained low for another 5–10 min (Fig. 4D). The Δ[Ca2+]i response to 0% O2 after 40 min of AICAR exposure was 264.8 ± 22.9 nM (n=15) vs. 330.0 ± 37.3 nM (n=15) approximately 10 min after drug and DMSO washout (NS). Subsequent perfusion with 20 mM KCl solution quickly and reversibly elevated [Ca2+]i (Fig 4D).
In glomus cells perfused with 500 µM A769662 for 30 min, the levels of [Ca2+]i were low (< 100 nM) at the beginning of the measurement and remained low for another 5–10 min in the presence of the drug (Fig. 4E). In these cells exposed to A769662 for 40 min, [Ca2+]i increased to 273.1 ± 20.9 nM (n=8) in response to a brief exposure to 0% O2. When the same cells were perfused with drug-free, control solution for 10 min, the [Ca2+]i response to 0% O2 was significantly higher (406.2 ±27.5 nM; n=8) than that obtained in the presence of A769662 (p < 0.05; Fig. 4E). The concentration of A769662 used in figure 4E was five-fold of that used in the experiments of figure 4B and required a higher concentration of DMSO (0.5%) to dissolve the drug. Therefore, we tested the effect of 0.5% DMSO alone on [Ca2+]i response to 0% O2. The increase in [Ca2+]i in response to 0% O2 after a 40 min exposure to 0.5% DMSO alone was 252.8 ± 19.3 nM (n=28). After washout of DMSO for 10 min, the increase in [Ca2+] response to 0% O2 was 352.9 ± 21.3 nM (n=28; p < 0.05), suggesting that a high concentration of DMSO (0.5%) causes an inhibition of the [Ca2+]i response to hypoxia (Fig. 4F). Thus, after prolonged exposure to AICAR and prolonged exposure to a five-fold higher concentration of A769662, baseline [Ca2+]i remained low and glomus cells retained a brisk [Ca2+]i response to hypoxia. Together, the results of our experiments suggest that AMPK does not mediate the hypoxia-induced increase in [Ca2+]i.
4. Discussion
One of the important questions regarding the mechanism of O2 sensing by chemoreceptors relates to how hypoxia inhibits K+ channels to cause excitation of glomus cells. Various signals such as reactive oxygen species, carbon monoxide and hydrogen sulfide have been proposed, but the pathway involving AMPK has been gaining attention. The reason for this is that AICAR was found to inhibit the outward K+ current and also inhibit the hypoxia-induced elevation of [Ca2+]i. Furthermore, Compound C was found to block these effects of AICAR (Wyatt et al., 2007). Thus, available evidence supports the role of AMPK as a major signaling component of hypoxia-induced excitation of glomus cells.
TASK is an O2-sensitive background K+ channel whose inhibition is believed to cause depolarization of rat glomus cells. Our initial goal was to study the effect of AMPK-induced phosphorylation of TASK on channel kinetics, and therefore to provide additional proof in support of the role of AMPK in TASK inhibition using a method of assessing the background K+ current different from that used earlier (Wyatt et al., 2007). To our surprise, our findings were not consistent with the AMPK hypothesis, and we are led to conclude that an AMPK-independent mechanism is involved in hypoxia-induced inhibition of TASK in glomus cells. At present, we do not have evidence for another signaling pathway for the hypoxia-induced excitation of glomus cells. Nevertheless, we believe that our finding of the lack of effect of AMPK on glomus cell TASK channel activity and [Ca2+]i is important for carotid body oxygen sensing research, and should help to better focus our efforts on other mechanisms.
4.1. Activators and inhibitor of AMPK have no effect on TASK
AICAR activates AMPK after conversion in the cell to an AMP-mimetic molecule (5-aminoimidazole-4-carboxamide-1-D-ribofuranosyl or AICAR monophosphate), whereas A769662 activates AMPK directly (Goransson et al., 2007). Both AICAR (1 mM) and A769662 (0.1 mM) inhibited BK expressed in HEK293 cells (Ross et al., 2011). The maximal effect of A769662 on BK occurred within 2–3 min, compared to ~8 min with AICAR. Regardless of the time of onset of their action, our data show that the two AMPK activators that stimulate AMPK via different mechanisms have no significant effect on TASK activity in isolated glomus cells. At the whole-cell level, AICAR was found to inhibit a Ba2+-sensitive K+ current that was assumed to be TASK-like (Wyatt et al., 2007). However, TASK is only partially sensitive to Ba2+, and thus the molecular identities of the K+ channels that gave rise to the small Ba2+-sensitive K+ current are not clearly defined. It could consist of several K+ channels, including TASK-1/3, BK, HERG, Kv and other background K+ channels such as TASK-2 and TREK (Kim, 2013). We believe that the use of cell-attached patches that contain only TASK channels represents another useful method for studying the modulation of TASK activity in intact cells.
The lack of effect of AICAR and A769662 on TASK activity cannot be due to their inability to diffuse through the plasma membrane, as the two agents are highly membrane-permeable molecules that have been used in many studies to activate AMPK (Guo et al., 2009; Mace et al., 2008). We think that there is no particular reason that AICAR and A769662 would not diffuse into isolated glomus cells, as other membrane-permeable DMSO-soluble molecules (such as methanandamide) can easily enter these cells. Although the ~10 min treatment of glomus cells with these agents may be considered a short duration, the 2- to 3-hr treatment should have definitely stimulated AMPK, as both AICAR and A769662 phosphorylated AMPKα in PC12 cells after 1 hr of treatment. We also found phosphorylation of AMPKα after 30 min of treatment in all three trials.
It remains possible that AICAR and A769662 entered the glomus cells but failed to stimulate AMPK in these cells. We were not able to test the effect of these two agents on phosphorylation of AMPK in the carotid body due to the small tissue size. Even if the phosphorylation assay shows a positive response in carotid bodies, the results would be ambiguous because the carotid body contains many different types of cells. Isolation of pure glomus cells for phosphorylation assay is not feasible at present. We performed immunocytochemistry in isolated glomus cells using a commercially available antibody to the phosphorylated form of AMPKα, but the results were not clear due to non-specific binding.
Additional evidence that AMPK does not affect TASK is the strong inhibition of TASK by hypoxia in glomus cells incubated with Compound C, which blocks the phosphorylation of AMPKα by AICAR. Although Compound C is a highly non-specific drug that inhibits many kinases, it has been shown to inhibit AMPK with a K1/2 of ~0.2 µM in a biochemical assay (Bain et al., 2007). Our phosphorylation experiments clearly show that incubating cells with Compound C (40 µM) blocks AICAR-induced phosphorylation of AMPK. We cannot prove at this time that AICAR and Compound C actually altered the phosphorylated state of AMPK in our glomus cell preparations. However, it seems very likely that Compound C at this high concentration blocked AMPK activity in all types of cells, especially after several hours of incubation. Nevertheless, there remains a possibility that AICAR and A769662 did not cause phosphorylation of AMPK in our glomus cell preparations and therefore did not affect TASK activity and [Ca2+].
In an earlier study, whole-cell currents of TASK-1, TASK-3 and TASK-1/3 expressed in HEK293 cells were unaffected by 2 mM AICAR (Kreneisz et al., 2009), consistent with our findings in glomus cells. In previous work, when glomus cells were dialyzed with a pipette solution containing millimolar ATP, hypoxia could still reduce the whole-cell K+ current (Lopez-Barneo et al., 1988). It is likely that the [AMP]/[ATP] ratio would remain relatively constant or only briefly altered in these cells due to continuous dialysis of the cell interior with the pipette solution. Such consideration also suggests that AMPK is unlikely to be involved in the hypoxia-induced inhibition of K+ current, although the degree of contribution by TASK to the whole-cell current has not been determined.
4.2. Modulators of AMPK do not affect resting Em and [Ca2+]i
AICAR has been reported to depolarize glomus cells and elevate [Ca2+]i within ~10 min after treatment (Wyatt et al., 2007). Despite our repeated efforts, we found no significant depolarization by AICAR and A769662 in glomus cells even after 2–3 hr of treatment. The depolarization of glomus cells produced by AICAR observed earlier has been attributed mainly to inhibition of BK (Wyatt et al., 2007). Therefore, the lack of BK activity in our glomus cells at rest could explain why AICAR failed to depolarize the cells. If so, our finding that hypoxia can strongly depolarize and elevate [Ca2+]i, either in the presence or absence of AMPK activators, provide evidence that TASK is a major target of hypoxia, at least in our glomus cell preparations that show little or no BK activity at rest. To resolve the question of the role of AMPK in hypoxia-induced depolarization and changes in [Ca2+]i, it may be necessary to determine what causes BK to be active at rest in some studies and not active in others.
It is truly difficult to know why there is a discrepancy in the results from our study and that of Wyatt et al., particular when the cells are from the same species and cell preparations seem similar. As described below, we considered further the following potential reasons for the different findings on the role of AMPK on glomus cell O2 sensing. (a) Different strains of rat: we used the Sprague-Dawley strain, but Wyatt et al. did not mention the rat strain in their paper. Therefore, we cannot judge whether the difference is due to the strain of the rat used in the study. (b) Age of rats: we used 14–18 day old rats. Wyatt et al. used 10–21 day old rats. Therefore, the age of the rats should not be a factor. (c) Recording of channel current and [Ca2+]: for electrophysiology, we used cell-attached patches whereas Wyatt et al. used whole-cell current recording. Hypoxia produced clear and strong inhibition of TASK in cell-attached patches near the resting Em, but AICAR and A769662 showed no effect on TASK (our study). Under whole-cell conditions (Wyatt et al. study), AICAR reduced the current near the resting Em (-100 mV to −40 mV) by ~1–2 pA, but the identity of the channel that was inhibited is not known. (d) Pipette/perfusion solutions (electrophysiology): For bath perfusion solution, we used HCO3−, whereas Wyatt et al. used HEPES as the buffering chemical. We do not think this should make a difference. If anything, we should see a stronger response in HCO3−-buffered solution than in HEPES-buffered solution, because earlier work showed that HCO3− is important for O2 sensing by carotid body glomus cells (Panisello and Donnelly, 1998; Shirahata and Fitzgerald, 1991). With cell-attached patches, the cell interior is maintained unchanged and not dialyzed as in whole-cell current recording. Although this could potentially affect the responses of glomus cells to hypoxia and AMPK activators, the cell-attached method is more “undisturbed and intact” than whole-cell conditions.
For [Ca2+]i experiments, we do not see methodological differences, and thus cannot offer an explanation as to why the results are different. In the present study, we were not able to examine the effects of compound C on the hypoxia response because pilot experiments on the glomus cell response to hypoxia revealed artifact when using compound C with Fura-2. In dissociated glomus cells, at the excitation wavelengths used for Fura-2, compound C shows strong autofluorescence when excited by both 340 nm and 380 nm UV light. This has a large, artifactual effect on the 340/380 fluorescence ratio, and markedly reduces the ratio whenever compound C is present, leading to erroneous [Ca2+]i measurements (Kim et al., 2013). Wyatt et al., used Fura-2 for [Ca2+]i measurements but did not discuss the potential for artifact introduced by compound C.
Wyatt et. al. did not mention whether DMSO was used in their study. In our studies, 1 mM AICAR and 0.1 mM A769662 solutions contained 0.1% DMSO, which had no effect on TASK activity. The higher concentration of A769662 used in Fig. 4E required 0.5% DMSO, which may have mildly reduced the [Ca2+]i response to hypoxia. This likely explains the small increase in amplitude of the hypoxia response 10 min after DMSO washout (Fig. 4E and F). Our results clearly show that even after a prolonged exposure to a high concentration of A769662, the baseline [Ca2+]i was unaffected and glomus cells remained briskly responsive to hypoxia. Future research by other laboratories on this topic should hopefully resolve the contradictory findings on the role of AMPK in hypoxia-induced excitation of glomus cells.
4.3. Potential mechanisms for hypoxia-induced inhibition of TASK
If AMPK does not mediate the inhibition of TASK by hypoxia in our glomus cell preparations, then what signal is involved? Hemeoxygenase-2 (HO-2) and its product carbon monoxide (CO) as a potential sensor and signal in hypoxia-induced excitation of glomus cells have been a topic of great interest because CO is an activator of BK (Jaggar et al., 2005; Williams et al., 2004). Thus, a reduction of [CO] by hypoxia would decrease BK activity and cause cell depolarization. At present, the effect of CO on TASK activity is not yet known. Another potential signal is hydrogen sulfide (H2S) that is endogenously formed by cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE). Intracellular [H2S] is increased by hypoxia as a result of reduced mitochondrial breakdown of H2S (Olson, 2011; Prabhakar, 2012). According to recent studies, H2S inhibits BK and TASK, and increases carotid sinus afferent nerve activity (Buckler, 2012; Li et al., 2010; Peng et al., 2010; Telezhkin et al., 2010). Thus, H2S could serve as a hypoxic signal that inhibits TASK. Because H2S is an inhibitor of cytochrome oxidase, it may be reducing the TASK current indirectly via inhibition of mitochondrial oxidative phosphorylation (Buckler, 2012). It has been reported that CO limits the generation of H2S by acting on CSE (Peng et al., 2010). Thus, the interaction between HO-2/CO and CSE/H2S systems during normoxia and hypoxia may be important for regulating the K+ currents such as TASK and BK, as discussed recently (Kemp and Telezhkin, 2013). H2O2 and superoxide radical do not inhibit TASK activity (Papreck et al., 2012), indicating that NADPH oxidase and mitochondrial generation of reactive oxygen species are probably not the signals.
A mechanism proposed by Buckler and colleagues is that the reduction of [ATP]i during acute hypoxia is responsible for the decrease in TASK activity (Varas et al., 2007; Williams and Buckler, 2004). This mechanism is quite plausible because TASK activity is dependent on cytosolic [ATP]. Thus, in inside-out patches when ATP is removed, TASK activity falls markedly, and addition to ATP restores TASK activity (Williams and Buckler, 2004). Currently, it is not possible to test whether [ATP] near the cell membrane has indeed decreased sufficiently to reduce TASK activity within a few seconds of hypoxia, and this issue remains unresolved at this time.
AMPK has been proposed as a key mediator of hypoxia-induced excitation of carotid body glomus cells.
Activators of AMPK showed no effect on TASK K+ channel activity, membrane potential and intracellular Ca2+ concentration in isolated glomus cells.
Our results suggest that AMPK does not affect the background K+ current or mediate the hypoxic-induced excitation of isolated carotid body glomus cells
Acknowledgments
This work was funded by grant awards to D. Kim (NIH), J.L. Carroll (NIH) and D. Kang (Korean Research Foundation # 2010-0024258).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. TASK-like potassium channels and oxygen sensing in the carotid body. Respir Physiol Neurobiol. 2007;157:55–64. doi: 10.1016/j.resp.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012;463:743–754. doi: 10.1007/s00424-012-1089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. The Journal of physiology. 1998;513((Pt 3)):819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Dallas ML, Scragg JL, Wyatt CN, Ross F, Hardie DG, Evans AM, Peers C. Modulation of O(2) sensitive K (+) channels by AMP-activated protein kinase. Adv Exp Med Biol. 2009;648:57–63. doi: 10.1007/978-90-481-2259-2_6. [DOI] [PubMed] [Google Scholar]
- Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007;282:32549–32560. doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M, Radu CG, Cloughesy TF, Mischel PS. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci U S A. 2009;106:12932–12937. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circulation research. 2005;97:805–812. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp PJ, Telezhkin V. Oxygen sensing by the carotid body: is it all just rotten eggs? Antioxidants & redox signaling. 2013 doi: 10.1089/ars.2013.5377. [DOI] [PubMed] [Google Scholar]
- Kim D. K(+) channels in O(2) sensing and postnatal development of carotid body glomus cell response to hypoxia. Respir Physiol Neurobiol. 2013;185:44–56. doi: 10.1016/j.resp.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Cavanaugh EJ, Kim I, Carroll JL. Heteromeric TASK-1/TASK-3 is the major oxygen-sensitive background K+ channel in rat carotid body glomus cells. The Journal of physiology. 2009;587:2963–2975. doi: 10.1113/jphysiol.2009.171181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Papreck JR, Kim I, Donnelly DF, Carroll JL. Changes in oxygen sensitivity of TASK in carotid body glomus cells during early postnatal development. Respir Physiol Neurobiol. 2011;177:228–235. doi: 10.1016/j.resp.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Fite JL, Kim D, Donnelly DF, Carroll JL. NAD(P)H autofluorescence induction by compound C in rat carotid chemoreceptor cells. Experimental Biology, 2013;396 (abstract) [Google Scholar]
- Kreneisz O, Benoit JP, Bayliss DA, Mulkey DK. AMP-activated protein kinase inhibits TREK channels. The Journal of physiology. 2009;587:5819–5830. doi: 10.1113/jphysiol.2009.180372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, Rong W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxidants & redox signaling. 2010;12:1179–1189. doi: 10.1089/ars.2009.2926. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Lopez-Lopez JR, Urena J, Gonzalez C. Chemotransduction in the carotid body: K+ current modulated by PO2 in type I chemoreceptor cells. Science. 1988;241:580–582. doi: 10.1126/science.2456613. [DOI] [PubMed] [Google Scholar]
- Mace OJ, Woollhead AM, Baines DL. AICAR activates AMPK and alters PIP2 association with the epithelial sodium channel ENaC to inhibit Na+ transport in H441 lung epithelial cells. The Journal of physiology. 2008;586:4541–4557. doi: 10.1113/jphysiol.2008.158253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- Olson KR. Hydrogen sulfide is an oxygen sensor in the carotid body. Respir Physiol Neurobiol. 2011 doi: 10.1016/j.resp.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Ortega-Saenz P, Pardal R, Garcia-Fernandez M, Lopez-Barneo J. Rotenone selectively occludes sensitivity to hypoxia in rat carotid body glomus cells. The Journal of physiology. 2003;548:789–800. doi: 10.1113/jphysiol.2003.039693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Saenz P, Pascual A, Piruat JI, Lopez-Barneo J. Mechanisms of acute oxygen sensing by the carotid body: lessons from genetically modified animals. Respir Physiol Neurobiol. 2007;157:140–147. doi: 10.1016/j.resp.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Panisello JM, Donnelly DF. Chemotransduction by carotid body chemoreceptors is dependent on bicarbonate currents. Respiration physiology. 1998;112:265–281. doi: 10.1016/s0034-5687(98)00035-8. [DOI] [PubMed] [Google Scholar]
- Papreck JR, Martin EA, Lazzarini P, Kang D, Kim D. Modulation of K2P3.1 (TASK-1), K2P9.1 (TASK-3), and TASK-1/3 heteromer by reactive oxygen species. Pflugers Arch. 2012;464:471–480. doi: 10.1007/s00424-012-1159-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C, Wyatt CN, Evans AM. Mechanisms for acute oxygen sensing in the carotid body. Respir Physiol Neurobiol. 2010;174:292–298. doi: 10.1016/j.resp.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR. O2 sensing at the mammalian carotid body: why multiple O2 sensors and multiple transmitters? Exp Physiol. 2006;91:17–23. doi: 10.1113/expphysiol.2005.031922. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Carbon monoxide (CO) and hydrogen sulfide (H(2)S) in hypoxic sensing by the carotid body. Respir Physiol Neurobiol. 2012 doi: 10.1016/j.resp.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FA, Rafferty JN, Dallas ML, Ogunbayo O, Ikematsu N, McClafferty H, Tian L, Widmer H, Rowe IC, Wyatt CN, Shipston MJ, Peers C, Hardie DG, Evans AM. Selective expression in carotid body type I cells of a single splice variant of the large conductance calcium- and voltage-activated potassium channel confers regulation by AMP-activated protein kinase. J Biol Chem. 2011;286:11929–11936. doi: 10.1074/jbc.M110.189779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahata M, Fitzgerald RS. The presence of CO2/HCO3- is essential for hypoxic chemotransduction in the in vivo perfused carotid body. Brain research. 1991;545:297–300. doi: 10.1016/0006-8993(91)91301-g. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Telezhkin V, Brazier SP, Cayzac SH, Wilkinson WJ, Riccardi D, Kemp PJ. Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir Physiol Neurobiol. 2010;172:169–178. doi: 10.1016/j.resp.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Varas R, Wyatt CN, Buckler KJ. Modulation of TASK-like background potassium channels in rat arterial chemoreceptor cells by intracellular ATP and other nucleotides. The Journal of physiology. 2007;583:521–536. doi: 10.1113/jphysiol.2007.135657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasicko MJ, Sterni LM, Bamford OS, Montrose MH, Carroll JL. Resetting and postnatal maturation of oxygen chemosensitivity in rat carotid chemoreceptor cells. The Journal of physiology. 1999;514((Pt 2)):493–503. doi: 10.1111/j.1469-7793.1999.493ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BA, Buckler KJ. Biophysical properties and metabolic regulation of a TASK-like potassium channel in rat carotid body type 1 cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L221–L230. doi: 10.1152/ajplung.00010.2003. [DOI] [PubMed] [Google Scholar]
- Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- Wyatt CN, Buckler KJ. The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type I cells. The Journal of physiology. 2004;556:175–191. doi: 10.1113/jphysiol.2003.058131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt CN, Mustard KJ, Pearson SA, Dallas ML, Atkinson L, Kumar P, Peers C, Hardie DG, Evans AM. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J Biol Chem. 2007;282:8092–8098. doi: 10.1074/jbc.M608742200. [DOI] [PMC free article] [PubMed] [Google Scholar]