

Abstract
We present Oqtans, an open-source workbench for quantitative transcriptome analysis, that is integrated in Galaxy. Its distinguishing features include customizable computational workflows and a modular pipeline architecture that facilitates comparative assessment of tool and data quality. Oqtans integrates an assortment of machine learning-powered tools into Galaxy, which show superior or equal performance to state-of-the-art tools. Implemented tools comprise a complete transcriptome analysis workflow: short-read alignment, transcript identification/quantification and differential expression analysis. Oqtans and Galaxy facilitate persistent storage, data exchange and documentation of intermediate results and analysis workflows. We illustrate how Oqtans aids the interpretation of data from different experiments in easy to understand use cases. Users can easily create their own workflows and extend Oqtans by integrating specific tools. Oqtans is available as (i) a cloud machine image with a demo instance at cloud.oqtans.org, (ii) a public Galaxy instance at galaxy.cbio.mskcc.org, (iii) a git repository containing all installed software (oqtans.org/git); most of which is also available from (iv) the Galaxy Toolshed and (v) a share string to use along with Galaxy CloudMan.
Contact: vipin@cbio.mskcc.org, ratschg@mskcc.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Technological advance in large-scale sequencing has revolutionized molecular biology. Its application to profiling the transcriptome, the total complement of cellular RNA, called RNA-seq, provides an unmatched dynamic range for expression quantification and single base pair resolution for the discovery of new transcripts (Mortazavi et al., 2008). However, analyzing these complex data to their full potential requires computational frameworks.
Here, we present Oqtans, the online platform for quantitative RNA-seq data analysis (online since 2010). Its integration into the Galaxy framework ensures transparent and reproducible computational analyses. Oqtans provides a Galaxy interface to many recently developed RNA-seq analysis tools, and this way considerably extends the standard repertoire of the Galaxy toolbox (usegalaxy.org). To reach non-expert users and experienced developers, we provide the Oqtans tool suite in five incarnations: (i) as a cloud machine image (see cloud.oqtans.org for a demo), (ii) as a public Galaxy instance at galaxy.cbio.mskcc.org, (iii) as a git repository (oqtans.org/git); most of these tools are moreover available from (iv) the Galaxy Toolshed and (v) a preconfigured share string to launch Galaxy CloudMan using sharing instance functionality.
2 RESULTS
Oqtans provides a versatile analysis workbench for RNA-seq data comprising tools suitable for basic and advanced analysis tasks (see Supplementary Table S1 for a current list of Oqtans tools and Supplementary Table S2 for supported file formats). Their modular organization within the Galaxy framework allows advanced users to easily customize and extend analysis workflows.
We showcase Oqtans capabilities in use cases for which all data, parameters, intermediate output and final results are made public on a Page in our Galaxy cloud instance (see oqtans.org/usecases).
As a first use case, we wanted to identify annotated genes that were differentially expressed between male and female Drosophila melanogaster fruit flies [using data from (Daines et al., 2011)]. This analysis requires three major steps: read alignment, quantification and enrichment analysis (Fig. 1A and B). The chosen Oqtans tools were combined in a workflow (Supplementary Fig. S1).
Fig. 1.

Schematic workflows of the Oqtans use cases. (A) The general steps needed to perform the analysis. (B) Tools included in Oqtans used for differential expression and GO term enrichment analysis (use case 1). The same workflow in the Galaxy instance is shown in Supplementary Figure S1
After starting an Oqtans cloud instance in Amazon Web Service EC2 (machine image ami-65376a0c) and importing the RNA-seq read data from the NCBI short read archive, we aligned these to the reference genome. Oqtans currently offers three tools for spliced alignments of short reads, Tophat (Trapnell et al., 2009), STAR (Dobin et al., 2013) and PALMapper (Jean et al., 2010). Subsequently, we determined genes that were differentially expressed in males and females using DESeq, which tests read counts for statistically significant differences (Anders and Huber, 2010).
To determine enriched Gene Ontology (GO) terms among differentially expressed genes, we supplied the gene list to topGO (Alexa et al., 2006), which we integrated into Oqtans. Its graphical output highlights expression differences in genes annotated with the functions ‘reproduction’ and ‘sex determination’, as is expected for this comparison between male and female fruit fly transcriptomes (see Supplementary Fig. S3).
The whole experiment excluding short read alignment requires ∼10 min of compute time. Duration of the alignment depends on the number and size of compute nodes that can be allocated for this task (20 min in our setup with 19 ‘4× large memory’ instances on Amazon Web Service).
Uniquely within Oqtans and through the benefits of the Galaxy framework, we can directly compare integrated software tools on the same input data. This is of great value for a researchers who are looking for the most appropriate and accurate algorithm to analyze their newly generated data. For instance, for de novo transcript prediction, the accuracy of the alignments is particularly important. We demonstrate this in a comparison of the accuracy of introns predicted from spliced alignments against the genome annotation generated by TopHat and PALMapper (Fig. 2A and see Supplementary Section S3 for details). Although alignment accuracy may have a negligible effect on the detection of differentially expressed annotated genes, it becomes crucial for de novo inference of transcripts (isoforms). Owing to the high resolution provided by RNA-seq, the discovery of novel transcript isoforms from these data has been a prime analysis goal. In Görnitz et al. (2011), the authors compared the accuracy of transcript inference by combining different read alignment programs (PALMapper, TopHat) with different transcript predictors (margin-based Transcript Identification Method, Cufflinks). All tools used in this example are integrated into Oqtans and can be easily combined in workflows to reproduce this and similar comparisons (Fig. 2B) (see Section S3 at Supplementary Material for more details).
Fig. 2.
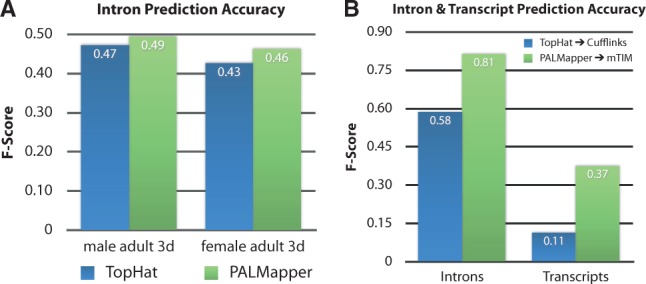
(A) Performance comparison of two alignment programs integrated in Oqtans, evaluated on the data from the use case in terms of intron accuracy (see Supplementary Fig. S3 for details). Such comparative evaluations are made easy, since the replicability assertion of the Galaxy Oqtans setup ensures otherwise identical comparisons. (B) Performance comparison from Görnitz et al. (2011), where PALMapper and TopHat alignments are processed with the de novo transcript inference tools mTIM and Cufflinks, again demonstrating the value of Oqtans for comparisons of analysis tool
3 DISCUSSION
As high-throughput genome and transcriptome sequencing becomes routine in many laboratories around the world, there is an increasing demand for standardized data analysis. Directly associated with this need are accessibility, transparency and persistency of analysis pipelines. As a Galaxy web server, Oqtans brings us closer to these goals (Schultheiss, 2011) for the important task of RNA-seq data analysis by providing easy access to state-of-the-art analysis tools to a wide audience. Importantly, while profiting from many free software development efforts, its user friendly interface abstracts from programming languages and operating systems, and thus enables even inexperienced users to rapidly analyze their RNA-seq data.
Supplementary Material
ACKNOWLEDGEMENT
The authors acknowledge support provided by the Galaxy developer team.
Funding: Max Planck Society, the German Research Foundation (RA1894/1-1 and RA1894/2-1) and the Memorial Sloan-Kettering Cancer Center (MSKCC).
Conflict of Interest: GR, SJS are stakeholders in computomics.com, which offers related bioinformatics data analysis services.
REFERENCES
- Alexa A, et al. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daines B, et al. The Drosophila melanogaster transcriptome by paired-end RNA sequencing. Genome Res. 2011;21:315–324. doi: 10.1101/gr.107854.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görnitz N, et al. mTiM: margin-based transcript mapping from RNA-seq. In: Alkan C, editor. RECOMB Sattelite Workshop on Massively Parallel Sequencing. Vol. 12. London: BMC Bioinformatics; 2011. [Google Scholar]
- Jean G, et al. RNA-seq read alignments with PALMapper. Curr. Protoc. Bioinform. 2010;32:6.1–6.37. doi: 10.1002/0471250953.bi1106s32. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, et al. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Schultheiss SJ. Ten simple rules for providing a scientific web resource. PLoS Comput. Biol. 2011;7:e1001126. doi: 10.1371/journal.pcbi.1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. TopHat: discovering splice junctions with RNA-seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.