

Abstract
Motivation: Metabolic reaction maps allow visualization of genome-scale models and high-throughput data in a format familiar to many biologists. However, creating a map of a large metabolic model is a difficult and time-consuming process. MetDraw fully automates the map-drawing process for metabolic models containing hundreds to thousands of reactions. MetDraw can also overlay high-throughput ‘omics’ data directly on the generated maps.
Availability and implementation: Web interface and source code are freely available at http://www.metdraw.com.
Contact: papin@virginia.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The number of genome-scale metabolic models has increased greatly in recent years (Oberhardt et al., 2009). An accompanying expansion in available algorithms for analyzing these models in the context of high-throughput data (Lewis et al., 2012) has created the need for tools to visualize large models and datasets. Cytoscape (Shannon et al., 2003) and similar graph-drawing software can visualize arbitrary biological networks. However, the resulting node-and-edge graphs are visually distinct from the more familiar ‘metabolic map’ layout where lines show the flow of reactants coming together before branching into products. Only a few genome-scale metabolic reconstructions include a manually curated visualization. However, producing these maps is difficult and time-consuming, and the maps are rarely updated as the model is revised.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2012) and other metabolic pathway databases provide visualizations of indexed reactions, but these maps exclude reactions not in the database (such as transport or species-specific reactions). Metabolic modeling software packages such as the COBRA Toolbox (Schellenberger et al., 2011) and CellNetAnalyzer (Klamt et al., 2007) can overlay flux and gene expression data on reaction maps. Although both packages allow users to modify existing maps, no software package is capable of assembling a complete genome-scale metabolic map de novo from a set of reactions.
2 FEATURES
We present MetDraw, an online tool and software package for automatically generating a reaction map for genome-scale metabolic reconstructions. MetDraw also allows users to visualize metabolomic, reaction flux and gene/protein expression data directly on the resulting maps. Maps are created from Systems Biology Markup Language (SBML) model files and exported as Scalable Vector Graphics (SVG) images. This widely accepted image format allows users to easily customize details of the final maps with image editing software. Although the map creation process is completely automated, several features allow users to control the drawing output.
2.1 Input
MetDraw begins with a valid SBML file of the metabolic reconstruction. The SBML file can be uploaded to the MetDraw Web site (http://www.metdraw.com), or the software can be run on a local computer. MetDraw allows compartmentalized models with transport reactions spanning multiple compartments. For optimal layouts, the SBML file should also contain subsystem assignments in the ‘notes’ section of several reactions. These designations are used to partition the smaller subgraphs that can be visualized more easily.
Metabolites are designated as ‘major’ or ‘minor’ depending on the number of reactions involving each species. Major metabolites are drawn once in each compartment with arrows denoting how the species is produced or consumed by each reaction. Minor, or currency, metabolites are those that appear in many reactions, e.g. high-energy phosphates, water, protons and common metabolic cofactors. Minor metabolites are redrawn for each reaction rather than being drawn once per subsystem and shared by multiple reactions. The removal of minor metabolites reduces much of the visual clutter caused by these highly connected species. MetDraw identifies minor metabolites by thresholding metabolite/reaction participation counts. Optionally, MetDraw will export a list of reaction counts for each metabolite and allow the user to designate the minor metabolites manually.
2.2 Layout
If subsystem designations are available for any reactions, MetDraw partitions the corresponding reactions. Small subsystems or subsystems that share nearly all reactions with other subsystems are merged to preserve information flow in the final map. Subsystems and unclassified reactions are then placed into compartments. Transport reactions, reactions with reactants in more than one compartment, are identified and separated for layout across the compartment boundaries.
The final layout uses the widely used Graphviz (Gansner and North, 2000) software. MetDraw converts the reactions to a series of edges and nodes using the Graphviz DOT language. Unlike other graph-drawing programs for biological networks, MetDraw inserts invisible nodes and edges and uses multiple graph elements to create a final layout that more closely resembles a classical biochemical map (Fig. 1A).
Fig. 1.
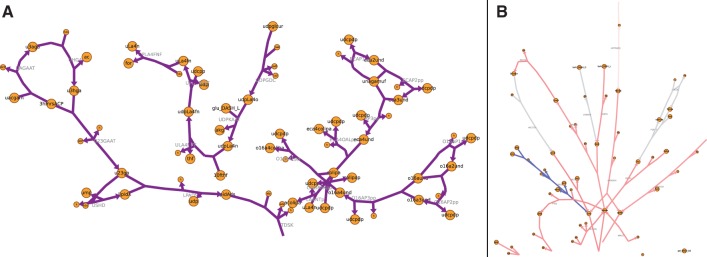
Visualization of the Escherichia coli metabolic model described in Orth et al. (2011). The map was created by MetDraw from the SBML model file without additional user input. (A) Pathways from lipopolysaccharide biosynthesis and recycling pathways, enlarged to show detail. The complete network visualization is available as Supplementary Figure S1. (B) Visualization of Gibbs free energy change for reactions (ΔGr) in the alternative carbon metabolism subsystem. Coloring was applied by MetDraw. Blue (red) indicates positive (negative) ΔGr. The complete map is available as Supplementary Figure S2
Even after identifying minor metabolites and partitioning the reactions by subsystem, the resulting graph may still contain several overlapping edges that clutter the map. MetDraw attempts to alleviate these problem areas by identifying major metabolites that are more highly connected than other metabolites in the same subsystem. These metabolites are ‘cloned’ and redrawn several times in the subsystem to spread the layout and remove overlapping reactions. The cloned metabolites are connected with a dashed line to aid viewing.
Compartments are bounded by a box and labeled in the final image. Subsystems can either be bounded in a similar manner or left free for tighter packing of the compartment. Transport reactions are added to visualize mass flow between compartments.
Additional details on the MetDraw layout algorithm are included in the Supplementary Methods.
2.3 Output and data visualization
By default, MetDraw exports an SVG image of the reconstruction. This format is compatible with MetDraw’s data visualization features. MetDraw can also export visualization in any other format supported by Graphviz, including PDF, PNG and EPS. Final changes to the layout can be made with a vector graphic editing program. Users can freely add graphical elements or move metabolites and reactions; the resulting images can still be used to visualize data with MetDraw.
MetDraw allows visualization of fluxomic, metabolomic and gene/protein expression data on metabolic maps. MetDraw accepts a text file containing numerical data for each metabolite or reaction over several conditions. These data are normalized, applied to a colormap and overlayed on a previously generated metabolic map (Fig. 1B). If several conditions are given in the same data file, MetDraw creates a separate image for each condition using the same color scale. These images can be combined for visual comparisons and to animate transient conditions.
3 IMPLEMENTATION
MetDraw is written in Python 2.7 and runs on Linux, Microsoft Windows and Mac OS X. MetDraw requires Graphviz 2.28 or later. An online interface is available at http://www.metdraw.com.
Supplementary Material
ACKNOWLEDGEMENT
The authors thank Jennifer Bartell, Phillip Yen and Kevin D’Auria for their helpful suggestions on the layout aesthetics.
Funding: National Institutes of Health (R01 GM088244 to J.P.) and National Science Foundation graduate research fellowship (to P.A.J.).
Conflict of Interest: none declared.
REFERENCES
- Gansner ER, North SC. An open graph visualization system and its applications to software engineering. Softw. Pract. Exp. 2000;30:1203–1233. [Google Scholar]
- Kanehisa M, et al. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klamt S, et al. Structural and functional analysis of cellular networks with cellnetanalyzer. BMC Syst. Biol. 2007;1:2. doi: 10.1186/1752-0509-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NE, et al. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 2012;10:291–305. doi: 10.1038/nrmicro2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhardt MA, et al. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 2009;5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, et al. A comprehensive genome-scale reconstruction of Escherichia coli metabolism–2011. Mol. Syst. Biol. 2011;7:535. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J, et al. Quantitative prediction of cellular metabolism with constraint-based models: the cobra toolbox v2.0. Nat. Protoc. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}